

Abstract
The activity of daptomycin (DAP) against methicillin-resistant Staphylococcus aureus (MRSA) is enhanced in the presence of subinhibitory concentrations of antistaphylococcal β-lactam antibiotics by an undefined mechanism. Given the variability in the penicillin-binding protein (PBP)-binding profiles of different β-lactam antibiotics, the purpose of this study was to examine the relative enhancement of DAP activity against MRSA by different β-lactam antibiotics to determine if a specific PBP-binding profile is associated with the ability to enhance the anti-MRSA activity of DAP. We determined that both broad- and narrow-spectrum β-lactam antibiotics known to exhibit PBP1 binding demonstrated potent enhancement of DAP anti-MRSA activity, whereas β-lactam antibiotics with minimal PBP1 binding (cefoxitin, ceftriaxone, cefaclor, and cefotaxime) were less effective. We suspect that PBP1 disruption by β-lactam antibiotics affects pathways of cell division in S. aureus that may be a compensatory response to DAP membrane insertion, resulting in DAP hypersusceptibility.
INTRODUCTION
The presence of subinhibitory concentrations of β-lactam antibiotics has been shown to increase daptomycin (DAP) activity against both DAP-susceptible and DAP-nonsusceptible (DNS) methicillin-resistant Staphylococcus aureus (MRSA) strains (1, 2). The mechanisms of this are not fully understood, but it has been attributed to a β-lactam-induced increase in the binding of DAP to bacterial cell membranes (3, 4). These observations fall in line with the observed DAP–β-lactam “seesaw effect” whereby S. aureus frequently gains susceptibility to β-lactams upon the acquisition of the DNS phenotype (5). Collectively, these in vitro observations have been translated to antimicrobial therapy combinations to successfully manage difficult-to-treat MRSA infections (2, 3, 5, 6).
Most in vivo studies have focused on three antistaphylococcal β-lactams, oxacillin, nafcillin (NAF), and ceftaroline, the latter because of its unique anti-MRSA properties among the β-lactam classes. However, no studies have compared the relative differences among the various β-lactam classes in their DAP-potentiating properties at therapeutically relevant concentrations.
The potentiation of DAP activity obtained by the addition of a β-lactam antibiotic may provide a straightforward way to enhance anti-MRSA therapy against both DAP-susceptible and DNS MRSA. Achieving a bacteriostatic DAP area under the concentration-time curve (AUC)/MIC ratio against S. aureus (438 ± 67) is challenging at a normal dose of 6 mg/kg (AUC = 705 ± 67) when the organism's DAP MIC is 2 μg/ml or greater (7). Dose escalation to 8 or 10 mg/kg improves the likelihood of attaining this target parameter against strains with elevated MICs but results in a higher minimum drug concentration in serum (Cmin), which has been associated with an increased risk of creatine phosphokinase elevation (8). High-dose DAP therapy is of particular concern in obese patients, where DAP AUCs are reported to be 30% greater than in nonobese matched controls (9).
In contrast to increasing pharmacokinetic parameters via dose escalation, pharmacodynamic enhancement of DAP with β-lactam antibiotics reduces the effective DAP MIC and therefore may increase the AUC/MIC ratio without increasing the DAP Cmin. The results of this study suggest that some β-lactams are more effective than others in potentiating the anti-MRSA activity of DAP and this difference may be associated with the relative affinity for penicillin-binding protein 1 (PBP1).
MATERIALS AND METHODS
Five pairs of MRSA isolates from five different patients were examined in this study, including the paired isolates from the index case of the clinical series utilizing DAP plus an antistaphylococcal β-lactam against refractory bacteremia (3). Each pair represents clinically sequential DAP-susceptible and DNS MRSA isolates identified as isogenic by whole-genome sequencing (3, 4, 10). The single nucleotide polymorphisms (SNPs) are displayed in Table 1. The DAP susceptibility of each isolate was determined by broth microdilution (11) in the presence or absence of static concentrations of nine different β-lactam antibiotics corresponding to the maximum, average, and minimum free concentrations (fCmax, fCavg, and fCmin, respectively) of that agent in serum (12–18). All antibacterials were purchased as commercial agents, except cefuroxime and cefotaxime (CTX), which were purchased from Sigma-Aldrich (St. Louis, MO). The selection of β-lactam antibiotics was made to capture a wide variety of β-lactam classes and PBP-binding profiles (Table 2).
Table 1.
Mutational differences between DAP-susceptible and DNS isogenic strain pairs identified by whole-genome sequencing
| Isolate pair and position | Mutationa | Gene | Function | Reference |
|---|---|---|---|---|
| D592/D712b | ||||
| 1440962 | T to C | mprF | Phosphatidylglycerol lysyltransferase | 4 |
| 1608916 | A to G | srrB | Respiratory response protein | |
| 1960627 | G to A | NA,d noncoding | ||
| 2448257 | C to A | Transcriptional regulator | ||
| 2592882 | G to A | Endo-1,4-beta-glucanase | ||
| J01/J03b | ||||
| 42283 | C to T | Mobile element protein | This study | |
| 530079 | T deletion | NA, indel, noncoding | ||
| 605255 | T to C | rpoB | DNA-directed RNA polymerase beta subunit | |
| 832922 | T to C | secA | Protein export cytoplasm protein SecA ATPase RNA helicase | |
| 1440974 | C to T | mprF | Phosphatidylglycerol lysyltransferase | |
| 2724219 | 6-bp deletion | NA, indel in frame, hypothetical protein | ||
| O325/O510b | ||||
| 20183 | A to G | Phosphoesterase, DHH family protein | This study | |
| 901046 | A to G | NA, noncoding | ||
| 946004 | G to A | Phage tail length tape measure protein | ||
| 1442416 | T to C | mprF | Phosphatidylglycerol lysyltransferase | |
| 2219610 | T to C | cls | Cardiolipin synthetase | |
| JKD6000/JKD6001c | ||||
| 24673 | G to A | walR | Response regulator | 10 |
| 1217720 | G to A | NA, noncoding | ||
| 2006385 | T to A | Putative phage protein | ||
| 2142758 | G to A | dUTPase | ||
| 2142774 | T to G | dUTPase | ||
| 2150689 | T to C | Acetyltransferase | ||
| 2151557 | C to T | NA, noncoding | ||
| JKD6004/JKD6005,c 25010 | A to G | walR | Response regulator | 10 |
Parent-to-child strain.
Reference genome S. aureus Mu50.
Reference genome S. aureus JKD6008.
NA, not applicable.
Table 2.
PBP selectivity and pharmacokinetic parameters of selected β-lactam antibiotics used in this study
| Drug | Proposed dosagea | t1/2(h)b | Cmax (μg/ml) | Protein binding (%) | fCmax (μg/ml) | PBP selectivity | Reference |
|---|---|---|---|---|---|---|---|
| AMP | 2,000 mg q4h i.v. | 1.2 | 97 | 18 | 80 | Nonselective | 13 |
| NAF | 2,000 mg q4h i.v. | 1 | 40 | 87 | 5 | Nonselective | 14 |
| TZP | 3,375 mg q4h i.v. | 0.8/0.8 | 213/29 | 30/30 | 150/20 | Nonselective | 15 |
| CFZ | 2,000 mg q8h i.v. | 2.8 | 404 | 94 | 26 | Nonselective | 16 |
| FEP | 2,000 mg q8h i.v. | 2.3 | 130 | 20 | 105 | Nonselective | 12 |
| MEM | 1,000 mg q8h i.v. | 1 | 112 | 2 | 110 | PBP1 | Product literature |
| CRO | 2,000 mg daily i.v. | 6 | 280 | 93 | 20 | PBP2 | Product literature |
| CTX | 2,000 mg q4h i.v. | 1.1 | 200 | 40 | 120 | PBP2 | 18 |
| CEC | 500 mg q8h p.o. | 1 | 17 | 25 | 13 | PBP3 | Product literature |
| FOX | 1,000 mg q4h i.v. | 0.8 | 115 | 73 | 31 | PBP4 | 17 |
q4h, every 4 h; i.v., intravenously; p.o., peroral.
t1/2, half-life.
Time-kill curves were determined with all DNS strains at 37°C in calcium-supplemented (50 μg/ml) Mueller-Hinton broth containing DAP at 0.5 times the MIC and a β-lactam antibiotic alone and in combination. The concentration of each β-lactam antibiotic was chosen to reflect the fCavg achieved in serum during a typical dosing interval, which was selected with the formula fCavg = (fCmax − fCmin)/2.
The rationale for using this test concentration was to approximate a clinically relevant concentration that would be achieved regardless of variation in the dose, volume of distribution, or patient-dependent clearance of β-lactam antibiotics (17, 19). Also, the fCavg would not overestimate the effect that could occur with fCmax, which is achieved for only a brief period in patients because of the short half-lives of most β-lactams. In addition to the fCavg, we also determined NAF and meropenem (MEM) kill curves at estimated concentrations consistent with continuous infusion therapy (fC at steady state [fCss], 1.9 and 12.9 μg/ml, respectively), which is of growing interest to improve efficacy (20–22). Time-kill experiments were performed in duplicate with a starting inoculum of 1 × 106 CFU/ml and 24 h of antibiotic exposure.
All statistical analyses were performed with GraphPad Prism software (version 5.0a; GraphPad Software, Inc.). The antibacterial activity (CFU/ml) of each regimen was compared by t test for two-group analyses and by two-way analysis of variance with Tukey's post hoc test for analyses of more than two groups. A P value of ≤0.05 was considered significant.
RESULTS
DAP susceptibilities with and without a β-lactams at their fCavgs are shown in Table 3. For all strains, the DAP MIC was lowered in the presence of all of the β-lactam antibiotics tested. The DAP MIC for DNS MRSA was decreased to the susceptible range, consistent with prior data (23). The high fCavgs of some β-lactam antibiotics alone (piperacillin-tazobactam [TZP], cefepime [FEP], MEM, and CTX) inhibited the growth of certain strains, and therefore, DAP enhancement could not be evaluated. Although strains showed a reduction in the DAP MIC in the presence of β-lactams, the magnitude of this effect was heterogeneous. For example, although strains J03 and D712 have the same DAP MIC, the reduction in the DAP MIC in the presence of a β-lactam antibiotic was consistently greater for J03 than for D712. To provide the greatest therapeutic benefit, an ideal β-lactam antibiotic would maintain the ability to potentiate DAP activity at all concentrations throughout the dosing interval, including its fCmin. Broth microdilution DAP susceptibility testing was repeated for paired strains D592 and D712 in the presence of a static concentration corresponding to the fCmin of each β-lactam antibiotic. As shown in Table 4, each β-lactam was still partially effective in reducing the DAP MIC at the equivalent of its fCmin.
Table 3.
MICs of DAP for study strain pairs in the presence of β-lactam antibiotics at their fCavgsa
| Strain | DAP MIC (μg/ml) with: |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| No β-lactam | AMP (44)b | NAF (2.7) | TZP (88) | CFZ (16) | FEP (57) | MEM (55) | CRO (12) | CTX (65) | CEC (6.5) | FOX (16) | |
| Pair 1 | |||||||||||
| O325 | 0.13 | 0.06 | 0.06 | NGc | 0.03 | 0.03 | NG | 0.06 | 0.13 | 0.06 | 0.03 |
| O510 | 1 | 0.13 | 0.25 | NG | 0.25 | 0.25 | NG | 0.25 | 0.25 | 0.25 | 0.5 |
| Pair 2 | |||||||||||
| J01 | 0.25 | 0.03 | 0.03 | NG | NG | NG | NG | 0.01 | NG | 0.06 | 0.01 |
| J03 | 2 | 0.13 | 0.13 | NG | NG | NG | NG | 0.13 | NG | 0.13 | 0.25 |
| Pair 3 | |||||||||||
| D592 | 0.5 | 0.13 | 0.13 | 0.16 | 0.13 | 0.13 | 0.03 | 0.13 | 0.13 | 0.13 | 0.13 |
| D712 | 2 | 0.5 | 0.5 | NG | 0.5 | 0.5 | NG | 1 | 0.5 | 1 | 1 |
| Pair 4 | |||||||||||
| JKD6000 | 0.25 | 0.06 | 0.06 | 0.01 | 0.06 | 0.06 | NG | 0.02 | 0.02 | 0.02 | 0.01 |
| JKD6001 | 1 | 0.06 | 0.13 | 0.13 | 0.06 | 0.06 | NG | 0.13 | 0.06 | 0.25 | 0.13 |
| Pair 5 | |||||||||||
| JKD6004 | 0.5 | 0.06 | 0.13 | NG | 0.13 | 0.13 | NG | 0.13 | 0.13 | 0.13 | 0.13 |
| JKD6005 | 1 | 0.06 | 0.13 | NG | 0.13 | 0.13 | NG | 0.13 | 0.25 | 0.13 | 0.25 |
Although some DNS strains may appear DAP sensitive on the basis of the broth microdilution MIC results presented in the no-β-lactam control column, confirmation by Etest consistently demonstrated an MIC of >1 μg/ml for all DNS strains, a discrepancy that has also been noted by other groups (40).
Values in parentheses are fCavgs.
NG, no growth observed at 24 h in any well, including the control containing the β-lactam antibiotic alone.
Table 4.
DAP MICs for study strains in the presence of β-lactam antibiotics at their fCmaxs, fCavgs, and fCmins
| β-Lactam | β-Lactam fCmax | DAP MIC for strain: |
β-Lactam fCavg | DAP MIC for strain: |
β-Lactam fCmin | DAP MIC for strain: |
|||
|---|---|---|---|---|---|---|---|---|---|
| D592 | D712 | D592 | D712 | D592 | D712 | ||||
| AMP | 80a | 0.13 | 0.5 | 44 | 0.13 | 0.5 | 8 | 0.13 | 1 |
| NAF | 5 | 0.13 | 0.5 | 2.7 | 0.13 | 0.5 | 0.33 | 0.13 | 1 |
| TZP | 170 | NGb | NG | 88 | 0.16 | NG | 5 | 0.13 | 1 |
| CFZ | 26 | 0.13 | 1 | 16 | 0.13 | 0.5 | 6 | 0.13 | 1 |
| FEP | 105 | 0.13 | 0.25 | 57 | 0.13 | 0.5 | 9 | 0.13 | 1 |
| MEM | 110 | NG | NG | 55 | 0.03 | NG | 0.43 | 0.13 | 1 |
| CRO | 20 | 0.13 | 1 | 12 | 0.13 | 1 | 4 | 0.13 | 1 |
| CTX | 120 | 0.13 | 0.5 | 65 | 0.13 | 0.5 | 9.6 | 0.13 | 1 |
| CEC | 13 | 0.13 | 1 | 6.5 | 0.13 | 1 | 0.05 | 0.25 | 1 |
| FOX | 31 | 0.13 | 1 | 16 | 0.13 | 1 | 1 | 0.25 | 1 |
All values are in micrograms per milliliter.
NG, no growth.
While DAP broth microdilution is useful to assess the potential of β-lactam antibiotics to enhance DAP susceptibility, information regarding time-kill kinetics and antibacterial killing is not captured in this assay. To better assess bactericidal synergy, DNS MRSA strains were evaluated in 24-h killing assays in the presence of 0.5 times the DAP MIC, the fCavgs of the study β-lactam antibiotics, or a combination of both. Figure 1 displays the results of each DAP–β-lactam combination for all five strain combinations tested. Overall, β-lactams binding PBP1 produced greater killing when combined with DAP than those with a PBP2-to-PBP4 preference (−1.50 versus 1.73 Δlog CFU/ml, respectively; P = 0.001). Notably, PBP1-selective MEM plus DAP had the highest mean killing. The representative bacterial kill curves for well-characterized DNS strain D712 with all of the DAP–β-lactam combinations tested are shown in Fig. 2 and 3. For this isolate, DAP anti-MRSA activity was enhanced the most by PBP1-selective MEM, followed by PBP-nonspecific TZP and FEP, resulting in 4 to 5 log10 killing at 24 h. DAP plus PBP-nonspecific ampicillin (AMP), NAF, or CFZ resulted in 2 to 3 log10 killing at 24 h (Fig. 2). Of great interest, however, was that β-lactams with minimal affinity for PBP1 (PBP2-selective ceftriaxone [CRO] and CTX, PBP3-selective cefaclor [CEC], and PBP4-selective cefoxitin [FOX]) all did not enhance DAP killing of MRSA strain D712 (Fig. 3; P < 0.001 versus PBP1-selective and -nonspecific β-lactams).
Fig 1.
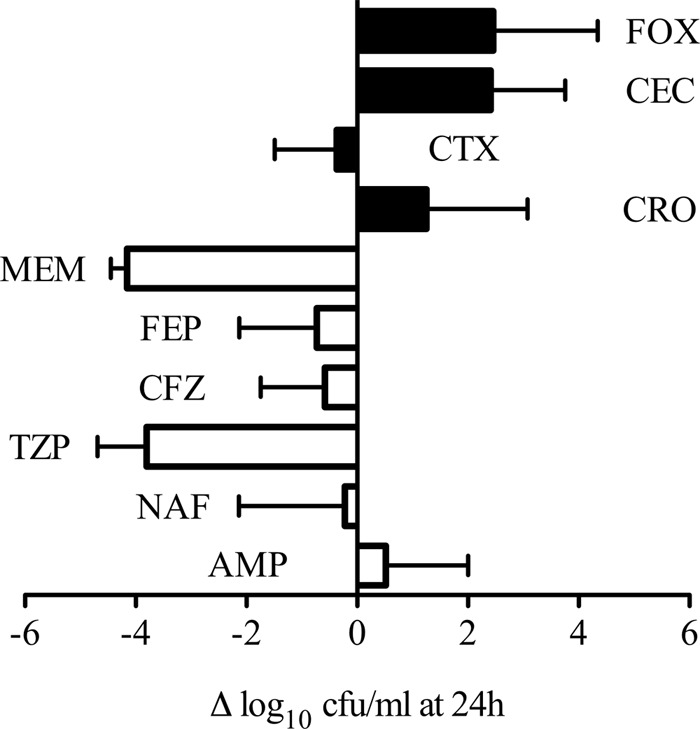
Activity of DAP in combination with β-lactam antibiotics against all five DNS MRSA strains tested. The change in the number of CFU/ml from time zero is presented as the mean with the standard error of the mean of the five strains. White bars, β-lactam antibiotics that preferentially bind to PBP1; black bars, β-lactam antibiotics that do not have PBP1 binding preference. Collectively, the mean activity in the PBP1 group was significantly greater than that in the non-PBP1 group (−1.50 versus 1.73 log CFU/ml, respectively; P = 0.001).
Fig 2.

Activity of DAP alone and in combination with β-lactam antibiotics that bind to PBP1 against DNS MRSA strain D712. Dotted line, growth control; white circles, DAP; white squares, β-lactam; black diamonds, DAP plus a β-lactam. DAP was included at 1 μg/ml (equivalent to 0.5 times the measured MIC for strain D712); β-lactam antibiotics were included at the fCavg of the doses listed in Table 2. MEM inset: activity detailed in the first 4 h that displays increased potency with MEM plus DAP after just 1 h of exposure.
Fig 3.

Activity of DAP alone and in combination with β-lactam antibiotics that do not target PBP1 against DNS MRSA strain D712. Dotted line, growth control; white circles, DAP; white squares, β-lactam; black diamonds, DAP plus a β-lactam. DAP was included at 1 μg/ml (equivalent to 0.5 times the measured MIC for strain D712); β-lactam antibiotics were included at the fCavg of the doses listed in Table 2.
In order to test the efficacy with which NAF or MEM continuous infusion potentiates DAP, DNS MRSA strain D712 was grown for 24 h in the presence of 0.5 times the DAP MIC and the predicted fCss of NAF analogous to a 12-g daily continuous NAF infusion (1.9 μg/ml) or a 3-g daily continuous MEM infusion (12.9 μg/ml) (Fig. 4). Growth in DAP plus NAF at the fCss was statistically indistinguishable from growth at the fCmax and clearly superior to growth at the fCmin (P < 0.001). Similarly, bacterial killing with DAP plus MEM at the fCss was superior to that of DAP plus MEM at the fCmin, but the fCss produced both less rapid killing and a lesser extent of killing than the fCmax. On the basis of these results, it is apparent that continuous infusion of NAF may provide an important benefit compared to intermittent infusion over the entire dosing interval to improve DAP killing, while the rapid effects of the fCmax with intermittent infusion of MEM may provide important therapeutic enhancements.
Fig 4.
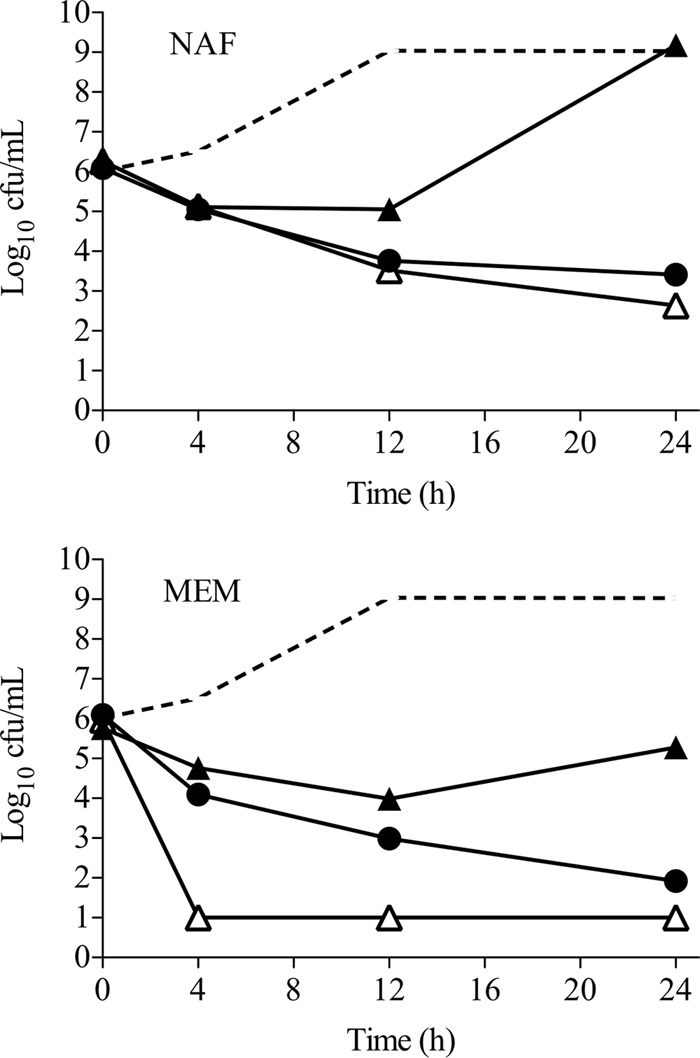
Activity of DAP at 0.5 times the MIC in combination with NAF or MEM at the fCmax (white triangles), fCss (black circles), or fCmin (black triangles) against strain D712. Continuous infusion of NAF, represented by the fCss, appears to provide better DAP enhancement than intermittent infusion throughout a dosing interval, while intermittent infusion of MEM is rapidly more bactericidal with DAP. Dotted line, growth control.
DISCUSSION
Although β-lactam antibiotics have no clinically relevant antimicrobial effects against MRSA as single agents, recent data show that (i) they improve DAP activity against MRSA, (ii) they potentiate cationic host defense peptide killing of MRSA, and (iii) they prevent the emergence of DAP resistance (23, 24). However, important gaps remain in the knowledge about the combination of DAP and β-lactams necessary to optimize their clinical use, including the relative potency of the multiple agents within this class, which we evaluated in this study.
DAP synergy was most pronounced with β-lactam antibiotics known to preferentially bind PBP1, whereas β-lactam antibiotics with preferential binding to PBP2, PBP3, or PBP4 showed significantly less synergy. The mutational differences among the DNS strains identified by whole-genome sequencing (Table 1) represent a variety of genetic polymorphisms, including mprF, rpoB, cls, and walR, associated with the DNS phenotype (10, 25, 26) and may contribute to the observed differences in sensitization among the strains. It is notable that DNS strains with SNPs in genes involved in membrane function (walR [yycF] in JKD6001 and JKD6005) and cardiolipin synthesis (cls in O510) showed considerably less synergy with β-lactams than DNS strains without these mutations (D712 and J03). SNPs in walR and cls are considered to be cumulative mutations leading to further reduced DAP susceptibility upon continued exposure (27, 28). One isolate, J03, displayed synergy with any β-lactam regardless of its affinity for PBP. Interestingly, PBP-nonselective agents were also synergistic with DAP against this isolate, but specifically PBP1 selective MEM had the most rapid and sustained activity with DAP. We hypothesize that the clear differences observed in the DAP-enhancing activities of β-lactam antibiotics may be related to differential PBP binding, specifically, the degree of binding to PBP1.
S. aureus produces four distinct PBPs (PBP1 to PBP4), each of which is a pharmacodynamic target of β-lactams to various degrees, while MRSA is able to produce a fifth PBP (PBP2a) with reduced affinity for standard β-lactams. All PBPs are able to catalyze transpeptidation of bacterial peptidoglycan, while only PBP2 is able to catalyze transglycosylation (29). Our hypothesis that PBP specificity is associated with the ability to potentiate DAP is based on the fact that β-lactam antibiotics that had poor DAP potentiation have less relative PBP1 binding and higher binding specificity for alternative PBPs (i.e., CRO [PBP2], CTX [PBP2], CEC [PBP3], and FOX [PBP4]). In contrast, carbapenems like MEM, which showed the greatest DAP potentiation, have high selection for PBP1 binding (30, 31).
These data provide a very significant link to our further understanding of the DAP–β-lactam synergy phenomenon, as they direct attention to PBP1 specifically. The role of PBP1 in S. aureus is relatively poorly characterized, but it is appreciated that the C-terminal end carries the transpeptidase activity whereas the N-terminal end, while possessing no enzymatic activity, is important in cell division and septum formation (32). The facts that PBP1 of S. aureus is homologous to PBP3 of Escherichia coli (33) and PBP3 in E. coli has been shown to interact with critical proteins of the divisome of that organism (34) provide additional clues that interference with PBP1 activity with PBP1-selective β-lactams may render MRSA more susceptible to other antibiotics that influence the divisome of MRSA, such as DAP (35). Pogliano et al. hypothesized that DAP insertion into the membrane triggers what may be a compensatory response to recruit proteins involved in synthesis and arrangement of the cell surface architecture, as occurs in septum formation with cell division, at sites of DAP insertion (35). Interference with this compensatory response by inhibition of cell division proteins such as PBP1 may induce profound vulnerability to DAP-mediated bacterial killing. This effect may be independent of the ability of β-lactams to reduce the S. aureus surface charge, resulting in enhanced DAP susceptibility (3). Future studies could use S. aureus modified to express PBP1 or antibodies to PBP1 to examine the role of PBP1 blockade in the enhancement of DAP activity (36).
Exposure to β-lactams has also been shown to alter bacterial gene regulation, leading to diverse physiological effects. Binding of β-lactam to PBP1 by oxacillin (nonselective) or imipenem (selective) triggers the global regulator SarA, resulting in increased Panton-Valentine leukocidin toxin expression. However, this induction did not occur upon β-lactam binding to alternative PBPs, implying a PBP1-specific effect (30). Both of these agents do show excellent affinity for PBP1. Exposure to subinhibitory β-lactam concentrations has also been associated with increased production of hemolysins, nucleases, and fibronectin-binding proteins, although this effect was not definitively shown to be mediated by PBP binding. Of interest, the β-lactam selected for the study of hemolysin induction was FOX, which has minimal affinity for PBP1 but rather is strongly selective for PBP4 (37). This suggests that the panel of toxins produced by S. aureus may be influenced on the basis of the identity of the PBP inhibited by β-lactams.
In addition to altering toxin production by MRSA, PBP disruption has a well-documented direct influence on susceptibility to cationic antimicrobial peptides in Streptococcus agalactiae (38). This is particularly relevant as both DAP and cationic antimicrobial peptides exert their antimicrobial effects via membrane disruption and resistance to cationic peptides often coincides with cross-resistance to DAP (39). In S. agalactiae, loss of PBP1a (homologous to PBP2 in S. aureus) resulted in enhanced killing by host antimicrobial peptides (38). Thus, inactivation of specific PBPs either through mutation or pharmacologically can alter cell signaling and influence toxin production and susceptibility to innate immunity. Interestingly, this effect is not appreciated under standard susceptibility testing conditions, where toxin production and antimicrobial peptide activity are not assessed.
Another unexplored effect with DAP is the impact of the β-lactam infusion type on MRSA activity. Recently, there has been increased interest in providing antistaphylococcal β-lactams as a continuous infusion (20, 21) to provide a consistent level of antibiotic exposure in the treatment of infections with susceptible organisms. If the DAP-enhancing activity of NAF or MEM could be optimized throughout the dosing interval, this combination would be highly beneficial both on a theoretical basis and for in vivo evidence of efficacy. Our results indicate that NAF at its fCss enhances DAP killing similar to that of NAF at its fCmax and better than at its fCmin, but intermittent infusion of MEM may be more beneficial than continuous infusion because of its rapid killing in the first few hours. It will be important to determine if continuous infusion of NAF or MEM along with other β-lactam antibiotics presented in this study with DAP provides clinical treatment outcomes better than those attained with standard infusions.
Although the novel evaluation of β-lactam antibiotics at the fCmax and fCmin in this study provides the first relevant comparative in vivo data, in vivo animal and clinical studies are needed to confirm these relationships to further guide the use of DAP combined with β-lactam antibiotics in complex MRSA infections. These studies are also limited in that they examined only a small group of MRSA isolates for DAP–β-lactam synergy. It remains unknown if our findings of β-lactam selectivity in DNS S. aureus apply to DAP-susceptible strains, which were not studied in similar detail. However, Table 2 indicates that β-lactams do further enhance DAP activity against susceptible MRSA strains.
In summary, we have shown for the first time that β-lactams have different potencies in enhancing DAP-mediated MRSA killing and that this may be driven by interference with PBP1 on the basis of the relative selectivity of binding by different β-lactams to the different PBPs of S. aureus. These findings further categorize the identified “seesaw” effect in which S. aureus with reduced DAP activity correspondingly has enhanced susceptibility to oxacillin and potent DAP–β-lactam synergy both in vitro and in an animal endocarditis model (1). More research is needed to evaluate these combinations at the clinical level, as well as basic research to further characterize the effects of these different β-lactams on the process of S. aureus cell division.
ACKNOWLEDGMENT
We thank Benjamin Howden for providing two of the strain pairs used in this study.
Footnotes
Published ahead of print 29 July 2013
REFERENCES
- 1.Rand KH, Houck HJ. 2004. Synergy of daptomycin with oxacillin and other beta-lactams against methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 48:2871–2875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rose WE, Schulz LT, Andes D, Striker R, Berti AD, Hutson PR, Shukla SK. 2012. Addition of ceftaroline to daptomycin after emergence of daptomycin-nonsusceptible Staphylococcus aureus during therapy improves antibacterial activity. Antimicrob. Agents Chemother. 56:5296–5302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dhand A, Bayer AS, Pogliano J, Yang SJ, Bolaris M, Nizet V, Wang G, Sakoulas G. 2011. Use of antistaphylococcal beta-lactams to increase daptomycin activity in eradicating persistent bacteremia due to methicillin-resistant Staphylococcus aureus: role of enhanced daptomycin binding. Clin. Infect. Dis. 53:158–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Werth BJ, Sakoulas G, Rose WE, Pogliano J, Tewhey R, Rybak MJ. 2013. Ceftaroline increases membrane binding and enhances the activity of daptomycin against daptomycin-nonsusceptible vancomycin-intermediate Staphylococcus aureus in a pharmacokinetic/pharmacodynamic model. Antimicrob. Agents Chemother. 57:66–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang SJ, Xiong YQ, Boyle-Vavra S, Daum R, Jones T, Bayer AS. 2010. Daptomycin-oxacillin combinations in treatment of experimental endocarditis caused by daptomycin-nonsusceptible strains of methicillin-resistant Staphylococcus aureus with evolving oxacillin susceptibility (the “seesaw effect”). Antimicrob. Agents Chemother. 54:3161–3169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moise PA, Amodio-Groton M, Rashid M, Lamp KC, Hoffman-Roberts HL, Sakoulas G, Yoon MJ, Schweitzer S, Rastogi A. 2013. Clinical outcomes of daptomycin with and without concomitant beta-lactams in patients with Staphylococcus aureus bacteremia and mild to moderate renal impairment: a multicenter evaluation. Antimicrob. Agents Chemother. 57:1192–1200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Safdar N, Andes D, Craig WA. 2004. In vivo pharmacodynamic activity of daptomycin. Antimicrob. Agents Chemother. 48:63–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bhavnani SM, Rubino CM, Ambrose PG, Drusano GL. 2010. Daptomycin exposure and the probability of elevations in the creatine phosphokinase level: data from a randomized trial of patients with bacteremia and endocarditis. Clin. Infect. Dis. 50:1568–1574 [DOI] [PubMed] [Google Scholar]
- 9.Dvorchik BH, Damphousse D. 2005. The pharmacokinetics of daptomycin in moderately obese, morbidly obese, and matched nonobese subjects. J. Clin. Pharmacol. 45:48–56 [DOI] [PubMed] [Google Scholar]
- 10.Howden BP, McEvoy CR, Allen DL, Chua K, Gao W, Harrison PF, Bell J, Coombs G, Bennett-Wood V, Porter JL, Robins-Browne R, Davies JK, Seemann T, Stinear TP. 2011. Evolution of multidrug resistance during Staphylococcus aureus infection involves mutation of the essential two component regulator WalKR. PLoS Pathog. 7:e1002359. 10.1371/journal.ppat.1002359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Clinical and Laboratory Standards Institute 2011. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically; approved standard, 11th ed. Clinical and Laboratory Standards Institute, Wayne, PA [Google Scholar]
- 12.Barbhaiya RH, Forgue ST, Gleason CR, Knupp CA, Pittman KA, Weidler DJ, Movahhed H, Tenney J, Martin RR. 1992. Pharmacokinetics of cefepime after single and multiple intravenous administrations in healthy subjects. Antimicrob. Agents Chemother. 36:552–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Clarke JT, Libke RD, Ralph ED, Luthy RP, Kirby WM. 1974. Human pharmacokinetics of BL-P1654 compared with ampicillin. Antimicrob. Agents Chemother. 6:729–733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leonard SN, Rolek KM. 2013. Evaluation of the combination of daptomycin and nafcillin against vancomycin-intermediate Staphylococcus aureus. J. Antimicrob. Chemother. 68:644–647 [DOI] [PubMed] [Google Scholar]
- 15.Mattoes HM, Capitano B, Kim MK, Xuan D, Quintiliani R, Nightingale CH, Nicolau DP. 2002. Comparative pharmacokinetic and pharmacodynamic profile of piperacillin/tazobactam 3.375G Q4H and 4.5G Q6H. Chemotherapy 48:59–63 [DOI] [PubMed] [Google Scholar]
- 16.Douglas A, Udy AA, Wallis SC, Jarrett P, Stuart J, Lassig-Smith M, Deans R, Roberts MS, Taraporewalla K, Jenkins J, Medley G, Lipman J, Roberts JA. 2011. Plasma and tissue pharmacokinetics of cefazolin in patients undergoing elective and semielective abdominal aortic aneurysm open repair surgery. Antimicrob. Agents Chemother. 55:5238–5242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goodwin CS, Raftery EB, Goldberg AD, Skeggs H, Till AE, Martin CM. 1974. Effects of rate of infusion and probenecid on serum levels, renal excretion, and tolerance of intravenous doses of cefoxitin in humans: comparison with cephalothin. Antimicrob. Agents Chemother. 6:338–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Esmieu F, Guibert J, Rosenkilde HC, Ho I, Le Go A. 1980. Pharmacokinetics of cefotaxime in normal human volunteers. J. Antimicrob. Chemother. 6(Suppl A):83–92 [DOI] [PubMed] [Google Scholar]
- 19.Hagihara M, Wiskirchen DE, Kuti JL, Nicolau DP. 2012. In vitro pharmacodynamics of vancomycin and cefazolin alone and in combination against methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 56:202–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dulhunty JM, Roberts JA, Davis JS, Webb SA, Bellomo R, Gomersall C, Shirwadkar C, Eastwood GM, Myburgh J, Paterson DL, Lipman J. 2013. Continuous infusion of Beta-lactam antibiotics in severe sepsis: a multicenter double-blind, randomized controlled trial. Clin. Infect. Dis. 56:236–244 [DOI] [PubMed] [Google Scholar]
- 21.Hughes DW, Frei CR, Maxwell PR, Green K, Patterson JE, Crawford GE, Lewis JS., II 2009. Continuous versus intermittent infusion of oxacillin for treatment of infective endocarditis caused by methicillin-susceptible Staphylococcus aureus. Antimicrob. Agents Chemother. 53:2014–2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pea F, Viale P, Cojutti P, Furlanut M. 2012. Dosing nomograms for attaining optimum concentrations of meropenem by continuous infusion in critically ill patients with severe gram-negative infections: a pharmacokinetics/pharmacodynamics-based approach. Antimicrob. Agents Chemother. 56:6343–6348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mehta S, Singh C, Plata KB, Chanda PK, Paul A, Riosa S, Rosato RR, Rosato AE. 2012. β-Lactams increase the antibacterial activity of daptomycin against clinical methicillin-resistant Staphylococcus aureus strains and prevent selection of daptomycin-resistant derivatives. Antimicrob. Agents Chemother. 56:6192–6200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Berti AD, Wergin JE, Girdaukas GG, Hetzel SJ, Sakoulas G, Rose WE. 2012. Altering the proclivity towards daptomycin resistance in methicillin-resistant Staphylococcus aureus using combinations with other antibiotics. Antimicrob. Agents Chemother. 56:5046–5053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Peleg AY, Miyakis S, Ward DV, Earl AM, Rubio A, Cameron DR, Pillai S, Moellering RC, Jr, Eliopoulos GM. 2012. Whole genome characterization of the mechanisms of daptomycin resistance in clinical and laboratory derived isolates of Staphylococcus aureus. PLoS One 7:e28316. 10.1371/journal.pone.0028316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jones T, Yeaman MR, Sakoulas G, Yang SJ, Proctor RA, Sahl HG, Schrenzel J, Xiong YQ, Bayer AS. 2008. Failures in clinical treatment of Staphylococcus aureus infection with daptomycin are associated with alterations in surface charge, membrane phospholipid asymmetry, and drug binding. Antimicrob. Agents Chemother. 52:269–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bayer AS, Schneider T, Sahl HG. 2013. Mechanisms of daptomycin resistance in Staphylococcus aureus: role of the cell membrane and cell wall. Ann. N. Y. Acad. Sci. 1277:139–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mishra NN, Rubio A, Nast CC, Bayer AS. 2012. Differential adaptations of methicillin-resistant Staphylococcus aureus to serial in vitro passage in daptomycin: evolution of daptomycin resistance and role of membrane carotenoid content and fluidity. Int. J. Microbiol. 2012:683450. 10.1155/2012/683450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reed P, Veiga H, Jorge AM, Terrak M, Pinho MG. 2011. Monofunctional transglycosylases are not essential for Staphylococcus aureus cell wall synthesis. J. Bacteriol. 193:2549–2556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dumitrescu O, Choudhury P, Boisset S, Badiou C, Bes M, Benito Y, Wolz C, Vandenesch F, Etienne J, Cheung AL, Bowden MG, Lina G. 2011. Beta-lactams interfering with PBP1 induce Panton-Valentine leukocidin expression by triggering sarA and rot global regulators of Staphylococcus aureus. Antimicrob. Agents Chemother. 55:3261–3271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Davies TA, Page MG, Shang W, Andrew T, Kania M, Bush K. 2007. Binding of ceftobiprole and comparators to the penicillin-binding proteins of Escherichia coli, Pseudomonas aeruginosa, Staphylococcus aureus, and Streptococcus pneumoniae. Antimicrob. Agents Chemother. 51:2621–2624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pereira SF, Henriques AO, Pinho MG, de Lencastre H, Tomasz A. 2009. Evidence for a dual role of PBP1 in the cell division and cell separation of Staphylococcus aureus. Mol. Microbiol. 72:895–904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sauvage E, Kerff F, Terrak M, Ayala JA, Charlier P. 2008. The penicillin-binding proteins: structure and role in peptidoglycan biosynthesis. FEMS Microbiol. Rev. 32:234–258 [DOI] [PubMed] [Google Scholar]
- 34.Tormo A, Ayala JA, de Pedro MA, Aldea M, Vicente M. 1986. Interaction of FtsA and PBP3 proteins in the Escherichia coli septum. J. Bacteriol. 166:985–992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pogliano J, Pogliano N, Silverman JA. 2012. Daptomycin-mediated reorganization of membrane architecture causes mislocalization of essential cell division proteins. J. Bacteriol. 194:4494–4504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pereira SF, Henriques AO, Pinho MG, de Lencastre H, Tomasz A. 2007. Role of PBP1 in cell division of Staphylococcus aureus. J. Bacteriol. 189:3525–3531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kuroda H, Kuroda M, Cui L, Hiramatsu K. 2007. Subinhibitory concentrations of beta-lactam induce haemolytic activity in Staphylococcus aureus through the SaeRS two-component system. FEMS Microbiol. Lett. 268:98–105 [DOI] [PubMed] [Google Scholar]
- 38.Hamilton A, Popham DL, Carl DJ, Lauth X, Nizet V, Jones AL. 2006. Penicillin-binding protein 1a promotes resistance of group B Streptococcus to antimicrobial peptides. Infect. Immun. 74:6179–6187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mishra NN, Bayer AS, Moise PA, Yeaman MR, Sakoulas G. 2012. Reduced susceptibility to host-defense cationic peptides and daptomycin coemerge in methicillin-resistant Staphylococcus aureus from daptomycin-naive bacteremic patients. J. Infect. Dis. 206:1160–1167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fuchs PC, Barry AL, Brown SD. 2001. Evaluation of daptomycin susceptibility testing by Etest and the effect of different batches of media. J. Antimicrob. Chemother. 48:557–561 [DOI] [PubMed] [Google Scholar]