

Abstract
GSK2336805 is an orally bioavailable hepatitis C virus (HCV) inhibitor working through an NS5A-mediated mechanism. This first-time-in-human study was conducted to assess the safety, tolerability, pharmacokinetics, metabolism, and efficacy of GSK2336805 in healthy subjects and subjects infected with HCV genotype 1. We performed a three-part, randomized, double-blind, placebo-controlled study in 46 healthy subjects and 23 HCV-infected subjects. After an overnight fast, healthy subjects received GSK2336805 as 10 mg, 30 mg, 30 mg plus food, and 60 mg in a single dose and 10 mg (7 days), 30 mg (7 days), and 75 mg (14 days) in a once-daily multiple dose. Subjects with HCV received GSK2336805 as a 1- to 120-mg single dose. In subjects with HCV, reductions in HCV RNA were observed within 4 h and a single dose of GSK2336805 of ≥10 mg resulted in a statistically significant ≥2-log reduction in HCV RNA compared with placebo at 24 h postdose. GSK2336805 was readily absorbed in all subjects, and the half-life (t1/2) was suitable for once-daily dosing. Administration of GSK2336805 with food had no effect on plasma GSK2336805 exposure; however, absorption was delayed, with a median tmax (time to maximum concentration of drug in serum) of 4.5 versus 2.0 h. Twenty subjects who received GSK2336805 experienced mild to moderate adverse events; none were serious. GSK2336805 was well tolerated and exhibited rapid, significant antiviral activity after a single dose in HCV-infected subjects. These results support the conduct of further studies evaluating GSK2336805 administered once daily for longer durations in combination with peginterferon, ribavirin, and other direct-acting antivirals. (This study has been registered at ClinicalTrials.gov under registration no. NCT01277692.)
INTRODUCTION
Hepatitis C virus (HCV) is the leading cause of cirrhosis, liver failure, and primary hepatocellular carcinoma and the primary indication for liver transplantation (1). Of the six major HCV genotypes, genotype 1 (GT-1) is the most prevalent and is associated with the highest rate of treatment failure (2–4).
Administration of two recently approved HCV nonstructural protein 3 (NS3) serine protease inhibitors, telaprevir and boceprevir, with the standard treatment of peginterferon and ribavirin led to sustained viral response rates of 75% and 68%, respectively (5–8). However, increased side effects, especially skin rash and anemia, are still problematic (9, 10). Therefore, antiviral agents with novel modes of action are necessary for an all-oral combination therapy to further improve sustained viral response rates and reduce side effects (11).
NS5A is essential for HCV replication, and NS5A inhibitors have shown potent anti-HCV activity relative to interferon-ribavirin regimens in clinical trials (12–15). GSK2336805 is an orally bioavailable NS5A inhibitor with selective activity against GT-1a and GT-1b subtypes in HCV replicon systems (50% effective concentration [EC50] for GT-1a, 58.5 pM; for GT-1b, 7.4 pM) in vitro (J. Walker, submitted for publication).
The current first-time-in-human study was conducted to evaluate safety, tolerability, pharmacokinetics, and metabolism of GSK2336805 in healthy and HCV-infected subjects and antiviral activity of GSK2336805 in HCV-infected subjects. The design consisted of a randomized, double-blind, placebo-controlled fusion of single and repeat doses of GSK2336805 monotherapy administered sequentially and in parallel. The study also explored the effect of a moderate-fat/caloric meal on single-dose pharmacokinetic endpoints in healthy subjects.
MATERIALS AND METHODS
Subjects.
The study population comprised healthy subjects and subjects with chronic HCV GT-1 infection. Males and females without childbearing potential, aged 18 to 65 years, with mean body mass indices of 18 to 35 kg/m2 for healthy subjects and 18.5 to 35.0 kg/m2 for HCV-infected subjects, were eligible. Subjects with chronic HCV (for ≥6 months) were eligible if they were treatment naive and noncirrhotic with HCV RNA levels of >100,000 IU/ml. Subjects were excluded if infected with human immunodeficiency virus, hepatitis B virus, or HCV (healthy subjects only). Healthy subjects with certain laboratory values less than the maximum upper limits of normal were eligible: aspartate aminotransferase (AST), <45 U/liter; alanine aminotransferase (ALT), <50 U/liter; alkaline phosphatase, <188 U/liter; bilirubin, <1.3 mg/dl; and creatinine, <1.46 mg/dl. HCV-infected subjects with an ALT level of ≤3× the upper limit of normal (<150 U/liter) were eligible.
All subjects provided written informed consent prior to screening. The study was approved by local ethics review committees and conducted in accordance with the guidelines of the International Conference on Harmonisation (ICH) of Technical Requirements for Registration of Pharmaceuticals for Human Use Good Clinical Practice.
Study design.
This randomized, double-blind, three-part, dose-escalation study (protocol no. HAI114885, ClinicalTrials.gov no. NCT01277692) was conducted from November 2010 to May 2011 at three U.S. sites. The primary endpoint was to evaluate the safety, tolerability, pharmacokinetics, and metabolism following single and repeat oral administration of GSK2336805 in healthy subjects and single administration in HCV-infected subjects and subsequent antiviral activity. The secondary endpoint was to evaluate food effect on a single dose of GSK2336805 in healthy subjects. Subjects received a treatment allocation number and were assigned according to the randomization schedule provided by GlaxoSmithKline. The study team, subjects, and site personnel (except the pharmacokineticist, virologist, pharmacist, and designer) were blinded to subject randomization throughout the study. Doses were escalated following review of safety, pharmacokinetic (all subjects), and antiviral activity (HCV-infected subjects only) data in earlier study cohorts. Bayesian prediction methods estimated exposure at the next dose level and the maximum dose to be administered; predefined stopping criteria were utilized. Subjects received a single morning dose of GSK2336805 during each study period following an overnight fast of at least 10 h and for 4 h after dosing on days when serial pharmacokinetic samples were obtained. In part 1, two alternating cohorts of 16 total healthy subjects received GSK2336805 or placebo. Cohort A1 included four subjects, and cohort A2 included the four subjects from cohort A1 plus an additional four subjects who received GSK2336805 or placebo, while cohort B included 8 subjects who received GSK2336805 or placebo with or without food. Subjects in the food effect cohort of part 1 were fed a moderate-fat meal (approximately 30% fat/669 cal) 30 min prior to dosing. Part 2 consisted of 30 healthy subjects who received repeat doses up to 14 days of GSK2336805 or placebo, and part 3 consisted of 23 HCV-infected subjects who received a single dose of GSK2336805 or placebo. Study design and dose information are presented in Fig. 1.
Fig 1.

Schematic representation of study design.
Safety analysis.
Adverse events (AEs) were reported from first dose to follow-up and coded using the Medical Dictionary for Regulatory Activities (MedDRA; version 14.0). Vital signs, clinical laboratory testing, and 12-lead electrocardiograms were assessed at screening, selected time points during the study, and follow-up. All subjects were evaluated with a Holter monitor for 24 h at screening. Adverse events and clinical laboratory toxicities were graded according to the protocol-defined toxicity criteria based on the 2009 DAIDS Therapeutic Research Program's Table for Grading Severity of Adult and Pediatric Adverse Experiences (16).
Pharmacokinetic analysis.
In parts 1 and 3, serial blood samples for GSK2336805 pharmacokinetic analysis were collected predose and at 0.25, 0.5, 0.75, 1, 1.5, 2, 3, 4, 6, 8, 10, 12, 16, 24, 36, and 48 h postdose. In part 2, serial blood samples were collected predose and at 0.5, 1, 1.5, 2, 2.5, 3, 4, 6, 8, 10, 12, 16, and 24 h postdose on days 1, 7, and 14 (75-mg cohort only). Single predose samples were taken on the mornings of days 3 through 6 from all cohorts and days 9 through 13 from the 75-mg cohort. GSK2336805 was extracted from 75 μl of plasma by protein precipitation with acetonitrile containing an isotopically labeled internal standard, [13C6]GSK2336805, and analyzed by high-performance liquid chromatography with tandem mass spectrometry (HPLC-MS/MS) using a TurboIonSpray (Applied Biosystems/MDS Sciex, Canada) interface and multiple reaction monitoring (analyte m/z 797 to 539, internal standard m/z 803 to 545). The lower limit of quantification for GSK2336805 was 0.1 ng/ml, with a higher limit of quantification of 100 ng/ml. Samples above the higher limit of quantification were diluted 2-, 5-, 10-, or 20-fold. The maximum intra- and interday run precision values observed were 8.8% and 8.5%, respectively.
Pharmacokinetic parameters determined from the GSK2336805 plasma concentration-actual time data for subjects in each active treatment group included area under the concentration-time curve from time zero to 24 h postdose (AUC0–24), AUC from time zero extrapolated to infinity (AUC0–∞), maximum observed concentration (Cmax), time of occurrence of Cmax (tmax), concentration at 24 h after dose (C24), apparent oral clearance (CL/F), apparent oral volume of distribution (V/F), and terminal-phase half-life (t1/2).
Metabolite identification.
Time-proportional pooled plasma extracts and urine were analyzed by HPLC-MS using an LTQ-Orbitrap Hybrid mass spectrometer (ThermoScientific, USA) equipped with an HP1100 HPLC (Agilent, USA) to characterize and estimate the amount of metabolites in plasma and urine following administration of GSK2336805. Definitive structures were determined using a combination of 1H and 13C nuclear magnetic resonance (NMR) correlation experiments (700 MHz; Avance II TCI probe) and nuclear Overhauser effect experiments on isolates.
Clinical virology assessments.
Hepatitis C virus RNA viral load was measured using a Cobas TaqMan HCV test (Roche, United Kingdom) at screening and predose and from samples collected within the first 24 h and up to 336 h following single-dose administration of GSK2336805 (or until viral load returned to baseline levels). Blood samples for genotypic and phenotypic analysis were taken predose and 12, 24, 48, 72, and 96 h postdose, continuing until viral load returned to baseline levels. Viral genotype was determined at baseline and nadir (maximum change). Phenotypic data were evaluated in samples from HCV-infected subjects who had mutations on NS5A known to cause resistance to GSK2336805. Genotypic and phenotypic analyses were performed by Monogram BioSciences, Inc., USA.
Statistical analysis.
Sample size was based on feasibility. No formal calculation was performed to determine sample size. Food effect on GSK2336805 pharmacokinetics was assessed in part 1 using analysis of variance (ANOVA). Dose proportionality in GSK2336805 AUC0–∞, AUC0–24, C24, and Cmax was evaluated in all parts by a power model analysis and ANOVA using loge-transformed pharmacokinetic data for each dose. In part 2, accumulation was evaluated by comparing AUC0–24, Cmax, and C24 on day 7 and day 1 for each dose and by comparing day 14 and day 1 for the 75-mg dose.
Hepatitis C virus RNA data were summarized using log10-tranformed data. Change from baseline of plasma HCV RNA measured at 24 h and nadir was compared between each active treatment and placebo using analysis of covariance. A mixed-effects linear model was fitted to plasma HCV RNA change from baseline during 24 h by treatment, with baseline plasma HCV RNA, treatment, and hour fitted as fixed effects and subject fitted as a random effect. Relationships between pharmacokinetic parameters (AUC0–24, Cmax, and C24) and pharmacodynamic measures (reduction in log10 IU/ml plasma HCV RNA to 24 h from baseline) were explored with linear and maximum effect (Emax) models for GT-1a- and GT-1b-infected subjects combined. Model selection was based on the lowest Akaike information criterion value.
RESULTS
Demographics and subject disposition.
All healthy subjects completed the study in part 1 (n = 16) and part 2 (n = 30). In part 3, 20 of the 23 enrolled HCV-infected subjects completed and three were lost at follow-up (Fig. 1; Table 1). Of the three, one subject (10 mg) completed all assessments through 48 h postdosing and two subjects (30 mg, placebo) completed all assessments up to day 8.
Table 1.
Subject disposition and baseline characteristicsa
| Study part and patient group (n) | No. of subjects completing trial (%) | Mean age, yr (SD) | Sex, no. (%) |
Mean BMI, kg/m2 (SD) | Race, n (%) |
Mean baseline viral load, log10 IU/ml (SD) | ||
|---|---|---|---|---|---|---|---|---|
| Female | Male | Caucasian | Non-Caucasian | |||||
| Part 1, SD, healthy subjects | ||||||||
| 10-mg SD (3) | 3 (100) | 41.0 (20.0) | 1 (33) | 2 (67) | 25.8 (2.6) | 3 (100) | 0 | |
| 30-mg SD (6) | 6 (100) | 42.2 (10.2) | 2 (33) | 4 (67) | 26.6 (3.1) | 5 (83) | 1 (17) | |
| 60-mg SD (6) | 6 (100) | 40.8 (16.6) | 2 (33) | 4 (67) | 26.3 (1.8) | 6 (100) | 0 | |
| 30-mg SD + food (6) | 6 (100) | 42.2 (10.2) | 2 (33) | 4 (67) | 26.6 (3.1) | 5 (83) | 1 (17) | |
| Total SD active (12) | 12 (100) | 41.5 (13.2) | 4 (33) | 8 (67) | 26.5 (2.4) | 11 (92) | 1 (8) | |
| Total SD placebo (4) | 4 (100) | 30.3 (14.9) | 1 (25) | 3 (75) | 23.8 (3.6) | 4 (100) | 0 | |
| Part 2, RD, healthy subjects | ||||||||
| 10-mg RD (8) | 8 (100) | 40.8 (15.0) | 3 (38) | 5 (63) | 27.5 (2.2) | 7 (88) | 1 (13) | |
| 30-mg RD (8) | 8 (100) | 40.6 (13.3) | 1 (13) | 7 (88) | 26.8 (2.9) | 7 (88) | 1 (13) | |
| 75-mg RD (8) | 8 (100) | 34.8 (14.6) | 0 | 8 (100) | 25.1 (3.1) | 8 (100) | 0 | |
| Total RD active (24) | 24 (100) | 38.7 (14.0) | 4 (17) | 20 (83) | 26.5 (2.8) | 22 (92) | 2 (8) | |
| Total RD placebo (6) | 6 (100) | 40.3 (19.0) | 1 (17) | 5 (83) | 25.5 (3.1) | 5 (83) | 1 (17) | |
| Part 3, SD, subjects with HCV | ||||||||
| 1-mg SD (3) | 3 (100) | 54.7 (6.7) | 0 | 3 (100) | 29.0 (5.6) | 3 (100) | 0 | 6.17 (0.22) |
| 10-mg SD (5) | 4 (80) | 46.8 (3.5) | 1 (20) | 4 (80) | 26.1 (3.6) | 5 (100) | 0 | 5.74 (0.42) |
| 30-mg SD (1) | 0 | 56.0 | 0 | 1 (100) | 23.9 | 1 (100) | 0 | 6.71 |
| 60-mg SD (5) | 5 (100) | 52.6 (3.6) | 2 (40) | 3 (60) | 29.9 (6.5) | 5 (100) | 0 | 6.07 (0.67) |
| 120-mg SD (3) | 3 (100) | 41.7 (5.0) | 0 | 3 (100) | 27.6 (5.4) | 2 (67) | 1 (33) | 6.44 (0.46) |
| Total SD active (17) | 15 (88) | 49.5 (6.3) | 3 (18) | 14 (82) | 27.9 (5.0) | 16 (94) | 1 (6) | NA |
| Total SD placebo (6) | 5 (83) | 53.7 (5.7) | 0 | 6 (100) | 25.6 (3.5) | 6 (100) | 0 | 6.11 (0.59) |
Abbreviations: SD, single dose; RD, repeat dose; HCV, hepatitis C virus; BMI, body mass index; NA, not applicable.
Safety.
All AEs were mild or moderate in intensity, except for one severe ALT elevation in part 3 not considered related to study drug; all resolved by the end of the study. No deaths, serious AEs, or withdrawals from the study due to AEs occurred. The most frequently reported AE was headache (n = 6) (Table 2). Liver-related AEs with elevated levels of ALT and AST without elevated levels of bilirubin not considered related to study drug were experienced by four HCV-infected subjects who received GSK2336805 at 1 mg (grade 2) or 10 or 60 mg or placebo (all grade 3). All four subjects had elevated levels of ALT and AST at screening or prior to dosing but were within inclusion criteria for the study (ALT, ≤3× upper limit of normal). There were no apparent dose-dependent trends in AEs and no changes in vital signs, telemetry, or electrocardiogram values.
Table 2.
Summary of most frequently reported adverse eventsa
| Preferred term | No. (%) of events |
|||||
|---|---|---|---|---|---|---|
| Part 1, healthy subjects |
Part 2, healthy subjects |
Part 3, subjects with HCV |
||||
| Total SD active (n = 12) | Total SD placebo (n = 4) | Total RD active (n = 24) | Total RD placebo (n = 6) | Total SD active (n = 17) | Total SD placebo (n = 6) | |
| Adverse event | 1 (8) | 1 (25) | 9 (38) | 1 (17) | 10 (59) | 2 (33) |
| Headache | 1 (8) | 0 | 2 (8) | 0 | 2 (12) | 1 (17) |
| Dizziness | 0 | 0 | 0 | 0 | 3 (18) | 0 |
| ALT increased | 0 | 0 | 0 | 0 | 2 (12) | 1 (17) |
| AST increased | 0 | 0 | 0 | 0 | 2 (12) | 1 (17) |
| Diarrhea | 0 | 1 (25) | 1 (4) | 0 | 0 | 0 |
| Upper RTI | 0 | 0 | 2 (8) | 0 | 0 | 0 |
ALT, alanine aminotransferase; AST, aspartate aminotransferase; RTI, respiratory tract infection; SD, single dose; RD, repeat dose.
Pharmacokinetics.
Following oral administration of single and repeat doses of GSK2336805 to fasted healthy subjects, the median tmax occurred 1.5 to 3 h after dosing and geometric mean t1/2 values ranged from 7 to 10 h (Table 3). Based on the power model assessment, plasma GSK2336805 AUC and Cmax increased in a dose-proportional manner following single-dose administration but increased in a greater-than-dose-proportional manner following repeat dosing. Plasma concentrations reached steady state by day 5 following repeat dose administration. Accumulation ratios (day 7 versus day 1) were estimated to be 0.97 to 1.19 for AUC0–24, 0.83 to 1.20 for Cmax, and 1.13 to 1.41 for C24 across the dose range evaluated. Coadministration of GSK2336805 at 30 mg with a 30% fat/669-kcal meal had no effect on plasma exposure (AUC0–24 and Cmax); however, absorption was delayed—median tmax was 4.5 h with food compared with 2.0 h in the fasted state.
Table 3.
Summary of plasma GSK2336805 pharmacokinetic parameters (geometric mean, 95% confidence interval)c
| Dose, mg (n) | AUC0–24 (ng · h/ml) | Cmax (ng/ml) | tmaxa (h) | C24 (ng/ml) | CL/Fb (liters/h) | V/Fb (liters) | t1/2b (h) |
|---|---|---|---|---|---|---|---|
| Part 1, healthy subjects (single dose) | |||||||
| 10 (3) | 692 (391, 1,224) | 93 (44, 198) | 3.0 (1.0, 3.0) | 7.2 (4.0, 13.0) | 12.9 (7.5, 22.2) | 152 (54, 422) | 8.1 (4.9, 13.4) |
| 30 (6) | 2,423 (855, 6,870) | 266 (78, 908) | 2.0 (1.5, 4.0) | 40.8 (15.0, 111.2) | 9.9 (3.5, 28.0) | 140 (47, 415) | 9.8 (7.2, 13.3) |
| 60 (6) | 5,460 (2,950, 10,104) | 784 (457, 1,345) | 1.8 (1.5, 2.0) | 68.5 (36.1, 130.0) | 9.8 (5.4, 18.1) | 100 (53, 191) | 7.1 (6.0, 8.3) |
| 30 + food (6) | 2,393 (1,300, 4,405) | 271 (155, 476) | 4.5 (1.5, 6.1) | 46.5 (20.0, 108.2) | 9.8 (4.7, 20.3) | 129 (70, 238) | 9.2 (6.5, 13.0) |
| Part 2, healthy subjects (repeat dose) (n = 8) | |||||||
| 10 (day 1) (8) | 501 (295, 854) | 72 (42, 124) | 2.5 (1.5, 4.0) | 5.7 (3.4, 9.5) | |||
| 10 (day 7) (8) | 485 (279, 841) | 59 (32, 112) | 2.0 (1.5, 6.0) | 7.1 (4.5, 11.3) | 20.6 (11.9, 35.8) | 215 (115, 402) | |
| 30 (day 1) (8) | 1,369 (805, 2,330) | 185 (104, 330) | 1.7 (1.2, 3.0) | 15.9 (10.2, 25.0) | |||
| 30 (day 7) (8) | 1,516 (914, 2,516) | 190 (104, 348) | 2.5 (2.0, 4.0) | 17.9 (12.8, 25.0) | 19.8 (11.9, 32.8) | 172 (92, 322) | |
| 75 (day 1) (8) | 5,801 (3,598, 9,352) | 752 (444, 1,272) | 1.5 (1.0, 4.0) | 62.7 (36.3, 108.3) | |||
| 75 (day 7) (8) | 6,917 (3,938, 12,149) | 904 (497, 1,643) | 2.2 (1.5, 4.0) | 88.7 (54.8, 143.4) | 10.8 (6.2, 19.0) | 112 (58, 216) | |
| 75 (day 14) (8) | 6,161 (3,466, 10,950) | 753 (434, 1,308) | 2.7 (1.5, 3.0) | 72.6 (39.3, 134.3) | 12.2 (7.0, 21.6) | 151 (69, 330) | |
| Part 3, subjects with HCV (single dose) | |||||||
| 1 (3) | 14.4 (6, 33) | 2 (1, 3) | 2.0 (1.0, 4.0) | 0.2 (0.04, 1.0) | 60.9 (23.5, 157.6) | 646 (294, 1,420) | 7.4 (4.7, 11.5) |
| 10 (5) | 196 (89, 434) | 27 (13, 55) | 2.0 (1.5, 3.00) | 2.5 (1.0, 6.1) | 44.5 (20.3, 97.4) | 537 (227, 1,269) | 8.4 (7.1, 9.8) |
| 30 (1) | 551 | 65 | 4.0 | 8.1 | 46.5 | 556 | 8.30 |
| 60 (5) | 2,533 (1,751, 3,664) | 408 (286, 582) | 1.5 (1.0, 2.0) | 22.7 (16.5, 31.2) | 21.6 (15.6, 29.7) | 235 (136, 405) | 7.6 (5.8, 9.9) |
| 120 (3) | 5,952 (1,788, 19,808) | 999 (296, 3,376) | 1.5 (1.5, 4.1) | 43.4 (3.0, 659.7) | 18.2 (4.9, 66.8) | 265 (84, 839) | 10.1 (8.5, 12.1) |
Data are presented as median (range).
t1/2, CL/F, and V/F could not be reliably estimated with 24-h pharmacokinetic data on day 1 of part 2.
Abbreviations: AUC0–24, area under the concentration-time curve from time zero to 24 h postdose; AUC0–τ, area under the concentration-time curve over the dosing interval; Cmax, maximum observed concentration; tmax, time of occurrence of Cmax; C24, concentration at 24 h after dose; CL/F, apparent oral clearance; V/F, apparent oral volume of distribution; t1/2, terminal-phase half-life.
Subjects infected with HCV had tmax and t1/2 values similar to those of healthy subjects (Table 3) but had higher CL/Fs, higher V/Fs, and 40 to 75% lower geometric mean AUCs and Cmaxs at the common single doses studied (Table 3). The power model assessment revealed a greater-than-dose-proportional increase in AUC, Cmax, and C24 for GSK2336805 over the dose range of 1 to 120 mg. A subsequent ANOVA pairwise dose proportionality assessment also revealed a greater-than-dose-proportional increase in plasma exposures of GSK2336805 between 1 mg and 10 mg and at doses of ≥60 mg.
Metabolite identification.
GSK2336805 was estimated to account for >95% of total drug-related material in time-proportionally pooled plasma extracts from healthy and HCV-infected subjects (Fig. 2). Minor GSK2336805 metabolites, M10 (formed by oxidation) and M11 (formed by ketal hydrolysis and reduction), were present at levels of <1% following single dosing; estimated levels were higher for M10, <5%, following repeat dosing. Unchanged GSK2336805, M10, and M11 were identified in the urine of healthy subjects following single-dose administration of GSK2336805. No previously uncharacterized metabolites were identified (unpublished data).
Fig 2.

Structural summary and estimated plasma exposure of drug-related material for GSK2336805 and its metabolites identified in pooled plasma extracts in all subjects after administration of GSK2336805.
Antiviral activity.
In part 3, mean HCV RNA change from baseline measured at 24 h after a single dose of GSK2336805 was −0.996 log10 IU/ml for 1 mg, −2.22 log10 IU/ml for 10 mg, −2.18 log10 IU/ml for 30 mg, −2.84 log10 IU/ml for 60 mg, −2.95 log10 IU/ml for 120 mg, and +0.04 log10 IU/ml for placebo. The HCV RNA levels reached nadir between 24 and 48 h for all doses, followed by a return to baseline. A dose-dependent decrease in viral load was observed; the largest two doses showed the largest declines. At 24 h postdose, statistically significant decreases in HCV RNA were observed for all active treatment groups compared with placebo (P < 0.001) (Fig. 3); at 48 h postdose, statistically significant decreases were observed for GSK2336805 at 10 mg, 30 mg, 60 mg, and 120 mg (P ≤ 0.002).
Fig 3.
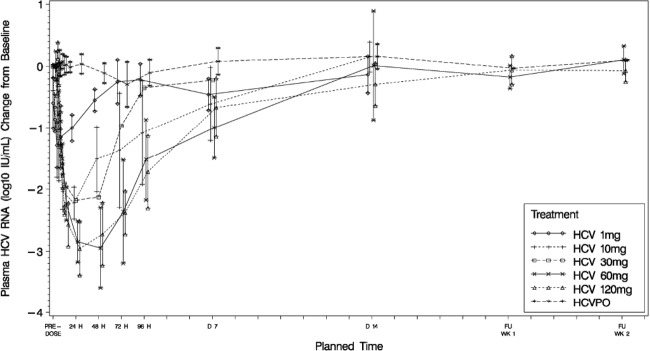
Mean change from baseline in plasma HCV RNA (log10 IU/ml) by treatment and time following single-dose administration of GSK2336805 in HCV-infected subjects (part 3).
Greater antiviral activity was associated with higher GSK2336805 plasma exposure. The relationship between reduction in log10 IU/ml of plasma HCV RNA from baseline to 24 h and AUC0–24 was explored using linear and various Emax models [viral load change from baseline = Emax × AUCγ/(EPK50γ + AUCγ)]. The Emax fixed to 3.2 with estimated γ (Hill factor) provided the lowest Akaike information criterion value; this was considered to be the best model. The estimated value that produced 50% of Emax (EPK50) was 51.86 (standard error [SE], 13.3; P = 0.001) ng · h/ml with an estimated γ of 0.524 (SE, 0.067; P < 0.0001) (Fig. 4). No obvious differences in HCV RNA decrease were observed between subjects with GT-1a and those with GT-1b. However, the numbers of subjects with GT-1b were small (one subject in the 10-mg group and one in the 60-mg group).
Fig 4.
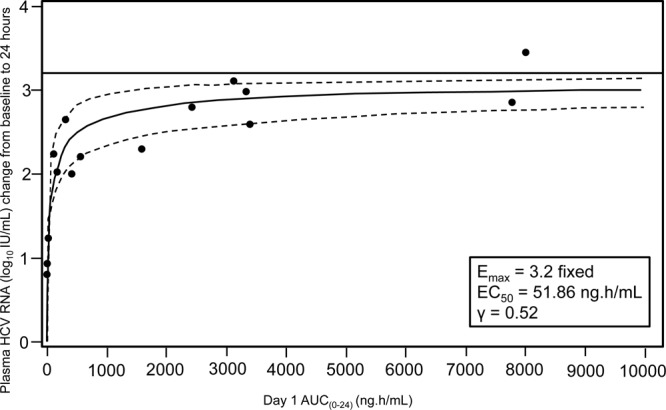
Plot showing the relationship between change in log10 IU/ml of plasma HCV RNA from baseline to 24 h and day 1 AUC0–24. Lines represent the best fitted line (solid) and 95% confidence interval (dashed). All subjects with HCV GT-1a and GT-1b were included.
Antiviral resistance.
Genotypic analysis of samples from HCV-infected subjects showed that NS5A mutations known to impact the in vitro potency of GSK2336805 were present in nadir samples for 13 out of 23 subjects and were not observed in the predose sample (Table 4) (one subject had received placebo). Phenotypic analysis of these samples revealed that 10 of the 13 subjects analyzed had phenotypic resistance, with GSK2336805 potency reduced between 5- and >22-fold from baseline. Ultradeep sequencing of additional time points from these subjects showed that two subjects had a low level of baseline resistance and nine subjects had detectable levels of resistance mutations persisting for at least 7 days after receiving GSK2336805 (see Table S1 in the supplemental material).
Table 4.
Number and percentage of subjects with HCV with GSK2336805 in vitro resistant mutants present after dosing with GSK2336805
| Dose, mg (n) | No. with amino acid change from baseline genotype: |
||||
|---|---|---|---|---|---|
| M28 | Q30 | L/I31 | P32 | Y93 | |
| 1a (3) | 0 | 0 | 0 | 0 | 0 |
| 10 (5) | 2 (40),b M→M/Tc | 2 (40), Q→Q/R | 1 (20), I→I/V; 1 (20), L→L/M | 1 (20), P→P/L | 0 |
| 30 (1) | 1 (100), M→M/T | 0 | 1 (100), L→L/M | 0 | 0 |
| 60 (5) | 3 (60), M→M/T | 2 (40), Q→Q/R; 2 (40), Q→Q/R/H | 1 (20), L→L/M; 1 (20), L→M/V | 1 (20), P→P/L | 1 (20), Y→Y/H |
| 120 (3) | 3 (100), M→M/T | 1 (33), Q→Q/R; 1 (33), Q→Q/R/H | 3 (100), L→L/M | 0 | 0 |
At 1 mg, no mutations were detected in any of the residues known to impact GSK2336805 potency.
n (%) = number of subjects with the amino acid populations listed (% of cohort with the amino acid changes).
Amino acid detected at baseline is listed to the left of the arrow; amino acid(s) detected after exposure to GSK2336805 is listed to the right of the arrow.
DISCUSSION
GSK2336805 exhibited antiviral activity after a single dose, a pharmacokinetic profile consistent with once-daily dosing, and favorable safety and tolerability in this first-time-in-human study. Reductions in HCV RNA were observed within 4 h for all dose groups, and the level of decline and length of time required to return to baseline were dose dependent. The concentration of GSK2336805 24 h postdosing was above the GT-1a protein-adjusted EC90 (PAEC90; 0.35 ng/ml) value for all doses except the 1-mg dose. Dose ranging showed that maximal reductions in HCV RNA were seen at GSK2336805 doses of ≥60 mg where mean C24s were greater than 60× PAEC90.
Mutations in NS5A known to cause in vitro resistance were observed in the nadir samples of 13 of the 23 subjects. Phenotypic resistance was detected in 10 of the 13 nadir samples, confirming that the in vitro resistance patterns translated in vivo. Further genotypic analysis of samples from these subjects showed that two subjects had a low level of baseline resistant mutants that persisted through the study period, suggesting that they were a stable part of the quasispecies. Both subjects had a >2-log10 reduction in viral load, so the presence of the mutations at baseline did not prevent a robust response to GSK2336805.
GSK2336805 was readily absorbed in all subjects and had a relatively low apparent oral clearance compared with liver blood flow. The t1/2 was suitable for once-daily dosing, with plasma concentrations being maintained well above the in vitro-derived, protein-adjusted EC90 for at least 24 h in HCV-infected subjects receiving doses of ≥10 mg. GSK2336805 exposures increased in a slightly greater-than-dose-proportional manner across the dose ranges, and minimal accumulation was noted following repeat dose administration in healthy subjects, consistent with the half-life and 24-hour dosing interval.
Absorption and elimination rate characteristics (tmax, tlag, and t1/2) of GSK2336805 were similar in all subjects. However, HCV-infected subjects tended to have higher CL/F, higher V/F, and 2- to 4-fold-lower GSK2336805 AUC0–∞ and Cmaxs than did healthy subjects. The apparent population differences in GSK2336805 exposures were unexpected and could not be explained by differences in gender, race, or smoking status. Lower plasma exposures in HCV-infected subjects and/or hepatically impaired subjects have recently been reported for NS5A inhibitors daclatasvir and GS-5885 (15, 17, 18) and the HCV protease inhibitor telaprevir (19). For daclatasvir and telaprevir, the lower exposures were attributed to a higher free (unbound) fraction of these agents in the plasma of HCV-infected and hepatically impaired individuals than in the healthy control subjects. Plasma protein binding of GSK2336805 was not measured in the present study, and the underlying mechanism for the population difference remains unknown. Of note, the metabolite characterization did not reveal any substantial differences between healthy and HCV-infected subjects.
Clinical laboratory findings were restricted to elevated ALT and AST. The elevations were not considered by the investigator to be related to GSK2336805, as all four subjects had elevated levels of ALT and AST at screening.
GSK2336805 in both healthy and HCV-infected subjects in our study allowed early evaluation of safety, pharmacokinetics, and antiviral activity of GSK2336805 while exposing a limited number of HCV-infected subjects. Each part of the study defined the dose range where single doses of GSK2336805 produced rapid and significant antiviral activity in treatment-naive HCV GT-1 subjects. These results support progression to phase II studies evaluating GSK2336805 once-a-day dosing for longer durations in combination with peginterferon, ribavirin, and other direct-acting antivirals.
Supplementary Material
ACKNOWLEDGMENTS
This study was funded by GlaxoSmithKline. All authors are employed by GlaxoSmithKline.
We thank the subjects and staff who participated in the study and the investigators from the three research sites: Sandra Willsie, Steven Glass, and William O'Riordan. We thank W. Hardesty and D. Wagner for providing technical expertise and helpful discussions and Mi Xie, Ermias Woldu, and Stephanie Van Horn for their contributions to the UDS of the clinical samples. Justin Cook and Susan Reijntjes provided writing and editorial support to the development of the manuscript and were paid for these services by GlaxoSmithKline.
Footnotes
Published ahead of print 29 July 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.00910-13.
REFERENCES
- 1.Lauer GM, Walker BD. 2001. Hepatitis C virus infection. N. Engl. J. Med. 345:41–52 [DOI] [PubMed] [Google Scholar]
- 2.Sievert W, Altraif I, Razavi HA, Abdo A, Ahmed EA, Alomair A, Amarapurkar D, Chen CH, Dou X, El Khayat H, Elshazly M, Esmat G, Guan R, Han KH, Koike K, Largen A, McCaughan G, Mogawer S, Monis A, Nawaz A, Piratvisuth T, Sanai FM, Sharara AI, Sibbel S, Sood A, Suh DJ, Wallace C, Young K, Negro F. 2011. A systematic review of hepatitis C virus epidemiology in Asia, Australia and Egypt. Liver Int. 31(Suppl 2):61–80 [DOI] [PubMed] [Google Scholar]
- 3.Cornberg M, Razavi HA, Alberti A, Bernasconi E, Buti M, Cooper C, Dalgard O, Dillion JF, Flisiak R, Forns X, Frankova S, Goldis A, Goulis I, Halota W, Hunyady B, Lagging M, Largen A, Makara M, Manolakopoulos S, Marcellin P, Marinho RT, Pol S, Poynard T, Puoti M, Sagalova O, Sibbel S, Simon K, Wallace C, Young K, Yurdaydin C, Zuckerman E, Negro F, Zeuzem S. 2011. A systematic review of hepatitis C virus epidemiology in Europe, Canada and Israel. Liver Int. 31(Suppl 2):30–60 [DOI] [PubMed] [Google Scholar]
- 4.Kershenobich D, Razavi HA, Sánchez-Avila JF, Bessone F, Coelho HS, Dagher L, Gonçales FL, Quiroz JF, Rodriguez-Perez F, Rosado B, Wallace C, Negro F, Silva M. 2011. Trends and projections of hepatitis C virus epidemiology in Latin America. Liver Int. 31(Suppl 2):18–29 [DOI] [PubMed] [Google Scholar]
- 5.Jacobson IM, McHutchison JG, Dusheiko G, Di Bisceglie AM, Reddy KR, Bzowej NH, Marcellin P, Muir AJ, Ferenci P, Flisiak R, George J, Rizzetto M, Shouval D, Sola R, Terg RA, Yoshida EM, Adda N, Bengtsson L, Sankoh AJ, Kieffer TL, George S, Kauffman RS, Zeuzem S, Study Team ADVANCE 2011. Telaprevir for previously untreated chronic hepatitis C virus infection. N. Engl. J. Med. 364:2405–2416 [DOI] [PubMed] [Google Scholar]
- 6.Poordad F, McCone J, Jr, Bacon BR, Bruno S, Manns MP, Sulkowski MS, Jacobson IM, Reddy KR, Goodman ZD, Boparai N, DiNubile MJ, Sniukiene V, Brass CA, Albrecht JK, Bronowicki JP, SPRINT-2 Investigators 2011. Boceprevir for untreated chronic HCV genotype 1 infection. N. Engl. J. Med. 364:1195–1206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.US Food and Drug Administration 2011. Approval of Incibek (telaprevir), a direct acting antiviral drug (DAA) to treat hepatitis C virus (HCV). US Food and Drug Administration, Silver Spring, MD: http://www.fda.gov/ForConsumers/ByAudience/ForPatientAdvocates/ucm256328.htm [Google Scholar]
- 8.US Food and Drug Administration 2011. Approval of Victrelis (boceprevir), a direct acting antiviral drug (DAA) to treat hepatitis C virus (HCV). US Food and Drug Administration, Silver Spring, MD: http://www.fda.gov/ForConsumers/ByAudience/ForPatientAdvocates/ucm255413.htm [Google Scholar]
- 9.Vertex Pharmaceuticals Inc. 2011. Incivek (telaprevir) package insert. Vertex Pharmaceuticals Inc., Cambridge, MA [Google Scholar]
- 10.Merck & Co, Inc. 2011. Victrelis (boceprevir) package insert. Merck & Co, Inc., Whitehouse Station, NJ [Google Scholar]
- 11.De Clercq E. 2012. The race for interferon-free HCV therapies: a snapshot by the spring of 2012. Rev. Med. Virol. 22:392–411 [DOI] [PubMed] [Google Scholar]
- 12.Gao M, Nettles RE, Belema M, Snyder LB, Nguyen VN, Fridell RA, Serrano-Wu MH, Langley DR, Sun JH, O'Boyle DR, II, Lemm JA, Wang C, Knipe JO, Chien C, Colonno RJ, Grasela DM, Meanwell NA, Hamann LG. 2010. Chemical genetics strategy identifies an HCV NS5A inhibitor with a potent clinical effect. Nature 465:96–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nettles RE, Gao M, Bifano M, Chung E, Persson A, Marbury TC, Goldwater R, DeMicco MP, Rodriguez-Torres M, Vutikullird A, Fuentes E, Lawitz E, Lopez-Talavera JC, Grasela DM. 2011. Multiple ascending dose study of BMS-790052, a nonstructural protein 5A replication complex inhibitor, in patients infected with hepatitis C virus genotype 1. Hepatology 54:1956–1965 [DOI] [PubMed] [Google Scholar]
- 14.Pol S, Ghalib RH, Rustgi VK, Martorell C, Everson GT, Tatum HA, Hézode C, Lim JK, Bronowicki JP, Abrams GA, Bräu N, Morris DW, Thuluvath PJ, Reindollar RW, Yin PD, Diva U, Hindes R, McPhee F, Hernandez D, Wind-Rotolo M, Hughes EA, Schnittman S. 2012. Daclatasvir for previously untreated chronic hepatitis C genotype-1 infection: a randomised, parallel-group, double-blind, placebo-controlled, dose-finding, phase 2a trial. Lancet Infect. Dis. 12:671–677 [DOI] [PubMed] [Google Scholar]
- 15.Lawitz E, Gruener D, Hill JM, Marbury T, Moorehead L, Mathias A, Cheng G, Link JO, Wong KA, Mo H, McHutchison JG, Brainard DM. 2012. A phase 1, randomized, placebo-controlled, 3-day, dose-ranging study of GS-5885, an NS5A inhibitor, in patients with genotype 1 hepatitis C. J. Hepatol. 57:24–31 [DOI] [PubMed] [Google Scholar]
- 16.National Institute of Allergy and Infectious Diseases (NIAID) 2004. Table for grading the severity of adult and pediatric adverse events, version 1.0 Division of Acquired Immunodeficiency Syndrome (DAIDS), National Institute of Allergy and Infectious Diseases (NIAID), Bethesda, MD [Google Scholar]
- 17.Bifano M, Sevinsky H, Persson A, Chung E, Wind-Rotolo M, Hwang C, Nettles R, Grasela D, Marbusy T, DeMicco M, Bertz R. 2011. Single-dose pharmacokinetics of daclatasvir (DCV; BMS-790052) in subjects with hepatic impairment compared with healthy subjects. Hepatology 54(Suppl S1):1004A [Google Scholar]
- 18.Lawitz E, Gruener D, Hill J, Marbury T, Komjathy S, DeMicco M, Murillo A, Jenkin F, Kim K, Simpson J, Aycock M, Mathias A, Yang C, Dowdy E, Liles M, Cheng G, Mondou E, Link J, Ohmstede C, Bannister R, McHutchison J, Brainard D. 2011. Three-day, dose-ranging study of the HCV NS5A inhibitor GS-5885, abstr 1219. 46th Annu. Meet. Eur. Assoc. Study Liver, Berlin, Germany, 30 March to 3 April 2011 [Google Scholar]
- 19.Adiwijaya B, Chandorkar G, van Heeswijk R, McNair L, Kwo PY, Gordon S, Garg V. 2011. Effect of mild and moderate hepatic impairment on telaprevir pharmacokinetics, abstr PK_1. 6th Int. Workshop Clin. Pharmacol. Hepatitis Ther. Cambridge, MA [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.