

Abstract
The 5′ terminal nucleotide sequence of a gene is often a bottleneck in recombinant protein production. The ifn-α2bS gene is poorly expressed in Escherichia coli unless a translocation signal sequence (pelB) is fused to the 5′ end of the gene. A combined in silico and in vivo analysis reported here further indicates that the ifn-α2bS 5′ coding sequence is suboptimal for efficient gene expression. ifn-α2bS therefore presents a suitable model gene for describing properties of 5′ fusions promoting expression. We show that short DNA sequences corresponding to the 5′ end of the highly expressed celB gene, whose protein product is cytosolic, can functionally replace pelB as a 5′ fusion partner for efficient ifn-α2bS expression. celB fusions of various lengths (corresponding to a minimum of 8 codons) led to more than 7- and 60-fold stimulation of expression at the transcript and protein levels, respectively. Moreover, the presence of a celB-based fusion partner was found to moderately reduce the decay rate of the corresponding transcript. The 5′ fusions thus appear to act by enhancing translation, and bound ribosomes may accordingly contribute to increased mRNA stability and reduced mRNA decay. However, other effects, such as altered protein stability, cannot be excluded. We also developed an experimental protocol that enabled us to identify improved variants of the celB fusion, and one of these (celBD11) could be used to additionally increase ifn-α2bS expression more than 4-fold at the protein level. Interestingly, celBD11 also stimulated greater protein production of three other medically important human genes than the wild-type celB fragment.
INTRODUCTION
The current availability of strong and well-characterized promoter systems practically guarantees sufficient transcript amounts for production of recombinant proteins, provided that downstream expression processes are effective. However, this is frequently not the case (1, 2). A central control point in expression of bacterial genes is the transcript's 5′ terminal end that contains the start codon, which defines both the start of translation and the reading frame, and the Shine-Dalgarno (SD) sequence within the 5′ untranslated region (5′-UTR), which facilitates 16S rRNA-specific ribosome binding (3). The encompassing ribosomal binding site (RBS) is usually defined as a segment of mRNA sterically protected by the ribosome against RNase digestion and consists of about 15 to 25 nucleotides on each side of the start codon (4, 5).
Multiple lines of evidence have suggested that structural features of the RBS quantitatively control the efficiency of translation by modulating ribosome binding (6–9). Although less has been published about sequence structural features in the 5′ coding region than those in the 5′-UTR, several studies have directly or indirectly implicated the importance of folding free energy at the beginning of a coding sequence (10–12) and documented the interplay between the SD, the initiation codon, and the 5′ coding region in translation initiation (13–16). In addition to the role in translation initiation, structural features such as stable 5′-terminal stem-loops strongly affect half-lives of bacterial mRNAs (17–19) by facilitating protection from RNase E-mediated degradation in Escherichia coli (20, 21). Also, ribosome binding to the RBS typically appears to protect mRNAs from RNase attack (22, 23), although such protection does not always lead to more protein product (24, 25). A dual function of the ribosomal protein S1 represents a possible link between translation and mRNA degradation in controlling gene expression. This essential mRNA binding protein has been described to have a role in the control of both translation and mRNA stability, presumably due to an overlap of its binding site with the RNase E cleavage sites upstream of the SD sequence (26–29).
We previously showed that the codon-optimized ifn-α2bS gene, encoding the medically important cytokine alpha interferon 2b (IFN-α2b), is poorly expressed in E. coli, unless a translocation signal sequence like pelB is used as its 5′ fusion partner (30). The use of the strong Pm promoter (30–32) should ensure that sufficient amounts of transcripts are produced, an assumption that is in agreement with our recent finding that relatively high levels of ifn-a2bS transcript can be reached also in the absence of the pelB signal sequence (33). The pelB fusion partner is therefore likely involved in the stimulation of processes downstream of transcription and not transcription per se. Generally, short translocation signal sequences have previously been shown to have a high positive impact on the expression level (34–36), but it is not clear to what extent the translocation process itself is involved in this stimulation.
By using ifn-α2bS as a model, we here report an investigation of the importance of the length of the fusion partner and to what extent translocation is required to achieve high protein production levels. We also developed a procedure for identification of improved fusion partners via random mutagenesis of an oligonucleotide sequence that does not represent a translocation signal. The results demonstrate that such improved variant sequences can be identified, and by using a selected example, we also show that it quite significantly stimulated the expression of all tested target genes.
MATERIALS AND METHODS
Biological materials, DNA manipulation, and growth conditions.
Standard DNA manipulations, E. coli cultivation, and basic expression studies were performed as described previously (37). Kanamycin (50 mg/liter) or ampicillin (200 mg/liter) was added as the selection marker when appropriate. For expression experiments, induction of the XylS-Pm system was done by adding m-toluic acid to a final concentration of 0.5 mM. E. coli strains and plasmids used in this study are listed in Table 1. Custom PCR primers and oligonucleotides (see Table S1 in the supplemental material) were supplied by Eurofins MWG Operon or Sigma-Aldrich Co. Spiked oligonucleotide mixtures used for combinatorial library construction were supplied by MedProbe AS. DNA sequencing was performed by Eurofins MWG Operon.
Table 1.
Bacterial strains and plasmids
| Strain or plasmid | Descriptiona | Reference/source |
|---|---|---|
| E. coli strains | ||
| DH5α | General cloning and expression host | Bethesda Research Laboratories |
| RV308 | Production strain | ATCC 31608 |
| Plasmids | ||
| pIFN30S | RK2-based vector expressing the IFN-α2bS–c-Myc–His6 fusion protein from the XylS-Pm expression cassette; Apr; 8.8 kb | 30 |
| pIFN30SpelB | RK2-based vector expressing the PelB–IFN-α2bS–c-Myc–His6 fusion protein from the XylS-Pm expression cassette; Apr; 8.9 kb | 30 |
| pLB11 | RK2-based expression vector containing the XylS-Pm promoter system and celB as a reporter gene for Pm; Kmr; 9.0 kb | 37 |
| pIFN30Seq_X | pIFN30S derivatives containing ifn-α2bS synonymous variants; X corresponds to seq_01, seq_03 seq_09, seq_74, seq_75, seq_81, or seq_98 | This study |
| pIFNcelBN | pIFN30SpelB derivative where the pelB signal sequence was exchanged with various celB fragments, N = 3, 5, 6, 7, 8, 9, 10, 15, 20, 23, 25, 30, 38, or 69; N denotes no. of codons of a fusion partner | This study |
| pGM29 | RK2-based vector expressing the GM-CSF–c-Myc–His6 fusion protein from the XylS-Pm expression cassette; Apr; 8.7 kb | 30 |
| pGM29ompA | RK2-based vector expressing the OmpA–GM-CSF–c-Myc–His6 fusion protein from the XylS-Pm expression cassette; Apr; 8.8 kb | 30 |
| pGM29celB23 | pGM29ompA derivative where the ompA signal sequence was exchanged with the celB23 fragment | This study |
| pMA-T-G-CSF | pMA vector (GeneArt; Invitrogen) with the g-csf insert, provided by Vectron Biosolutions; Apr; 3.0 kb | Unpublished |
| pMA-T-TNF-α1a | pMA vector (GeneArt; Invitrogen) with the tnf-α1a insert, provided by Vectron Biosolutions; Apr; 3.0 kb | Unpublished |
| pG-CSFNF | Derivative of pIFN30S expressing G-CSF–c-Myc–His6 fusion protein from the XylS-Pm expression cassette; Apr; 8.9 kb | This study |
| pG-CSFcelB23 | Derivative of pIFNcelB23 expressing the CelB23–G-CSF–c-Myc–His6 fusion protein from the XylS-Pm expression cassette; Apr; 9.0 kb | This study |
| pTNFNF | Derivative of pIFN30S expressing the TNF-α1a–c-Myc–His6 fusion protein from the XylS-Pm expression cassette; Apr; 8.8 kb | This study |
| pTNFcelB23 | Derivative of pIFNcelB23 expressing the CelB23–TNF-α1a–c-Myc–His6 fusion protein from the XylS-Pm expression cassette; Apr; 8.9 kb | This study |
| pBSP1bla | RK2-based expression vector containing the XylS-Pm promoter system and bla as a reporter gene; Kmr; 9.5 kb | 40 |
| pJN100 | Conjugative shuttle expression vector with the pANT1202-derived SnpR (LysR-like protein)-activated snpA promoter; Arr; 6.5 kb | 49 |
| pVK61 | pBSP1 derivative in which the bla reporter gene was replaced by the aac(3)-IV gene; Kmr; 7.9 kb | This study |
| pVK65 | pVK61 derivative with introduced NcoI site at the 5′ terminal of the aac(3)-IV gene; Kmr; 7.9 kb | This study |
| pARcelB23 | Derivative of pVK65 expressing the celB23–aac(3)-IV fusion gene; Kmr; 8.0 kb | This study |
| pARcelBD11 | Derivative of pARcelB23 in which the NdeI-NcoI fragment encoding celB23 was replaced by oligonucleotides encoding the D11 mutant; Kmr; 8.0 kb | This study |
Apr, ampicillin resistance; Kmr, kanamycin resistance; Arr, apramycin resistance.
Vector construction.
The construction of pIFN30Sy_X vectors (X = 01, 03, 09, 54, 74, 75, 81, 82, 88, 98; X denotes the corresponding ifn-α2bS synonymous variant) was based on the pIFN30S plasmid. The DNA fragment containing the ifn-α2bS gene was in each case PCR amplified by using pIFN30S plasmid DNA and one of 10 variant primer pairs of INFSy_X.F and TMB8goi.R. Specific synonymous mutations in the ifn-α2bS 5′ coding sequence were always introduced by the forward primer (INFSy_X.F). The resulting PCR products were NdeI-NotI digested and inserted into the corresponding sites of pIFN30S, replacing the original 501-bp fragment with the ifn-α2bS DNA sequence.
pIFNcelBN vectors (N = 3, 5, 6, 7, 8, 10, 15, 20, 23, 25, 30, or 38; N denotes the number of codons) were made by replacing the 60-bp NdeI-NcoI fragment in pIFN30SpelB, containing the pelB sequence, with annealed oligonucleotides corresponding to the 5′ terminal end of the celB gene. pIFNcelB69 was constructed by PCR amplification of the first 69 codons of celB, using the pLB11 vector as the DNA template (primer pair PmUTR.F and celB69NcoI.R). The resulting DNA fragment was digested with NdeI and NcoI and ligated into the corresponding sites of pIFN30SpelB. Vector pGM29celB23 was constructed in an analogous way, by replacing the 69-bp NdeI-NcoI fragment from pGM29ompA, containing the ompA sequence, with annealed oligonucleotides corresponding to the first 23 codons of celB.
Plasmids pG-CSFNF and pTNFNF are pIFN30S derivatives constructed by exchanging the NcoI-NotI fragment that encodes ifn-α2bS by the coding regions of g-csfS and tnf-α1aS genes, respectively. The DNA fragments encoding g-csfS and tnf-α1aS were generated by NdeI-NotI digestion of pMA-T-G-CSF and pMA-T-TNF-α1a plasmids, respectively. For construction of the pG-CSFcelB23 and pTNFcelB23 vectors, the g-csfS and tnf-α1aS coding regions were PCR amplified from plasmid pMA-T-G-CSF and pMA-T-TNF-α1a, respectively, by using the primer pairs G-CSF–NcoI.F/VectronGOI.R and TNF-NcoI.F/VectronGOI.R, respectively. The resulting DNA fragments were digested with NcoI and NotI and ligated into corresponding sites of the pIFNcelB23 plasmid, replacing the ifn-α2bS coding region.
The plasmid pARcelB23, used for construction of the celB23 mutant library, was based on the vector pBPS1bla. In order to introduce an NcoI site at the beginning of the aac(3)-IV gene, aac(3)-IV was PCR amplified with the primer pair accNcoI.F and acc3IV.R from pJN100. The resulting PCR product was NdeI-EcoRI digested and introduced into the corresponding sites of pBPS1bla, replacing the bla coding region and generating plasmid pVK65. The DNA sequence corresponding to celB23 was introduced as annealed oligonucleotides into the NdeI-NcoI site of pVK65 generating the pARcelB23 vector. Plasmid pARcelBD11 was generated by replacing the NdeI-NcoI fragment containing the celB23 sequence in pARcelB23 with the D11 variant sequence as annealed oligonucleotides. Vectors pIFNcelBD11, pGM29celBD11, pG-CSFcelBD11, and pTNFcelBD11 were created in an analogous way from plasmid pIFNcelB23, pGM29celB23, pG-CSFcelB23, and pTNFcelB23, respectively.
Construction and screening of the combinatorial library in the celB23 fragment.
A strategy involving doped synthetic oligonucleotides was used for introducing random mutations in the celB23 sequence, similarly to the protocol previously described for the Pm promoter and the 5′-UTR region (38, 39). Synthetic oligonucleotides were designed to constitute a double-stranded DNA fragment with NdeI- and NcoI-compatible ends when annealed for subsequent easy cloning into the pARcelB23 vector. Four different nucleotide mixtures, shown in the oligonucleotide sequences by the numbers 1 to 4, were used to synthesize doped oligonucleotide mixtures. The probability of keeping the original base in each position was set to 79%, and the other accepted bases were introduced at equal frequencies. The mixtures used were the following: 1: 79% A, 7% C, 7% G, 7% T; 2: 7% A, 79% C, 7% G, 7% T; 3: 7% A, 7% C, 79% G, 7% T; 4: 7% A, 7% C, 7% G, 79% T. A randomized celBR library was generated by replacing the original celB23 sequence with an oligonucleotide mixture defined by 5′-TATG2221321411322214443223321132233423142233122342443421141423123222433C-3′. The noncoding strand was kept complementary to the original celB23 sequence (5′-CATGGCCAGGGCGTCGATATTGACAAGACGGTCCGGATCGACCGGCTTGCCGGCAAATGGGCTTATGCTGGGCA-3′). The oligonucleotides were annealed as described before (40) and ligated into the NdeI-NcoI-digested and calf intestinal alkaline phosphatase-treated plasmid pARcelB23. The ligation mixture was transferred into DH5α using kanamycin as selection. Approximately 150,000 transformants were mixed to constitute the library.
Screening for variant celB sequences resulting in increased apramycin tolerance levels of the host cells was performed essentially as previously described (40), except that apramycin was used for selection instead of ampicillin.
Measurements of mRNA decay by inducer washout and qRT-PCR analysis.
Recombinant cultures were grown and induced as described above in biological materials, DNA manipulation, and growth conditions. After 90 min of continuous growth (30°C, 200 rpm), the cultures were concentrated by rapid filtration through a Millipore EZ-Pak membrane filter (Millipore) with the use of a vacuum pump. The filter with harvested cells was washed with 10 ml of phosphate-buffered saline (137 mmol/liter NaCl, 2.7 mmol/liter KCl, 8.1 mmol/liter Na2HPO4 · 2H2O, 1.76 mmol/liter KH2PO4 [pH 7.4]), and the cells were resuspended in fresh 30°C prewarmed LB medium without inducer. The culture growth was maintained for another 10 to 30 min. Samples for quantitative reverse transcription-PCR (qRT-PCR) analysis were collected directly after filter transfer and cell resuspension and at several time points after transfer. Immediately after collection, each sample was treated with the stabilizing RNAprotect cell reagent (Qiagen). Total RNA isolation, cDNA preparation, and qRT-PCR were performed as described previously (38). Oligonucleotides used for transcript quantification were the previously used qRT-PCR primers for ifn-α2bS and gm-csf (33), the APR69-263F/APR69-327R primer pair [the acc(3)-IV gene], and the primer pair used to amplify a fragment from the 16S rRNA gene that was applied as a normalizer (38). All experiments were repeated at least twice, and measurements were carried out with a minimum of three technical replicates. Computational estimation of relative decay rates was performed as described in reference 33.
Quantification of recombinant proteins by SDS-PAGE/Western blot analysis.
For analysis of recombinant protein production levels, exponentially growing cells were induced with m-toluic acid as described above, and cell growth was continued for 5 h. Qualitative detection of recombinant proteins was performed by using SDS-PAGE and Western blotting essentially as previously described (41), except that direct detection with HisProbe-horseradish peroxidase (HRP) (Thermo Scientific) was applied for His-tagged proteins. Crude extracts were prepared by sonication four times each for 90 s, with 30-s cooling periods (Branson sonifier, 30% duty control, 3 output control). Total protein concentration was determined with the Bio-Rad detergent-compatible protein assay (Bio-Rad Laboratories, Hercules, CA, USA) as described by the manufacturers, and the maximum protein concentration of a sample of cell lysate was ∼20 to 25 μg/μl. The maximum volume of a protein gel well was 15 μl, making the maximum capacity of a protein gel well ∼300 to 375 μg. Signals were developed by using Pierce ECL Western blot substrate for chemiluminescent detection according to the manufacturer's instructions. Chemiluminescence was detected by the ChemiDoc XRS imaging system and analyzed by Image Lab 4.0 software (Bio-Rad Laboratories.)
Construction of the in silico library of ifn-α2bS and bioinformatic sequence analysis and in silico analysis of 5′ fusion sequences.
The combinatorial library of synonymous ifn-α2bS sequence variants was created with the use of the Python programming tool by allowing all possible synonymous codon substitutions except for very rare codons (less than 5% frequency of usage) in the first eight codons after the ATG start site. Each synonymous variant was subsequently ranked with respect to (i) the sum of the codon usage frequency in the eight variable codons (defined as the codon usage index) and (ii) the free folding energy of the −32 to +30 region (where +1 corresponds to the A of the ATG translation initiation codon). The −32 to −1 region is the Pm promoter-associated 5′-UTR (sequence shown in reference 39), and the +1 to +30 region represents the first 10 codons of the gene coding sequence. Codon usage frequencies were calculated from coding regions of all genes in the E. coli K-12 genome obtained from the Ensembl project databases (http://www.ensembl.org). To calculate the free folding energies, we used the hybrid-ss-min program from the UNAfold package (42). Predictions of translation initiation rates were carried out as previously described (37) by the reverse engineering tool of the RBS calculator (43).
In order to characterize the nature of 5′ terminal sequences, the translated sequence of ifn-α2bS or the celB-based fusion/pelB signal sequence added to the 5′ terminal of the ifn-α2bS gene was submitted to the SignalP 4.0 server (http://www.cbs.dtu.dk/services/SignalP/) (44), which predicts the presence and location of signal peptide cleavage sites in amino acid sequences. The predicted discrimination score (D-score) was used to discriminate signal peptides from nonsignal peptides. The PelB–IFN-α2b protein served as a positive control (D-score, 0.881), the IFN-α2b protein served as a negative control (D-score, 0.157), the default cutoff value discriminating a signal peptide from a nonsignal peptide was 0.570, and the D-scores of fusion proteins CelB23–IFN-α2b and CelBD11–IFN-α2b were predicted to be 0.109 and 0.180, respectively.
RESULTS AND DISCUSSION
Rationally designed synonymous variants of the ifn-α2bS 5′ coding sequence can lead to strong stimulation of transcript accumulation without a corresponding increase in protein production.
To test whether predictions based on bioinformatics analyses could be used to achieve high-level expression of ifn-α2bS without interfering with the native amino acid sequence, a procedure involving three steps was used. First, we generated in silico a combinatorial synonymous library of the first 9 codons of ifn-α2bS that after exclusion of very rare codons represented 83.3% of all possible combinations of synonymous codon substitutions (see Materials and Methods). Next, the resulting 7,680 sequences were bioinformatically sorted with respect to minimum folding free energy and codon usage of the 5′ region (Fig. 1). A few common characteristics, such as an unstructured mRNA sequence near the RBS and frequent occurrence of rare codons at the 5′ end, have been suggested to positively influence translation (10, 45). We, therefore, as the last step, selected several ifn-α2bS sequence variants with either the highest folding free energy (seq_09, seq_74, seq_81, seq_98) or a low codon usage index (see Materials and Methods) of the 5′ end (seq_01, seq_03, seq_75) and then analyzed ifn-α2bS expression for all these in vivo. The corresponding expression constructs pIFN30Seq_X (Table 1), together with the wild-type plasmid pIFN30S and positive-control pIFN30SpelB, were established in E. coli DH5α and used for determination of both mRNA (qRT-PCR) and protein levels (Western blotting). The results showed that none of the variants led to a stimulation of protein production that was sufficient for detection. Moreover, as deduced from the amount of total cell protein needed for obtaining a visible detection pattern on a Western blot (Table 2), the protein production levels of ifn-α2bS synonymous variants (and the wild type) were at least 60-fold lower than for the plasmid construct in which the ifn-α2bS gene starts with the in-frame fusion partner pelB.
Fig 1.

In silico combinatorial library of the 5′ coding sequence of ifn-α2bS plotted as a function of minimum free folding energy and the codon usage index (defined as a sum of frequencies of synonymous codon usage in E. coli). Sequences that have been experimentally characterized are marked with arrows. ifn-α2bS, original gene (30); seq_01 to seq_98, selected synonymous variants for expression studies. Expression of none of the tested ifn-α2bS synonymous variants could be detected at the protein level, although some variants (indicated in dark-shaded boxes) could increase the transcript levels around 8-fold compared to that of ifn-α2bS.
Table 2.
5′ Coding sequence characteristics together with corresponding transcript and protein levels for 8 synonymous codon variants of the ifn-α2bS gene, when expressed from the XylS-Pm system in the DH5α strain
| Gene name | mRNA coding sequence | Transcript levela | IFN-α2b protein detectionb |
|---|---|---|---|
| ifn-α2bS | ATGTGCGATCTGCCGCAGACCCATAGC | 1.01 ± 0.12 | BDL |
| seq_01 | .....T..C..T..C..A..A..CTCA | 6.81 ± 1.72 | BDL |
| seq_03 | .....T..CT....C..A..A..CTCA | 4.74 ± 0.35 | BDL |
| seq_09 | ........C..T..C..A.....CTCA | 1.25 ± 0.10 | BDL |
| seq_74 | ........C..T..C..A..A..CTCA | 2.11 ± 0.29 | BDL |
| seq_75 | .....T..CT.A..A..A..A..CTCA | 7.27 ± 0.78 | BDL |
| seq_81 | ........C.....C..A......... | 0.88 ± 0.18 | BDL |
| seq_98 | .........T.A..C..A.....CTCA | 8.19 ± 1.91 | BDL |
| pelBc | ATGAAATACCTATTGCCTACGGCAGCC | 6.07 ± 0.87 | 1–5 μg |
All values are relative to the ifn-α2bS transcript level, which is arbitrarily set to 1.
Limits of Western blot detection when total cell protein was used as the sample. BDL, below detection limit (>300 μg). One to five micrograms of total protein was sufficient to detect the PelB–IFN-α2b fusion protein by Western blotting (see Fig. 5), while using the maximum capacity of a protein gel (300 μg) was not sufficient to detect the protein production of any of the ifn-α2bS gene variants.
The pelB-ifn-α2bS fusion served as a positive control in the expression experiments.
Interestingly, four out of seven ifn-α2bS synonymous variants resulted in much higher levels of accumulated transcripts (over 8-fold, see seq_98) than the original gene. The three variants generating the most transcripts (seq_01, seq_75, seq_98) also performed similarly to or even better than the pelB–ifn-α2bS fusion, with respect to the amounts of transcript generated (Table 2). It is in itself intriguing that variants at the 5′ end of the coding sequence can lead to such a strong stimulation at the transcript level, which in principle could be caused by enhanced mRNA stability, by increased rate of transcription, or by a combination of both. Protection of mRNA from degradation by improved ribosome binding and/or translation has been described (22, 23), but in this case it seems less likely since protein production was still very inefficient. We could not rule out that the amount of protein produced from this particular gene under the conditions used is not solely limited by the amount of transcript but might be also affected by some sequence features negatively influencing processes downstream of transcription.
The results reported above indicate that bioinformatics predictions can give an indication of the limiting steps in bacterial gene expression. In a search for variants of the ifn-α2bS 5′ coding sequence that would result in an expression level comparable to or higher than that obtained by a pelB–ifn-α2bS fusion, a physical library which statistically corresponds to the one generated in silico could in principle have been generated and screened. However, such an approach would require a very laborious screening program, and we therefore instead focused on analyzing the nature of 5′ fusion partners that are able to stimulate expression at the protein level.
A nonsignal sequence leads to stimulation of ifn-α2bS expression when used as the 5′ terminal fusion partner.
The translocation function of the PelB peptide could potentially be important for efficient protein production, as also other translocation signals, such as OmpA and consensus signal peptide (CSP), have been described to stimulate expression levels of recombinant proteins (41, 46). To indirectly analyze the role of protein translocation in ifn-α2bS expression, we substituted the pelB signal with the 5′ part of the celB gene (length similar to that of pelB), whose protein product (phosphoglucomutase) is cytoplasmic (47). We further confirmed by the SignalP prediction tool (44) that the celB 5′ terminal does not have properties of a signal sequence (see Materials and Methods). Another reason for selecting this particular gene is that it has been previously shown to be very efficiently expressed from the XylS-Pm system (31). An initial test showed that such a fusion improves protein production of ifn-α2bS to a similar extent as pelB (data not shown), suggesting that translocation is not required for ifn-α2bS expression. We therefore continued with a more systematic analysis of several variant celB-based 5′ fusion partners.
Initially, we explored to what extent the length of a celB-based fusion influences ifn-α2bS expression at the protein level. This was done by constructing 13 different plasmid constructs expressing different celBN–ifn-α2bS fusions, in which N (3, 5, 6, 7, 8, 10, 15, 20, 23, 25, 30, 38 and 69) denotes the number of codons from celB. Determination of the protein production levels in the corresponding DHα strains showed that 5′ celB fusions have to include a minimum of 8 codons to lead to detectable protein production (Fig. 2). For sequences longer than 8 codons, the protein levels appeared to be positively correlated with fusion partner length of up to 20 to 25 codons. An exception from this expression pattern was observed for the celB fusion of 10 codons that was less efficient than any other celB fusion longer than 8 codons. The reason for this is unknown, but it illustrates how minor differences in the 5′ coding region can have a strong effect on the gene expression level. All celB-based fusions longer than 20 codons also seemed to function equally well as pelB, meaning that ifn-α2bS expression at the protein level is again at least 60-fold better with the 5′ celB fusion partner than without it. In conclusion, besides being independent of translocation, the celB-based 5′ fusion partner can strongly stimulate protein production of the ifn-α2bS model gene, with the corresponding levels varying significantly over a certain threshold value (represented here by the 5′ fusion length of 8 codons).
Fig 2.
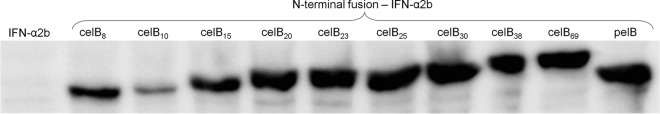
Expression of celBN–ifn-α2bS (N = 8, 10, 15, 20, 23, 25, 30, 38, or 69 codons) fusion genes at the protein level, as determined by SDS-PAGE/Western blot analysis of corresponding E. coli DH5 cell extracts. HisProbe-HRP was used for specific detection of the fusion proteins. The same amount of total protein (50 μg) was loaded in all wells. There was no detectable protein when the ifn-α2bS gene was expressed without a fusion partner and when celB fusions were shorter than 8 codons. The IFN-α2b production level when the pelB sequence is used is shown in the rightmost column. The size of the IFN-α2b–c-Myc–His6 protein complex is 22.4 kDa, the size of PelB–IFN-α2b–c-Myc–His6 is 24.4 kDa, and for CelBN–IFN-α2b–c-Myc–His6 it varies between 23.3 kDa (N = 8 amino acids) and 29.9 kDa (N = 69 amino acids).
celB-based 5′ fusion partners increase ifn-α2bS transcript levels more than 7-fold and also lead to more stable mRNA.
As presented in Table 2, the transcript amounts of pelB–ifn-α2bS are enhanced about 6-fold (compared to those of ifn-α2bS), but in contrast to the synonymous variants of ifn-α2bS, this increase is associated with a much stronger effect at the protein level. A corresponding analysis of the transcript amounts produced from 11 different celBN–ifn-α2bS variants showed an increase in transcript accumulation from around 5- to 7-fold for 5′ celB fusions containing from 8 to 69 codons (Table 3). Shorter celB-based sequences of 3 and 5 codons did not lead to more transcripts, consistent with what was also observed at the protein levels (Fig. 2). However, the observed increase in transcript amounts did not directly reflect the multifold stimulation at the protein levels displayed by celB fusions longer than 8 codons.
Table 3.
Transcript amounts for celBN–ifn-α2bS fusions when expressed from the XylS-Pm system in corresponding DH5α strains

All values are relative to the ifn-α2bS transcript level (no fusion), which is arbitrarily set to 1.
Unlike the synonymous mutations in the ifn-α2bS sequence, the DNA sequences of the celB-based 5′ fusions appear to stimulate both transcript accumulation and downstream processes, ultimately resulting in high-level protein production. The stimulation at the level of transcripts may potentially be an indirect effect of improved translation by possibly protecting the transcripts from degradation. To experimentally test this idea, a newly developed noninvasive technique for monitoring mRNA stability, based on washout of the transcriptional inducer (33), was employed, using strains DH5α(pIFNcelB25) and DH5α(pIFN30S) (Fig. 3). Subsequent mathematical fitting (developed together with the inducer washout method) estimated the decay rates of ifn-α2bS and celB25–ifn-α2bS mRNAs to be 0.32 (95% confidence interval [CI] = 0.26 to 0.38) and 0.17 (95% CI = 0.13 to 0.20), respectively. Even though this difference appears to be significant, it can hardly alone explain the multifold expression differences observed at the protein level. Therefore, the use of 5′ fusion partners presumably modulates translation of ifn-α2bS, while the improved mRNA stability can be a secondary effect of the improved translation. The increase in accumulated transcripts is then a consequence of this stabilization, although some effect on transcription as well as stabilization of the fusion transcript per se cannot be excluded. In addition, the fusion might also act through enhancing the protein stability by modulating the N-end rule proteolytic pathway (48).
Fig 3.

Determination of transcript decay kinetics by the inducer washout method in DH5α cells (see Materials and Methods for details); both ifn-a2bS and celB25–ifn-a2bS transcript amounts at time point 0 are arbitrarily set to 1 in order to discriminate transcript decay rates. Error bars show the deviation between two biological recurrences. The figure plotted in the upper right corner represents the same data, only in this case all transcript amounts are relative to the ifn-a2bS transcript amount at time zero, arbitrarily set to one. Solid lines represent the best fit to the data, calculated according to the methodology described in reference 33. RQ, relative quantification; au, arbitrary units.
Development of a selection system for direct identification of 5′ fusion partners optimized for high-level protein production.
Generation and selection of mutant libraries of short nucleotide sequences have been established as an effective approach to identify variants that improve recombinant gene expression, as described previously for the Pm promoter, its UTR, and the CSP translocation signal sequence (38–40). All of these previous studies used the bla gene (encoding β-lactamase) as a powerful screening tool that can report the expression levels through the corresponding ampicillin tolerance levels of the host cells. β-Lactamase is translocated into the periplasm, and the enzyme is providing host resistance/tolerance only if export takes place. In a search for candidate genes that could be used as reporters decoupled from a translocation process, we found that the apramycin resistance gene [aac(3)-IV; encoding cytoplasmic aminoglycoside-(3)-acetyltransferase IV] might be a good choice.
One requirement of this approach is that the AAC(3)-IV protein, with a celB fusion partner at the N-terminal end, retains its enzymatic activity. This was tested with 11 5′ fusion partners of various lengths (containing 3, 5, 8, 10, 15, 20, 23, 25, 30, 38, or 69 codons from the 5′ end of celB). The corresponding DNA sequences were fused in frame to aac(3)-IV under the control of Pm, and the resulting apramycin tolerance levels of the host cells were determined. The tolerance was found to increase with the length of the celB fusion, differing up to 10-fold between DH5α(pARcelB3) and DH5α(pARcelB69), under induced conditions (50 μM m-toluate). The strain displaying the highest apramycin tolerance under induced conditions while keeping low background when uninduced (induction ratio of 10) was found to be DH5α(pARcelB23).
To establish whether apramycin tolerance levels of DH5α(pARcelB23) display a proportional response to a change in the expression level of the aac(3)-IV reporter, DH5α(pARcelB23) cells were plated on solid medium supplemented with increasing amounts of inducer (0 to 2,000 μM). In this way, only transcriptional stimulation through the XylS-Pm system, which displays a continuous response to various inducer concentrations (44), was expected to occur. The result showed that the apramycin tolerance of the host increased as a growing function of the inducer concentration and therefore confirmed a direct relationship between antibiotic tolerance of the host and the corresponding reporter gene expression (Fig. 4). Based on this result, we considered the apramycin-based selection system competent for identifying sequence-optimized 5′ fusion partners that can be used to promote recombinant protein production.
Fig 4.
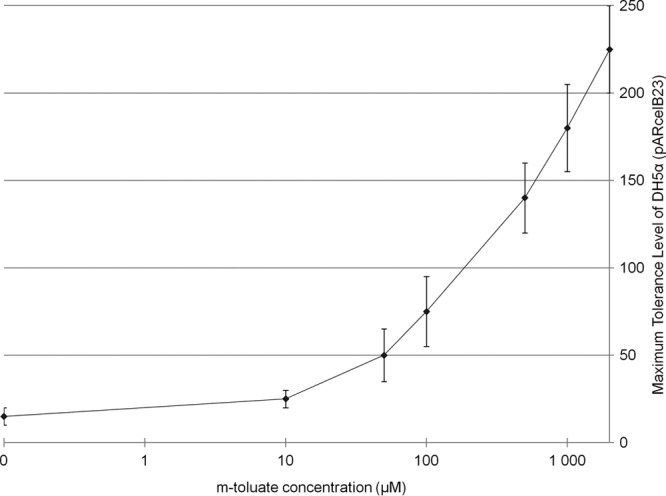
Apramycin tolerance level of DH5α(pARcelB23) presented as a log 10 function of increasing concentration of the XylS-Pm expression system inducer (m-toluate). LB medium (100 μl supplemented with 50 mg/liter kanamycin) in a 96-well microtiter plate (Nunc) was inoculated with DH5α(pARcelB23) cell culture and incubated at 30°C overnight. The cells were then diluted twice by a 96-pin replicator into new microtiter plates with 100 μl LB medium in each well and subsequently spotted onto l-agar with m-toluate and apramycin at the concentrations indicated (μg/ml). The plates were incubated at 30°C for 2 days.
Additionally, up to 4-fold stimulation of protein production of ifn-α2bS can be achieved by an optimized 5′ celB fusion.
A library of randomized DNA sequences based on celB23 was generated by doped oligonucleotide mutagenesis, allowing all the relevant bases to vary. Screening of this library [termed celBR-aac(3)-IV] led to identification of 26 candidates with up to a 20-fold increase in apramycin tolerance of the host (selection of 14 sequences, which conferred the highest apramycin tolerance to the host cell, is shown in Table 4) compared to that of the original DH5α(pARcelB23) strain.
Table 4.
Sequence and expression characteristics of celB23 fusion and its variants identified from screening of the celBR–aac(3)-IV library
| Name | 5′ Fusion partner DNA sequence | Translated amino acid sequence | Max Arr tolerancea | aac(3)-IV transcript levelb |
|---|---|---|---|---|
| celBc | ATGCCCAGCATAAGCCCATTTGCCGGCAAGCCGGTCGATCCGGACCGTCTTGTCAATATC | MPSISPFAGKPVDPDRLVNI | 1.00d ± 0.33 | 1.00 ± 0.35 |
| D2 | ...T.T.CT..T................................................ | .ST................. | 16.67 ± 1.67 | 1.29 ± 0.19 |
| D3 | ...T.......T.T.A.....T...................................... | .S..IT.S............ | 16.67 ± 1.67 | 1.32 ± 0.11 |
| D4 | .....A.......T.......A....T..........T.......G.....C........ | ....I..T....V..G.L.. | 16.67 ± 2.50 | 0.78 ± 0.24 |
| D5 | ...T....................C...........T....C..G............... | .S......R...Y.E..... | 11.33 ± 0.83 | 1.13 ± 0.12 |
| D7 | ...T...CT.A.........C.......G...............T...T..T........ | .STK.....R......FF.. | 16.67 ± 1.67 | 0.70 ± 0.18 |
| D9 | ...T...TA....T.............................................. | .SI.I............... | 16.67 ± 1.33 | 1.08 ± 0.10 |
| D10 | ...A.A.......A...T.A.T...................................... | .T..N.YS............ | 12.00 ± 0.67 | 1.44 ± 0.08 |
| D11 | ...T.T.......A.......A...............T.......T.....T......C. | .S..N..T....V..C.F.T | 20.00 ± 2.50 | 4.74 ± 0.54 |
| D13 | ...A.A....A.....A.A..A........................A...A.....A... | .T.K.QIT.......H..K. | 11.67 ± 1.33 | 0.80 ± 0.18 |
| D15 | ...T.........T......................C....................... | .S..I.......H....... | 12.50 ± 1.33 | 0.89 ± 0.07 |
| D17 | ...T..........TG...G........................A............... | .S...AC.......E..... | 9.17 ± 0.83 | 1.39 ± 0.08 |
| D19 | ...T............A...........T.......C...AC..A....C.......... | .S...Q...M..HHE.P... | 14.17 ± 1.65 | 1.84 ± 0.13 |
| D20 | ...T........................................................ | .S.................. | 13,33 ± 1.17 | 2.41 ± 0.13 |
| D21 | ...T.........A..T........................................... | .S..NL.............. | 14.17 ± 1.50 | 1.45 ± 0.10 |
Max Arr tolerance is the maximum apramycin concentration tolerated by DH5α host cells (induction with 0.05 mM m-toluate). All values are relative to the tolerance level of the wild-type strain DH5α(pARcelB23), which is arbitrarily set to 1 and corresponds to 0.06 g/liter.
All values are relative to the celB23–aac(3)-IV transcript level, which is arbitrarily set to 1.
Only the region where random mutations had occurred is shown (first 60 nucleotides/first 20 amino acids).
The apramycin resistance level of the wild-type strain DH5α(pARcelB23) (0.06 ± 0.02 g/liter) slightly differs from the level given in Fig. 4 (0.05 ± 0.02 g/liter; 0.05 mM m-toluate induction). This is because the experiments represent the average of different biological replicates (each experiment is performed as two individual biological replicates with a minimum of 4 technical replicates).
Among the selected celB variants, the average number of point mutations was 5.6, and they were typically randomly distributed throughout the celB coding region. A second codon mutation (CCC to TCC or TCT, proline to serine) was found in 11 variants and observed to cause a major effect in the final expression level (see variant D20). We envisioned that the observed stimulation of host apramycin tolerance might also in some cases be caused by enhanced specific catalytic activity of the CelB-AAC(3)-IV fusion protein, without being associated with higher levels of protein production. Such improvement might be based on changes in the sequence of the 5′ fusion and/or on changes in the coding sequence of the reporter. Because our goal was to select a 5′ fusion variant that affects expression in general (theoretically through changes in mRNA stability, translation, or protein stability), hypothetical candidates that improve specific catalytic activity of the reporter protein (thus acting in a protein-specific manner) would be irrelevant in the current study. They could have been eliminated by Western analysis, but relevant antibodies are unfortunately not directly available. We therefore instead prescreened the 14 mutants listed in Table 4 by relative quantification of aac(3)-IV transcript amounts, to eliminate those with potentially improved catalytic activity. The qRT-PCR analysis showed that several fusion partners led to increased transcript levels, and particularly for celBD11, an approximate 5-fold improvement relative to that of celB23 was observed (Table 4).
The celBD11 fusion partner was selected as the most promising candidate and analyzed for its potential to increase protein production of ifn-α2bS. We confirmed by the SignalP prediction tool that celBD11 does not exhibit properties of a signal sequence (see Materials and Methods). Determination of protein levels in E. coli production strains RV308 harboring pIFNcelBD11, pIFNcelB23, or pIFN30S plasmids showed that, compared to celB23 and pelB, the celBD11 fusion improves ifn-α2bS expression at the protein level about 4- and 2-fold, respectively (Fig. 5). Since pelB has been shown to be useful for industrial-level production of the IFN-α2b protein from XylS-Pm (30), we conclude that the selection protocol for improved fusion partners reported here has a significant potential for improvement of recombinant gene expression. It also appears likely that a more extensive screening of promising candidates would yield even more potent candidates than celBD11.
Fig 5.

Western blot detection of the IFN-α2b protein when the respective gene was expressed either with pelB, celB23 (23 indicates the number of codons), or the optimized celBD11 fusion. Expression of ifn-a2bS without any fusion did not lead to detectable protein (IFN-α2b). The genes were expressed in E. coli production strain RV308. Cell samples were lysed by sonication, and the total crude cell extracts were subjected to analysis. HisProbe-HRP was used for specific detection of the fusion proteins. For IFN-α2b proteins containing pelB, celB23, or celBD11 fusions, dilution series of decreasing total protein amounts were loaded; 1 corresponds to 10 μg. The size of PelB–IFN-α2b–c-Myc–His6 is 24.4 kDa and of CelB23–IFN-α2b–c-Myc–His6 and CelBD11–IFN-α2b–c-Myc–His6 is 24.9 kDa, while the protein complex without an N-terminal fusion peptide is 22.4 kDa.
The celBD11 fusion partner appears to have a generally positive effect on recombinant protein production.
We could not exclude that some context dependency would limit the usefulness of the celBD11 fusion partner, and we therefore also tested its performance with respect to its ability to stimulate production of three other poorly expressed heterologous genes of human origin: granulocyte colony-stimulating factor (G-CSF), granulocyte macrophage colony-stimulating factor (GM-CSF), and tumor necrosis factor α1a (TNF-α1a). The protein production levels in strain RV308 showed that celB23 by itself is very effective; for gm-csf and g-csf, it raised the protein production level from undetectable to a clearly visible protein band (Fig. 6). The protein level was further increased when using the optimized celBD11, ranging from about 2-fold (gm-csf and tnf-α1a) to 4-fold (g-csf) compared to the effect of celB23.
Fig 6.
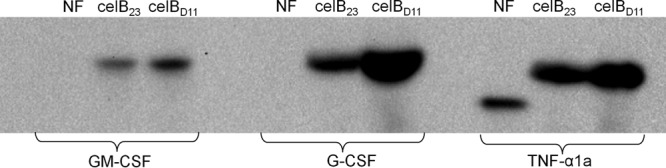
Characterization of gm-csf, g-csfS, and tnf-α1aS expression at the protein level by Western blotting. The gene constructs containing either the celB23 fusion partner or its optimized version, celBD11, or no 5′-terminal fusion (NF) were expressed in E. coli RV308. Cells were lysed by sonication, and the total crude cell extracts were subjected to analysis. HisProbe-HRP was used for specific detection of the fusion proteins, and the same amount of total protein (50 μg) was loaded in all wells. The size of GM-CSF–c-Myc–His6 is 17.5 kDa, of CelB23–GM-CSF–c-Myc–His6 and CelBD11–GM-CSF–c-Myc–His6 is 20.1 kDa, of G-CSF–c-Myc–His6 is 21.7 kDa, of CelB23–G-CSF–c-Myc–His6 and CelBD11–G-CSF–c-Myc–His6 is 24.3 kDa, of TNF-α1a–c-Myc–His6 is 20.1 kDa, and of CelB23–TNF-α1a–c-Myc–His6 and CelBD11–TNF-α1a–c-Myc-His6 is 22.7 kDa.
Concluding remarks.
Fusion partners commonly used to stimulate recombinant protein production are often also protein translocation signals, but the results reported here indicate that translocation is not needed for the fusion partner to give rise to the desired effect. However, the length is critical, and in the model system used here, a minimum of eight codons were found to be required. The study also demonstrated that a fusion partner could simply be selected from the 5′ end of a highly expressed gene (celB), giving rise to a stimulation of expression similar to that of the well-established pelB signal sequence, also originating from a well-expressed gene (30). Interestingly, the celB sequence could be further improved via random mutagenesis combined with the strong selection method developed in this study. By introducing a protease cleavage site allowing the fusion partner to be cleaved off after production, the methodology described here should be applicable for the improvement of many protein production processes. Even though the use of fusion partners seems to work for expression of several different proteins, the underlying mechanisms are not clear. The nucleotide sequence at the 5′ terminus may represent a problem for the initial steps in translation, but not if it is moved further into the coding sequence by the 5′ fusion partner. An alternative explanation is that the proteins are extremely unstable in E. coli but are somehow protected from degradation by the fusion partner.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by Research Council of Norway grant no. 182672/I40.
We thank Adrian E. Naas and Magnus Leithaug for their contribution to the project during their Master of Science studies at the Department of Biotechnology, NTNU, Trondheim, Norway. We also thank Vectron Biosolutions for providing plasmids pMA-T-G-CSF and pMA-T-TNF-α1a.
Footnotes
Published ahead of print 23 August 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.01676-13.
REFERENCES
- 1. Huang CJ, Lin H, Yang X. 2012. Industrial production of recombinant therapeutics in Escherichia coli and its recent advancements. J. Ind. Microbiol. Biotechnol. 39:383–399 [DOI] [PubMed] [Google Scholar]
- 2. Sørensen HP, Mortensen KK. 2005. Advanced genetic strategies for recombinant protein expression in Escherichia coli. J. Biotechnol. 115:113–128 [DOI] [PubMed] [Google Scholar]
- 3. Simonetti A, Marzi S, Jenner L, Myasnikov A, Romby P, Yusupova G, Klaholz B, Yusupov M. 2009. A structural view of translation initiation in bacteria. Cell. Mol. Life Sci. 66:423–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kozak M. 2005. Regulation of translation via mRNA structure in prokaryotes and eukaryotes. Gene 361:13–37 [DOI] [PubMed] [Google Scholar]
- 5. Hüttenhofer A, Noller H. 1994. Footprinting mRNA-ribosome complexes with chemical probes. EMBO J. 13:3892–3901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. de Smit MH, van Duin J. 1990. Secondary structure of the ribosome binding site determines translational efficiency: a quantitative analysis. Proc. Natl. Acad. Sci. U. S. A. 87:7668–7672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Schauder B, McCarthy JEG. 1989. The role of bases upstream of the Shine-Dalgarno region and in the coding sequence in the control of gene expression in Escherichia coli: translation and stability of mRNAs in vivo. Gene 78:59–72 [DOI] [PubMed] [Google Scholar]
- 8. Barrick D, Villanueba K, Childs J, Kalil R, Schneider TD, Lawrence CE, Gold L, Stormo GD. 1994. Quantitative analysis of ribosome binding sites in E. coli. Nucleic Acids Res. 22:1287–1295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Marzi S, Myasnikov AG, Serganov A, Ehresmann C, Romby P, Yusupov M, Klaholz BP. 2007. Structured mRNAs regulate translation initiation by binding to the platform of the ribosome. Cell 130:1019–1031 [DOI] [PubMed] [Google Scholar]
- 10. Kudla G, Murray AW, Tollervey D, Plotkin JB. 2009. Coding-sequence determinants of gene expression in Escherichia coli. Sci. 324:255–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Seo SW, Yang J, Jung GY. 2009. Quantitative correlation between mRNA secondary structure around the region downstream of the initiation codon and translational efficiency in Escherichia coli. Biotechnol. Bioeng. 104:611–616 [DOI] [PubMed] [Google Scholar]
- 12. Cèbe R, Geiser M. 2006. Rapid and easy thermodynamic optimization of the 5′-end of mRNA dramatically increases the level of wild type protein expression in Escherichia coli. Protein Expr. Purif. 45:374–380 [DOI] [PubMed] [Google Scholar]
- 13. Stenström CM, Jin H, Major LL, Tate WP, Isaksson LA. 2001. Codon bias at the 3′-side of the initiation codon is correlated with translation initiation efficiency in Escherichia coli. Gene 263:273–284 [DOI] [PubMed] [Google Scholar]
- 14. Stenström CM, Isaksson LA. 2002. Influences on translation initiation and early elongation by the messenger RNA region flanking the initiation codon at the 3′ side. Gene 288:1–8 [DOI] [PubMed] [Google Scholar]
- 15. Gonzalez de Valdivia EI, Isaksson LA. 2004. A codon window in mRNA downstream of the initiation codon where NGG codons give strongly reduced gene expression in Escherichia coli. Nucleic Acids Res. 32:5198–5205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Brock JE, Paz RL, Cottle P, Janssen GR. 2007. Naturally occurring adenines within mRNA coding sequences affect ribosome binding and expression in Escherichia coli. J. Bacteriol. 189:501–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Emory SA, Bouvet P, Belasco JG. 1992. A 5′-terminal stem-loop structure can stabilize mRNA in Escherichia coli. Genes Dev. 6:135–148 [DOI] [PubMed] [Google Scholar]
- 18. Bricker AL, Belasco JG. 1999. Importance of a 5′ stem-loop for longevity of papA mRNA in Escherichia coli. J. Bacteriol. 181:3587–3590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Arnold TE, Yu J, Belasco JG. 1998. mRNA stabilization by the ompA 5′ untranslated region: two protective elements hinder distinct pathways for mRNA degradation. RNA 4:319–330 [PMC free article] [PubMed] [Google Scholar]
- 20. Deana A, Celesnik H, Belasco JG. 2008. The bacterial enzyme RppH triggers messenger RNA degradation by 5′ pyrophosphate removal. Nature 451:355–358 [DOI] [PubMed] [Google Scholar]
- 21. Celesnik H, Deana A, Belasco JG. 2007. Initiation of RNA decay in Escherichia coli by 5′ pyrophosphate removal. Mol. Cell 27:79–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Vytvytska O, Moll I, Kaberdin VR, von Gabain A, Bläsi U. 2000. Hfq (HF1) stimulates ompA mRNA decay by interfering with ribosome binding. Genes Dev. 14:1109–1118 [PMC free article] [PubMed] [Google Scholar]
- 23. Iost I, Dreyfus M. 1995. The stability of Escherichia coli lacZ mRNA depends upon the simultaneity of its synthesis and translation. EMBO J. 14:3252–3261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wagner LA, Gesteland RF, Dayhuff TJ, Weiss RB. 1994. An efficient Shine-Dalgarno sequence but not translation is necessary for lacZ mRNA stability in Escherichia coli. J. Bacteriol. 176:1683–1688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Matsunaga J, Simons EL, Simons RW. 1997. Escherichia coli RNase III (rnc) autoregulation occurs independently of rnc gene translation. Mol. Microbiol. 26:1125–1135 [DOI] [PubMed] [Google Scholar]
- 26. Kaberdin VR, Bläsi U. 2006. Translation initiation and the fate of bacterial mRNAs. FEMS Microbiol. Rev. 30:967–979 [DOI] [PubMed] [Google Scholar]
- 27. Delvillani F, Papiani G, Deho G, Briani F. 2011. S1 ribosomal protein and the interplay between translation and mRNA decay. Nucleic Acids Res. 39:7702–7715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Komarova AV, Tchufistova LS, Dreyfus M, Boni IV. 2005. AU-rich sequences within 5′ untranslated leaders enhance translation and stabilize mRNA in Escherichia coli. J. Bacteriol. 187:1344–1349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Komarova AV, Tchufistova LS, Supina EV, Boni IV. 2002. Protein S1 counteracts the inhibitory effect of the extended Shine-Dalgarno sequence on translation. RNA 8:1137–1147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sletta H, Tøndervik A, Hakvåg S, Aune TEV, Nedal A, Aune R, Evensen G, Valla S, Ellingsen TE, Brautaset T. 2007. The presence of N-terminal secretion signal sequences leads to strong stimulation of the total expression levels of three tested medically important proteins during high-cell-density cultivations of Escherichia coli. Appl. Environ. Microbiol. 73:906–912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Blatny JM, Brautaset T, Winther-Larsen HC, Karunakaran P, Valla S. 1997. Improved broad-host-range RK2 vectors useful for high and low regulated gene expression levels in Gram-negative bacteria. Plasmid 38:35–51 [DOI] [PubMed] [Google Scholar]
- 32. Winther-Larsen HC, Josefsen KD, Brautaset T, Valla S. 2000. Parameters affecting gene expression from the Pm promoter in Gram-negative bacteria. Metab. Eng. 2:79–91 [DOI] [PubMed] [Google Scholar]
- 33. Kucharova V, Strand TA, Almaas E, Nass AE, Brautaset T, Valla S. 2013. Non-invasive analysis of recombinant mRNA stability in Escherichia coli by a combination of transcriptional inducer wash-out and qRT-PCR. PLoS One 8:e66429. 10.1371/journal.pone.0066429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Power PM, Jones RA, Beacham IR, Bucholtz C, Jennings MP. 2004. Whole genome analysis reveals a high incidence of non-optimal codons in secretory signal sequences of Escherichia coli. Biochem. Biophys. Res. Commun. 322:7. [DOI] [PubMed] [Google Scholar]
- 35. Zalucki YM, Gittins KL, Jennings MP. 2008. Secretory signal sequence non-optimal codons are required for expression and export of β-lactamase. Biochem. Biophys. Res. Commun. 366:135–141 [DOI] [PubMed] [Google Scholar]
- 36. Zalucki YM, Power PM, Jennings MP. 2007. Selection for efficient translation initiation biases codon usage at second amino acid position in secretory proteins. Nucleic Acids Res. 35:5748–5754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Berg L, Kucharova V, Bakke I, Valla S, Brautaset T. 2012. Exploring the 5′-UTR DNA region as a target for optimizing recombinant gene expression from the strong and inducible Pm promoter in Escherichia coli. J. Biotechnol. 158:224–230 [DOI] [PubMed] [Google Scholar]
- 38. Bakke I, Berg L, Aune TE, Brautaset T, Sletta H, Tøndervik A, Valla S. 2009. Random mutagenesis of the PM promoter as a powerful strategy for improvement of recombinant-gene expression. Appl. Environ. Microbiol. 75:2002–2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Berg L, Lale R, Bakke I, Burroughs N, Valla S. 2009. The expression of recombinant genes in Escherichia coli can be strongly stimulated at the transcript production level by mutating the DNA-region corresponding to the 5′-untranslated part of mRNA. Microb. Biotechnol. 2:379–389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Heggeset TMB, Kucharova V, Nærdal I, Valla S, Sletta H, Brautaset T. 2013. Combinatorial mutagenesis and selection of improved signal sequences and their application for high level production of heterologous proteins in Escherichia coli. Appl. Environ. Microbiol. 79:559–568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sletta H, Nedal A, Aune TEV, Hellebust H, Hakvåg S, Aune R, Ellingsen TE, Valla S, Brautaset T. 2004. Broad-host-range plasmid pJB658 can be used for industrial-level production of a secreted host-toxic single-chain antibody fragment in Escherichia coli. Appl. Environ. Microbiol. 70:7033–7039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Markham NR, Zuker M. 2008. UNAFold: software for nucleic acid folding and hybridization, p 3–31 Bioinformatics: structure, function and applications, vol 453 Springer, New York, NY: [DOI] [PubMed] [Google Scholar]
- 43. Salis HM, Mirsky EA, Voigt CA. 2009. Automated design of synthetic ribosome binding sites to control protein expression. Nat. Biotech. 27:946–950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Petersen TN, Brunak S, von Heijne G, Nielsen H. 2011. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat. Meth. 8:785–786 [DOI] [PubMed] [Google Scholar]
- 45. Tuller T, Carmi A, Vestsigian K, Navon S, Dorfan Y, Zaborske J, Pan T, Dahan O, Furman I, Pilpel Y. 2010. An evolutionarily conserved mechanism for controlling the efficiency of protein translation. Cell 141:344–354 [DOI] [PubMed] [Google Scholar]
- 46. Choi JH, Lee SY. 2004. Secretory and extracellular production of recombinant proteins using Escherichia coli. Appl. Microbiol. Biotechnol. 64:625–635 [DOI] [PubMed] [Google Scholar]
- 47. Brautaset T, Standal R, Fjærvik E, Valla S. 1994. Nucleotide sequence and expression analysis of the Acetobacter xylinum phosphoglucomutase gene. Microbiology 140:1183–1188 [DOI] [PubMed] [Google Scholar]
- 48. Varshavsky A. 1996. The N-end rule: functions, mysteries, uses. Proc. Natl. Acad. Sci. U. S. A. 93:12142–12149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Nikodinovic J, Priestley ND. 2006. A second generation snp-derived Escherichia coli–Streptomyces shuttle expression vector that is generally transferable by conjugation. Plasmid 56:223–227 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.