

Abstract
Pneumocystis jirovecii is an opportunistic pathogen that causes serious pneumonia in immunosuppressed patients. Standard therapy and prophylaxis include trimethoprim (TMP)-sulfamethoxazole; trimethoprim in this combination targets dihydrofolate reductase (DHFR). Fourteen clinically observed variants of P. jirovecii DHFR were produced recombinantly to allow exploration of the causes of clinically observed failure of therapy and prophylaxis that includes trimethoprim. Six DHFR variants (S31F, F36C, L65P, A67V, V79I, and I158V) showed resistance to inhibition by trimethoprim, with Ki values for trimethoprim 4-fold to 100-fold higher than those for the wild-type P. jirovecii DHFR. An experimental antifolate with more conformational flexibility than trimethoprim showed strong activity against one trimethoprim-resistant variant. The two variants that were most resistant to trimethoprim (F36C and L65P) also had increased Km values for dihydrofolic acid (DHFA). The catalytic rate constant (kcat) was unchanged for most variant forms of P. jirovecii DHFR but was significantly lowered in F36C protein; one naturally occurring variant with two amino acid substitutions (S106P and E127G) showed a doubling of kcat, as well as a Km for NADPH half that of the wild type. The strongest resistance to trimethoprim occurred with amino acid changes in the binding pocket for DHFA or trimethoprim, and the strongest effect on binding of NADPH was linked to a mutation involved in binding the phosphate group of the cofactor. This study marks the first confirmation that naturally occurring mutations in the gene for DHFR from P. jirovecii produce variant forms of DHFR that are resistant to trimethoprim and may contribute to clinically observed failures of standard therapy or prophylaxis.
INTRODUCTION
Pneumocystis jirovecii is an opportunistic pathogen that has for decades been a leading cause of serious or fatal pneumonia in immunosuppressed patients, including those with AIDS, organ transplantation, or congenital immune deficiencies. The standard therapy and prophylaxis for pneumonia caused by P. jirovecii has been cotrimoxazole, which includes sulfamethoxazole and trimethoprim (TMP). Sulfamethoxazole targets dihydropteroate synthase (DHPS). Mutations leading to amino acid substitutions in DHPS have been linked to resistance to sulfamethoxazole (1). Mutations leading to amino acid substitutions in dihydrofolate reductase of Pneumocystis jirovecii have also been observed worldwide for over a decade, but attempts to link clinical outcomes to these mutations have been inconclusive (2–10).
The broadest array of variant forms of dihydrofolate reductase (DHFR) in clinical isolates of P. jirovecii was observed in patients in a European study (2) in which 16 nonsynonymous mutations in the gene for DHFR occurred (Table 1). In isolates from some patients, two alleles were observed, which suggested to the authors that coinfections with multiple genotypes of P. jirovecii existed. For example, the mutation leading to the amino acid substitution F36C appeared in one allele, and the mutation leading to the amino acid substitution L65P appeared in a second independent allele in P. jirovecii from the same patient (patient 7). True double mutations also occurred in this population; for example, the mutations producing the amino acid substitutions T14A and P26Q in P. jirovecii DHFR (pjDHFR) appeared in one allele in an isolate from patient 31, and the mutations producing the amino acid substitutions S106P and E127G appeared in one allele in an isolate from patient 33. Additionally, the isolate from patient 33 also had a mutation giving rise to the single variant R170G. Similarly, double variants were also observed in single alleles in P. jirovecii from patient 32 (M52I E63G and T144A K171E). The authors of this study concluded that patients who had received prophylaxis with an antifolate (trimethoprim or pyrimethamine) were more likely to harbor P. jirovecii isolates containing variant DHFR (6/15) than patients who had received no prophylaxis or prophylaxis with dapsone, atovaquone, or pentamidine (2/18). These authors noted that mutations creating amino acid substitutions in DHFR did occur in patients who failed prophylaxis with pyrimethamine or sulfadoxine but five of the seven patients also harbored mutations in dihydropteroate synthase (DHPS). Thus, a linkage of DHFR mutations to failure of prophylaxis was suggested but not proven by this study.
Table 1.
Amino acid substitutions in DHFR found in clinical isolates of Pneumocystis jirovecii
| Type of change in DHFR | Specific amino acid substitution(s)a | Reference(s) |
|---|---|---|
| Single amino acid substitutions | L13S | 6 |
| N23S | 6 | |
| S31F | 6 | |
| F36Cb | 2 | |
| S37T | 2 | |
| M52L | 6 | |
| R59G | 5 | |
| L65Pb | 2 | |
| A67V | 3–6 | |
| V79I | 2 | |
| D153V | 2 | |
| I158V | 2 | |
| C166Y | 3, 4 | |
| R170Gc | 2 | |
| Y197L | 2 | |
| Double amino acid substitutions arising from a single allele | T14A P26Q | 2 |
| M52I E63Gd | 2 | |
| T144A K171Ed | 2 | |
| S106P E127Gc | 2 |
P. jirovecii DHFR variants containing the amino acid substitutions shown in boldface type were produced recombinantly in quantities sufficient for kinetic analysis. The following additional DHFR variants were created as single amino acid substitutions from observed clinical double variants: S106P, T144A, and M52I. The following additional DHFR variants were created as double amino acid substitutions from observed clinical single variants: F36C L65P, A67V C166Y, and R59G A67V. (All of the additional DHFR variants listed were available in sufficient quantities for testing except for the M52I variant.)
Mutations leading to the amino acid substitutions F36C and L65P were observed in two independent DHFR alleles in P. jirovecii from a single patient.
Mutations leading to the double substitution S106P E127G in P. jirovecii DHFR were observed in one allele; a second allele in the same patient coded for the single substitution R107G.
Two separate alleles, each creating double amino acid substitutions in P. jirovecii DHFR, were observed in a single patient.
A study of P. jirovecii isolates from 32 patients in Japan observed two independent mutated alleles (Table 1; amino acid substitutions A67V and C166Y) of DHFR from P. jirovecii (3, 4). The authors of this study noted that these mutations did not align with regions in other forms of DHFR that were connected to trimethoprim or pyrimethamine resistance. Clinically, these two patients had not received prophylaxis with either drug before presenting with Pneumocystis pneumonia (PCP), and both were successfully treated with cotrimoxazole.
A study from South Africa independently reported the mutation leading to amino acid substitution A67V and the previously unknown R59G substitution in DHFR from P. jirovecii (5). These patients had not received prophylaxis with antifolates before developing Pneumocystis pneumonia. Resistance to clinical agents was suggested by these authors to be more likely linked to changes in DHPS than in DHFR.
A study of patients in Portugal (6) reported the known P. jirovecii DHFR A67V variant as well as four additional previously unknown variants: L13S, N23S, S31F, and M52L (Table 1). There was no evidence of mixed infections with P. jirovecii of different genotypes. This study specifically evaluated the relationship between failure of prophylaxis with cotrimoxazole and the presence of polymorphisms in DHFR but found no linkage. These authors also noted that 17 of the 35 patients with DHFR polymorphisms (both synonomous and nonsynonomous) were successfully treated for PCP with cotrimoxazole.
Studies from other sites have failed to find nonsynonomous mutations in DHFR from P. jirovecii, suggesting failure of therapy is not uniquely linked to mutations in DHFR (7–10). Although DHPS mutations apparently contribute to clinical failure by conferring resistance to sulfamethoxazole (1, 2), the appearance of mutations in both DHPS and DHFR in clinical isolates and the apparently more frequent development of DHFR mutations among patients who did receive prophylaxis that included an inhibitor of DHFR suggest a selection pressure is exerted on DHFR by exposure to trimethoprim or pyrimethamine (2). Therefore, the question remains of whether mutations in the gene for DHFR create resistance to trimethoprim and thus may also contribute to therapeutic failure. This study is designed to address this question at the molecular level.
MATERIALS AND METHODS
Construction and expression of mutant Pneumocystis jirovecii DHFR.
Mutations were introduced into the cDNA of pjDHFR, and the entire coding sequence was verified by the Roswell Park Cancer Center (Buffalo, NY). DNA oligonucleotides were obtained from Integrated DNA Technologies (Coralville, IA) and used without further purification. Plasmid DNA was purified using the plasmid minikit (Qiagen). Mutagenesis was performed using the QuikChange site-directed mutagenesis kit (Stratagene) following manufacturer's protocol: 50-μl reactions with a general PCR template: 1 cycle of 95°C for 30 s, followed by 16 cycles of 95°C for 30 s, and 55°C for 30 s (with the annealing temperature modified according to the melting temperature [Tm] of the respective primer pair) and 68°C for 1 min. The primer sequences are as follows: N23S, forward, 5′GGCTTGAAAAGTGATCTTCC3′, and reverse, 5′GGAAGATCACTTTTCAAGCC3′; T14A, forward, 5′CGTTGCATTGGCATTATCTCG3′, and reverse, 5′CGAGATAATGCCAATGCAACG3′; S31F, forward, 5′GGAAATTGAAGTTTGATATGATG3′, and reverse, 5′CATCATATCAAACTTCAATTTCC3′; F36C, forward, 5′GATGTTTTGCAGTCGAGTTAC3′, and reverse, 5′GTAACTCGACTGCAAAACATC3′; S37T, forward, 5′GTCTGATATGATGTTTTTTAAGCGAGT3′, and reverse, 5′CCAGATGTAACTCGCTTAAAAAAC3′; L65P, forward, 5′GGGAAAGTCTTCCAGCTCATTCTAGG3′, and reverse, 5′CCTAGAATGAGCTGGAAGACTTTCCC3′; A67V, forward, 5′GGAAAGTCTTCCTGTTCATTCTAGG3′, and reverse, 5′CCTAGAATGAACAGGAAGACTTTCC3′; V79I, forward, 5′CGACTAATAATAACATTAATTCG3′, and reverse, 5′CGAATTAATGTTATTATTAGTCG3′; D153V, forward, 5′GTTGACTGTGTAGTATTTTTCCC3′, and reverse, 5′GGGAAAAATACTACACAGTCAAC3′; I158V, forward, 5′GATGTATTTTTCCCTGTTGATTTTTCG3′, and reverse, 5′CGAAAAATCAACAGGGAAAAATACAT3′; C166Y, forward, 5′GGAGTTCTCAGTCATATTTGCCTTGG3′, and reverse, 5′CCAAGGCAAATATGACTGAGAACTCC3′; R170G, forward, 5′GCCTTGGGGAAAGCAAGATC3′, and reverse, 5′GATCTTGCTTTCCCCAAGGC3′; T144A, forward, 5′CGTATTATAGCTGCTGTAATTC3′, and reverse, 5′GGATTACAGCAGCTATAATACG3′; K171E, forward, 5′CCTTGGAGAGAGCAAGATCATTCG3′, and reverse, 5′CGAATGATCTTGCTCTCTCCAAGG3′; P26Q, forward, 5′GATCTTCAGTGGAAATTGAAG3′, and reverse, 5′CTTCAATTTCCACTGAAGATC3′; E127G, forward, 5′GGTGGAGGGTTGTATAAGG3′, and reverse, 5′CCTTATACAACCCTCCACC3′; and S106P, forward, 5′GCACTATTGCCACAGATTTATG3′, and reverse, 5′CATAAATCTGTGGCAATAGTGC3′.
The original wild-type pjDHFR (pET-SUMO vector) (Invitrogen) was used for PCR and all subsequent mutagenesis experiments. Three double mutant proteins (T14A P26Q, T144A K171E, and S106P E127G) were created by using the parental template DNA having one confirmed single-residue mutation and using primers for the second desired mutation during PCR.
Expression and purification of DHFR.
The expression and purification of recombinant wild-type P. jirovecii DHFR and the clinically observed variants were carried out as previously described (11). Clinical isolates of P. jirovecii have shown amino acid substitutions at 22 unique sites in DHFR (the Met at position 52 was replaced by Leu or Ile in different patients); 15 of the observed variants were single amino acid substitutions, and four other variants included double amino acid substitutions in the same allele (Table 1). Of these, protein has been expressed for 11 of the single-amino-acid variant forms and three of the naturally occurring doubly substituted variants in quantities sufficient for kinetic study (Table 2). In addition, five single amino acid changes observed clinically were combined to form three double variants of amino acid substitutions not found in the clinical samples (F36C L65P, A67V C166Y, and R59G A67V) to evaluate the potential for synergy between combinations of these variations. Furthermore, two single variants that were part of naturally occurring double variants were expressed independently (S106P and T144A) to evaluate their individual contributions to the kinetic parameters measured for the naturally occurring double variants.
Table 2.
Kinetic properties of mutated DHFR matching clinical isolates of Pneumocystis jiroveciia
| DHFR form |
Ki, nM (n) |
Km, μM (n) |
kcat, s−1 (n) | ||
|---|---|---|---|---|---|
| TMP | OAAG324 | DHFA | NADPH | ||
| Wild type | 38 ± 6.5 (10) | 2.6 ± 0.3 (14) | 2.1 ± 0.2 (22) | 18 ± 1.4 (5) | 33 ± 2 (25) |
| Variants | |||||
| T14A P26Q | 107 ± 38 (6) | 8.5 ± 1.1 (5) | 6.6 ± 0.5 (2) | 42 ± 5.1 (2) | 44 ± 1 (2) |
| N23S | 44 ± 4.4 (9) | 4.8 ± 0.5 (10) | 7.2 ± 0.6† (12) | 25 ± 3 (2) | 28 ± 2 (12) |
| S31F | 419 ± 48† (6) | 13 ± 2† (6) | 11 ± 2† (8) | 17.1 (1) | 27 ± 3 (9) |
| F36C | 3800 ± 1200† (4) | 17.5 ± 3† (3) | 56 ± 20† (4) | 18.1 (1) | 2 ± 0.5† (4) |
| L65P | 849 ± 210† (4) | 32 ± 11.9† (2) | 54 ± 7.5† (2) | 32 ± 3.6 (2) | 27 ± 1 (2) |
| F36C L65Pb | 15100 ± 1990† (2) | 21 ± 4.9 (2) | 269 ± 109† (2) | 19 ± 3 (2) | 9 ± 3† (2) |
| S37T | 26 ± 4 (3) | 2.0 ± 0.2 (3) | 2 ± 0.3 (3) | 21 ± 1.3 (3) | 35 ± 3 (7) |
| A67V | 346 ± 86† (7) | 0.7 ± 0.1 (9) | 9 ± 1† (10) | 16 ± 2 (2) | 17 ± 2† (9) |
| A67V C166Yb | 140 ± 46† (6) | 7.6 ± 1.1 (9) | 4.3 ± 0.6 (11) | 20 ± 1.0 (2) | 26 ± 2 (11) |
| R59G A67Vb | 1710 ± 230† (3) | 216 ± 93† (3) | 39 ± 5† (2) | 219 ± 56† (2) | 5 ± 1† (2) |
| V79I | 135 ± 27† (7) | 8.0 ± 0.8† (9) | 4.2 ± 0.9 (7) | 27 ± 1.6 (2) | 37 ± 3 (7) |
| S106Pc | 59 ± 10 (7) | 12 ± 2.3† (6) | 4.9 ± 0.7 (5) | 22 ± 3.1 (3) | 46 ± 7 (3) |
| S106P E127G | 60 ± 13 (3) | 3.7 ± 0.4 (6) | 19 ± 2.1† (8) | 9.0 ± 1.1 (2) | 61 ± 2† (6) |
| T144Ac | 192 ± 32† (10) | 10 ± 2.5 (6) | 7.5 ± 3.0 (4) | 12.9 ± 1.2 (4) | 70 ± 12 (3) |
| T144A K171E | 71 ± 21 (3) | 30 ± 15† (3) | 5.9 ± 1.7 (5) | 16 ± 0.1 (2) | 20 ± 3 (3) |
| D153V | 113 ± 40 (8) | 24.1 (1) | 56 ± 12† (8) | 64 ± 11† (2) | 13 ± 1† (5) |
| I158V | 233 ± 64† (8) | 14 ± 3.0† (6) | 17 ± 3.0† (7) | 18 ± 1 (2) | 28 ± 3 (7) |
| C166Y | 154 ± 37 (2) | ND | 10 ± 3 (2) | 13 ± 2.1 (3) | 11 (1) |
| R170G | 46 ± 9 (6) | 4.5 ± 1.3 (5) | 5.9 ± 0.8 (3) | 35 ± 2.3 (3) | 63 ± 10 (3) |
Data are reported as means ± standard errors of the means, except for cases in which n = 2, for which the mean ± range is reported. †, statistically significantly different from the wild type (P < 0.05, Kruskal-Wallis and Dunn's test; InStat 3).
The individual mutations leading to these single substitutions were observed independently in clinical isolates, but the dual substitution produced recombinantly has not yet been observed in clinical samples.
These mutations have been observed in clinical samples also bearing other substitutions, but they have not been seen alone.
Kinetic analysis.
The reaction scheme for P. jirovecii DHFR is presumed to follow the sequence determined for Pneumocystis carinii DHFR (pcDHFR) and other forms of the enzyme, in which NADPH is bound before the substrate (dihydrofolic acid) and exchange of NADP+ for NADPH precedes release of the product tetrahydrofolate (12). Standard DHFR assays contained 41 nM sodium phosphate buffer at pH 7.4, 8.9 mM 2-mercaptoethanol, 150 mM KCl, and saturating concentrations of NADPH (117 μM) and dihydrofolic acid (DHFA). All components of the assay except DHFA were preincubated at 37°C before the reaction was initiated with the addition of DHFA. The progress of the reaction was measured by continuous recording of changing absorbance at 340 nM (conversion of NADPH to NADP+). Initial linear rates of enzyme activity were measured; rates were linear under standard conditions for 1 to 5 min, depending upon the enzyme form being assayed. Activity was linearly related to the protein concentration under the selected conditions of assay.
Km values were determined by holding either the substrate or cofactor at a constant, saturating concentration (determined for each variant) and varying the other factor over a range of concentrations. Km values were determined by fitting the data to the Michaelis-Menten equation with or without substrate inhibition, using nonlinear regression methods to select the best statistical fit (Prism 4.0). The value of kcat was determined from the Vmax value and the total enzyme concentration, Etot, by the equation kcat = Vmax/Etot. The enzyme concentration was determined by titration with methotrexate (11).
Ki values, which reflect binding of inhibitors to DHFR, were determined by measuring inhibition of the reaction at two to five different concentrations of the substrate (DHFA). For the competitive inhibitors TMP and OAAG324 (Fig. 1) in this study, Ki could be calculated from the equation Ki = IC50/(1 + S/Km), where IC50 is the 50% inhibitory concentration. Ki was also independently calculated by the method of Dixon (13). Increases in the value of Ki reflect impaired binding of inhibitor to the enzyme and therefore reflect resistance to the inhibitor. Statistical comparisons were performed with InStat 2.03, using conservative nonparametric tests because variances among groups were not always equal.
Fig 1.
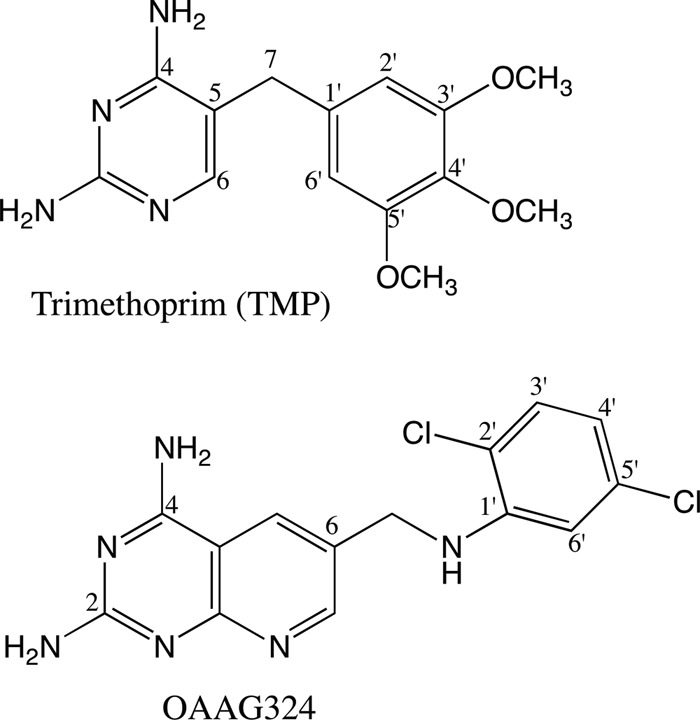
Schematics of the pjDHFR inhibitors trimethoprim and OAAG 324 used in this study.
Trimethoprim was retested against the wild-type P. jirovecii DHFR, alongside the variant forms, as a control; by avoiding historical control values, time was eliminated as a variable from the comparisons. Testing was also performed with the experimental antifolate OAAG324 (Fig. 1) to assess whether resistance to one antifolate was likely to confer resistance to the class. OAAG324 was chosen as the experimental antifolate because it differed significantly in structure from trimethoprim (14) and the compound was already known to be longer and more flexible than trimethoprim, such that the dichlorophenyl ring can adopt alternate conformations within the active site (15).
Structural models of P. jirovecii DHFR.
The deduced structure of P. jirovecii DHFR was calculated by homology modeling to the crystal structure of the P. carinii DHFR ternary complex with methotrexate and NADPH (15–17). The homology data for the pjDHFR complex with OAAG324 (Fig. 1) and NADPH reveals that this compound is displaced above the plane of the pyrido[2,3-d]pyrimidine ring compared to the crystal structure data (15). Similar modeling of pjDHFR with TMP and NADPH is consistent with the crystal structure of pcDHFR with TMP and NADPH (18). Computer models of the variants observed in P. jirovecii isolated from immunosuppressed patients were derived by substitution of the amino acids shown in Table 1.
RESULTS
Steady-state kinetic parameters.
Among the 14 clinically observed variant forms of P. jirovecii DHFR studied, over 10-fold increases in experimentally determined Ki values for trimethoprim relative to wild-type P. jirovecii DHFR were seen with three variants: S31F, F36C, and L65P (Table 2). Among these DHFR variants, the F36C protein showed a Ki value for trimethoprim 100-fold higher than that of the native P. jirovecii DHFR, which would confer a greatly decreased ability of this form of DHFR to bind trimethoprim.
Of the remaining naturally occurring variant forms of P. jirovecii DHFR, five (A67V, V79I, D153V, I158V, and C166Y) showed Ki values for trimethoprim 3- to 9-fold greater than that of the wild-type enzyme. Two singly substituted natural variants, the N23S and S37T proteins, had Ki values for trimethoprim that were indistinguishable from the wild type (Table 2). Three naturally occurring doubly substituted variants (T14A P26Q, S106P E127G, and T144A K171E) tended to have higher Ki values for trimethoprim than the wild type, but the changes were not statistically significant (Table 2).
In order to answer the question of whether the apparent resistance of certain naturally occurring variant forms of P. jirovecii DHFR to trimethoprim affected all classes of DHFR inhibitors equally, we also tested these variants against the experimental antifolate OAAG324 (Fig. 1), which is, like trimethoprim, a competitive inhibitor of P. jirovecii DHFR (14). The N23S, S37T, R170G, and S106P E127G variants were just as sensitive as the wild type to OAAG324, the same pattern seen with trimethoprim; however, the A67V variant showed a greater sensitivity to OAAG324 than did wild-type DHFR, a pattern very different from the 9-fold increase in Ki for trimethoprim seen with the A67V protein. Among the natural variants, only the L65P and T144A K171E proteins showed a greater than 10-fold increase in Ki value for OAAG324 relative to the wild type.
The apparent coexistence of two separate alleles for P. jirovecii DHFR in the same patient (2) would seem to set up the possibility for recombination to produce double mutants. The detection of naturally occurring true double mutants confirmed that by some process, double mutations were in fact being generated. These facts led us to produce double mutants that have not yet been confirmed in nature as a way to anticipate possible outcomes of future recombinant events in the organism. Thus, the doubly substituted F36C L65P, R59G A67V, and A67V C166Y variants were created and tested (Table 2). The F36C L65P variant showed extreme resistance to trimethoprim, with a Ki value 400-fold higher than that of the wild type; this doubly substituted variant had a Ki value for trimethoprim that was 4-fold higher than that for the F36C variant alone and 18-fold higher than that for the L65P variant alone, suggesting additivity or synergy between the two substitutions. Likewise, the R59G A67V variant had a Ki value for trimethoprim five times higher than that of the singly substituted A67V variant. In contrast, the A67V C166Y variant had a Ki for trimethoprim similar to that of the C166Y protein, which was lower than the Ki for the A67V variant, suggesting that in this case the effect of substitution at position 67 was offset by the substitution at position 166.
The kinetics toward the substrate and cofactor were examined for each DHFR variant and compared to wild-type values (Table 2). Eight of the 14 clinically observed variants tested had statistically significantly elevated Km values for DHFA, the amount ranging from 3-fold (N23S) to 27-fold (F36C); only the S37T variant was identical to the wild type. Small apparent increases in Km for the T14A P26Q, V79I, T144A K171E, C166Y, and R170G variants were not statistically significant. Elevation in the Km value for DHFA reflects reduced binding of the substrate, and as might be expected, the two clinically observed variants with the greatest resistance to the competitive inhibitor trimethoprim (F36C and L65P) also showed strong increases in the Km for DHFA. The artificially created F36C L65P double mutant showed the strongest effects on DHFA binding, with an increase in Km for DHFA of 128-fold.
Fewer variants had significant changes in the Km value for NADPH (Table 2). The clinically observed variant D153V had a Km value over three times higher than that of the wild type, but the R59G A67V variant had a Km value for NADPH more than 10 times higher than that of the wild type, indicating a significant impairment in binding this essential cofactor.
The value of kcat (measured as the ratio of Vmax for DHFA/Etot in the presence of saturating concentrations of NADPH) was significantly lowered from wild-type values for the clinically observed F36C, A67V, and D153V variants. The doubly substituted variants not found in clinical samples (F36C L65P and R59G A67V) also showed lower kcat values than the wild type, but the A67V C166Y variant was similar to the wild type. Ten of the clinically observed variants (T14A P26Q, N23S, S31F, L65P, S37T, V79I, T144A K171E, I158V, C166Y, and R170G) also had kcat values similar to the wild type, but the clinically observed doubly substituted S106P E127G variant showed an interesting increase in kcat relative to the wild type (Table 2).
Models of ligand interactions among variant forms of Pneumocystis jirovecii DHFR.
Although a crystal structure is not yet available for the DHFR from P. jirovecii, a simulated structure has been produced by homology modeling based on the crystal structure of P. carinii DHFR (15). Using this type of constructed model for P. jirovecii DHFR, the positions of the various amino acid substitutions evaluated in this study may be located within the protein (Fig. 2).
Fig 2.
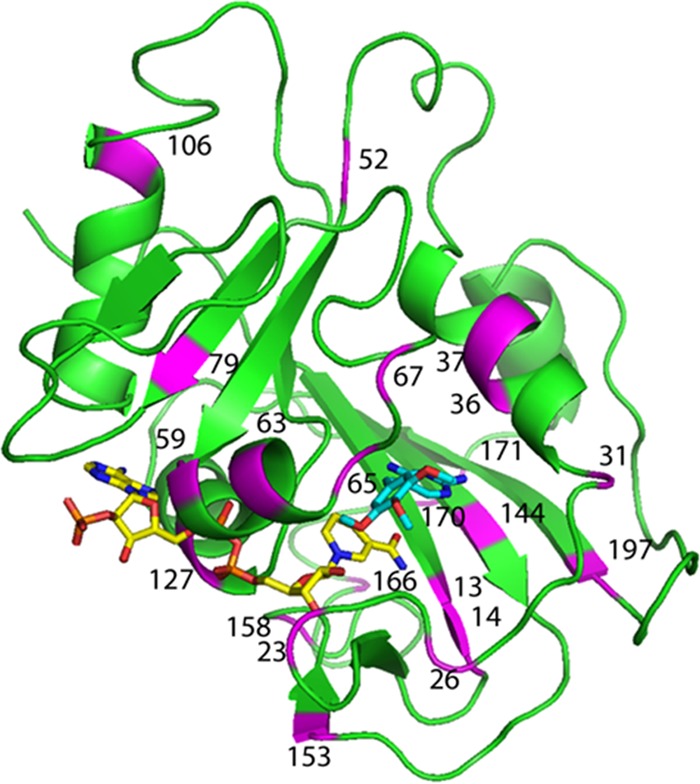
Homology model of Pneumocystis jirovecii DHFR (green), with NADPH (yellow) and trimethoprim (cyan). The locations of the 22 individual sites where amino acid substitutions have been observed in clinical samples (2–6) are indicated in violet and by the residue number.
The naturally occurring L65P and F36C single variants both had profound effects on the ability of the variant DHFR to bind to trimethoprim, as reflected by the greatly increased Ki value for trimethoprim. These effects are consistent with the structural placement of the variant amino acids in the wall of the pocket binding trimethoprim (or the substrate DHFA) in P. jirovecii DHFR (Fig. 3). Because both variants potentially alter space within the pocket binding trimethoprim, the model may explain the additive effects seen with the constructed double mutant. The profound effects on the binding of trimethoprim, an inhibitor competitive with DHFA, suggested that alterations in the kinetics of DHFA should also exist, and those were observed; these results are again consistent with the position of the variants in the wall of the pocket that would bind DHFA. OAAG324 is longer and more flexible than trimethoprim which may explain why the binding of this compound was less affected by these mutations than was binding to trimethoprim (15). Structural data for the complex of OAAG324 with human DHFR revealed the antifolate conformation was flexible and that the dichlorophenyl ring could occupy a position as shown in Fig. 4 and an alternate conformation in which the dichlorophenyl ring was near the cofactor binding site (15).
Fig 3.
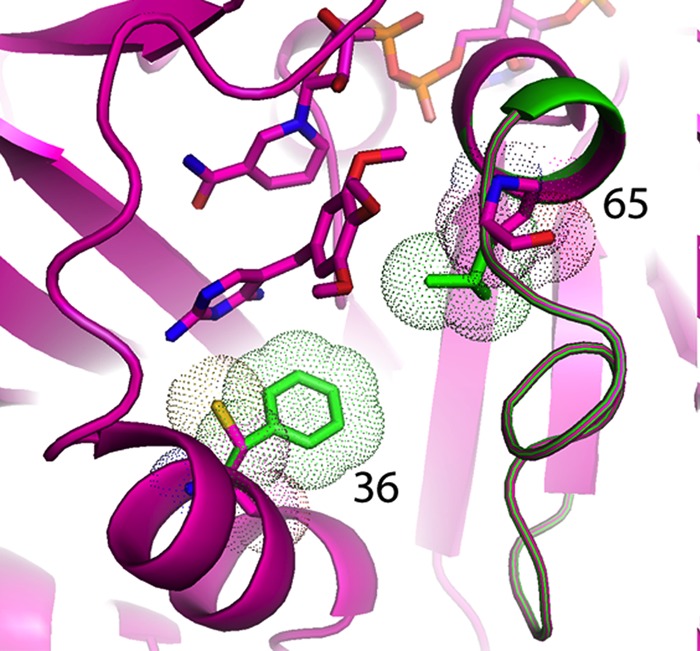
Model of pjDHFR with trimethoprim and NADPH showing the location of the F36C and L65P variants. The natural residues at positions 36 and 65 are shown in green.
Fig 4.
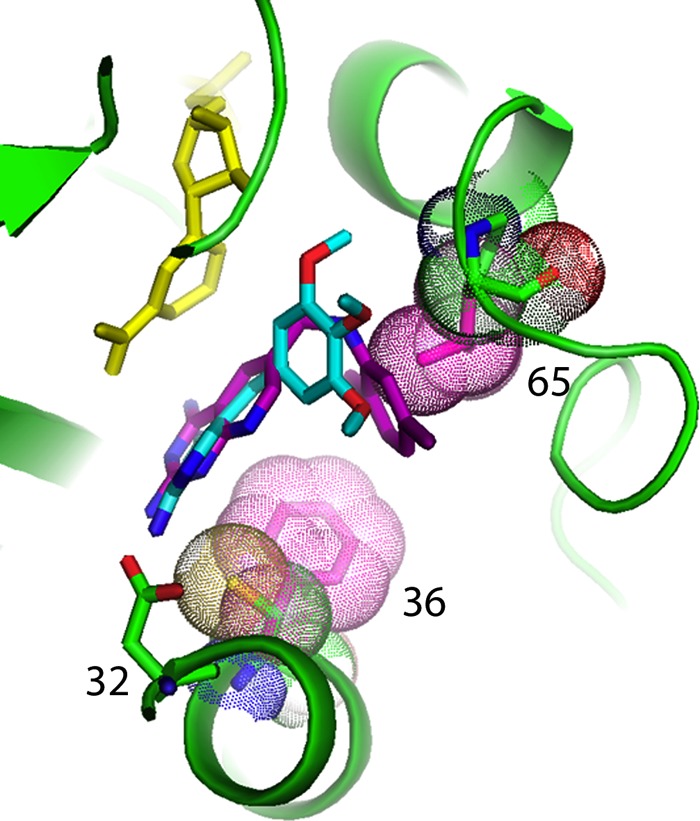
Model of the ternary complex of pjDHFR (green) with trimethoprim (cyan) and NADPH (yellow) showing the fit of OAAG324 (purple) and its interactions with the variants F36C and L65P, shown with their van der Waals surface dots (natural residues are in red, variants in green). Also labeled is Asp32. Note that the native L65 comes into close contact with OAAG324, whereas the P65 variant is further away.
The R59G A67V variant, which has not been observed in clinical isolates, showed lowered binding of trimethoprim, OAAG324, DHFA, and NADPH (elevated Ki and Km values), as well as a lower value for the catalytic rate constant, kcat (Table 2). The amino acid substitution at position 67, which is on a loop pointing away from the active site (Fig. 5), had modest effects alone but in combination with the substitution of glycine for arginine at position 59 had a much more profound change on the kinetics of the enzyme. Position 59 is within a region of the enzyme that interacts with the phosphate of NADPH, as is evident both from the crystal structure of P. carinii DHFR with TMP and NADPH (18) and from the homology model of P. jirovecii DHFR (Fig. 6). No interaction can take place when glycine is at position 59; thus, the observed large increase in the Km for NADPH in the R59G A67V variant is consistent with the impaired ability to bind the cofactor NADPH predicted by the structural models.
Fig 5.

Model of pjDHFR with trimethoprim and NADPH showing the location of the A67V and R59G variants highlighted with their van der Waals surfaces.
Fig 6.
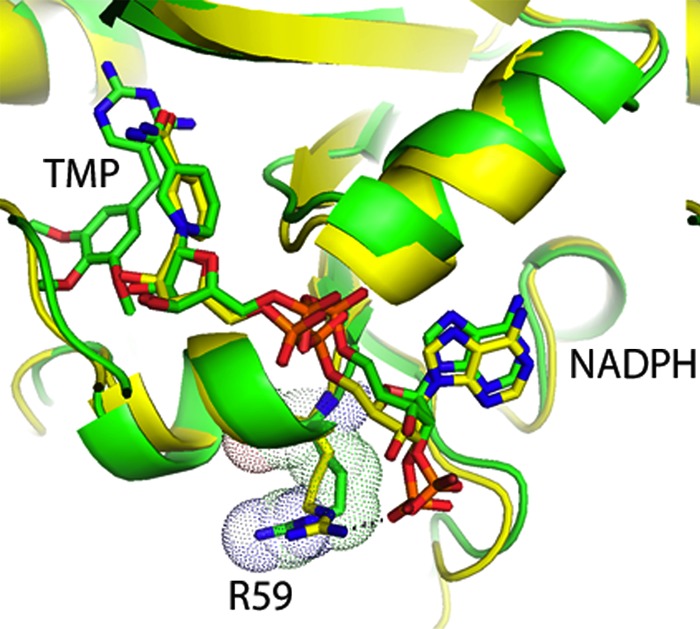
Model of pjDHFR with trimethoprim and NADPH (green) superimposed on the crystal structure of pcDHFR (yellow) (18) with NADPH showing the position of R59 (with their van der Waals surfaces) in the two structures and their interactions with the phosphate of the NADPH.
The amino acid in position 127 is also implicated in binding to NADPH: the hydrogen bond that may occur when glutamic acid is in that position cannot be formed when mutation places a glycine at that position (Fig. 7). The prediction of decreased NADPH binding was not borne out, in that the naturally occurring doubly substituted S106P E127G variant showed no statistical difference in the Km for NADPH from the wild-type enzyme. The amino acid substitution S106P puts proline at bottom of the helical turn, which tightens the turn, but it is far from the active site (Fig. 7) and alone has little effect on the binding of NADPH (Table 2). How these two amino acid substitutions might interact to explain the kinetic observations would require that the singly substituted E127G variant be produced and analyzed.
Fig 7.
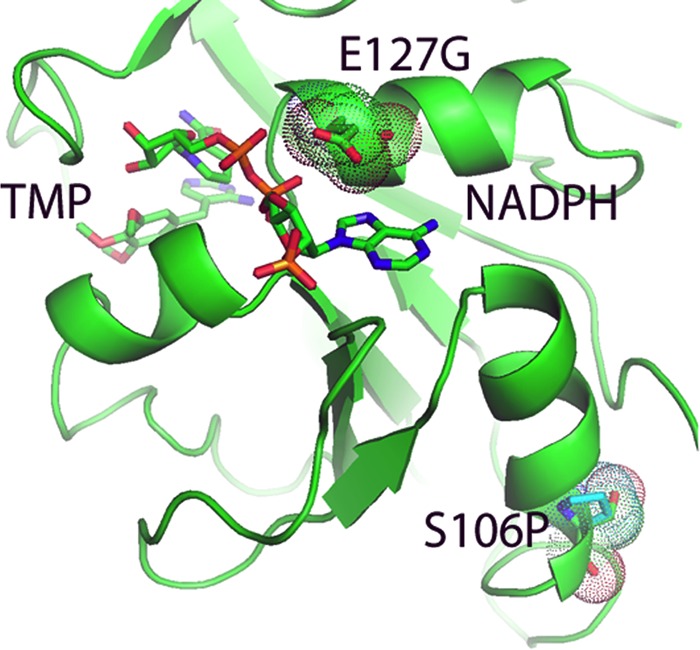
Model of pjDHFR with trimethoprim and NADPH showing the variants at E127G and S106P (highlighted with their van der Waals surfaces). Note the interaction of the E127 with the adenine ring of NADPH.
The amino acid at position 144 plays a role in binding trimethoprim through interactions with the hydroxyl group of the threonine usually found at that position (Fig. 8). Conversion of threonine to alanine prevents that interaction and lowers binding to trimethoprim (Table 2). This single mutation has not yet been reported in clinical samples, but the T144A K171E variant has been seen (2). In this doubly substituted variant, the loss of the large positively charged side chain of lysine (K) and the substitution of the smaller negatively charged side chain of glutamic acid (E) make the enzyme less resistant to trimethoprim but more resistant to OAAG324 (Table 2). It is not immediately obvious from the structure why binding of OAAG324 should be impaired in this doubly substituted variant, unless the approach to the pocket is impeded for the larger molecule (Fig. 9).
Fig 8.
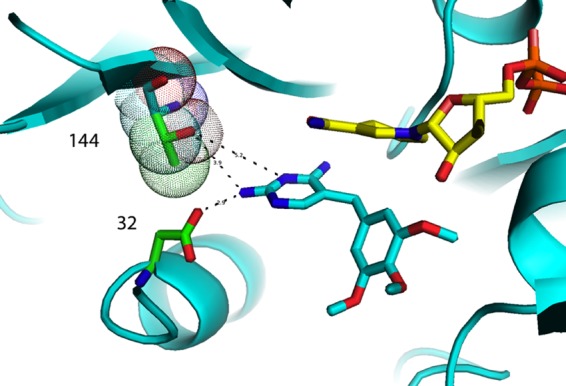
Interaction of variant T144A in the homology model for pjDHFR (cyan) with NADPH (yellow) and trimethoprim (cyan). The green dot surface residue is Thr144, and the cyan dot surface is A144. Asp32 is also shown.
Fig 9.
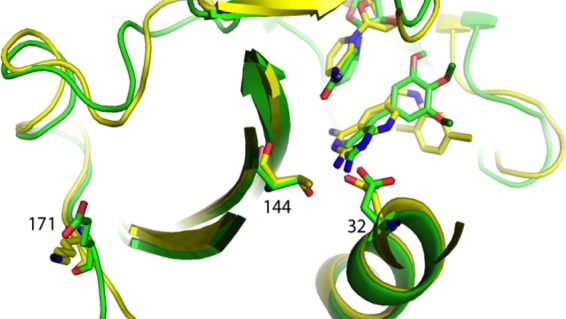
Comparison of homology models for the K171E T144A double variant for pjDHFR with NADPH and TMP (green) and pjDHFR modeled with the inhibitor OAAG324 and NADPH (yellow).
In two other cases, the structure suggested the possibility of strong effects on enzyme function with single amino acid substitutions, but these were not observed. For example, the N23S protein, a variant with kinetic properties very similar to wild type, is in loop 23 in pcDHFR (loop 20 in Escherichia coli), the loop that opens and closes over NADPH during the catalytic cycle (16). Likewise position 31 sits near the substrate binding site, but substitution of phenylalanine for serine at that site produced only modest effects on the Km values for substrate and cofactor while producing statistically significant resistance to both trimethoprim and OAAG324.
Most of the observed mutations in the gene for P. jirovecii DHFR produced amino acid substitutions in external regions of the enzyme (Fig. 2). Little could be predicted for the expected kinetic parameters of many of these variants (e.g., C166Y, I158V, D153V, S106P, A67V, and S37T), and indeed the changes observed were generally modest.
DISCUSSION
This study marks the first confirmation that mutations observed in the DHFR gene in P. jirovecii from clinical isolates produce variant forms of DHFR that have reduced sensitivity to trimethoprim, a key component of standard prophylaxis and therapy for infections caused by P. jirovecii. The fact that not all observed variants are linked to selected resistance to trimethoprim may argue that the clinical studies in part reflect the natural occurrence of polymorphisms in the gene for DHFR of P. jirovecii. Although it is known that Pneumocystis may be transmitted between patients, how much this process has contributed to the diversity of variant forms of P. jirovecii DHFR is not known (19–21).
The array of reported variant forms of P. jirovecii DHFR may be considered a snapshot of the distribution of mutations at the time the patients were originally seen: 1993 to 1996 (2), 1994 to 2000 (3, 4), 2000 to 2003 (5), and 1995 to 2004 (6). Therefore, it is logical to consider which of those variant forms of P. jirovecii DHFR possess qualities that would suggest they would survive in the absence of selective pressure exerted by antifolate prophylaxis. One measure of fitness could be the catalytic efficiency of the enzyme, usually measured as the ratio of kcat to Km for DHFA. The catalytic efficiency of wild-type P. jirovecii DHFR is 16 s−1 M−6. The clinically observed variants found most often in patients were the S37T protein (four patients in one study [2]) and the A67V protein (four patients in three studies [3–6]). The catalytic efficiency of the S37T variant was 17 s−1 M−6, almost identical to that of the wild-type enzyme; in contrast, the catalytic efficiency of the A67V variant was only 2 s−1 M−6. If a catalytic efficiency of at least 2 is required for persistence of a mutation in the population, then the N23S, S31F, V79I, R170G, T14A P26Q, S106P E127G, and T144A K171E variants might be predicted to persist, with calculated catalytic efficiencies ranging from 2.5 to 8.8 s−1 M−6. In contrast, those variants with very high Km values for DHFA and catalytic efficiencies at or below 1 (e.g., F36C, L65P, and D153V) might not be expected to persist unless strong selective pressure was being applied through exposure to trimethoprim. These predictions could be tested by genotyping clinical isolates of P. jirovecii collected after 2004, the last date of collection of the variants used in this study.
The study of naturally occurring or selected variants of DHFR from P. jirovecii is important for understanding the outcomes of current therapeutic strategies for treating human disease. Previous work demonstrated that prophylaxis with antifolates may be linked to an increased selection of variant forms of P. jirovecii DHFR within those patients, but these clinical studies were not able to show definitive linkage between the presence of these variant forms of DHFR and clinical outcomes (2, 6). Our results explain that observation: not all variant forms in those clinical samples in fact confer resistance to trimethoprim. Insufficient published clinical data exist to allow us to link individual patient outcomes to the presence of specific mutants. For example, 17 of 35 patients with polymorphisms in the gene for P. jirovecii DHFR were successfully treated with co-trimoxazole, but the outcomes for individual patients were not reported (6); of the five amino acid variants produced by these polymorphisms, we were able to study three (N23S, S31F, and A67V). Our results suggest that of these three variants, the S31F and A67V proteins might show clinical resistance to trimethoprim. However, a patient who carried the A67V P. jirovecii DHFR variant was successfully treated with co-trimoxazole (trimethoprim-sulfamethoxazole) (3). This patient was known to carry native dihydropteroate synthase (DHPS), the target enzyme for sulfamethoxazole, and thus the therapy may have succeeded as essentially monotherapy with sulfamethoxazole. It is interesting to speculate on the potential for drug failure for patients who might carry variant forms of DHFR resistant to trimethoprim as well as a variant form of DHPS relatively resistant to sulfamethoxazole. For example, the patient from whom the trimethoprim-resistant L65P and F36C DHFR variants were derived also harbored a variant form of DHPS known to confer a degree of resistance to sulfamethoxazole (2); unfortunately, no clinical outcome data were available for this patient.
One interesting implication of this study is that the patterns of resistance seem to be unique to each antifolate. Both trimethoprim and OAAG324 are competitive inhibitors of P. jirovecii DHFR, but they are different enough in structure that they reveal subtle changes in the binding site with several variant forms of P. jirovecii DHFR. For example, the F36C L65P double mutant showed no added resistance over the single mutants toward OAAG324, but there appeared to be additive or synergistic effects on trimethoprim binding. Likewise, the A67V variant showed statistically significant resistance to trimethoprim (an increase in Ki), but the Ki for OAAG324 is statistically lower (Mann-Whitney nonparametric test) than with the wild type, suggesting improved binding of OAAG324.
Finally, it is interesting to note that some but not all of the sites for mutations observed in P. jirovecii DHFR are also sites for mutations seen in pathogenic bacteria that are resistant to trimethoprim. For example, the S31F amino acid substitution that produces a roughly 10-fold increase in Ki for TMP in P. jirovecii DHFR is in the same relative position as the A26T amino acid substitution seen in the DHFR from Escherichia coli selected for trimethoprim resistance in a laboratory setting (22). In E. coli, this substitution produced about an 18-fold increase in Kd for trimethoprim, measured by extinction of tryptophan fluorescence of the pure protein; this substitution also lowered the Km values for DHFA and NADPH by half and increased the kcat about 8-fold. In contrast, the S31F substitution in P. jirovecii DHFR increased the Km for DHFA but had only minor effects on the Km for NADPH or kcat. Thus, each DHFR form showed different kinetic responses to amino acid changes at the same site that conferred resistance to trimethoprim.
Other amino acid substitutions observed in P. jirovecii DHFR tend to cluster in regions where amino acid substitutions also occur in trimethoprim-resistant pathogenic bacteria. For example, a helical region that includes an amino acid involved in binding folates occurs in P. jirovecii DHFR between amino acids 30 and 38; the S31F and F36C substitutions that we demonstrated confer trimethoprim resistance to P. jirovecii DHFR fall in this region, but neither changes the amino acid directly involved in folate binding (aspartate at position 32). The corresponding helical region (amino acids 27 to 33) in Staphylococcus aureus DHFR contains the H30N substitution observed in strains of the bacteria that are resistant to trimethoprim (23). The corresponding helical region in E. coli DHFR also spans residues 27 to 33 in the protein, a region that includes the W30R substitution that confers resistance to trimethoprim (22); another substitution linked to trimethoprim resistance, A26T, occurs just at the beginning of the helical region. As with the DHFR from P. jirovecii, none of these amino acid substitutions directly affects the aspartate residue in this region implicated in folate binding.
The amino acid substitution of alanine for threonine at position 144 in P. jirovecii DHFR, as discussed earlier in this section, affects binding to folates and folate analogs. In the sequence T(H/V)I at positions 144 to 146 (P. jirovecii DHFR numbering), both T and I are conserved in the DHFR proteins from E. coli, S. aureus, and Streptococcus pneumoniae. Trimethoprim-resistant E. coli shows a substitution of valine for isoleucine (I115V) at this site in DHFR. Trimethoprim-resistant S. pneumoniae shows a mutation that affects the center residue in this triad (H120Q), although substitutions at positions 100 (I100L) and 135 (L135F) (S. pneumoniae numbering) are more likely to contribute most of the trimethoprim resistance observed (24, 25).
Mutations in regions involved in NADPH binding are also found not only in trimethoprim-resistant P. jirovecii DHFR but also in trimethoprim-resistant DHFR from pathogenic bacteria. For example, one NADPH binding site corresponds to amino acids 59 to 61 (RKT) in P. jirovecii DHFR. Amino acid substitutions L65P and A67V in P. jirovecii DHFR flank this NADPH binding region, as does the amino acid substitution H45R in trimethoprim-resistant E. coli DHFR (22). Another NADPH binding site corresponding to amino acids 124 to 127 in P. jirovecii DHFR shows amino acid substitutions conferring trimethoprim resistance in S. pneumoniae (I100L) (24) and in E. coli (I94L) (22), but the only amino acid substitution in that region in P. jirovecii DHFR was observed in a doubly substituted variant (S106P E127G) that was not remarkable resistant to TMP.
In summary, this study is the first to confirm that mutations leading to key amino acid substitutions in DHFR from P. jirovecii may produce high-level resistance to trimethoprim, a component of standard prophylaxis and therapy for infections caused by P. jirovecii. The inability of prior studies to clearly link mutations in the gene for DHFR to clinical resistance to co-trimoxazole (trimethoprim-sulfamethoxazole) is explained by our observation that not all observed amino acid substitutions in P. jirovecii DHFR found in clinical samples in fact confer resistance to trimethoprim. The prediction based upon the results of this study is that those patients with P. jirovecii DHFR bearing amino acid substitution S31F, F36C, L65P, or A67V (and possibly V79I and I158V) would be resistant to clinically administered trimethoprim and the success or failure of therapy would rest on the ability of the coadministered sulfamethoxazole to inhibit its target dihydropteroate synthase (DHPS).
ACKNOWLEDGMENTS
V.C. thanks E. Steward and N. F. Mustafa for their efforts to clone and express the recombinant pjDHFR clinical variants.
This research was supported in part by grants from the National Institutes of Health GM51670 (V.C.), CA09885 (A.G.), and AI098458 (A.G.). Significant portions of this work were also supported by Biomedical Research Grant funding from the Indiana University School of Medicine (S.F.Q.).
Footnotes
Published ahead of print 29 July 2013
REFERENCES
- 1.Iliades P, Meshnick SR, Macreadie IG. 2005. Mutations in the Pneumocystis jirovecii DHPS gene confer cross-resistance to sulfa drugs. Antimicrob. Agents Chemother. 49:741–748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nahimana A, Rabodonirina M, Bille J, Francioli P, Houser PM. 2004. Mutations of Pneumocystis jirovecii dihydrofolate reductase associated with failure of prophylaxis. Antimicrob. Agents Chemother. 48:4301–4305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Takahashi T, Endo T, Nakamura T, Sakashita H, Kimura K, Ohnishi K, Kitamura Y, Iwamoto A. 2002. Dihydrofolate reductase gene polymorphisms in Pneumocystis carinii f. sp. Hominis in Japan. J. Med. Microbiol. 51:510–515 [DOI] [PubMed] [Google Scholar]
- 4.Takahashi T. 2009. Mutations of drug target molecules in Pneumocystis jirovecii isolates and future investigations. Jpn. J. Med. Mycol. 50:67–73 [DOI] [PubMed] [Google Scholar]
- 5.Robberts FJL, Chalkley LJ, Weyer K, Goussard P, Liebowitz LD. 2005. Dihydropteroate synthase and novel dihydrofolate reductase gene mutations in strains of Pneumocystis jirovecii from South Africa. J. Clin. Microbiol. 43:1443–1444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Costa MC, Esteves F, Antunes F, Matos O. 2006. Genetic characterization of the dihydrofolate reductase gene of Pneumocystis jirovecii isolates from Portugal. J. Antimicrob. Chemother. 58:1246–1249 [DOI] [PubMed] [Google Scholar]
- 7.Ma L, Borio L, Masur H, Kovacs JA. 1999. Pneumocystis carinii dihydropteroate synthase but not dihydrofolate reductase gene mutations correlate with prior trimethoprim-sulfamethoxazole or dapsone use. J. Infect. Dis. 180:1969–1978 [DOI] [PubMed] [Google Scholar]
- 8.Siripattanapipong S, Leelavoova S, Mungthin M, Worapong J, Tan-Ariya P. 2008. Study of DHPS and DHFR genes of Pneumocystis jirovecii in Thai HIV-infected patients. Med. Mycol. 46:389–392 [DOI] [PubMed] [Google Scholar]
- 9.Esteves F, Gaspar J, De Sousa B, Antunes F, Mansinho K, Matos O. 2011. Clinical relevance of multiple single-nucleotide polymorphisms in Pneumocystis jirovecii pneumonia: development of a multiplex PCR-single-base-extension methodology. J. Clin. Microbiol. 49:1810–1815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Munoz C, Zuluaga A, Restrepo A, Tobon A, Cano LE, Gonzalez A. 2012. Molecular dignosis and detection of Pneumocystis jirovecii DHPS and DHFR genotypes in respiratory specimens from Colombian patients. Diagn. Microbiol. Infect. Dis. 72:204–213 [DOI] [PubMed] [Google Scholar]
- 11.Cody V, Pace J, Makin J, Piraino J, Queener SF, Rosowsky A. 2009. Correlations of inhibitor kinetics for Pneumocystis jirovecii and human dihydrofolate reductase with structural data for human active site mutant enzyme complexes. Biochemistry 48:1702–1711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Margosiak SA, Appleman JR, Santi DV, Blakley RL. 1993. Dihydrofolate reductase from the pathogenic fungus Pneumocystis carinii: catalytic properties and interaction with antifolates. Arch. Biochem. Biophys. 305:499–508 [DOI] [PubMed] [Google Scholar]
- 13.Segel IH. 1975. Enzyme kinetics: behavior and analysis of rapid equilibrium and steady-state enzyme systems. Wiley-Interscience, New York, NY [Google Scholar]
- 14.Gangjee A, Adair OO, Queener SF. 2003. Synthesis and biological evaluation of 2,4-diamino-6-(arylaminomethyl)pyrido[2,3-d]-pyrimidines as inhibitors of Pneumocystis carinii and Toxoplasma gondii dihydrofolate reductase and as antiopportunistic infection and antitumor agents. J. Med. Chem. 46:5074–5082 [DOI] [PubMed] [Google Scholar]
- 15.Cody V, Pace J, Queener SF, Adair OO, Gangjee A. 2013. Kinetic and structural analysis for potent antifolate inhibition of Pneumocystis jirovecii, Pneumocystis carinii, and human dihydrofolate reductase and their active-site variants. Antimicrob. Agents Chemother. 57:2669–2677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cody V, Galitsky N, Rak D, Luft JR, Pangborn W, Queener SF. 1999. Ligand induced conformational changes in the crystal structures of Pneumcystis carinii dihydrofolate reductase complexes with folate and NADP+. Biochemistry 38:4303–4312 [DOI] [PubMed] [Google Scholar]
- 17.Cody V, Freindorf M, Furlani T, Queener SF, Gangjee A. 2009. Correlations of the kinetic properties and computational QM/MM modeling of potent dihydrofolate reductase inhibitors, abstr 2458. Abstr. 100th Annu. Meet. Am. Assoc. Cancer Researchers [Google Scholar]
- 18.Champness JN, Achari A, Ballantine SP, Bryant PK, Delves CJ, Stammers DK. 1994. The structure of Pneumocystis carinii dihydrofolate reductase to 1.9Å resolution. Structure 2:915–924 [DOI] [PubMed] [Google Scholar]
- 19.Ripamonti C, Orenstein A, Kutty G, Huang L, Schuhegger R, Sing A, Fantoni G, Atzori C, Vinton C, Huber C, Conville PS, Kovacs JA. 2009. Restriction fragment length polymorphism typing demonstrates substantial diversity among Pneumocystis jirovecii isolates. J. Infect. Dis. 200:1616–1622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Choukri F, Menotti J, Sarfati C, Lucet JC, Nevez G, Garin YJF, Derouin F, Totet A. 2010. Quantification and spread of Pneumocystis jirovecii in the surrounding air of patients with Pneumocystis pneumonia. Clin. Infect. Dis. 51:259–265 [DOI] [PubMed] [Google Scholar]
- 21.Hauser PM, Nahimana A, Taffe P, Weber R, Francioli P, Bille J, Rabodonirina M. 2010. Interhuman transmission as a potential key parameter for geographical variation in the prevalence of Pneumocystis jirovecii dihydropteroate synthase mutations. Clin. Infect. Dis. 51:e28–e33 [DOI] [PubMed] [Google Scholar]
- 22.Watson M, Liu JW, Ollis D. 2007. Directed evolution of trimethoprim resistance in Escherichia coli. FEBS J. 274:2661–2671 [DOI] [PubMed] [Google Scholar]
- 23.Dale GE, Broger C, D'Arcy A, Hartman PG, DeHoogt R, Jolidon S, Kompis I, Labhardt AM, Langen H, Locher H, Page MGP, Stuber D, Then RL, Wipf B, Oefner C. 1997. A single amino acid substitution in Staphylococcus aureus dihydrofolate reductase determines trimethoprim resistance. J. Mol. Biol. 266:23–30 [DOI] [PubMed] [Google Scholar]
- 24.Maskell JP, Sefton AM, Hall LMC. 2001. Multiple mutations modulate the function of dihydrofolate reductase in trimethoprim-resistant Streptococcus pneumoniae. Antimicrob. Agents Chemother. 45:1104–1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pikis A, Donkersloot JA, Rodriguez WJ, Keith JM. 1998. A conservative amino acid mutation in the chromosomal dihydrofolate reductase confers trimethoprim resistance in Streptococcus pneumoniae. J. Infect. Dis. 178:700–706 [DOI] [PubMed] [Google Scholar]