

Abstract
Deleobuvir (BI 207127) is an investigational oral nonnucleoside inhibitor of hepatitis C virus (HCV) NS5B RNA polymerase. Antiviral activity, virology, pharmacokinetics, and safety were assessed in HCV genotype 1-infected patients receiving 5 days' deleobuvir monotherapy. In this double-blind phase 1b study, treatment-naive (TN; n = 15) and treatment-experienced (TE; n = 45) patients without cirrhosis received placebo or deleobuvir at 100, 200, 400, 800, or 1,200 mg every 8 h (q8h) for 5 days. Patients with cirrhosis (n = 13) received deleobuvir at 400 or 600 mg q8h for 5 days. Virologic analyses included NS5B genotyping and phenotyping of individual isolates. At day 5, patients without cirrhosis had dose-dependent median HCV RNA reductions of up to 3.8 log10 (with no placebo response); patients with cirrhosis had median HCV RNA reductions of approximately 3.0 log10. Three patients discontinued due to adverse events (AEs). The most common AEs were gastrointestinal, nervous system, and skin/cutaneous tissue disorders. Plasma exposure of deleobuvir was supraproportional at doses ≥ 400 mg q8h and approximately 2-fold higher in patients with cirrhosis than in patients without cirrhosis. No virologic breakthrough was observed. NS5B substitutions associated with deleobuvir resistance in vitro were detected in 9/59 patients; seven encoded P495 substitutions, including P495L, which conferred 120- to 310-fold-decreased sensitivity to deleobuvir. P495 variants did not persist in follow-up without selective drug pressure. Deleobuvir monotherapy was generally well tolerated and demonstrated dose-dependent antiviral activity against HCV genotype 1 over 5 days.
INTRODUCTION
The current standard of care for chronic hepatitis C virus (HCV) genotype 1 (GT1) infection involves triple combination therapy with a direct-acting antiviral (DAA) agent plus pegylated interferon alfa (IFN-α) and ribavirin (PegIFN-RBV) (1, 2). DAAs currently approved for the treatment of HCV GT1 are the NS3/4A protease inhibitors (PIs) telaprevir (3, 4) and boceprevir (5, 6). Either drug, combined with PegIFN-RBV, produces sustained virologic response rates significantly greater than those seen with PegIFN-RBV alone in treatment-naive (TN) and treatment-experienced (TE) patients (1, 7–13). However, telaprevir and boceprevir are associated with various side effects (7–13), which add to the adverse-effect burden of RBV (anemia) and PegIFN (fatigue, flu-like symptoms, rash, neuropsychiatric impairment, and hemotoxicity) (14). Furthermore, the parenteral administration of PegIFN can be inconvenient and unacceptable to some patients. For patients who are IFN intolerant or for whom IFN is contraindicated, there is currently no alternative approved treatment.
IFN-free treatment may be a future option for patients with HCV GT1. This is likely to involve the combination of PIs with other antiviral agents because trials of PI monotherapy have shown high rates of treatment failure and the emergence of resistant variants (15). Results from in vitro and in vivo studies have suggested that combining DAAs with different modes of action and nonoverlapping resistance profiles, such as NS5B or NS5A polymerase inhibitors and NS3/4A PIs, could overcome these limitations (16). The validity of this approach was demonstrated in patients infected with HCV GT1 in a phase 1b study combining an NS3/4A PI (danoprevir; RG7227) and a nucleoside NS5B RNA polymerase inhibitor (mericitabine; RG7128) (17).
Deleobuvir (BI 207127) is a nonnucleoside inhibitor of HCV NS5B RNA polymerase that reversibly and noncovalently binds to thumb-pocket 1 (18). The median 50% effective concentration (EC50) values in cell-based HCV subgenomic replicons are 23 nM for GT1a and 11 nM for GT1b (19). In preclinical in vitro studies, amino acid substitutions in the thumb-pocket 1 at key positions P495, P496, and V499A were associated with decreased susceptibility to deleobuvir but did not change sensitivity to other DAA classes or to IFN-α (19). Similar observations have been reported for other thumb-pocket 1 nonnucleoside NS5B inhibitors, showing P495 and P496 substitutions selected in vitro at high concentrations (10 times and 20 times the EC50) and V499 variants at low concentrations (6 times the EC50) (16).
This article describes a phase 1b multiple rising-dose study to assess the safety, pharmacokinetic (PK) parameters, and antiviral effect of deleobuvir monotherapy for 5 days in patients with chronic HCV GT1. TE patients were included because those with prior failure of PegIFN-RBV are potential candidates for new agents.
MATERIALS AND METHODS
Study design.
TN or TE patients without cirrhosis or TE patients with compensated cirrhosis were eligible. NS5B genotyping and phenotyping were performed on individual clinical isolates at baseline and during the study to investigate their sensitivity to deleobuvir.
Patients without cirrhosis were randomized to deleobuvir in a double-blinded manner at rising doses of 100 mg, 200 mg, 400 mg, 800 mg, and 1,200 mg every 8 h (q8h) (n = 9 planned in each dosage group) or administered a matching placebo (randomization of 3:1 between active treatment and placebo, so n = 3 planned to receive placebo in each dosage group) for 5 days, with no stratification for TNs/TEs. Patients with cirrhosis received open-label deleobuvir at 400 mg q8h (n = 8) or 600 mg (n = 5) q8h for 5 days; there was no placebo group for patients with cirrhosis. The decision to proceed to the next dose level was based on acceptable safety and tolerability of the current dose as determined by the study sponsor's data monitoring committee.
Patients.
Eligible patients were male or female, aged 18 to 70 years, with confirmed chronic HCV GT1 infection. Deleobuvir had shown activity against HCV GT1a and GT1b in vitro; therefore, patients with either subgenotype were eligible. All patients had an HCV RNA level ≥ 100,000 IU/ml at screening. The TE group included previous null responders, partial responders, and relapsers. The presence or absence of cirrhosis was confirmed by liver biopsy or transient elastography (Fibroscan > 12.5 kPa). Patients were excluded if they had hepatitis B or human immunodeficiency virus coinfection, concurrent liver disease other than HCV, past treatment with any experimental polymerase inhibitor, planned or concurrent use of any other approved or investigational pharmacological therapy, or current drug or alcohol abuse. Patients were also excluded if they had hyperbilirubinemia, abnormal hematologic or laboratory values at screening, or concurrent disease considered clinically significant by the investigator. The study was carried out in accordance with the Declaration of Helsinki and International Conference on Harmonization guidelines. All patients gave written informed consent before participation.
Laboratory methods. (i) HCV RNA level.
Plasma HCV RNA levels were measured by the Roche Cobas TaqMan HPS (version 2) assay (linear range, 25 to 390,000,000 IU/ml; lower limit of detection, approximately 10 to 20 IU/ml). Blood samples were taken for HCV RNA analysis 5 min before the morning dose on each day of treatment. To measure the time course of the initial response to deleobuvir and HCV RNA levels after treatment, samples were also taken 2, 4, 8, 10, 16, and 18 h after the first dose on day 1 and 2, 4, 8, 12, 16, 24, and 48 h after the last morning dose of day 5. Samples were also taken on day 14 for posttreatment analysis.
(ii) Genotyping and NS5B sequencing.
HCV genotype was determined using the INNO-LiPA (version 1) test at screening and confirmed post hoc by NS5B region sequencing and phylogenetic analyses. Blood samples were taken for NS5B genotyping at screening, baseline, and day 6 and day 14. Viral RNA was extracted as previously described (20) and processed with HCV-specific primers to amplify the NS5B region. The lower limit of detection of the reverse transcriptase PCR amplification method restricted the analysis to samples with HCV RNA ≥ 1,000 IU/ml. NS5B sequences were determined by population sequencing (and clonal sequencing on the cohort without cirrhosis who had ≥3 log10 HCV RNA reductions during deleobuvir treatment) using BigDye Terminator (version 3.1) and an ABI 3100 Genetic Analyzer (Applied Biosystems). An NS5B subfragment (spanning codons 422 to 587) was cloned into a vector, and 80 to 95 transformed colonies were used to generate the clonal sequences. All changes to the nucleotide sequences from reference sequences for GT1a (GenBank accession no. AF009606) or GT1b (GenBank accession no. AJ238799) were noted, with particular attention paid to substitutions at 40 key NS5B amino acids known to confer resistance to nonnucleoside and nucleoside NS5B inhibitors, including amino acids 495, 496, and 499 (16, 21, 22).
(iii) NS5B phenotyping.
The NS5B amplicons were ligated into HCV replicon shuttle vectors containing a luciferase reporter gene to generate NS5B chimeric replicons. The reconstituted plasmid DNA was used to generate HCV subgenomic replicon RNA transcripts from this plasmid. These were transfected into Huh-7.5 cells, and luciferase activity was measured as a marker for HCV RNA replication. Serial dilutions of inhibitor were used to determine the EC50 for inhibition of HCV RNA replication.
(iv) PK analyses.
Blood samples were taken for pharmacokinetic (PK) analysis 5 min before the morning dose on each day of treatment. In addition, samples were taken frequently for PK analysis on day 1 and day 5. Plasma levels of deleobuvir were determined by a high-performance liquid chromatography, tandem mass spectrometry assay. PK parameters were calculated using noncompartmental analysis.
Statistical analysis.
The primary analysis was of the proportion of patients who achieved a virologic response (VR), defined as a ≥1 log10 reduction from baseline in serum HCV RNA level at any time from the start of administration to day 5. Secondary endpoints included PK parameters, safety assessments, and the time-dependent change in the HCV RNA level from baseline to day 7. No formal hypothesis testing was planned. Descriptive statistics were used to summarize the data.
RESULTS
Patient characteristics.
In total, 60 patients without cirrhosis (15 TN patients and 45 TE patients) were randomized and treated with deleobuvir or placebo; 13 patients with cirrhosis (12 TE patients and 1 TN patient enrolled by error but included in all analyses) received open-label deleobuvir. Baseline HCV RNA levels were similar across treatment groups. Some random variability was observed between dose groups for other baseline characteristics. Patients with cirrhosis tended to be older than those without cirrhosis (Table 1).
Table 1.
Baseline demographics of patients (randomized and treated)
| Patient characteristic | Value(s) for patients without cirrhosis treated with: |
Value(s) for patients with cirrhosis treated with: |
||||||
|---|---|---|---|---|---|---|---|---|
| Placebob (n = 14) | 100 mg deleobuvir (n = 9) | 200 mg deleobuvir (n = 9) | 400 mg deleobuvir (n = 9) | 800 mg deleobuvir (n = 9) | 1,200 mg deleobuvir (n = 10) | 400 mg deleobuvir (n = 8) | 600 mg deleobuvir (n = 5) | |
| Age (yrs, mean ± SD) | 47.2 ± 9.1 | 50.3 ± 11.9 | 51.7 ± 8.5 | 52.3 ± 8.7 | 47.3 ± 12.7 | 46.8 ± 8.6 | 55.4 ± 7.0 | 56.2 ± 8.1 |
| Body mass index (kg/m2, mean ± SD) | 24.7 ± 2.9 | 26.7 ± 3.6 | 24.9 ± 2.8 | 24.5 ± 1.8 | 26.2 ± 3.4 | 23.9 ± 3.1 | 25.8 ± 3.8 | 24.6 ± 1.4 |
| Baseline HCV RNA (mean log10 ± SD) | 6.39 ± 0.77 | 6.14 ± 0.55 | 6.49 ± 0.53 | 6.59 ± 0.51 | 6.35 ± 0.68 | 6.47 ± 0.47 | 6.52 ± 0.47 | 6.38 ± 0.39 |
| Sex (n) | ||||||||
| No. of males | 9 | 8 | 6 | 8 | 7 | 10 | 6 | 4 |
| No. of females | 5 | 1 | 3 | 1 | 2 | 0 | 2 | 1 |
| Time since HCV diagnosis (yrs, mean ± SD) | 10.4 ± 8.0 | 9.9 ± 7.2 | 9.0 ± 5.2 | 11.8 ± 7.7 | 8.1 ± 5.9 | 9.5 ± 10.0 | 15.3 ± 5.2 | 11.9 ± 7.2 |
| No. with indicated HCV genotype (by sequencing) | ||||||||
| Subtype 1a | 8 | 4 | 3 | 6 | 2 | 6 | 2 | 1 |
| Subtype 1b | 6 | 5 | 6 | 3 | 7 | 4 | 6 | 4 |
| No. PegIFN-RBV naive | 4 | 1 | 2 | 3 | 3 | 2 | 1 | |
| Prior PegIFN-RBV response | ||||||||
| No. whose data were missing/not assessable | 2 | 3 | 1 | 0 | 0 | 1 | 3c | 2 |
| No. of nonrespondersa | 3 | 5 | 3 | 4 | 1 | 3 | 3 | 3 |
| No. with breakthrough | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 |
| No. with relapse | 5 | 0 | 3 | 2 | 4 | 4 | 2 | 0 |
| Total | 10 | 8 | 7 | 6 | 6 | 8 | 8 | 5 |
The 19 nonresponders without cirrhosis were 6 null responders, 5 partial responders, and 8 nonresponders of unknown type. Of the 6 nonresponders with cirrhosis, there were 4 null responders, 1 partial responder, and 1 nonresponder of unknown type.
Patients were randomized 3:1 between active treatment and placebo; the data from all patients randomized to placebo are combined in the table.
Includes the PegIFN-RBV-naive patient.
HCV RNA level.
In patients without cirrhosis, deleobuvir produced a dose-related reduction in the HCV RNA level (Table 2). The VRs and changes in plasma HCV RNA levels were similar in TN and TE patients without cirrhosis. In patients with cirrhosis, the decrease in the HCV RNA level on day 5 was similar to that in patients without cirrhosis receiving deleobuvir doses ≥ 400 mg (Table 2). The median HCV RNA level declined rapidly during the first 24 h of treatment; at higher doses, there was a further slower decrease during day 2, with a plateau thereafter (Fig. 1). Overall, there was a greater decrease in the HCV RNA level in patients infected with HCV GT1b than in patients infected with GT1a (Fig. 1). The mean maximum reduction in HCV RNA in patients with HCV GT1b infection (>3 log10) was achieved at deleobuvir ≥ 400 mg, and yet in GT1a-infected patients the reduction in HCV RNA increased gradually with dose and did not exceed 3 log10 (Table 2). No virologic breakthrough was observed during treatment at any dose. The antiviral effect of deleobuvir persisted for at least 24 h after the last drug intake. Thereafter, HCV RNA levels slowly returned to baseline levels.
Table 2.
VR rates through day 5 and changes in HCV RNA level in dosage groups
| Parametera | Value(s) for patients without cirrhosis treated with: |
Value(s) for patients with cirrhosis treated with: |
||||||
|---|---|---|---|---|---|---|---|---|
| Placebo (n = 14) | 100 mg deleobuvir (n = 9) | 200 mg deleobuvir (n = 9) | 400 mg deleobuvir (n = 9) | 800 mg deleobuvir (n = 9) | 1,200 mg deleobuvir (n = 10) | 400 mg deleobuvir (n = 8) | 600 mg deleobuvir (n = 5) | |
| No. of patients with VR through day 5/total no. of patients (%) | ||||||||
| Overall | 2/14 (14.3) | 2/9 (22.2) | 5/9 (55.6) | 8/9 (88.9) | 8/9 (88.9) | 10/10 (100.0) | 7/8 (87.5)b | 5/5 (100.0) |
| TN | 2/4 (50.0) | 0/1 (0.0) | 1/2 (50.0) | 3/3 (100.0) | 3/3 (100.0) | 2/2 (100.0) | ||
| TE | 0/10 (0.0) | 2/8 (25.0) | 4/7 (57.1) | 5/6 (83.3) | 5/6 (83.3) | 8/8 (100.0) | ||
| Change in log10 HCV RNA on morning of day 5, mean ± SD (median) | ||||||||
| Overall | −0.04 ± 0.3 (−0.03) | −0.6 ± 0.7 (−0.4) | −1.1 ± 0.9 (−0.8) | −1.9 ± 1.3 (−1.3) | −3.1 ± 1.2 (−3.8) | −2.9 ± 0.8 (−3.2) | −2.6 ± 1.2 (−3.0)b | −3.2 ± 0.6 (−3.1) |
| TN | +0.1 ± 0.3 (+0.2) | —c | −0.4 ± 0.1 (−0.4) | −2.5 ± 1.4 (−2.5) | −2.9 ± 1.0 (−3.2) | −2.7 ± 0.9 (−2.7) | ||
| TE | −0.1 ± 0.2 (−0.1) | −0.6 ± 0.7 (−0.4) | −1.2 ± 1.0 (−0.9) | −1.6 ± 1.4 (−1.0) | −3.2 ± 1.4 (−3.9) | −3.0 ± 0.9 (−3.2) | ||
| By-patient maximal change in HCV RNA by genotype, mean ± SD (median) | ||||||||
| GT1a | −0.5 ± 0.5 (−0.2) (n = 8) | −0.7 ± 0.4 (−0.9) (n = 4) | −0.9 ± 0.1 (−0.9) (n = 3) | −1.3 ± 0.4 (−1.1) (n = 6) | −1.9 ± 1.3 (−1.9) (n = 2) | −2.8 ± 0.6 (−2.9) (n = 6) | −1.4 ± 0.8 (−1.4) (n = 2) | −2.4 (−2.4) (n = 1) |
| GT1b | −0.4 ± 0.3 (−0.3) (n = 6) | −0.9 ± 0.8 (−0.6) (n = 5) | −1.9 ± 1.3 (−1.9) (n = 6) | −3.5 ± 0.4 (−3.5) (n = 3) | −3.8 ± 0.9 (−4.1) (n = 7) | −3.8 ± 0.9 (−3.7) (n = 4) | −3.3 ± 0.6 (−3.4) (n = 6) | −3.7 ± 0.5 (−3.6) (n = 4) |
TE, treatment experienced; TN, treatment naive; VR, virologic response (≥1 log10 reduction from baseline in serum HCV RNA at any time from start of administration to day 5).
Includes one treatment-naive patient.
—, the one patient in this subgroup had a missing value.
Fig 1.
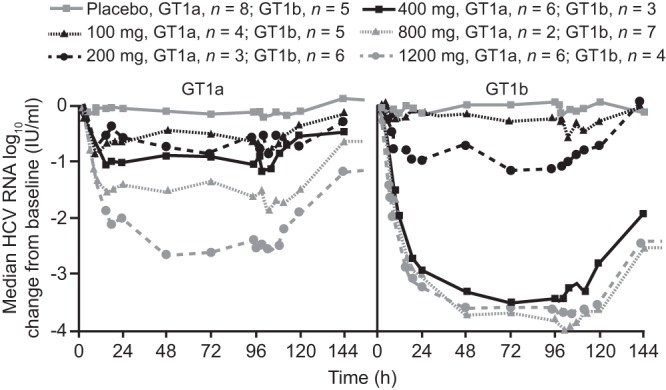
Median changes in the HCV RNA level for patients without cirrhosis according to deleobuvir dose and HCV genotype.
Virology. (i) In vitro susceptibility of HCV NS5B polymerase.
NS5B polymerases were phenotyped from 70 (30 GT1a and 40 GT1b) of the 73 patient baseline samples. Their susceptibilities to deleobuvir were distributed over a 2 log10 range, with geometric mean EC50 values of 26 nM for GT1a and 7.2 nM for GT1b (Table 3). These values were within a 2-fold range of the reference values for nonchimeric wild-type GT1a and GT1b isolates (23 nM and 11 nM, respectively). The geometric mean EC50 for IFN-α, used as a control for the intrinsic variability of the assay, was 0.19 IU/ml for both the GT1a and GT1b subtypes (Table 3).
Table 3.
Susceptibility of baseline sample-derived NS5B chimeric replicons to deleobuvir and interferon alfa
| Parameter | Deleobuvir EC50 (nM) |
IFN-α EC50 (IU/ml) |
||
|---|---|---|---|---|
| GT1a (n = 30) | GT1b (n = 40) | GT1a (n = 30) | GT1b (n = 40) | |
| Range | 1.2–153 | 0.4–98 | 0.12–0.38 | 0.13–0.3 |
| Geometric mean | 26 | 7.2 | 0.19 | 0.19 |
By population sequencing, baseline variants at codon 421 were detected in four patients with GT1a infection, one in each of the 200-mg, 400-mg (without cirrhosis), 800-mg, and 1,200-mg dose groups and one with GT1b infection (placebo group). Baseline variants at codon 499 were detected in 1 patient with GT1a (placebo group) and 13 patients with GT1b (3, 4, 4, and 2 patients in the 100-mg, 200-mg, 800-mg, and placebo groups, respectively). Mutations encoding amino acid substitutions at these positions were associated with EC50 values that were higher than those seen with other baseline samples from the same subtype (Table 4) as determined in the in vitro phenotyping assay. Notably, no baseline variants were detected at codon 495 or 496 by population sequencing.
Table 4.
EC50 values for deleobuvir at baseline for amplicons with and without mutations at codons 421 and 499
| Amplicon variant | n | EC50 (nM) |
||
|---|---|---|---|---|
| Geometric mean | Range | Fold changed | ||
| GT1a, wild-type A421 | 27 | 22 | 1.2–118 | |
| GT1a, A421V | 3a | 127 | 115–150 | 5.8 |
| GT1b, wild-type V499 | 28 | 4.3 | 0.4–31 | |
| GT1b, V499 variantsb | 12c | 24 | 6.7–98 | 5.6 |
| GT1a, wild-type A499 | 29 | 27 | 1.2–153 | |
| GT1a, A499T | 1 | 7.8 | ±1.5 | 0.3 |
Excluding 1 patient (deleobuvir at 400 mg) whose sample did not replicate in vitro and whose EC50 therefore could not be obtained.
The V499 variants included 6 V499A, 5 V499T, and 1 V499I.
Excluding 1 patient (deleobuvir at 200 mg) whose sample did not replicate in vitro and whose EC50 therefore could not be obtained. One GT1b patient (placebo) who had mutations A421V plus V499A at baseline but whose sample did replicate in vitro and whose EC50 therefore could not be obtained is excluded.
Wilcoxon rank-sum test for equality of mean baseline EC50 values for wild type compared to variant: P = 0.002, 0.0001, and 0.3 for the data in rows 2, 4, and 6, respectively.
(ii) Emergent NS5B variants.
NS5B amino acid changes relative to the baseline sequence at the three key thumb-pocket 1 residues (495, 496, and 499) were detected in 9 GT1b-infected patients in the deleobuvir treatment groups (Table 5). The predominant variants detected on day 6 encoded P495L or P495S (Table 5). P495L decreased sensitivity to deleobuvir 120- to 310-fold, and P495S decreased sensitivity 91-fold. V499A, a frequent baseline polymorphism, emerged in 3 patients, including 1 also with P495L variants and 1 with detection on day 14 but not day 6. V499A emerged only once as a predominant variant in one patient randomized to a low-dose group (200 mg) and decreased in vitro sensitivity to deleobuvir 3-fold. A NS5B T389S variant emerged in only one GT1a-infected patient in a low-dose group (400 mg [without cirrhosis]) and conferred a 3.2-fold shift in deleobuvir sensitivity. Substitutions at NS5B amino acid 389, a site proximal to the thumb-pocket 1 domain, were not detected in preclinical studies of BI 207127 (18) and have been shown to reduce sensitivity to benzimidazole-based pocket 1 inhibitors containing extra substituents with closer proximity to T389 7- to 13-fold (23). Chimeric replicons with all of these NS5B variants remained sensitive to IFN-α and to faldaprevir (data not shown).
Table 5.
Emerging NS5B variants at residues in the thumb-pocket 1 domain and fold change in deleobuvir susceptibility
| Dose group | Frequencya | HCV genotype | NS5B amino acid change | EC50 fold changeb |
|---|---|---|---|---|
| Placebo (n = 14) | 0/14 | |||
| 100 mg deleobuvir (n = 9) | 0/9 | |||
| 200 mg deleobuvir (n = 9) | 1/9 | 1b | V499A | 2.9 |
| 400 mg deleobuvir (n = 9) | 2/9 | 1b | P495[P/L] | 310 |
| 1a | T389S | 3.2 | ||
| 800 mg deleobuvir (n = 9) | 3/9 | 1b | P495[Q/L] | NA |
| 1b | P495S | ND | ||
| 1b | P495S | 91 | ||
| 1,200 mg deleobuvir (n = 10) | 1/10 | 1b | P495L | 120 |
| 400 mg deleobuvir; patients with cirrhosis (n = 8) | 1/8 | 1b | V499[A/V]c | 1.1 |
| 600 mg deleobuvir; patients with cirrhosis (n = 5) | 2/5 | 1b | P495[P/L], V499[A/V] | 11 |
| 1b | P495L | 280 |
The frequency is expressed as the number of patient samples with key amino acid changes relative to the baseline sequence. The data were obtained by population sequence analysis.
EC50 fold change relative to the baseline EC50 of the corresponding patient. NA, data not available (chimeric NS5B replicon did not replicate); ND, not determined.
V499[A/V] was not detected at day 6 and was detected only on day 14.
(iii) Clonal sequence analysis.
Samples with resistant variants that emerged at position(s) 495, 496, or 499 were also assessed by clonal sequence analysis at baseline, day 6, and day 14. In the two patients with a single P495L variant detected by population sequencing at day 6, the proportion of clones encoding this variant reached 72% (patient in the deleobuvir 400-mg group) and 100% (patient in the deleobuvir 1,200-mg group) of clones at day 6 but rapidly decreased to 0% and 5%, respectively, by day 14, with concomitant outgrowth of wild-type P495 (Fig. 2). However, in the latter patient, there was an increase in the levels of P495S (33% of clones) and V499A (5% of clones) at day 14. In one GT1b-infected patient (deleobuvir 200-mg group), V499A was detected in 10% of clones at baseline, in 98% at day 6, and 78% at day 14. Emergence of P496S variants was also detected in this patient (4% of clones at day 6), although none remained by day 14.
Fig 2.

Clonal sequence analysis at baseline, day 6, and day 14 of samples from a patient in the 400-mg-dose group (A), 1,200-mg-dose group (B), and 200-mg-dose group (C). HCV RNA at the 48-h time point for the patient whose results are presented in panel C could not be confirmed. WT, wild type.
Safety and tolerability. (i) Patients without cirrhosis.
Of the 46 patients without cirrhosis who received deleobuvir, 27 (58.7%) reported treatment-emergent adverse events (AEs); these were considered related to study medication in 22 of 46 patients (47.8%) (Table 6). The corresponding proportions in patients treated with placebo were 4/14 (28.6%) and 3/14 (21.4%). Serious or severe AEs were reported in three patients. One patient receiving the 800-mg dose developed moderate generalized erythema with facial involvement, leading to discontinuation before the last dose on day 5. This resolved within 2 days after discontinuation (with antihistamine therapy). In one patient receiving the 400-mg dose, deleobuvir was discontinued on day 3 due to apparent QT prolongation, although subsequent reassessment at a central electrocardiology laboratory indicated that this was not the case. One patient in the deleobuvir 200-mg group experienced arterial hypotension (considered related to treatment) on day 1; this resolved the same day without additional therapy.
Table 6.
Adverse events occurring overall and in >1 patient in any deleobuvir dosage group
| AE | No. (%) of patients with indicated treatment and AE(s) |
|||||||
|---|---|---|---|---|---|---|---|---|
| Without cirrhosis |
With cirrhosis |
|||||||
| Placebo (n = 14) | 100 mg deleobuvir (n = 9) | 200 mg deleobuvir (n = 9) | 400 mg deleobuvir (n = 9) | 800 mg deleobuvir (n = 9) | 1,200 mg deleobuvir (n = 10) | 400 mg deleobuvir (n = 8)a | 600 mg deleobuvir (n = 5) | |
| Total | 4 (28.6) | 2 (22.2) | 3 (33.3) | 7 (77.8) | 6 (66.7) | 9 (90.0) | 5 (62.5) | 4 (80.0) |
| GI disorder(s)b | 2 (14.3) | 2 (22.2) | 2 (22.2) | 5 (55.6) | 3 (33.3) | 8 (80.0) | 1 (12.5) | 3 (60.0) |
| Nervous system disorder(s)c | 3 (21.4) | 1 (11.1) | 2 (22.2) | 0 (0.0) | 2 (22.2) | 5 (50.0) | 2 (25.0) | 1 (20.0) |
| Skin/subcutaneousd | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (11.1) | 2 (22.2) | 5 (50.0) | 0 (0.0) | 1 (20.0) |
| Generale | 1 (7.1) | 0 (0.0) | 0 (0.0) | 3 (33.3) | 1 (11.1) | 4 (40.0) | 1 (12.5) | 2 (40.0) |
Includes one treatment-naive patient.
Included nausea, diarrhea, flatulence, and vomiting.
Included headache, hyperesthesia, and paresthesia.
Included erythema and rash.
Included fatigue, pain, gait disturbance, influenza-type illness, and irritability.
The most common categories of on-treatment AEs in patients receiving deleobuvir were gastrointestinal (GI) disorders (43.5% of patients [20/46]), nervous system disorders (21.7% [10/46], including headache in 15.2% [7/46]), and skin/cutaneous tissue disorders (17.4% [8/46]) (Table 6). These AEs were more common at higher deleobuvir doses. All GI and skin/subcutaneous tissue disorders were considered mild to moderate, and all resolved. No patient discontinued because of nervous system or GI AEs.
(ii) Patients with cirrhosis.
AEs were reported in 15.4% (2/13) of patients during screening and 69.2% (9/13) of patients during treatment (Table 5). No deaths, severe AEs, or serious AEs were reported. In one patient receiving the 400-mg dose, deleobuvir was discontinued on day 1 because of apparent QT prolongation, although subsequent reassessment at a central electrocardiology laboratory indicated that the QT interval had been overestimated at the trial site. The most common categories of AE were GI disorders (30.8% of patients [4/13]) and nervous system disorders (23.1% [3/13]). Skin/cutaneous tissue disorders were reported in one patient. There was some indication that GI disorders were dose related, although the numbers of patients were small.
(iii) Laboratory evaluations.
Laboratory parameters did not show any clinically relevant differences from baseline. There were no signs of hepatic, renal, or hematologic toxicity (see Table S1 in the supplemental material).
Pharmacokinetics.
Substantial variability between patients in PK parameters of deleobuvir was observed (with coefficients of variation > 60%). Mean plasma concentrations of deleobuvir decreased to below 1% of the maximum concentration within 40 h after last drug intake. The terminal elimination half-life values for deleobuvir were 3.7 to 5.3 h and 5.9 to 6.0 h in patients with and without cirrhosis, respectively (Table 7). Steady-state plasma concentrations of deleobuvir were reached by 24 h postdose at all dose levels. In patients without cirrhosis, plasma levels of deleobuvir exhibited supraproportional PK parameters at the steady state at doses ≥ 400 mg.
Table 7.
Selected PK parameters of deleobuvir at the steady state
| Parametera | Values for patients without cirrhosis treated with: |
Values for patients with cirrhosis treated with: |
|||||
|---|---|---|---|---|---|---|---|
| 100 mg deleobuvir (n = 9) | 200 mg deleobuvir (n = 9) | 400 mg deleobuvir (n = 8) | 800 mg deleobuvir (n = 7−8) | 1,200 mg deleobuvir (n = 10) | 400 mg deleobuvir (n = 7–8)b | 600 mg deleobuvir (n = 5) | |
| Geometric mean (% CV) | |||||||
| AUCτ,ss, ng · h/ml | 1,930 (79.0) | 4,130 (79.1) | 17,900 (37.1) | 43,500 (118) | 88,500 (83.3) | 36,800 (82.7) | 86,400 (78.1) |
| AUCτ,ss/dose, ng · h/ml/mg | 19.3 (79.0) | 20.7 (79.1) | 44.8 (37.1) | 54.4 (118) | 73.7 (83.3) | 92.1 (82.7) | 144 (78.1) |
| Cmax,ss, ng/ml | 391 (67.8) | 910 (80.3) | 3,780 (46.3) | 9,030 (103) | 16,500 (66.7) | 6,780 (76.7) | 15,400 (80.4) |
| Cmax,ss/dose, ng/ml/mg | 3.91 (67.8) | 4.55 (80.3) | 9.45 (46.3) | 11.3 (103) | 13.8 (66.7) | 17.0 (76.7) | 25.6 (80.4) |
| Cmin,ss, ng/ml | 109 (98.5) | 201 (90.9) | 1,150 (65.9) | 2,100 (247) | 6,630 (118) | 2,340 (110) | 6,920 (93.9) |
| Clearance, ml/min | 865 (79.0) | 806 (79.1) | 372 (37.1) | 319 (132) | 226 (83.3) | 181 (82.7) | 116 (78.1) |
| Median (min, max) | |||||||
| tmax,ss, h | 2.0 (1.0, 4.0) | 4.0 (1.0, 6.0) | 2.5 (1.0, 4.0) | 4.0 (2.0, 4.0) | 4.1 (1.0, 6.3) | 4.0 (1.0, 4.0) | 4.0 (0.5, 4.0) |
| t1/2,ss, h | 3.7 (2.6, 7.1) | 3.8 (2.2, 6.2) | 5.3 (3.7, 6.3) | 4.5 (2.5, 6.0) | 4.8 (4.2, 5.9) | 5.9 (3.3, 8.0) | 6.0 (5.6, 6.3) |
AUCτ,ss, area under the plasma concentration-time curve at the steady state; Cmax,ss, maximum plasma concentration at the steady state; Cmin,ss, minimum plasma concentration at the steady state; CV, coefficient of variance; tmax,ss, time to maximum plasma concentration at the steady state; t1/2,ss, plasma half-life at the steady state; τ, dosing interval (8 h).
Some values include data from one treatment-naive patient.
For patients with cirrhosis, plasma exposure of deleobuvir at the steady state was approximately 2-fold higher and clearance approximately 50% lower than in patients without cirrhosis at the same dose (400 mg q8h) (Table 7).
Pharmacokinetic/pharmacodynamic relationship.
To examine the relationship between deleobuvir plasma exposure and antiviral effect, PK parameters were plotted against HCV RNA reductions for all dose groups. The drop in the HCV RNA level at 24 h was loosely correlated (r2 = 0.55) with the deleobuvir trough plasma concentration at the steady state (Cmin,ss) (Fig. 3A). The inhibitory quotient (the ratio between the individual deleobuvir Cmin,ss values and phenotypically derived deleobuvir EC50 values for the corresponding baseline viral isolate), which is a widely used pharmacological parameter of antiviral activity (24), had an improved correlation (r2 = 0.75) (Fig. 3B).
Fig 3.
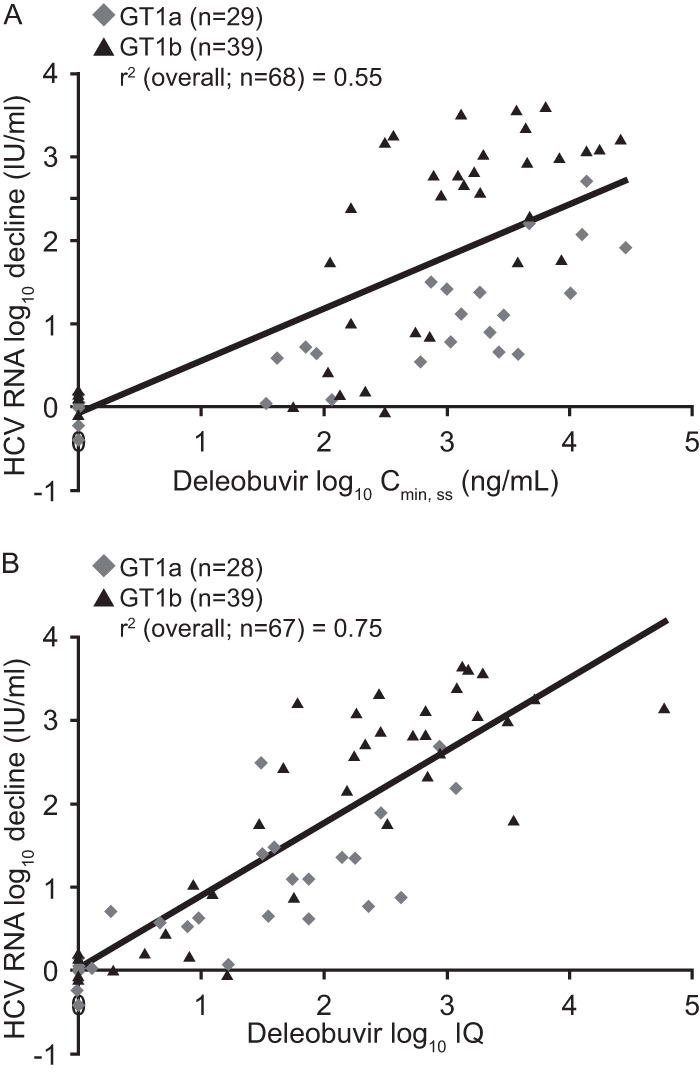
Initial HCV RNA decline at 24 h versus deleobuvir log10 Cmin,ss (A) and deleobuvir log10 inhibitory quotient (IQ) (B).
DISCUSSION
Deleobuvir, a nonnucleoside inhibitor of HCV replication, elicited a steep, dose-related decline in plasma HCV RNA levels in HCV-infected patients with or without cirrhosis. Deleobuvir was more potent against GT1b than against GT1a in terms of EC50 values and reductions in HCV RNA levels. Thus, selective drug pressure was lower against GT1a virus, which may have reduced the emergence of variants in the NS5B thumb-pocket 1 binding site in this subtype. Differences between GT1 subtypes in their sensitivity to NS5B thumb-pocket 1 inhibitors have been described previously (25). They are in part due to the subtype-specific polymorphism at amino acid 499—predominantly valine in GT1b and alanine in GT1a. Differences between GT1a and GT1b in antiviral efficacy and resistance have been observed for many HCV antiviral drugs (26–28).
Resistance-associated variants (RAVs) emerged less commonly during monotherapy with deleobuvir than had been previously reported for nonnucleoside analogs targeting other sites on the NS5B polymerase (16, 29). Half of the RAVs were at thumb-pocket 1 position 495; the large decreases in susceptibility to deleobuvir conferred by substitutions at this position are consistent with in vitro observations from studies of other nonnucleoside HCV NS5B thumb-pocket 1 inhibitors (16) and deleobuvir. The marked reduction in levels of P495L variants during follow-up without selective drug pressure and the low frequency of RAVs at position 496 (4% of clones in a single patient) suggest a lower fitness for P496 variants than for the wild type. This finding, coupled with the low fold changes in sensitivity associated with several substitutions that confer resistance to other NS5B polymerase inhibitors, suggests that deleobuvir may have an enhanced barrier to resistance compared with other members of this class. Importantly, RAVs that emerged in this trial were different from those reported with NS3/4A PIs (30, 31), reflecting their nonoverlapping resistance profiles.
PK assessments demonstrated supraproportional plasma exposure and progressive reductions in clearance with increasing dose of deleobuvir. At deleobuvir doses of ≥400 mg, the median reduction in HCV RNA levels appeared to reach a maximum in GT1b-infected patients but continued to gradually increase with dose in GT1a-infected patients. One explanation is that the reduction in viral load reached the maximum attainable in patients infected with GT1b, which is more sensitive to the drug, but higher doses would have been needed to reach this plateau for GT1a infection. Comparisons between groups should be made with caution because of the large variability in drug exposure levels, the small sample sizes, and, possibly, the differences between groups in baseline characteristics. Deleobuvir exposure was higher in patients with cirrhosis than in those without cirrhosis and, in both cases, higher than in healthy volunteers, in whom plasma exposure to deleobuvir is dose proportional (unpublished findings). These observations suggest a saturation of physiological clearance mechanisms at higher doses and reduced clearance of the drug in the presence of hepatic impairment.
Deleobuvir was generally well tolerated, and rates of discontinuation due to AEs were low. The most common types of event (mild to moderate GI, nervous system, and skin/subcutaneous reactions) have also been reported with PIs (7, 10, 12, 20, 30, 32, 33) and with other NS5B polymerase inhibitors under development (alone or in combination regimens) (34–37). Baseline and follow-up electrocardiographs revealed no clinically relevant findings.
The potency and resistance profile of deleobuvir in this study support its combination with other DAAs, such as an NS3/4A PI, as part of an IFN-free regimen. This could increase efficacy against HCV by inhibition of multiple steps in the viral replication cycle, suppress the emergence of resistant variants, and facilitate HCV treatment by all-oral regimens without the inconvenience and adverse effects of PegIFN.
IFN-free regimens combining deleobuvir with faldaprevir, with or without RBV, have shown promising results in TN patients in a phase 1b study (38) and in a phase 2b study that included patients with compensated cirrhosis (37). Despite the short terminal elimination half-lives in the present study, the latter trial indicated that twice-daily dosing of deleobuvir is effective when combined with faldaprevir and RBV (37).
Supplementary Material
ACKNOWLEDGMENTS
We thank Mireille Cartier for phylogenetic analyses and Genevieve Laflamme and Joe Scherer for help in NS5B phenotyping and supporting statistical analysis.
Medical writing assistance, supported financially by Boehringer Ingelheim, was provided by Richard Murphy and Catherine Elliott of Adelphi Communications Ltd. during the preparation of the manuscript.
Footnotes
Published ahead of print 15 July 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.00565-13.
REFERENCES
- 1.Ghany MG, Nelson DR, Strader DB, Thomas DL, Seeff LB. 2011. An update on treatment of genotype 1 chronic hepatitis C virus infection: 2011 practice guideline by the American Association for the Study of Liver Diseases. Hepatology 54:1433–1444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.European Association for the Study of the Liver 2011. EASL clinical practice guidelines: management of hepatitis C virus infection. J. Hepatol. 55:245–264 [DOI] [PubMed] [Google Scholar]
- 3.European Medicines Agency 2013. INCIVO™ (telaprevir). http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002313/WC500115529.pdf Accessed 21 June 2013
- 4.Vertex Pharmaceuticals Incorporated 2013. INCIVEK™ (telaprevir). Highlights of prescribing information. http://pi.vrtx.com/files/uspi_telaprevir.pdf Accessed 21 June 2013
- 5.Merck & Co 2013. VICTRELIS™ (boceprevir). Highlights of prescribing information. http://www.merck.com/product/usa/pi_circulars/v/victrelis/victrelis_pi.pdf Accessed 21 June 2013
- 6.European Medicines Agency 2013. Victrelis (boceprevir). http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/002332/human_med_001464.jsp&mid=WC0b01ac058001d124&jsenabled=true Accessed 21 June 2013
- 7.Bacon BR, Gordon SC, Lawitz E, Marcellin P, Vierling JM, Zeuzem S, Poordad F, Goodman ZD, Sings HL, Boparai N, Burroughs M, Brass CA, Albrecht JK, Esteban R. 2011. Boceprevir for previously treated chronic HCV genotype 1 infection. N. Engl. J. Med. 364:1207–1217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hézode C, Forestier N, Dusheiko G, Ferenci P, Pol S, Goeser T, Bronowicki JP, Bourliere M, Gharakhanian S, Bengtsson L, McNair L, George S, Kieffer T, Kwong A, Kauffman RS, Alam J, Pawlotsky JM, Zeuzem S. 2009. Telaprevir and peginterferon with or without ribavirin for chronic HCV infection. N. Engl. J. Med. 360:1839–1850 [DOI] [PubMed] [Google Scholar]
- 9.Jacobson IM, McHutchison JG, Dusheiko G, Di Bisceglie AM, Reddy KR, Bzowej NH, Marcellin P, Muir AJ, Ferenci P, Flisiak R, George J, Rizzetto M, Shouval D, Sola R, Terg RA, Yoshida EM, Adda N, Bengtsson L, Sankoh AJ, Kieffer TL, George S, Kauffman RS, Zeuzem S. 2011. Telaprevir for previously untreated chronic hepatitis C virus infection. N. Engl. J. Med. 364:2405–2416 [DOI] [PubMed] [Google Scholar]
- 10.Kwo PY, Lawitz EJ, McCone J, Schiff ER, Vierling JM, Pound D, Davis MN, Galati JS, Gordon SC, Ravendhran N, Rossaro L, Anderson FH, Jacobson IM, Rubin R, Koury K, Pedicone LD, Brass CA, Chaudhri E, Albrecht JK. 2010. Efficacy of boceprevir, an NS3 protease inhibitor, in combination with peginterferon alfa-2b and ribavirin in treatment-naive patients with genotype 1 hepatitis C infection (SPRINT-1): an open-label, randomised, multicentre phase 2 trial. Lancet 376:705–716 [DOI] [PubMed] [Google Scholar]
- 11.McHutchison JG, Everson GT, Gordon SC, Jacobson IM, Sulkowski M, Kauffman R, McNair L, Alam J, Muir AJ. 2009. Telaprevir with peginterferon and ribavirin for chronic HCV genotype 1 infection. N. Engl. J. Med. 360:1827–1838 [DOI] [PubMed] [Google Scholar]
- 12.Poordad F, McCone J, Jr, Bacon BR, Bruno S, Manns MP, Sulkowski MS, Jacobson IM, Reddy KR, Goodman ZD, Boparai N, DiNubile MJ, Sniukiene V, Brass CA, Albrecht JK, Bronowicki JP. 2011. Boceprevir for untreated chronic HCV genotype 1 infection. N. Engl. J. Med. 364:1195–1206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zeuzem S, Andreone P, Pol S, Lawitz E, Diago M, Roberts S, Focaccia R, Younossi Z, Foster GR, Horban A, Ferenci P, Nevens F, Mullhaupt B, Pockros P, Terg R, Shouval D, van Hoek B, Weiland O, Van Heeswijk R, De Meyer S, Luo D, Boogaerts G, Polo R, Picchio G, Beumont M, REALIZE Study Team 2011. Telaprevir for retreatment of HCV infection. N. Engl. J. Med. 364:2417–2428 [DOI] [PubMed] [Google Scholar]
- 14.Sulkowski MS, Cooper C, Hunyady B, Jia J, Ogurtsov P, Peck-Radosavljevic M, Shiffman ML, Yurdaydin C, Dalgard O. 2011. Management of adverse effects of Peg-IFN and ribavirin therapy for hepatitis C. Nat. Rev. Gastroenterol. Hepatol. 8:212–223 [DOI] [PubMed] [Google Scholar]
- 15.Sarrazin C, Zeuzem S. 2010. Resistance to direct antiviral agents in patients with hepatitis C virus infection. Gastroenterology 138:447–462 [DOI] [PubMed] [Google Scholar]
- 16.Kukolj G, McGibbon GA, McKercher G, Marquis M, Lefebvre S, Thauvette L, Gauthier J, Goulet S, Poupart MA, Beaulieu PL. 2005. Binding site characterization and resistance to a class of non-nucleoside inhibitors of the hepatitis C virus NS5B polymerase. J. Biol. Chem. 280:39260–39267 [DOI] [PubMed] [Google Scholar]
- 17.Gane EJ, Roberts SK, Stedman CA, Angus PW, Ritchie B, Elston R, Ipe D, Morcos PN, Baher L, Najera I, Chu T, Lopatin U, Berrey MM, Bradford W, Laughlin M, Shulman NS, Smith PF. 2010. Oral combination therapy with a nucleoside polymerase inhibitor (RG7128) and danoprevir for chronic hepatitis C genotype 1 infection (INFORM-1): a randomised, double-blind, placebo-controlled, dose-escalation trial. Lancet 376:1467–1475 [DOI] [PubMed] [Google Scholar]
- 18.Beaulieu PL, Coulombe R, Duan J, Fazal G, Godbout C, Hucke O, Jakalian A, Joly MA, Lepage O, Llinas-Brunet M, Naud J, Poirier M, Rioux N, Thavonekham B, Kukolj G, Stammers TA. 2013. Structure-based design of novel HCV NS5B thumb pocket 2 allosteric inhibitors with submicromolar gt1 replicon potency: discovery of a quinazolinone chemotype. Bioorg. Med. Chem. Lett. 23:4132–4140 [DOI] [PubMed] [Google Scholar]
- 19.Beaulieu PL, Anderson PC, Brochu C, Bos M, Cordingley M, Duan J, Garneau M, Lagrace L, Marquis M, McKercher G, Poupart B, Rancourt J, Stammers YS, Tsantrizos YS, Kukolj G. 2012. Preclinical characterization of the hepatitis C virus NS5B polymerase non-nucleoside inhibitor BI 207127. J. Hepatol. 56(Suppl 2):S321 [Google Scholar]
- 20.Manns MP, Bourliere M, Benhamou Y, Pol S, Bonacini M, Trepo C, Wright D, Berg T, Calleja JL, White PW, Stern JO, Steinmann G, Yong CL, Kukolj G, Scherer J, Boecher WO. 2011. Potency, safety, and pharmacokinetics of the NS3/4A protease inhibitor BI201335 in patients with chronic HCV genotype-1 infection. J. Hepatol. 54:1114–1122 [DOI] [PubMed] [Google Scholar]
- 21.Bruno R, Cima S, Maiocchi L, Sacchi P. 2011. Forthcoming challenges in the management of direct-acting antiviral agents (DAAs) for hepatitis C. Dig. Liver Dis. 43:337–344 [DOI] [PubMed] [Google Scholar]
- 22.Young KC, Lindsay KL, Lee KJ, Liu WC, He JW, Milstein SL, Lai MM. 2003. Identification of a ribavirin-resistant NS5B mutation of hepatitis C virus during ribavirin monotherapy. Hepatology 38:869–878 [DOI] [PubMed] [Google Scholar]
- 23.Delang L, Froeyen M, Herdewijn P, Neyts J. 2012. Identification of a novel resistance mutation for benzimidazole inhibitors of the HCV RNA-dependent RNA polymerase. Antiviral. Res. 93:30–38 [DOI] [PubMed] [Google Scholar]
- 24.Becker S, Fisher A, Flexner C, Gerber JG, Haubrich R, Kashuba AD, Luber AD, Piscitelli SC. 2001. Pharmacokinetic parameters of protease inhibitors and the Cmin/IC50 ratio: call for consensus. J. Acquir. Immune Defic. Syndr. 27:210–211 [DOI] [PubMed] [Google Scholar]
- 25.Beaulieu PL. 2009. Recent advances in the development of NS5B polymerase inhibitors for the treatment of hepatitis C virus infection. Expert Opin. Ther. Pat. 19:145–164 [DOI] [PubMed] [Google Scholar]
- 26.Chevaliez S, Bouvier-Alias M, Brillet R, Pawlotsky JM. 2009. Hepatitis C virus (HCV) genotype 1 subtype identification in new HCV drug development and future clinical practice. PLoS One 4:e8209. 10.1371/journal.pone.0008209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McCown MF, Rajyaguru S, Kular S, Cammack N, Najera I. 2009. GT-1a or GT-1b subtype-specific resistance profiles for hepatitis C virus inhibitors telaprevir and HCV-796. Antimicrob. Agents Chemother. 53:2129–2132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pawlotsky JM. 2009. Therapeutic implications of hepatitis C virus resistance to antiviral drugs. Therap. Adv. Gastroenterol. 2:205–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Powdrill MH, Bernatchez JA, Gotte M. 2010. Inhibitors of the hepatitis C virus RNA-dependent RNA polymerase NS5B. Viruses 2:2169–2195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lagacé L, White PW, Bousquet C, Dansereau N, Do F, Llinas-Brunet M, Marquis M, Massariol MJ, Maurice R, Spickler C, Thibeault D, Triki I, Zhao S, Kukolj G. 2012. In vitro resistance profile of the hepatitis C virus NS3 protease inhibitor BI 201335. Antimicrob. Agents Chemother. 56:569–572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Susser S, Vermehren J, Forestier N, Welker MW, Grigorian N, Fuller C, Perner D, Zeuzem S, Sarrazin C. 2011. Analysis of long-term persistence of resistance mutations within the hepatitis C virus NS3 protease after treatment with telaprevir or boceprevir. J. Clin. Virol. 52:321–327 [DOI] [PubMed] [Google Scholar]
- 32.Zeuzem S, Buggisch P, Agarwal K, Marcellin P, Sereni D, Klinker H, Moreno C, Zarski JP, Horsmans Y, Mo H, Arterburn S, Knox S, Oldach D, McHutchison JG, Manns MP, Foster GR. 2012. The protease inhibitor, GS-9256, and non-nucleoside polymerase inhibitor tegobuvir alone, with ribavirin, or pegylated interferon plus ribavirin in hepatitis C. Hepatology 55:749–758 [DOI] [PubMed] [Google Scholar]
- 33.Wedemeyer H, Jensen D, Herring R, Jr, Ferenci P, Ma M-M, Zeuzem S, Rodriguez-Torres M, Bzowej N, Pockros P, Vierling J, Ipe D, Munson ML, Chen Y-C, Najera I, Thommes J, PROPEL Investigators 2013. PROPEL: a randomized trial of mericitabine plus peginterferon alpha-2a/ribavirin therapy in treatment-naive HCV genotype 1/4 patients. Hepatology 58:524–537 [DOI] [PubMed] [Google Scholar]
- 34.Cooper C, Lawitz EJ, Ghali P, Rodriguez-Torres M, Anderson FH, Lee SS, Bedard J, Chauret N, Thibert R, Boivin I, Nicolas O, Proulx L. 2009. Evaluation of VCH-759 monotherapy in hepatitis C infection. J. Hepatol. 51:39–46 [DOI] [PubMed] [Google Scholar]
- 35.Poordad F, Lawitz E, Kowdley KV, Cohen DE, Podsadecki T, Siggelkow S, Heckaman M, Larsen L, Menon R, Koev G, Tripathi R, Pilot-Matias T, Bernstein B. 2013. Exploratory study of oral combination antiviral therapy for hepatitis C. N. Engl. J. Med. 368:45–53 [DOI] [PubMed] [Google Scholar]
- 36.Rodriguez-Torres M, Lawitz E, Conway B, Kaita K, Sheikh AM, Ghalib R, Adrover R, Cooper C, Silva M, Rosario M, Bourgault B, Proulx L, McHutchison J. 2010. Safety and antiviral activity of the HCV non-nucleoside polymerase inhibitor VX-222 in treatment-naive genotype 1 HCV-infected patients. J. Hepatol. 52:14S [Google Scholar]
- 37.Zeuzem S, Soriano V, Asselah T, Bronowicki J-P, Lohse AW, Müllhaupt B, Schuchmann M, Bourlière M, Buti M, Roberts SK, Gane EJ, Stern JO, Vinisko R, Kukolj G, Gallivan J-P, Böcher W-O, Mensa FJ. 2013. Faldaprevir and deleobuvir for HCV genotype 1 infection. N. Engl. J. Med. 369:630–639 [DOI] [PubMed] [Google Scholar]
- 38.Zeuzem S, Asselah T, Angus P, Zarski JP, Larrey D, Mullhaupt B, Gane E, Schuchmann M, Lohse A, Pol S, Bronowicki JP, Roberts S, Arasteh K, Zoulim F, Heim M, Stern JO, Kukolj G, Nehmiz G, Haefner C, Boecher WO. 2011. Efficacy of the protease inhibitor BI 201335, polymerase inhibitor BI 207127, and ribavirin in patients with chronic HCV infection. Gastroenterology 141:2047–2055 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.