

Abstract
According to the 2012 WHO global tuberculosis (TB) report (http://apps.who.int/iris/bitstream/10665/75938/1/9789241564502_eng.pdf), the death rate for tuberculosis was over 1.4 million patients in 2011, with ∼9 million new cases diagnosed. Moreover, the frequency of comorbidity with human immunodeficiency virus (HIV) and with diabetes is on the rise, increasing the risk of these patients for experiencing drug-drug interactions (DDIs) due to polypharmacy. Ethambutol is considered a first-line antituberculosis drug. Ethambutol is an organic cation at physiological pH, and its major metabolite, 2,2′-(ethylenediimino)dibutyric acid (EDA), is zwitterionic. Therefore, we assessed the effects of ethambutol and EDA on the function of human organic cation transporter 1 (hOCT1), hOCT2, and hOCT3 and that of EDA on organic anion transporter 1 (hOAT1) and hOAT3. Potent inhibition of hOCT1- and hOCT2-mediated transport by ethambutol (50% inhibitory concentration [IC50] = 92.6 ± 10.9 and 253.8 ± 90.8 μM, respectively) was observed. Ethambutol exhibited much weaker inhibition of hOCT3 (IC50 = 4.1 ± 1.6 mM); however, significant inhibition (>80%) was observed at physiologically relevant concentrations in the gastrointestinal (GI) tract after oral dosing. EDA failed to exhibit any inhibitory effects that warranted further investigation. DDI analysis indicated a strong potential for ethambutol interaction on hOCT1 expressed in enterocytes and hepatocytes and on hOCT3 in enterocytes, which would alter absorption, distribution, and excretion of coadministered cationic drugs, suggesting that in vivo pharmacokinetic studies are necessary to confirm drug safety and efficacy. In particular, TB patients with coexisting HIV or diabetes might experience significant DDIs in situations of coadministration of ethambutol and clinical therapeutics known to be hOCT1/hOCT3 substrates (e.g., lamivudine or metformin).
INTRODUCTION
Ethambutol dihydrochloride (EMB) (Fig. 1) is a potent antimycobacterial agent employed in the treatment of tuberculosis (TB) and Mycobacterium avium complex infections. The main pharmacological effect of EMB is to inhibit arabinosyl transferase, preventing the synthesis of arabinogalactan, which is a vital component of the mycobacterial cell wall. EMB, pyrazinamide, isoniazid, and rifampin comprise the four first-line antituberculosis drugs recommended by the World Health Organization (WHO) (1). EMB is used in the treatment of multidrug-resistant tuberculosis to prevent the emergence of isoniazid- or rifampin-resistant mycobacteria (2). In clinical practice, EMB use is associated with many adverse effects, including optic neuritis (reported in 1% to 5% of patients) (3) and reduced renal clearance of urate (reported in about 66% of patients) (4), the incidence of which is higher when EMB is concomitantly administered with pyrazinamide (5).
Fig 1.
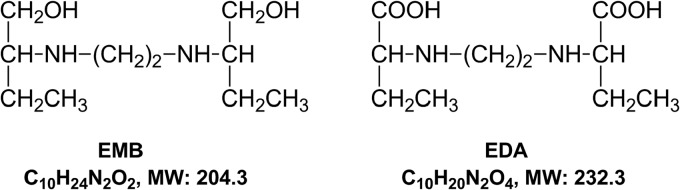
Chemical structures of ethambutol (EMB) and its dicarboxylic metabolite, 2,2′-(ethylenediimino)dibutyric acid (EDA). MW, molecular weight.
The in vivo pharmacokinetic properties of EMB have been well defined (6–8). In humans, EMB showed high bioavailability (∼80%) and low plasma protein binding (20 to 30%) (9). A peak serum concentration (Cmax) of ∼4.5 μg/ml was reported after oral dosing of 25 mg/kg (6). Only a small portion of EMB (8% to 15%) is metabolized in the liver to form the final dicarboxylic metabolite, 2,2′-(ethylenediimino)dibutyric acid (EDA) (Fig. 1) (10). Renal elimination is the major clearance mechanism for EMB, with 70 to 84% of an intravenous dose being excreted in the urine as the unchanged parent compound (9). The renal clearance rate (∼417 ml/min) indicates that active net tubular secretion, as well as glomerular filtration (120 ml/min), is involved (6). While EMB has been identified as a substrate of P-glycoprotein (11), based upon their chemical structures (Fig. 1) and physicochemical properties, EMB and EDA have the potential to be a substrate and/or inhibitor of organic cation transporters (OCTs) and/or organic anion transporters (OATs).
The organic cation/anion/zwitterion transporters belong to the solute carrier 22 (SLC22) family and in vivo mediate the absorption, distribution, and elimination of a broad variety of charged endogenous and exogenous organic substances (12–14). OATs and OCTs are widely expressed in many barrier organs, such as the intestine, kidney, liver, and brain (12–14). In the intestine (Fig. 2), human OCT1 (hOCT1) (SLC22A1) is expressed in the basolateral membrane and hOCT3 (SLC22A3) is targeted to the brush border membrane of enterocytes, where they mediate the cellular entry of cationic compounds (12). hOCT1 and hOCT3 are both expressed in the sinusoidal membrane (blood side) of hepatocytes (Fig. 2) and represent the first step in the hepatic excretion of many substances (12, 15). In the kidney (Fig. 2), hOCT2 (SLC22A2), hOCT3, hOAT1 (SLC22A6), and hOAT3 (SLC22A8) are each expressed on the basolateral sides of proximal tubule cells and extract drugs from the blood. hOCT1 mRNA has been detected; however, the protein has not been immunolocalized (12, 13). Hundreds of important clinical therapeutics are known substrates and/or inhibitors of OATs and OCTs, such as chemotherapeutics (e.g., cisplatin and paclitaxel), analgesics (e.g., morphine), cholesterol-lowering drugs (e.g., atorvastatin and pravastatin), antivirals (e.g., cidofovir and lamivudine), and antidiabetic agents (e.g., metformin) (12–14).
Fig 2.

Expression of the examined human OCTs and OATs (SLC22 family) in the intestine, liver, and kidney. Predicted (GI lumen and portal circulation) and clinically determined (systemic circulation) concentrations of EMB are indicated. Renal expression and targeting of hOCT1 remains controversial, with conflicting reports about its location in the literature; however, the rat Oct1 ortholog has been immunolocalized to the basolateral membrane of proximal tubule cells.
Recently, it was reported that TB infection is closely linked to both human immunodeficiency virus (HIV) and diabetes. According to the WHO, 10% of TB patients also suffer from diabetes and 13% of the ∼9 million TB patients newly diagnosed in 2011 were coinfected with HIV (1). Thus, the complexity of these disease treatment regimens (TB, TB-diabetes, or TB-HIV), in terms of polypharmacy, raises concerns regarding the potential for drug-drug interactions (DDIs) involving OCTs and/or OATs. The aim of the present study was to explore the inhibitory effects of EMB and its metabolite EDA on hOCT1, hOCT2, hOCT3, hOAT1, and hOAT3. Potent inhibition was further characterized by kinetic investigations to estimate IC50s, which were used to quantitatively evaluate the clinical DDI potential on these transporters during antituberculosis therapy.
MATERIALS AND METHODS
Chemicals.
Tritiated p-aminohippuric acid ([3H]PAH), estrone sulfate ([3H]ES), and 1-methyl-4-phenylpyridinium ([3H]MPP+) were purchased from PerkinElmer Life and Analytical Science (Waltham, MA). Unlabeled ES, MPP+, PAH, probenecid, and ethambutol dihydrochloride (EMB, ≥95% purity) were obtained from Sigma-Aldrich (St. Louis, MO). Quinine monohydrochloride dihydrate was purchased from Acros Organics (Fair Lawn, NJ). 2,2′-(Ethylenediimino)dibutyric acid (EDA; ≥99% purity) was purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA).
Tissue culture.
Derivation of stably transfected Chinese hamster ovary (CHO) cell lines expressing hOAT1 (CHO-hOAT1) and stably transfected human embryonic kidney 293 (HEK) cells expressing hOAT3 (HEK-hOAT3), hOCT1 (HEK-hOCT1), hOCT2 (HEK-hOCT2), or hOCT3 (HEK-hOCT3), as well as their corresponding empty vector-transfected background control cell lines, has been described previously (16–19). CHO-hOAT1 cells were maintained at 37°C with 5% CO2 in phenol red-free RPMI 1640 medium (Gibco-Invitrogen, Grand Island, NY) containing 10% serum, 1% penicillin/streptomycin, and 1 mg/ml G418. HEK-hOAT3 cells were maintained at 37°C with 5% CO2 in high-glucose Dulbecco's modified Eagle medium (DMEM; Mediatech, Inc., Herndon, VA) containing 10% serum, 1% penicillin/streptomycin, and 125 μg/ml hygromycin B. The HEK-hOCT1, HEK-hOCT2, and HEK-hOCT3 cell lines were maintained at 37°C with 5% CO2 in high-glucose DMEM containing 10% serum, 1% penicillin/streptomycin, and 600 μg/ml G418.
Cell accumulation assay.
The procedure for the cell accumulation assay has been described previously (20, 21). Briefly, cells were seeded into 24-well tissue culture plates at a density of 2 × 105 cells/well (without antibiotics) for 48 h. On the day of the cell transport experiment, the cells were equilibrated to serum-free conditions with transport buffer for 10 min (500 μl of Hanks' balanced salt solution containing 10 mM HEPES, pH 7.4). Equilibration buffer was replaced with 500 μl of fresh transport buffer containing 1 μM unlabeled substrate spiked with [3H]PAH (0.5 μCi/ml), [3H]ES (0.25 μCi/ml), or [3H]MPP+ (0.25 μCi/ml) in the presence or absence of test compounds. At the end of the incubation, the cells were quickly rinsed three times with ice-cold transport buffer and lysed. The radioactivity of cell lysate was quantified by liquid scintillation counting, and the uptake profile was normalized by the total protein content determined by the Bradford method. The intracellular accumulation of substrates was reported as picomoles of substrate per milligram total protein. All uptake data were corrected for background accumulation in corresponding empty vector-transfected control cells. Substrate concentration and accumulation time used for kinetic analyses were determined previously (12, 20). Kinetic calculations were performed using GraphPad Prism software version 5.0 (GraphPad Software Inc., San Diego, CA). The half-maximal inhibitory concentrations (IC50s) were calculated using nonlinear regression with the appropriate model. Results were confirmed by repeating all experiments at least three times with triplicate wells for each data point in every experiment.
Drug-drug interaction (DDI) index calculation.
The DDI index (22) was estimated as the ratio of the maximal unbound EMB concentration in biofluid (unbound Cmax) after therapeutic dosing of EMB divided by transporter-specific IC50s estimated by in vitro assay. A cutoff value of ≥0.1 is thought to indicate the need for a prospective in vivo pharmacokinetic study (22). Specifically, the predicted intestinal lumen concentration of EMB after oral dosing (25-mg/kg dose, 70-kg patient body weight, 250-ml volume) was used to estimate the DDI index for hOCT3 located in the apical (luminal) membranes of enterocytes, while the portal venous plasma concentration (presystemic circulation) of EMB was used to estimate DDI index values for hOCTs in the basolateral membranes of hepatocytes (hOCT1 and hOCT3) and/or enterocytes (hOCT1 and hOCT2), and the plasma concentration (systemic circulation) of EMB was used to estimate DDI index values for hOCT1, hOCT2, and hOCT3 in the basolateral membranes of renal proximal tubule cells (Fig. 2). The concentration of EMB in the intestinal lumen was estimated to be ∼34 mM, assuming that patients (70 kg) take EMB with 250 ml of water. From clinical reports, after oral dosing (25 mg/kg) the plasma Cmax of EMB was 22.02 μM, with about 20% being protein bound (6, 9, 23). Accordingly, the method of Ito et al. can be used to estimate drug concentrations in the portal vein (24): Cport,vn = Cmax + (ka × D)/(Qh × Fa), where Cport,vn, ka, D, Qh, and Fa represent concentration in the portal vein, absorption rate constant, dose, hepatic blood flow rate (∼1,200 ml/min), and fraction absorbed, respectively. For EMB, ka and Fa were reported as 0.48 h−1 and 80% (9, 23). Using an average patient body weight of 70 kg, the predicted portal venous blood concentration for EMB was ∼67 μM.
Statistical analysis.
The data in the figures and the raw uptake scores are expressed as means ± standard deviations (SD), while IC50 estimates are means ± standard errors of the means (SEM). Statistical differences were assessed using one-way analysis of variance (ANOVA) followed by post hoc analysis with Dunnett's t test (α = 0.05).
RESULTS
Inhibitory effects of EMB on hOCT1-, hOCT2-, and hOCT3-mediated MPP+ uptake.
The inhibitory effects of EMB were examined on three OCT paralogs, hOCT1, hOCT2, and hOCT3, using MPP+ as a prototypical substrate. Significant accumulation of MPP+ (∼33-fold) was observed in stably transfected hOCT1-expressing cells relative to empty-vector-transfected background control cells (132.6 ± 9.9 versus 4.0 ± 0.2 pmol/mg protein/10 min, respectively). The known hOCT inhibitor, quinine (200 μM), showed virtually complete inhibition of hOCT1-mediated MPP+ uptake (>90% inhibition) (Fig. 3A). Significant inhibition (66%) of hOCT1-mediated MPP+ transport by EMB (at 100 μM versus 1 μM MPP+) was observed (Fig. 3A). Subsequent dose-response (10−7 to 10−3 M EMB) studies were performed to derive the IC50 for EMB on hOCT1 (Fig. 4, top). The IC50 of EMB for hOCT1 was estimated as 92.6 ± 10.9 μM. Therefore, the DDI indices were calculated to gauge DDI potency in enterocytes, hepatocytes, and proximal tubule cells, using the EMB concentration in gastrointestinal (GI) biofluid, portal venous blood, or the systemic circulation, as appropriate (Fig. 2 and Table 1).
Fig 3.

Inhibition profiles of EMB for hOCT1, hOCT2, and hOCT3. (A) Inhibition of hOCT1-mediated MPP+ uptake by EMB and quinine (200 μM). (B) Inhibition of hOCT2-mediated MPP+ uptake by EMB and quinine (200 μM). (C) Inhibition of hOCT3-mediated MPP+ uptake by EMB and quinine (200 μM). The concentration of MPP+ was 1 μM, incubation time was 10 min, and data were corrected for nonspecific background. Values are means ± SD of triplicate values. ***, P < 0.001, determined by one-way ANOVA followed by Dunnett's t test.
Fig 4.
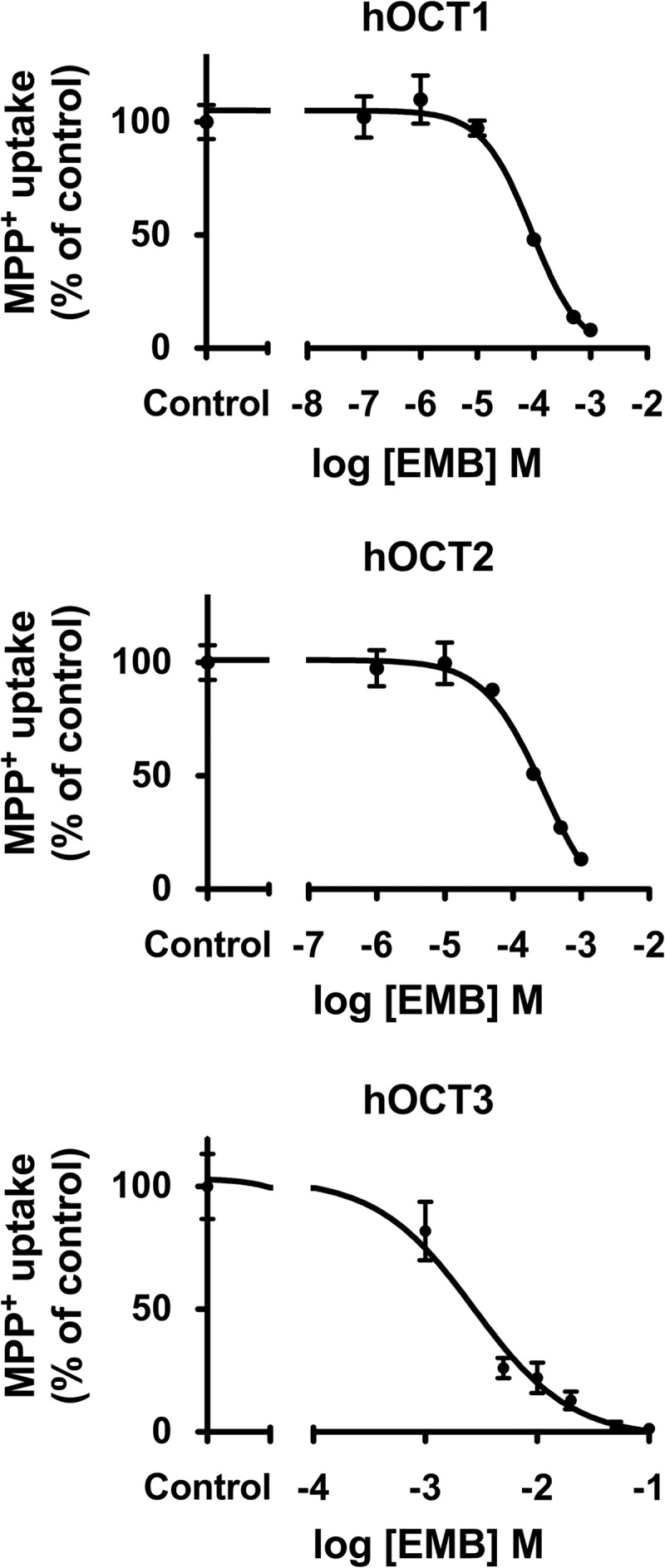
Dose-response curves for EMB on hOCT1, hOCT2, and hOCT3. Representative data showing 1-min uptake of MPP+ (1 μM) measured in HEK-hOCT1, HEK-hOCT2, and HEK-hOCT3 cells in the presence of increasing concentrations of EMB (10−7 to 10−1 M) are shown. Data were corrected for nonspecific background measured in the empty vector control cells and are means ± SD of triplicate values. IC50s were determined with nonlinear regression and the “log(inhibitor) versus response” model using GraphPad Prism software.
Table 1.
Estimated DDI index values for EMB on hOCT-mediated transport after an oral dose of 25 mg/kg
| hOCT | Drug-drug interaction indexa |
||
|---|---|---|---|
| GI tract | Liver | Kidney | |
| hOCT1 | 0.6 | 0.6 | 0.2b |
| hOCT2 | NEc | NE | 0.1 |
| hOCT3 | 8.3 | <0.1 | <0.1 |
Drug-drug interaction index is defined as the unbound concentration of drug divided by the drug IC50 for the transporter of interest. A DDI index value of >0.1 is thought to indicate the potential for clinically relevant DDIs.
Assuming basolateral targeting for hOCT1.
NE, transporter not expressed in this tissue.
We next examined the inhibitory effect of EMB on hOCT2-mediated transport. HEK-hOCT2 cells exhibited marked accumulation of MPP+ (93.0 ± 5.2 pmol/mg protein/10 min) compared to background control cells (4.0 ± 0.2 pmol/mg protein/10 min). Quinine (200 μM) completely blocked (>99%) hOCT2-mediated MPP+ uptake (Fig. 3B). The cell accumulation assay demonstrated that EMB at 100 μM significantly inhibited hOCT2-mediated MPP+ uptake (21%), and complete inhibition of hOCT2 transport activity (96%) was observed at 1 mM EMB. Kinetic studies were conducted to estimate the IC50 for EMB on hOCT2. Using EMB concentrations ranging from 10−6 to 10−3 M, the IC50 was estimated as 253.8 ± 90.8 μM (Fig. 4, middle). As shown in Table 1, the DDI index for hOCT2 expressed in kidney was estimated as 0.1.
Stably transfected hOCT3-expressing (HEK-hOCT3) cells also showed marked accumulation of MPP+ (∼30-fold) compared to empty vector-transfected background control cells (80.3 ± 11.3 versus 2.7 ± 0.1 pmol/mg protein/10 min). Such active transport was completely (99%) blocked by quinine at 200 μM (Fig. 3C). Though EMB failed to show significant inhibition on hOCT3 transport activity at 100 μM, marked inhibition was observed with increasing concentrations (Fig. 3C). EMB showed ∼50% and ∼80% inhibition at 1 and 10 mM, respectively. Further dose-response (10−3 to 10−1 M EMB) studies yielded an estimated IC50 of 4.1 ± 1.6 mM (Fig. 4, middle). Due to low EMB concentration in the presystemic (portal) and systemic circulation, DDI indices for hOCT3 in liver and kidney were negligible compared to the 0.1 threshold value (Table 1). However, the high concentration of EMB in the GI biofluid yielded an estimated DDI index around 8.3.
Inhibitory effects of EDA on hOAT (hOAT1 and hOAT3) and hOCT (hOCT1, hOCT2, and hOCT3) transport activity.
The zwitterionic dicarboxylic acid metabolite of EMB, EDA, was examined for interactions with hOAT1, hOAT3, hOCT1, hOCT2, and hOCT3 (Fig. 5). The standard substrate used for hOAT1 was PAH, and that for hOAT3 was ES. Stably transfected hOAT1-expressing (CHO-hOAT1) and hOAT3-expressing (HEK-hOAT3) cells showed 5-fold- and 3-fold-greater substrate accumulation than empty-vector-transfected background control cells, respectively (7.5 ± 1.6 versus 1.6 ± 0.2 pmol/mg protein/10 min for hOAT1 and 4.1 ± 0.2 versus 1.6 ± 0.1 pmol/mg protein/10 min for hOAT3). The known OAT inhibitor probenecid completely blocked (>99%) hOAT1- and hOAT3-mediated substrate uptake (Fig. 5). With the exception of hOAT3 (∼17% inhibition), EDA (100 μM) failed to produce any significant inhibitory effects. Despite the modest inhibition of hOAT3, for all tested transporters IC50s of EDA were expected to be greater than 100 μM, which greatly exceeds the EDA concentration in the systemic circulation, and further kinetic analysis was not performed.
Fig 5.

Inhibition profile of EDA on hOATs and hOCTs. (A) Inhibition of hOAT1-mediated PAH uptake by EDA (100 μM) and probenecid (1,000 μM). (B) Inhibition of hOAT3-mediated ES uptake by EDA (100 μM) and probenecid (1,000 μM). (C) Inhibition of hOCT1-mediated MPP+ uptake by EDA (100 μM) and quinine (200 μM). (D) Inhibition of hOCT2-mediated MPP+ uptake by EDA (100 μM) and quinine (200 μM). (E) Inhibition of hOCT3-mediated MPP+ uptake by EDA (100 μM) and quinine (200 μM). The concentration of substrates was 1 μM, incubation time was 15 min, and data were corrected for nonspecific background. Values are means ± SD of triplicate values. ***, P < 0.001, determined by one-way ANOVA followed by Dunnett's t test.
DISCUSSION
Although numerous antitubercular drugs have been developed since the 1940s, TB is still a prevalent disease and a common cause of death worldwide. Recently, the spread of multidrug-resistant TB strains (MDR-TB; estimated to account for 3.7% of new cases and 20% of previously treated cases) and the emergence of an extensively drug-resistant strain (XDR-TB; ∼9% of MDR-TB cases) that has proven virtually impossible to successfully treat have become new challenges to disease management (1, 25). The appearance of XDR-TB has been reported in 84 countries, and the threat of its spread is viewed as a significant enough health issue that the United Kingdom has enacted regulations obligating people traveling from regions where TB is prevalent to have a current chest X-ray in their possession upon arrival in order to be issued a visa (26). Isolation and therapy may be required for up to 2 years and can cost upwards of one million U.S. dollars per patient (26).
A further complication in TB therapy arises from the increasing clinical association of TB with diabetes and HIV, with TB becoming one of the leading causes of mortality among HIV-infected patients (27, 28). Indeed, of the ∼9 million new TB patients diagnosed in 2011, 13% were reported to be coinfected with HIV, and of the 1.4 million TB deaths, 430,000 were HIV associated (1). As a result, more TB patients need to be prescribed combination therapies to treat comorbidities or to minimize the development of drug resistance, making therapy management and compliance extremely complex and increasing the risk of patients' experiencing unintended drug-drug interactions. Indeed, there are a number of case reports regarding adverse events, including nephrotoxicity, associated with EMB use during antituberculosis therapy (29–32). In these patients, discontinuation of EMB resulted in improvement in renal function.
Competition for binding to membrane transporters is one mechanism resulting in DDIs, and while there is currently insufficient clinical data to conclusively determine transporter-mediated DDI involvement, it is possible that hOCT-mediated DDIs explain some of these cases. As such, potential interactions between antituberculosis agents and drug transporters have received increased attention. For example, rifampin was identified as an inhibitor of P-glycoprotein (ABCB1), OATP1B1 (SLCO1B1), and OAPT1B3 (SLCO1B3), whereas pyrazinamide was demonstrated to inhibit hURAT1 (SLC22A12) (33–35). EMB also was reported as a substrate for P-glycoprotein (11); however, information regarding potential interactions with other drug transporters, especially drug uptake transporters, is virtually nonexistent. Additionally, many anti-diabetic (metformin) and anti-HIV (lamivudine, raltegravir, tenofovir, and zalcitabine) agents have been identified as substrates and/or inhibitors of OCTs and OATs (12–14, 36–38). Therefore, in order to more effectively and safely manage the long-term therapies of these complicated patient populations, improved understanding of the interactions of these drugs with the transporters responsible for their absorption, distribution, and elimination is needed.
A number of preclinical and clinical studies have demonstrated the role of hOCT1, -2, and -3 in the disposition and efficacy of metformin, a hypoglycemic agent widely used for the treatment of type 2 diabetes mellitus (38–41). In humans, subjects carrying a mutated form of hOCT1 with reduced function exhibited significantly altered pharmacokinetic properties of metformin that manifested as increased area under the plasma concentration-time curve, increased Cmax, and reduced oral volume of distribution, as well as reduced glucose-lowering effects, i.e., loss of efficacy (40, 41). Given the predicted EMB portal vein concentration of 67 μM after oral dosing, the rank order of DDI potencies for hOCT1 was enterocytes (0.6) = hepatocytes (0.6) ≥ proximal tubule cells (0.2); all the DDI indexes are above the 0.1 threshold value (Fig. 2 and Table 1). These results indicated that 38% of hOCT1 transport activity might be inhibited in enterocytes and hepatocytes during routine EMB therapy, suggesting a strong potential for EMB-metformin interactions in TB-diabetes patients. The IC50 for hOCT3 was higher (4.1 ± 1.6 mM) than those for hOCT1 and hOCT2 (Fig. 4, bottom). However, given the expected high luminal GI tract concentration of EMB after oral dosing (∼34 mM after a 1,750-mg dose [25 mg/kg in a 70-kg patient] diluted in 250 ml GI fluid), hOCT3 may represent an important pathway for GI absorption of EMB due to its apical membrane localization in enterocytes (Fig. 2). Furthermore, the estimated DDI index of 8.3 (Fig. 4, bottom, and Table 1) is 83-fold higher than the DDI threshold value and is indicative of a marked potential for EMB to interfere (89% inhibition) with intestinal absorption of coadministered drugs that are hOCT3 substrates, including metformin.
As many anti-HIV agents are also hOCT substrates, the same DDI potential exists for polypharmacy in TB-HIV patients. Recently, expression of hOCT1 and hOCT2 mRNA was detected in CD4 cells isolated from patients with HIV (42). Therefore, EMB present in the systemic circulation also might reduce accumulation of anti-HIV agents in CD4 target cells that serve as a viral reservoir, resulting in loss of antiviral efficacy via inhibition of hOCT1 and/or hOCT2 function. Similarly, systemic EMB might impact hOCT1-mediated (DDI index = 0.2; ∼20% inhibition) and/or hOCT2-mediated (DDI index = 0.1; ∼9% inhibition) renal secretion.
hOCTs are also involved in the pharmacological action of platinum antineoplastics (e.g., cisplatin and oxaliplatin), raising the possibility for significant DDIs during chemotherapy in TB patients (43–46). Patients receiving cisplatin therapy frequently experience cumulative dose-dependent nephrotoxicity involving the proximal tubule (47–49). Moreover, treatment with inhibitors of hOCTs (e.g., tetraethylammonium and cimetidine) prevented accumulation of cisplatin in renal proximal tubule cells, and subsequently, cisplatin was identified as a competitive inhibitor of rat Oct2 (43, 50–53). Thus, coadministration of EMB and cisplatin or oxaliplatin might alter their oral absorption, as well as their associated hepatic and renal accumulation and toxicity.
In summary, our findings demonstrate the inhibitory effect of EMB on hOCT1, hOCT2, and hOCT3, while its zwitterionic dicarboxylic metabolite EDA failed to produce significant inhibition of hOCT1, -2, and -3, hOAT1, or hOAT3. EMB concentrations in the GI tract and portal vein after oral administration indicate the potential for marked DDIs in vivo for hOCT1 expressed in hepatocytes, enterocytes, and potentially renal proximal tubule cells. Human OCT3 in enterocytes exhibited the highest DDI index, suggesting that EMB might significantly alter cationic drug/nutrient absorption. Renal hOCT2 has a slight possibility of EMB DDIs. Future investigations encompassing in vivo DDI studies between EMB and known substrates for hOCT1, hOCT2, and hOCT3 appear to be necessary in order to optimize clinical safety and efficacy in these complex patient populations.
ACKNOWLEDGMENTS
We thank Tomas Cihlar for providing the CHO-hOAT1 and corresponding empty-vector-transfected background control cell lines.
Footnotes
Published ahead of print 5 August 2013
REFERENCES
- 1.WHO 2012. WHO global tuberculosis report 2012. WHO, Geneva, Switzerland: Available at http://apps.who.int/iris/bitstream/10665/75938/1/9789241564502_eng.pdf Accessed June 8, 2013 [Google Scholar]
- 2.Treatment Guidelines for The Medical Letter 2012. Drugs for tuberculosis. Treat. Guidel. Med. Lett. 10:29–36 [PubMed] [Google Scholar]
- 3.Sivakumaran P, Harrison AC, Marschner J, Martin P. 1998. Ocular toxicity from ethambutol: a review of four cases and recommended precautions. N. Z. Med. J. 111:428–430 [PubMed] [Google Scholar]
- 4.Postlethwaite AE, Bartel AG, Kelley WN. 1972. Hyperuricemia due to ethambutol. N. Engl. J. Med. 286:761–762 [DOI] [PubMed] [Google Scholar]
- 5.Khanna BK, Kumar J. 1991. Hyperuricemic effect of ethambutol and pyrazinamide administered concomitantly. Ind. J. Tuberc. 38:21–24 [Google Scholar]
- 6.Peloquin CA, Bulpitt AE, Jaresko GS, Jelliffe RW, Childs JM, Nix DE. 1999. Pharmacokinetics of ethambutol under fasting conditions, with food, and with antacids. Antimicrob. Agents Chemother. 43:568–572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hall RG, II, Swancutt MA, Meek C, Leff RD, Gumbo T. 2012. Ethambutol pharmacokinetic variability is linked to body mass in overweight, obese, and extremely obese people. Antimicrob. Agents Chemother. 56:1502–1507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhu M, Burman WJ, Starke JR, Stambaugh JJ, Steiner P, Bulpitt AE, Ashkin D, Auclair B, Berning SE, Jelliffe RW, Jaresko GS, Peloquin CA. 2004. Pharmacokinetics of ethambutol in children and adults with tuberculosis. Int. J. Tuberc. Lung Dis. 8:1360–1367 [PubMed] [Google Scholar]
- 9.Jonsson S, Davidse A, Wilkins J, Van der Walt JS, Simonsson US, Karlsson MO, Smith P, McIlleron H. 2011. Population pharmacokinetics of ethambutol in South African tuberculosis patients. Antimicrob. Agents Chemother. 55:4230–4237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Buyske DA, Peets E, Sterling W. 1966. Pharmacological and biochemical studies on ethambutol in laboratory animals. Ann. N. Y. Acad. Sci. 135:711–725 [DOI] [PubMed] [Google Scholar]
- 11.Hartkoorn RC, Chandler B, Owen A, Ward SA, Bertel Squire S, Back DJ, Khoo SH. 2007. Differential drug susceptibility of intracellular and extracellular tuberculosis, and the impact of P-glycoprotein. Tuberculosis (Edinb.) 87:248–255 [DOI] [PubMed] [Google Scholar]
- 12.Koepsell H, Lips K, Volk C. 2007. Polyspecific organic cation transporters: structure, function, physiological roles, and biopharmaceutical implications. Pharm. Res. 24:1227–1251 [DOI] [PubMed] [Google Scholar]
- 13.Wang L, Sweet DH. 2013. Renal organic anion transporters (SLC22 family): expression, regulation, roles in toxicity, and impact on injury and disease. AAPS J. 15:53–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.VanWert AL, Gionfriddo MR, Sweet DH. 2010. Organic anion transporters: discovery, pharmacology, regulation and roles in pathophysiology. Biopharm. Drug Dispos. 31:1–71 [DOI] [PubMed] [Google Scholar]
- 15.Cha SH, Kim HP, Jung NH, Lee WK, Kim JY, Cha YN. 2002. Down-regulation of organic anion transporter 2 mRNA expression by nitric oxide in primary cultured rat hepatocytes. IUBMB Life 54:129–135 [DOI] [PubMed] [Google Scholar]
- 16.Grundemann D, Hahne C, Berkels R, Schomig E. 2003. Agmatine is efficiently transported by non-neuronal monoamine transporters extraneuronal monoamine transporter (EMT) and organic cation transporter 2 (OCT2). J. Pharmacol. Exp. Ther. 304:810–817 [DOI] [PubMed] [Google Scholar]
- 17.Grundemann D, Schechinger B, Rappold GA, Schomig E. 1998. Molecular identification of the corticosterone-sensitive extraneuronal catecholamine transporter. Nat. Neurosci. 1:349–351 [DOI] [PubMed] [Google Scholar]
- 18.Ho ES, Lin DC, Mendel DB, Cihlar T. 2000. Cytotoxicity of antiviral nucleotides adefovir and cidofovir is induced by the expression of human renal organic anion transporter 1. J. Am. Soc. Nephrol. 11:383–393 [DOI] [PubMed] [Google Scholar]
- 19.Vanwert AL, Srimaroeng C, Sweet DH. 2008. Organic anion transporter 3 (Oat3/Slc22a8) interacts with carboxyfluoroquinolones, and deletion increases systemic exposure to ciprofloxacin. Mol. Pharmacol. 74:122–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang L, Sweet DH. 2013. Competitive inhibition of human organic anion transporters 1 (SLC22A6), 3 (SLC22A8) and 4 (SLC22A11) by major components of the medicinal herb Salvia miltiorrhiza (Danshen). Drug Metab. Pharmacokinet. 28:220–228 [DOI] [PubMed] [Google Scholar]
- 21.Wang L, Sweet DH. 2012. Potential for food-drug interactions by dietary phenolic acids on human organic anion transporters 1 (SLC22A6), 3 (SLC22A8), and 4 (SLC22A11). Biochem. Pharmacol. 84:1088–1095 [DOI] [PubMed] [Google Scholar]
- 22.Giacomini KM, Huang SM, Tweedie DJ, Benet LZ, Brouwer KL, Chu X, Dahlin A, Evers R, Fischer V, Hillgren KM, Hoffmaster KA, Ishikawa T, Keppler D, Kim RB, Lee CA, Niemi M, Polli JW, Sugiyama Y, Swaan PW, Ware JA, Wright SH, Yee SW, Zamek-Gliszczynski MJ, Zhang L. 2010. Membrane transporters in drug development. Nat. Rev. Drug Discov. 9:215–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Holdiness MR. 1984. Clinical pharmacokinetics of the antituberculosis drugs. Clin. Pharmacokinet. 9:511–544 [DOI] [PubMed] [Google Scholar]
- 24.Ito K, Iwatsubo T, Kanamitsu S, Ueda K, Suzuki H, Sugiyama Y. 1998. Prediction of pharmacokinetic alterations caused by drug-drug interactions: metabolic interaction in the liver. Pharmacol. Rev. 50:387–412 [PubMed] [Google Scholar]
- 25.Riccardi G, Pasca MR, Buroni S. 2009. Mycobacterium tuberculosis: drug resistance and future perspectives. Future Microbiol. 4:597–614 [DOI] [PubMed] [Google Scholar]
- 26.UK Health Protection Agency 2012. Tuberculosis in the UK: 2012 report. http://www.hpa.org.uk/webc/HPAwebFile/HPAweb_C/1317134913404 Accessed 8 June 2013
- 27.Faurholt-Jepsen D, Range N, PrayGod G, Jeremiah K, Faurholt-Jepsen M, Aabye MG, Changalucha J, Christensen DL, Krarup H, Witte DR, Andersen AB, Friis H. 2012. The role of diabetes on the clinical manifestations of pulmonary tuberculosis. Trop. Med. Int. Health 17:877–883 [DOI] [PubMed] [Google Scholar]
- 28.Pawlowski A, Jansson M, Skold M, Rottenberg ME, Kallenius G. 2012. Tuberculosis and HIV co-infection. PLoS Pathog. 8:e1002464. 10.1371/journal.ppat.1002464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Garcia-Martin F, Mampaso F, de Arriba G, Moldenhauer F, Martin-Escobar E, Saiz F. 1991. Acute interstitial nephritis induced by ethambutol. Nephron 59:679–680 [DOI] [PubMed] [Google Scholar]
- 30.Collier J, Joekes AM, Philalithis PE, Thompson FD. 1976. Two cases of ethambutol nephrotoxicity. Br. Med. J. 2:1105–1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stone WJ, Waldron JA, Dixon JH, Jr, Primm RK, Horn RG. 1976. Acute diffuse interstitial nephritis related to chemotherapy of tuberculosis. Antimicrob. Agents Chemother. 10:164–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pablo M, Gathe J, Varon J. 2013. Ethambutol-induced nephrotoxicity: case report and review of the literature. Crit. Care Shock 16:45–47 [Google Scholar]
- 33.Enomoto A, Kimura H, Chairoungdua A, Shigeta Y, Jutabha P, Cha SH, Hosoyamada M, Takeda M, Sekine T, Igarashi T, Matsuo H, Kikuchi Y, Oda T, Ichida K, Hosoya T, Shimokata K, Niwa T, Kanai Y, Endou H. 2002. Molecular identification of a renal urate anion exchanger that regulates blood urate levels. Nature 417:447–452 [DOI] [PubMed] [Google Scholar]
- 34.Vavricka SR, Van Montfoort J, Ha HR, Meier PJ, Fattinger K. 2002. Interactions of rifamycin SV and rifampicin with organic anion uptake systems of human liver. Hepatology 36:164–172 [DOI] [PubMed] [Google Scholar]
- 35.Schuetz EG, Schinkel AH, Relling MV, Schuetz JD. 1996. P-glycoprotein: a major determinant of rifampicin-inducible expression of cytochrome P4503A in mice and humans. Proc. Natl. Acad. Sci. U. S. A. 93:4001–4005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moss DM, Kwan WS, Liptrott NJ, Smith DL, Siccardi M, Khoo SH, Back DJ, Owen A. 2011. Raltegravir is a substrate for SLC22A6: a putative mechanism for the interaction between raltegravir and tenofovir. Antimicrob. Agents Chemother. 55:879–887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jung N, Lehmann C, Rubbert A, Knispel M, Hartmann P, van Lunzen J, Stellbrink HJ, Faetkenheuer G, Taubert D. 2008. Relevance of the organic cation transporters 1 and 2 for antiretroviral drug therapy in human immunodeficiency virus infection. Drug Metab. Dispos. 36:1616–1623 [DOI] [PubMed] [Google Scholar]
- 38.Kimura N, Okuda M, Inui K. 2005. Metformin transport by renal basolateral organic cation transporter hOCT2. Pharm. Res. 22:255–259 [DOI] [PubMed] [Google Scholar]
- 39.Nies AT, Hofmann U, Resch C, Schaeffeler E, Rius M, Schwab M. 2011. Proton pump inhibitors inhibit metformin uptake by organic cation transporters (OCTs). PLoS One 6:e22163. 10.1371/journal.pone.0022163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shu Y, Brown C, Castro RA, Shi RJ, Lin ET, Owen RP, Sheardown SA, Yue L, Burchard EG, Brett CM, Giacomini KM. 2008. Effect of genetic variation in the organic cation transporter 1, OCT1, on metformin pharmacokinetics. Clin. Pharmacol. Ther. 83:273–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shu Y, Sheardown SA, Brown C, Owen RP, Zhang S, Castro RA, Ianculescu AG, Yue L, Lo JC, Burchard EG, Brett CM, Giacomini KM. 2007. Effect of genetic variation in the organic cation transporter 1 (OCT1) on metformin action. J. Clin. Invest. 117:1422–1431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jung N, Lehmann C, Rubbert A, Schomig E, Fatkenheuer G, Hartmann P, Taubert D. 2013. Organic cation transporters OCT1 and OCT2 determine the accumulation of lamivudine in CD4 cells of HIV-infected patients. Infection 41:379–385 [DOI] [PubMed] [Google Scholar]
- 43.Pan BF, Sweet DH, Pritchard JB, Chen R, Nelson JA. 1999. A transfected cell model for the renal toxin transporter, rOCT2. Toxicol. Sci. 47:181–186 [DOI] [PubMed] [Google Scholar]
- 44.Ciarimboli G, Ludwig T, Lang D, Pavenstadt H, Koepsell H, Piechota HJ, Haier J, Jaehde U, Zisowsky J, Schlatter E. 2005. Cisplatin nephrotoxicity is critically mediated via the human organic cation transporter 2. Am. J. Pathol. 167:1477–1484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yokoo S, Yonezawa A, Masuda S, Fukatsu A, Katsura T, Inui K. 2007. Differential contribution of organic cation transporters, OCT2 and MATE1, in platinum agent-induced nephrotoxicity. Biochem. Pharmacol. 74:477–487 [DOI] [PubMed] [Google Scholar]
- 46.Yokoo S, Masuda S, Yonezawa A, Terada T, Katsura T, Inui K. 2008. Significance of organic cation transporter 3 (SLC22A3) expression for the cytotoxic effect of oxaliplatin in colorectal cancer. Drug Metab. Dispos. 36:2299–2306 [DOI] [PubMed] [Google Scholar]
- 47.Dobyan DC, Levi J, Jacobs C, Kosek J, Weiner MW. 1980. Mechanism of cis-platinum nephrotoxicity. II. Morphologic observations. J. Pharmacol. Exp. Ther. 213:551–556 [PubMed] [Google Scholar]
- 48.Goldstein RS, Mayor GH. 1983. Minireview. The nephrotoxicity of cisplatin. Life Sci. 32:685–690 [DOI] [PubMed] [Google Scholar]
- 49.Goldstein RS, Noordewier B, Bond JT, Hook JB, Mayor GH. 1981. cis-Dichlorodiammineplatinum nephrotoxicity: time course and dose response of renal functional impairment. Toxicol. Appl. Pharmacol. 60:163–175 [DOI] [PubMed] [Google Scholar]
- 50.Daley-Yates PT, McBrien DC. 1982. The mechanism of renal clearance of cisplatin (cis-dichlorodiammine platinum ii) and its modification by furosemide and probenecid. Biochem. Pharmacol. 31:2243–2246 [DOI] [PubMed] [Google Scholar]
- 51.Klein J, Bentur Y, Cheung D, Moselhy G, Koren G. 1991. Renal handling of cisplatin: interactions with organic anions and cations in the dog. Clin. Invest. Med. 14:388–394 [PubMed] [Google Scholar]
- 52.Nelson JA, Santos G, Herbert BH. 1984. Mechanisms for the renal secretion of cisplatin. Cancer Treat Rep. 68:849–853 [PubMed] [Google Scholar]
- 53.Osman NM, Litterst CL. 1983. Effect of probenecid and N′-methylnicotinamide on renal handling of cis-dichlorodiammineplatinum-II in rats. Cancer Lett. 19:107–111 [DOI] [PubMed] [Google Scholar]