

Abstract
Class D β-lactamases that hydrolyze carbapenems such as imipenem and doripenem are a recognized danger to the efficacy of these “last-resort” β-lactam antibiotics. Like all known class D carbapenemases, OXA-23 cannot hydrolyze the expanded-spectrum cephalosporin ceftazidime. OXA-146 is an OXA-23 subfamily clinical variant that differs from the parent enzyme by a single alanine (A220) inserted in the loop connecting β-strands β5 and β6. We discovered that this insertion enables OXA-146 to bind and hydrolyze ceftazidime with an efficiency comparable to those of other extended-spectrum class D β-lactamases. OXA-146 also binds and hydrolyzes aztreonam, cefotaxime, ceftriaxone, and ampicillin with higher efficiency than OXA-23 and preserves activity against doripenem. In this study, we report the X-ray crystal structures of both the OXA-23 and OXA-146 enzymes at 1.6-Å and 1.2-Å resolution. A comparison of the two structures shows that the extra alanine moves a methionine (M221) out of its normal position, where it forms a bridge over the top of the active site. This single amino acid insertion also lengthens the β5-β6 loop, moving the entire backbone of this region further away from the active site. A model of ceftazidime bound in the active site reveals that these two structural alterations are both likely to relieve steric clashes between the bulky R1 side chain of ceftazidime and OXA-23. With activity against all four classes of β-lactam antibiotics, OXA-146 represents an alarming new threat to the treatment of infections caused by Acinetobacter spp.
INTRODUCTION
Class D β-lactamases are divided into three categories based on their substrate specificities. Narrow-spectrum enzymes such as OXA-1, OXA-10, and OXA-46 bind and hydrolyze penicillins such as ampicillin and benzylpenicillin, as well as a few narrow-spectrum cephalosporins, including cephalothin and cephaloridine (1). Extended-spectrum β-lactamases (ESBL) have acquired the ability to hydrolyze expanded-spectrum cephalosporins, such as cefotaxime, ceftazidime, and cefepime (Fig. 1) (2). These enzymes (e.g., OXA-13 and -17) seem to arise when a small number of substitutions occur in the background of a narrow-spectrum parental enzyme (most often OXA-10). Carbapenem-hydrolyzing class D β-lactamases (CHDL) such as OXA-23, OXA-24/40, and OXA-48 provide resistance to carbapenems such as imipenem, meropenem, and doripenem (Fig. 1) through a weak hydrolytic activity toward those antibiotics (3). Because of the serious clinical threat posed by the second and third categories of class D β-lactamases, it has been remarked that to date, class D enzymes possessing both ESBL and CHDL properties have not been observed clinically (1).
Fig 1.

β-Lactam antibiotics. Structures of the penicillin ampicillin (1), the carbapenem doripenem (2), the monobactam aztreonam (3), and the cephalosporins ceftazidime (4), cefotaxime (5), and ceftriaxone (6).
Sequence alignments reveal that the class D carbapenemases can be classified into five subfamilies: OXA-23, OXA-24/40, OXA 48, OXA-51, and OXA-58 (1). OXA-23, reported in 1995 and initially referred to as ARI-1, was the first example of a class D enzyme conferring resistance to carbapenems (4, 5). OXA-24/40 was identified in 2000 in Spain (6). Since that time, the blaOXA-23 and blaOXA-24 genes have been identified with ever greater frequency as the causative mechanism of carbapenem resistance in nosocomial Acinetobacter baumannii infections across the world (3, 7). Much progress has been made in the last decade toward understanding the mechanism by which these CHDL enzymes accommodate and hydrolyze carbapenem substrates. Structural analyses of OXA-24/40 without (8) and with (9) doripenem bound revealed that a “hydrophobic bridge” (composed of Y112 and M223) across the top of the active site helps hold onto carbapenems that possess extended nonpolar side chains (e.g., doripenem and meropenem) while sterically occluding penicillins with bulky side chains (e.g., oxacillin). The extraordinarily tight carbapenem binding provided by this bridge (Km < 30 nM) likely compensates for the weak hydrolytic turnover rates of these enzymes, resulting in clinically significant resistance levels in high-threat organisms such as Acinetobacter baumannii. This bridge is not a universal feature of CHDLs: OXA-48 does not possess it, and its more open active site has been shown to bind carbapenems less tightly (and oxacillin more tightly) in comparison to OXA-24/40 (10). It has been proposed that a 6α-hydroxyethyl side chain of carbapenem substrates plays a key role in the hydrolysis mechanism of these drugs. The alcohol of this moiety either prevents activation of the hydrolytic water (in narrow-spectrum enzymes) (10, 11) or, when rotated to a different orientation, allows it to be activated (in CHDL enzymes) (9). Docquier and colleagues have demonstrated the importance of the loop between β-strands β5 and β6 for carbapenem turnover and proposed that it may promote hydrolysis by affecting the rotational orientation of the drug's hydroxyethyl group (10, 12).
A number of studies have shed light on the origin of ESBL activity in a number of clinical variants from the OXA-10 subfamily. Paetzel et al. noted that a number of such mutations (N73S, A124T, G157D, etc.) are clustered near a highly conserved tryptophan residue (W154; OXA-10 numbering) (13). Notably, substitution of the tryptophan itself, or deletion of a neighboring leucine, yields an enzyme that provides robust ceftazidime resistance (14). The most complete analysis of a class D ESBL involves OXA-145, which arose when L165 was deleted from the narrow-spectrum OXA-35 (15). X-ray crystallographic analysis showed that this deletion expands the active site of OXA-145 and likely makes room for the bulky oxyimino R1 side chain of ceftazidime (16).
Sequence analysis of β-lactamases indicated that a number of clinically isolated class D enzymes are single-residue variants of carbapenemase subfamily parental enzymes such as OXA-23 or OXA-24/40. One such variant is OXA-146, an OXA-23 subfamily member isolated in the vicinity of Anhui, China, in 2007 (GenBank accession number ACI28281). OXA-146 is identical to OXA-23 except for a duplication of A220, adding one residue to the loop between β-strands β5 and β6. As part of a study to investigate the effect of clinically observed mutations in the β5-β6 loop, we compared the kinetic properties of OXA-24/40, OXA-23, and OXA-146 (OXA-23 A220dup). We also describe here the crystal structures of OXA-23 and OXA-146, which provide tremendous insight into the importance of the β5-β6 loop in determining substrate specificity.
MATERIALS AND METHODS
Mutagenesis and protein purification.
The blaOXA-23 gene (17) was subcloned into the NdeI and XhoI restriction sites of the pET24a(+) expression vector such that residue H22 is the first residue after the initiator methionine. This vector was used as a template for mutagenesis using the PCR overlap extension method (18), leading to the insertion of an extra alanine between A220 and M221. The sequences of both genes were verified, and the plasmids were transformed into Escherichia coli BL21(DE3) cells. Both proteins were expressed and purified to homogeneity using a protocol identical to that previously used for the preparation of the related class D carbapenemase OXA-24/40 (9).
Kinetic analysis.
Steady-state kinetic analysis was carried out in 50 mM NaH2PO4, 25 mM NaHCO3 (pH 7.4) at 25°C in a Cary 100 UV-Vis spectrophotometer. Initial velocities were calculated using the following Δε (M−1 cm−1) values: ampicillin, −900 (λ = 235 nm); doripenem, −11,460 (λ = 297 nm); imipenem, −9,000 (λ = 300 nm); cefotaxime, −7,500 (λ = 260 nm); ceftriaxone, −7,800 (λ = 255 nm); ceftazidime, −6,900 (λ = 260 nm); aztreonam, −700 (λ = 297 nm). Values of Km and kcat were determined by nonlinear regression to the Michaelis-Menten equation. Slow substrates that bound with very high affinity were treated as inhibitors in a competition assay. A constant, subsaturating concentration of the reporter (ampicillin) was added to the cuvette with various concentrations of inhibitor. Absorption was measured at 235 nm, and velocities were measured in triplicate. The average velocities (v0) were then fitted to equation 1, where vu represents the reporter's uninhibited velocity and Kd app represents the inhibitor concentration that results in 50% vu.
| 1 |
The inhibitor's Km can be calculated using equation 2 (19).
| 2 |
X-ray crystallography.
OXA-23 crystals were grown by the hanging-drop vapor diffusion method in an 8-μl drop containing 4.5 mg/ml of the β-lactamase mixed 1:1 (vol/vol) with well buffer. For the wild-type crystals, 0.1 M sodium acetate (pH 5.0), 5% (wt/vol) polyglutamic acid (low-molecular-weight polymer), and 30% (vol/vol) polyethylene glycol (PEG) 550MME (monomethyl ether) was used as the well buffer. These crystals were harvested and soaked in a new well buffer containing 100 mM Tris (pH 7.1), 5% (wt/vol) polyglutamic acid (low-molecular-weight polymer), and 30% (vol/vol) PEG 550MME, 50 mM NaHCO3 before being flash cooled in liquid nitrogen.
The OXA-146 crystals were grown in well buffer containing 0.2 M sodium chloride, 0.1 M phosphate/citrate (pH 4.2), 20% (wt/vol) PEG 8000. Diffraction data were measured on the LS-CAT beamline (hutches 21-ID-D and 21-ID-G) at the Advanced Photon Source (Argonne, IL) at 100 K using a MarCCD detector (see Table 2). Reflections were indexed, integrated, and scaled using HKL-2000 (20). Molecular replacement for the determination of both the OXA-23 and OXA-146 structures was performed with Phaser (21). A Phyre2 (22)-generated homology model based on the OXA-24 structure was used as the initial phasing model and the OXA-23 structure (excluding all water molecules) was subsequently used as the initial phasing model for the determination of the OXA-146 structure. Refinement and electron density map calculations were done with REFMAC5 (23) in the CCP4 program suite (24). Manual rebuilding of the model was done with Coot (25).
Table 2.
Crystallographic data collection and refinement parametersa
| Parameter | OXA-23 | OXA-146 |
|---|---|---|
| Cell constants (Å [a, b, c] or ° [α, β, γ]) | a = 44.00, b = 46.53, c = 136.85 | a = 44.12, b = 46.66, c = 137.02 |
| α = β = γ = 90 | α = β = γ = 90 | |
| Space group | P21221 | P21221 |
| Resolution (Å) | 1.61 (1.67–1.61) | 1.20 (1.24–1.20) |
| No. of unique reflections | 37,261 | 83,827 |
| Total no. of reflections | 295,244 | 269,529 |
| Rmerge (%) | 7.2 (41.0) | 6.3 (53.5) |
| Completeness (%)b | 99.9 (100.0) | 94.4 (96.6) |
| <I/σI> | 14.8 (5.3) | 7.7 (1.9) |
| Resolution range for refinement (Å) | 136.85–1.61 | 50–1.20 |
| No. of protein residues | 243 | 244 |
| No. of water molecules | 255 | 286 |
| RMSD for bond lengths (Å) | 0.012 | 0.007 |
| RMSD for bond angles (°) | 1.574 | 1.128 |
| R factor (%) | 0.164 | 0.185 |
| Rfree (%)c | 0.192 | 0.201 |
| Avg B factor (Å2), protein atoms | 26.07 | 22.21 |
| Avg B factor (Å2), water molecules | 37.73 | 34.67 |
Values in parentheses are for the highest-resolution shell.
Fraction of theoretically possible reflections observed.
Rfree was calculated with 5% of reflections set aside randomly.
Protein data accession numbers.
The coordinates and structure factors for OXA-23 and OXA-146 have been deposited in the Protein Data Bank with accession numbers 4K0X and 4K0W, respectively.
RESULTS AND DISCUSSION
A sequence alignment of OXA-146 with two of the most clinically relevant CHDL enzymes, OXA-24 and OXA-23, shows that OXA-146 differs from OXA-23 by the duplication of a single alanine residue (A220) (Fig. 2). Interestingly, another OXA-23 subfamily variant (OXA-49; GenBank accession number AAP40270) contains this same alanine insertion, along with an additional K148E mutation. OXA-49 was also isolated in China but was reported 6 years earlier. In both variants, the alanine duplication occurs in the loop connecting β-strands β5 and β6, a structural element that has been implicated in acquisition of carbapenemase activity (12). We therefore wondered if the alanine duplication might have an effect on carbapenem hydrolysis.
Fig 2.

Multiple-sequence alignment of OXA-24, OXA-23, and OXA-146. OXA-23 and OXA-24 share 59% identity, and OXA-146 is identical to OXA-23 except for a duplication of A220.
We used steady-state kinetic analysis to determine the Km and kcat values for OXA-23, OXA-24/40, and OXA-146 for seven β-lactam substrates (ampicillin, imipenem, doripenem, cefotaxime, ceftriaxone, ceftazidime, and aztreonam) (Table 1). OXA-24/40 and OXA-23 share the core active site residues thought to be important for basic β-lactam binding and hydrolytic cleavage (S81, K84, S128, V130, W167, L168, K218, and R261; OXA-24/40 numbering) (8). Moreover, the bridge residues critical for carbapenem binding in OXA-24/40 (Y112 and M223) appear to be well conserved in OXA-23 (F110 and M221). Not surprisingly, the substrate profiles of the two enzymes for representative penicillin, cephalosporin, and carbapenem substrates are quite similar (Table 1). In particular, the Km for doripenem is less than 30 nM in both enzymes. Both have a relatively low turnover number for doripenem, though OXA-24 is a bit more active than OXA-23 (kcat values of 0.074 s−1 and 0.028 s−1, respectively).
Table 1.
Kinetic parameters for OXA-24, OXA-23, and OXA-146
| Enzyme and substrate | Km (μM) | kcat (s−1) | kcat/Km (μM−1 s−1) |
|---|---|---|---|
| OXA-24 | |||
| Ampicillin | 180 ± 20 | 480 ± 20 | 2.6 ± 0.3 |
| Imipenem | 0.752 ± 0.10 | 2.1 ± 0.04 | 2.8 ± 0.4 |
| Doripenem | 0.024 ± 0.003 | 0.074 ± 0.001 | 3.1 ± 0.4 |
| Cefotaxime | 750 ± 70 | 0.38 ± 0.01 | 0.00050 ± 0.00005 |
| Ceftriaxone | 114 ± 13 | 0.035 ± 0.001 | 0.00030 ± 0.00004 |
| Ceftazidime | < 0.01 | ||
| Aztreonam | >3,000 | ||
| OXA-23 | |||
| Ampicillin | 82 ± 9 | 460 ± 10 | 5.7 ± 0.6 |
| Imipenem | 0.204 ± 0.023 | 0.490 ± 0.01 | 2.4 ± 0.3 |
| Doripenem | 0.018 ± 0.002 | 0.028 ± 0.003 | 1.5 ± 0.3 |
| Cefotaxime | 340 ± 30 | 5.5 ± 0.1 | 0.016 ± 0.002 |
| Ceftriaxone | 3.7 ± 0.5 | 0.016 ± 0.001 | 0.0044 ± 0.0007 |
| Ceftazidime | < 0.01 | ||
| Aztreonam | 2,400 ± 140 | 0.24 ± 0.01 | 0.00010 ± 0.00001 |
| OXA-146 | |||
| Ampicillin | 16 ± 3 | 95 ± 3 | 6.0 ± 1 |
| Imipenem | 0.046 ± 0.01 | 0.043 ± 0.001 | 0.93 ± 0.18 |
| Doripenem | 0.0041 ± 0.0009 | 0.029 ± 0.001 | 7.1 ± 1.6 |
| Cefotaxime | 13 ± 1 | 2.8 ± 0.1 | 0.22 ± 0.02 |
| Ceftriaxone | 0.039 ± 0.012 | 0.222 ± 0.008 | 5.7 ± 1.7 |
| Ceftazidime | 160 ± 30 | 2.9 ± 0.1 | 0.018 ± 0.003 |
| Aztreonam | 2.18 ± 0.44 | 0.39 ± 0.01 | 0.18 ± 0.04 |
A significant trend differentiating the two enzymes involves a somewhat lower Km observed for OXA-23 with regard to ampicillin (82 μM versus 180 μM for OXA-24) and the third-generation cephalosporins cefotaxime (340 μM versus 750 μM) and ceftriaxone (3.7 μM versus 114 μM). Additionally, OXA-24/40 does not display detectable binding or hydrolytic activity toward the monobactam aztreonam, while OXA-23 has weak binding and turnover values for that drug (Km = 2400 μM; kcat = 0.24 s−1). Neither enzyme showed any turnover or binding (as measured by competition with a reporter substrate) of ceftazidime.
We were surprised that OXA-146, with an alanine duplication in a region known to affect carbapenem turnover, hydrolyzed doripenem with essentially the same efficacy as the parental OXA-23 enzyme. When we tested the variant for activity against cefotaxime and ceftriaxone, we found that the alanine insertion resulted in ∼20-fold and 100-fold decreases in Km, respectively, and a notable increase in kcat for ceftriaxone. Ampicillin also displayed a 5-fold decrease in Km. Most surprising was the fact that the alanine duplication allowed OXA-146 to bind ceftazidime with reasonably good affinity (Km = 160 μM) and a significant turnover rate (kcat = 2.9 s−1). The resulting kcat/Km of 0.018 μM−1 s−1 is equal to or greater than values observed for other class D or class A ESBL enzymes that yield “resistant” MIC values for ceftazidime (15, 26). Because ceftazidime and the monobactam aztreonam share the same bulky oxyimino side chain, we sought to determine if the alanine duplication enhanced hydrolysis of the latter. Indeed, the OXA-146 Km value for aztreonam is over 1,000-fold lower than that observed for OXA-23, while the kcat values for the two enzymes are similar. While we suspect that these lower Km values represent tighter binding affinity for these substrates, effects on catalytic rate constants may also contribute to the decrease observed.
In order to elucidate the mechanism for these gain-of-function effects observed with OXA-146, the structures of that enzyme and its parent enzyme OXA-23 were determined by X-ray crystallography. Both OXA-23 and OXA-146 crystals formed in the P21221 space group with one monomer in the asymmetric unit and yielded high-quality diffraction data sets (1.6 Å and 1.2 Å resolution, respectively) (Table 2). The final model of OXA-23 contained 243 amino acid residues, 255 water molecules, and two bicarbonate ions. The final OXA-146 model contained 234 residues, 283 water molecules, four bicarbonate ions, five sodium ions, one citrate molecule, and four ethylene glycol molecules. The citrate molecule of OXA-146 is bound in the active site, similar to what is seen for polycarboxylate ligands observed in other class D β-lactamase structures. It overlaps strongly with the tartrate molecule observed in OXA-46 (3IF6), as both ligands interact with the arginine that typically stabilizes the C3/C4 carboxylate of β-lactam substrates (R260 in OXA-23) (27). It also partially overlaps a citrate molecule in OXA-10 (2WGV) (28). The presence of citrate in the active site of OXA-146 strengthens the case for using polycarboxylates as scaffolds for inhibitor design (29). Quality of the final models was analyzed with MolProbity (30). For OXA-23, 98% of the residues were in the favored region, with no outliers of the Ramachandran plot, and for OXA-146, 96% of residues were in the favored region, with no outliers.
Three-dimensional structural alignments of OXA-23 and OXA-24 (PDB 3PAE) show very strong structural similarity for the overall topological main-chain trace (RMSD for Cα atoms = 0.471 Å) (Fig. 3A), as well as the key active-site residue side chains. Electron density maps of the active site show that K82 is fully carboxylated and the resulting carbamate moiety is stabilized by hydrogen bonds with W165 and S79 (Fig. 3B). Of particular note is the tight overlap of the serine nucleophile (S79), V128, and W165 between OXA-23 and OXA-24 (Fig. 3C). The root mean square deviation (RMSD) for all atoms of eight key active site residues (S79, S126, V128, W165, L166, K216, G218, and R259) is 0.325 Å, indicating high conservation of structure.
Fig 3.

Comparison of OXA-23 and OXA-24 structures. (A) Structural alignment of the α-carbon traces of OXA-23 (cyan) and OXA-24 (yellow; PDB accession no. 3PAE). Also shown are residues that form the bridge over the top of the active site (F110/M221 in OXA-23 and Y112/M223 in OXA-24). (B) 2Fo-Fc electron density maps of key active-site residues of OXA-23 contoured to 1.0 σ showing full carboxylation of K82. (C) Structural alignment of OXA-23 (cyan) and OXA-24 (yellow) active-site residues showing the high degree of overlap (OXA-24 structure 3PAE contains a K84D substitution).
In addition to the conservation observed for the core catalysis machinery, the structural alignment shows that the two enzymes possess a nearly identical hydrophobic bridge across the top of the active site (Fig. 3A). We interpret this observation to mean that this conservation is responsible for the highly similar substrate kinetic profiles for these two enzymes. The presence of the bridge also clearly distinguishes OXA-23 and OXA-24/40 from OXA-48, which has a more open active site (10). The bridge has been shown to be important for tight carbapenem binding (8, 9) and suggests that both OXA-23 and OXA-24/40 use the same mechanism to achieve tight substrate binding in order to compensate for a fairly low turnover rate for those substrates.
In one notable instance, there is a significant difference between the OXA-23 and OXA-24/40 active-site structures. There is a deviation in the backbone trace between the enzymes in the area of the loop connecting β-strands β5 and β6 (Fig. 3A). In OXA-24/40, the presence of glycine at position 224 allows a close approach of this loop to V167 across the active site. In OXA-23, however, this position is occupied by aspartate (D222), and that residue's β-carbon pushes the β5-β6 loop back 1.9 Å (as measured by the distance between the Cα atoms of D222 in OXA-23 and G224 in OXA-24). A220 and I225 in OXA-23 are both bulkier than their sequence homologues in OXA-24 (G218 and V223) and may thus contribute to the altered position of the loop as well. This backbone deviation likely explains the modest advantage in binding affinity observed for OXA-23 compared to OXA-24/40 with respect to ampicillin, cefotaxime, ceftriaxone, and aztreonam. The loop area is where one would expect a close approach from the R1 side chains of these drugs (31, 32), and the larger area provided by the OXA-23 loop position would likely accommodate those groups better.
Given that OXA-146 differs from OXA-23 by only a single alanine insertion, it is not surprising that the two structures are nearly identical: the RMSD for Cα atoms is 0.187 Å. The alanine insertion, however, does cause a striking local deviation of the β5-β6 loop. First, the second alanine takes the position normally occupied by the bridge methionine (M221 in OXA-23, now M222), pushing the latter residue one position into the β5-β6 loop (Fig. 4A and B). In this position, the methionine side chain is displaced 4.1 Å from its OXA-23 location (distance measured between β-carbons) and no longer bridges the active site. The side chain of M222 not only is laterally shifted but also rotates ∼90° from its OXA-23 orientation and could not possibly interact with Phe110. Additionally, the extra residue extends the length of the β5-β6 loop, causing it to move 2.0 to 2.5 Å further away from the core of the enzyme. The loop deviation between OXA-23 and OXA-146 is mostly limited to the region of M222 and D223 in the latter, with strong structural alignment fully resuming within the next two residues (I224 and K225 in OXA-146) as the chain prepares to enter the protein core as strand β6 (Fig. 4B).
Fig 4.

Structure of the loop between β strands β5 and β6. (A) Stereodiagram showing OXA-146 residues 218 to 228 with 2Fo-Fc electron density maps contoured to 1.0 σ. Residue V228 is shown in multiple conformations. (B) Structural alignment of the same loop region from OXA-146 (green) and OXA-23 (cyan).
We note that the side chain of the active site lysine K82 of OXA-146 adopts a more extended conformation than that seen for OXA-23, in which the lysine curves back toward the catalytic serine. Additionally, K82 remains uncarboxylated, a condition that is almost certainly due to the low pH of the crystallization buffer (pH 4.2). The loss of the carbamate moiety at low pH has been documented numerous times both for crystallization experiments and in solution (8, 33, 34). In its uncarboxylated and extended form, the side chain K82 would clash with the canonical structure of the S126-V128 loop (S115-V117 in OXA-10), and this loop in our OXA-146 structure deviates from its form seen in OXA-23 (this paper) and OXA-24 (8, 9).
Residues 105 to 114 were not included in the final OXA-146 model due to poor electron density observed in that region. We have observed disorder and poor-quality density for this loop in many OXA-23 and OXA-23 mutant structures that we have determined, with the key exception being the wild-type OXA-23 structure described above (other structures not shown). We also note that the Phe110 and Met221 residues of the wild-type OXA-23 “bridge” do not interact with each other (6.0 Å between the closest atoms), in contrast to the Tyr112/Met223 bridge of OXA-24 (3.4 Å between the closest atoms; PDB accession no. 3PAE). It is therefore highly unlikely that the disorder of residues 105 to 114 contributes to the loop deviations we observe in the β5-β6 loop.
In order to see if these loop deviations in OXA-146 might be responsible for the increase in ceftazidime binding, we superposed the structures of OXA-23, OXA-146, and the closely related β-lactam sensor protein BlaR1 with ceftazidime bound as an acyl adduct (32). As seen in Fig. 5A, the bulky Y-shaped R1 oxyimino side chain of the ceftazidime would likely clash sterically with OXA-23 in two places. Foremost, the R1 carboxy group of the drug (unique to ceftazidime and aztreonam) is incompatible with the location of the side chain of the bridge methionine M221. The replacement of that side chain by the duplicated alanine naturally alleviates that steric clash. Next, the aminothiazole ring of the ceftazidime side chain would likely clash with the main chain of the β5-β6 loop, most notably in the area of D222. It is easy to see in this model that the extension of this loop away from the active site would be expected to greatly lessen this steric incompatibility. These same two structural changes in OXA-146 would be expected to have a similar interaction with aztreonam, which has the same side chain as ceftazidime. It is interesting, however, that gains of activity against ceftazidime and aztreonam do not always occur together in class D ESBLs (e.g., OXA-14) (32, 35).
Fig 5.
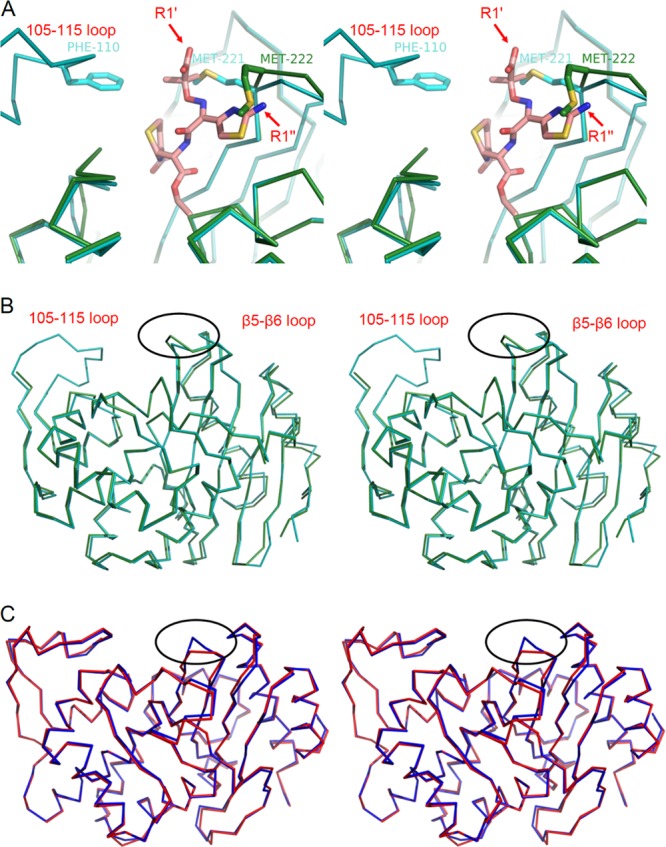
Overlaid structures showing the β5-β6 loop structural alteration. (A) Stereodiagram of OXA-146 (green) and OXA-23 (cyan) after alignment with the β-lactam sensor protein BlaR1 with ceftazidime bound as an acyl intermediate (PDB accession no. 1XKZ). The ceftazidime serine acyl moiety is shown in salmon, and the rest of the BlaR1 protein is not shown. The labels R1′ and R1″ indicate the carboxyl group and aminothiazole of the R1 side chain, respectively. The OXA-23 bridge methionine (M221) is shown in only one of its two alternative conformations, and the equivalent residue in OXA-146 (M222) is shown in its new shifted position after the insertion of the additional alanine (A221). (B) Stereodiagram showing the position of the β5-β6 loop deviation (circled) in the context of the full structures of OXA-23 and OXA-146. (C) Stereodiagram showing an alignment of SHV-1 (red; PDB accession no. 1SHV) and SHV-2 (blue; PDB accession no. 1N9B). The position of a similar loop deviation resulting in ceftazidime activity is shown (circled).
As noted before, most ESBL mutations in class D occur in the OXA-10 subfamily and cluster in an area near the carbamate-stabilizing tryptophan (W154; OXA-10 numbering). This tryptophan is found in the substrate-selecting “omega loop,” which makes contact with the β5-β6 loop. Deletion of the leucine next to this tryptophan, or substitution for the tryptophan itself, has been shown to yield enzymes that confer ceftazidime resistance (15). Such mutations are known to increase the flexibility of the omega loop and may enhance the substrate selectivity function of that structural element (28). These mutations are also known to decrease the stability of the carbamate carboxylation of K70 (in OXA-10), leading to a loss of penicillinase activity (15, 28). OXA-146 maintains good ampicillinase activity, suggesting that gain-of-function mutations in the β5-β6 loop do not acquire ESBL activity by a similar mechanism.
There is one previous example of a mutation in the β5-β6 loop of a class D β-lactamase leading to increased activity against extended-spectrum cephalosporins. OXA-163 is a variant of the carbapenemase OXA-48 in which 4 amino acids from that loop have been deleted (36). While this deletion leads to increases in turnover for ceftazidime and aztreonam, there is also a notable loss of activity against carbapenems such as imipenem and ertapenem (36).
An increase in hydrolysis of ceftazidime brought about by conformational changes of the β5-β6 loop has also been observed in other classes. The G238S substitution increases ceftazidimase activity in the SHV and TEM subfamilies of class A β-lactamases (37, 38). The crystal structure of SHV-2 (SHV-1 with a G238S substitution; PDB accession no. 1N9B) shows that the increased bulk of the serine side chain moves the main chain of the next 2 to 3 residues back ∼1 to 2 Å, creating a larger active site that would be expected to better accommodate the bulky aminothiazole ring of ceftazidime (39). Overlays of SHV-1/2 and OXA-23/146 reveal that this widened active site occurs in the same spot in both proteins, suggesting that this mode of ESBL property acquisition is broadly shared (Fig. 5B and C). A class C β-lactamase from Enterobacter cloacae (GC1) gains extended spectrum activity through a three-amino-acid insertion in an active site loop (40). This insertion occurs in a different loop (the “omega loop”), but like the mutations found in OXA-146 and SHV-2, it results in a widened active site (41). A pentapeptide duplication in the omega loop of SHV-1 (creating SHV-16) also results in a significant increase in ceftazidime resistance (42).
It is likely the alanine duplication results in a similar expansion of substrate selection for OXA-49, which contains that insertion as well as an additional missense mutation (K148E). The second substitution resides on the surface of the protein away from the active site (∼20 Å from the catalytic serine) and may simply represent a random genetic drift. Alternatively, this mutation may provide some additional advantageous kinetic effect or act as a global stabilizer of protein structure, as seen with the L201P substitution of TEM-1 (43).
OXA-23 and OXA-24 together are responsible for a large part of the very dangerous and still emerging threat of carbapenem resistance in Acinetobacter species. Our observation here that a single residue insertion in OXA-23 leads to a gain of efficiency in breaking down ceftazidime and aztreonam, with no loss of activity toward carbapenem substrates, shows that this threat continues to grow.
ACKNOWLEDGMENTS
This research was supported by National Institutes of Health grant R15AI082416-01 (D.A.L.), R15AI094489 (R.A.P.) and R01AI072219 and R01AI063517 (R.A.B.), as well as by the Veterans Affairs Merit Review Program (to R.A.B.). Funding was also provided by the U.S. Department of Energy Joint Genome Institute through the Office of Science of the U.S. Department of Energy under contract no. DE-AC02-05CH11231. Use of the Advanced Photon Source, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the U.S. DOE under contract no. DE-AC02-06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor (Grant 085P1000817).
Footnotes
Published ahead of print 22 July 2013
REFERENCES
- 1.Poirel L, Naas T, Nordmann P. 2010. Diversity, Epidemiology, and Genetics of Class D β-lactamases. Antimicrob. Agents Chemother. 54:24–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Naas T, Poirel L, Nordmann P. 2008. Minor extended-spectrum β-lactamases. Clin. Microbiol. Infect. 14(Suppl 1):42–52 [DOI] [PubMed] [Google Scholar]
- 3.Walther-Rasmussen J, Hoiby N. 2006. OXA-type carbapenemases. J. Antimicrob. Chemother. 57:373–383 [DOI] [PubMed] [Google Scholar]
- 4.Scaife W, Young HK, Paton RH, Amyes SG. 1995. Transferable imipenem-resistance in Acinetobacter species from a clinical source. J. Antimicrob. Chemother. 36:585–586 [DOI] [PubMed] [Google Scholar]
- 5.Donald HM, Scaife W, Amyes SG, Young HK. 2000. Sequence analysis of ARI-1, a novel OXA β-lactamase, responsible for imipenem resistance in Acinetobacter baumannii 6B92. Antimicrob. Agents Chemother. 44:196–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bou G, Oliver A, Martinez-Beltran J. 2000. OXA-24, a novel class D β-lactamase with carbapenemase activity in an Acinetobacter baumannii clinical strain. Antimicrob. Agents Chemother. 44:1556–1561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Evans BA, Hamouda A, Amyes SG. 2013. The rise of carbapenem-resistant Acinetobacter baumannii. Curr. Pharm. Des. 19:223–238 [PubMed] [Google Scholar]
- 8.Santillana E, Beceiro A, Bou G, Romero A. 2007. Crystal structure of the carbapenemase OXA-24 reveals insights into the mechanism of carbapenem hydrolysis. Proc. Natl. Acad. Sci. U. S. A. 104:5354–5359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schneider KD, Ortega CJ, Renck NA, Bonomo RA, Powers RA, Leonard DA. 2011. Structures of the class D carbapenemase OXA-24 from Acinetobacter baumannii in complex with doripenem. J. Mol. Biol. 406:583–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Docquier JD, Calderone V, De Luca F, Benvenuti M, Giuliani F, Bellucci L, Tafi A, Nordmann P, Botta M, Rossolini GM, Mangani S. 2009. Crystal structure of the OXA-48 β-lactamase reveals mechanistic diversity among class D carbapenemases. Chem. Biol. 16:540–547 [DOI] [PubMed] [Google Scholar]
- 11.Schneider KD, Karpen ME, Bonomo RA, Leonard DA, Powers RA. 2009. The 1.4 Å crystal structure of the class D β-lactamase OXA-1 complexed with doripenem. Biochemistry 48:11840–11847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.De Luca F, Benvenuti M, Carboni F, Pozzi C, Rossolini GM, Mangani S, Docquier JD. 2011. Evolution to carbapenem-hydrolyzing activity in noncarbapenemase class D β-lactamase OXA-10 by rational protein design. Proc. Natl. Acad. Sci. U. S. A. 108:18424–18429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Paetzel M, Danel F, de Castro L, Mosimann SC, Page MG, Strynadka NC. 2000. Crystal structure of the class D β-lactamase OXA-10. Nat. Struct. Biol. 7:918–925 [DOI] [PubMed] [Google Scholar]
- 14.Danel F, Hall LM, Livermore DM. 1999. Laboratory mutants of OXA-10 β-lactamase giving ceftazidime resistance in Pseudomonas aeruginosa. J. Antimicrob. Chemother. 43:339–344 [DOI] [PubMed] [Google Scholar]
- 15.Hocquet D, Colomb M, Dehecq B, Belmonte O, Courvalin P, Plésiat P, Meziane-Cherif D. 2011. Ceftazidime-hydrolysing β-lactamase OXA-145 with impaired hydrolysis of penicillins in Pseudomonas aeruginosa. J. Antimicrob. Chemother. 66:1745–1750 [DOI] [PubMed] [Google Scholar]
- 16.Meziane-Cherif D, Bonnet R, Haouz A, Plésiat P, Courvalin P. 2011. Structure of OXA-145, a ceftazidime hydrolysing class D β-lactamase with impaired hydrolysis of penicillins, abstr C1-599. Abstrs. 51st Intersci. Conf. on Antimicrob. Agents Chemother., Chicago, IL [Google Scholar]
- 17.Hujer KM, Hujer AM, Hulten EA, Bajaksouzian S, Adams JM, Donskey CJ, Ecker DJ, Massire C, Eshoo MW, Sampath R, Thomson JM, Rather PN, Craft DW, Fishbain JT, Ewell AJ, Jacobs MR, Paterson DL, Bonomo RA. 2006. Analysis of antibiotic resistance genes in multidrug-resistant Acinetobacter sp. isolates from military and civilian patients treated at the Walter Reed Army Medical Center. Antimicrob. Agents Chemother. 50:4114–4123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Higuchi R, Krummel B, Saiki RK. 1988. A general method of in vitro preparation and specific mutagenesis of DNA fragments: study of protein and DNA interactions. Nucleic Acids Res. 16:7351–7367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Drawz SM, Babic M, Bethel CR, Taracila M, Distler AM, Ori C, Caselli E, Prati F, Bonomo RA. 2010. Inhibition of the class C β-lactamase from Acinetobacter spp.: insights into effective inhibitor design. Biochemistry 49:329–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Otwinowski Z, Minor W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276:307–326 [DOI] [PubMed] [Google Scholar]
- 21.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. 2007. Phaser crystallographic software. J. Appl. Crystallogr. 40:658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kelley LA, Sternberg MJ. 2009. Protein structure prediction on the Web: a case study using the Phyre server. Nat. Protoc. 4:363–371 [DOI] [PubMed] [Google Scholar]
- 23.Murshudov GN, Vagin AA, Dodson EJ. 1997. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53:240–255 [DOI] [PubMed] [Google Scholar]
- 24.CCP4 (Collaborative Computational Project Number 4) 1994. The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D 50:760–763 [DOI] [PubMed] [Google Scholar]
- 25.Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60:2126–2132 [DOI] [PubMed] [Google Scholar]
- 26.Wang X, Minasov G, Shoichet BK. 2002. Evolution of an antibiotic resistance enzyme constrained by stability and activity trade-offs. J. Mol. Biol. 320:85–95 [DOI] [PubMed] [Google Scholar]
- 27.Docquier JD, Benvenuti M, Calderone V, Giuliani F, Kapetis D, De Luca F, Rossolini GM, Mangani S. 2010. Crystal structure of the narrow-spectrum OXA-46 class D β-lactamase: relationship between active-site lysine carbamylation and inhibition by polycarboxylates. Antimicrob. Agents Chemother. 54:2167–2174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baurin S, Vercheval L, Bouillenne F, Falzone C, Brans A, Jacquamet L, Ferrer JL, Sauvage E, Dehareng D, Frère JM, Charlier P, Galleni M, Kerff F. 2009. Critical role of tryptophan 154 for the activity and stability of class D β-lactamases. Biochemistry 48:11252–11263 [DOI] [PubMed] [Google Scholar]
- 29.Beck J, Vercheval L, Bebrone C, Herteg-Fernea A, Lassaux P, Marchand-Brynaert J. 2009. Discovery of novel lipophilic inhibitors of OXA-10 enzyme (class D β-lactamase) by screening amino analogs and homologs of citrate and isocitrate. Bioorg. Med. Chem. Lett. 19:3593–3597 [DOI] [PubMed] [Google Scholar]
- 30.Chen VB, Arendall WB, III, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. 2010. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66:12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vercheval L, Bauvois C, di Paolo A, Borel F, Ferrer JL, Sauvage E, Matagne A, Frère JM, Charlier P, Galleni M, Kerff F. 2010. Three factors that modulate the activity of class D β-lactamases and interfere with the post-translational carboxylation of Lys70. Biochem. J. 432:495–504 [DOI] [PubMed] [Google Scholar]
- 32.Birck C, Cha JY, Cross J, Schulze-Briese C, Meroueh SO, Schlegel HB, Mobashery S, Samama JP. 2004. X-ray crystal structure of the acylated β-lactam sensor domain of BlaR1 from Staphylococcus aureus and the mechanism of receptor activation for signal transduction. J. Am. Chem. Soc. 126:13945–13947 [DOI] [PubMed] [Google Scholar]
- 33.Golemi D, Maveyraud L, Vakulenko S, Samama JP, Mobashery S. 2001. Critical involvement of a carbamylated lysine in catalytic function of class D β-lactamases. Proc. Natl. Acad. Sci. U. S. A. 98:14280–14285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pernot L, Frénois F, Rybkine T, L'Hermite G, Petrella S, Delettré J, Jarlier V, Collatz E, Sougakoff W. 2001. Crystal structures of the class D β-lactamase OXA-13 in the native form and in complex with meropenem. J. Mol. Biol. 310:859–874 [DOI] [PubMed] [Google Scholar]
- 35.Danel F, Hall LM, Gur D, Livermore DM. 1995. OXA-14, another extended-spectrum variant of OXA-10 (PSE-2) β-lactamase from Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 39:1881–1884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Poirel L, Castanheira M, Carrër A, Rodriguez CP, Jones RN, Smayevsky J, Nordmann P. 2011. OXA-163, an OXA-48-related class D β-lactamase with extended activity toward expanded-spectrum cephalosporins. Antimicrob. Agents Chemother. 55:2546–2551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hujer AM, Hujer KM, Bonomo RA. 2001. Mutagenesis of amino acid residues in the SHV-1 β-lactamase: the premier role of Gly238Ser in penicillin and cephalosporin resistance. Biochim. Biophys. Acta 1547:37–50 [DOI] [PubMed] [Google Scholar]
- 38.Cantu C, III, Huang W, Palzkill T. 1996. Selection and characterization of amino acid substitutions at residues 237–240 of TEM-1 β-lactamase with altered substrate specificity for aztreonam and ceftazidime. J. Biol. Chem. 271:22538–22545 [DOI] [PubMed] [Google Scholar]
- 39.Nukaga M, Mayama K, Hujer AM, Bonomo RA, Knox JR. 2003. Ultrahigh resolution structure of a class A β-lactamase: on the mechanism and specificity of the extended-spectrum SHV-2 enzyme. J. Mol. Biol. 328:289–301 [DOI] [PubMed] [Google Scholar]
- 40.Nukaga M, Haruta S, Tanimoto K, Kogure K, Taniguchi K, Tamaki M, Sawai T. 1995. Molecular evolution of a class C β-lactamase extending its substrate specificity. J. Biol. Chem. 270:5729–5735 [DOI] [PubMed] [Google Scholar]
- 41.Crichlow GV, Kuzin AP, Nukaga M, Mayama K, Sawai T, Knox JR. 1999. Structure of the extended-spectrum class C β-lactamase of Enterobacter cloacae GC1, a natural mutant with a tandem tripeptide insertion. Biochemistry 38:10256–10261 [DOI] [PubMed] [Google Scholar]
- 42.Arpin C, Labia R, Andre C, Frigo C, El Harrif Z, Quentin C. 2001. SHV-16, a β-lactamase with a pentapeptide duplication in the omega loop. Antimicrob. Agents Chemother. 45:2480–2485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Marciano DC, Pennington JM, Wang X, Wang J, Chen Y, Thomas VL, Shoichet BK, Palzkill T. 2008. Genetic and structural characterization of an L201P global suppressor substitution in TEM-1 β-lactamase. J. Mol. Biol. 384:151–164 [DOI] [PMC free article] [PubMed] [Google Scholar]