

Abstract
In recent years, representatives of the Bacteroidetes have been increasingly recognized as specialists for the degradation of macromolecules. Formosa constitutes a Bacteroidetes genus within the class Flavobacteria, and the members of this genus have been found in marine habitats with high levels of organic matter, such as in association with algae, invertebrates, and fecal pellets. Here we report on the generation and analysis of the genome of the type strain of Formosa agariphila (KMM 3901T), an isolate from the green alga Acrosiphonia sonderi. F. agariphila is a facultative anaerobe with the capacity for mixed acid fermentation and denitrification. Its genome harbors 129 proteases and 88 glycoside hydrolases, indicating a pronounced specialization for the degradation of proteins, polysaccharides, and glycoproteins. Sixty-five of the glycoside hydrolases are organized in at least 13 distinct polysaccharide utilization loci, where they are clustered with TonB-dependent receptors, SusD-like proteins, sensors/transcription factors, transporters, and often sulfatases. These loci play a pivotal role in bacteroidetal polysaccharide biodegradation and in the case of F. agariphila revealed the capacity to degrade a wide range of algal polysaccharides from green, red, and brown algae and thus a strong specialization of toward an alga-associated lifestyle. This was corroborated by growth experiments, which confirmed usage particularly of those monosaccharides that constitute the building blocks of abundant algal polysaccharides, as well as distinct algal polysaccharides, such as laminarins, xylans, and κ-carrageenans.
INTRODUCTION
Representatives of the phylum Bacteroidetes have been found in various marine habitats. They constitute an important part of the marine heterotrophic bacterioplankton in shallow coastal waters (1) and the open ocean (2, 3) and an important part of benthic marine microbial communities in sediments (4). Marine members of the Bacteroidetes seem to be largely specialized for the degradation of biopolymers, such as proteins and polysaccharides (2, 5–11). In particular, members of the bacteroidetal class Flavobacteria have been associated with the degradation of marine high-molecular-weight (HMW) particulate organic matter (POM), since they are often enriched on detritus and colonize surfaces of living organisms, such as corals (12, 13), algae (12–21), and higher plants (22). The latter is facilitated by the ability of many Bacteroidetes to move by gliding and to form biofilms. Bacteroidetes are known to use dedicated transport systems for the uptake of macromolecule decomposition products, such as the bacteroidetal starch utilization system (Sus) and the related TonB-dependent transporter (TBDT) system. Many Bacteroidetes are characterized by high gene copy numbers of TonB-dependent receptors (TBDRs), which bind extracellular substrates before TBDT-mediated uptake (10, 23).
The flavobacterial genus Formosa was first described by Ivanova et al. in 2004 (17). As of now, three Formosa species have been described with valid names, F. algae KMM 3553T (17), F. agariphila KMM 3901T (24), and F. spongicola A2T (25), with the latter still listed as “unclassified Flavobacteriaceae bacterium A2” in some public databases. F. algae KMM 3553T was isolated from an enrichment of a degrading thallus of the brown alga Fucus evanescens at the Kuril Islands in the Pacific Ocean. F. agariphila KMM 3901T was obtained from the Troitsa Bay in the Gulf of Peter the Great from the Sea of Japan from a specimen of the shallow water green alga Acrosiphonia sonderi, and F. spongicola A2T was isolated from the marine sponge Hymeniacidon flavia from the coast of Jeju Island, South Korea. Formosa-related species have furthermore been found in seawater and the feces-rich effluent water of a fish farming pond in Qingdao (China) (26), in hydrocarbon-contaminated seawater of Ushuaia (Argentina) (27), and in the bacterioplankton off the coast of the North Sea island Helgoland during a diatom-dominated algal bloom (28). Most of these habitats indicate a preference of Formosa species for complex organic matter. Recently the first draft genome sequence of a Formosa isolate (Formosa sp. AK20), a pelagic strain that was isolated from seawater offshore of Kochi (India), has become available. Based on the available partial 16S rRNA sequence, this strain might be more distant from the validly named Formosa species (Fig. 1).
Fig 1.

Maximum-likelihood tree of 16S rRNA genes calculated using the program RAxML v7.0.3 (79) in Arb v5.19 (80), showing the phylogenetic position of F. agariphila KMM 3901T. The 16S rRNA sequence of Owenweeksia hongkongensis UST20020801T (NCBI accession number AB125062) was used as an outgroup.
F. agariphila KMM 3901T exhibits an alga-associated lifestyle. Algae are in large part composed of polysaccharides, for instance, as constituents of their extra- and intracellular matrices, cell walls, and storage compounds. In marine algae, polysaccharides can amount to ∼70% of the algal dry weight (29). Many of these polysaccharides do not occur in land plants, such as, for example, anionic sulfated and/or carboxylated polysaccharides (e.g., agars, carrageenans, fucoidans, ulvans, and alginic acid). Hence, the capability to degrade such marine polysaccharides constitutes an important trait of marine alga-associated heterotrophic bacteria.
Polysaccharide degradation genes in the Bacteroidetes are frequently organized in larger operon or regulon structures (8) that have been termed polysaccharide utilization loci (PULs) (30). These PULs typically encode a tandem of a SusC-like TBDR and a SusD-like protein, sensors/transcriptional regulators (often characteristic hybrid two-component systems [6, 31]), transporters, sometimes sulfatases, and carbohydrate-active enzymes (CAZymes) (32). The latter are enzymes for polysaccharide binding/recognition, degradation, and synthesis/modification that can be classified in carbohydrate-binding modules (CBMs), glycoside hydrolases (GHs), polysaccharide lyases (PLs), carbohydrate esterases (CEs), and glycosyltransferases (GTs). PULs are thought to comprise genes for the decomposition of distinct polysaccharides and are likely coregulated, e.g., coinduced by one of the multiple polysaccharides (or their components) that typically cooccur in nature. Thus, PUL analysis can provide valuable information on the spectrum of polysaccharides that an organism can potentially degrade and can thereby reveal information on its ecological niche. This has been exemplarily demonstrated for various human gut Bacteroidetes that decompose plant polysaccharides that humans cannot degrade themselves (e.g., see references 30 and 33 to 35).
In order to elucidate such ecological niche adaptations with respect to polysaccharide degradation in the alga-associated F. agariphila strain KMM 3901T, we sequenced its genome, performed an in-depth CAZyme and PUL analysis, and interrelated these data with results from growth experiments on various mono-, di-, and polysaccharides.
MATERIALS AND METHODS
Growth conditions and DNA isolation.
An actively growing culture of F. agariphila KMM 3901T was obtained from the Leibnitz Institute DSMZ-German Collection of Microorganisms and Cell Cultures (DSM-15362). These cells were grown in ZoBell′s marine broth 2216 (Difco, Detroit, MI, USA) at 21°C. DNA was extracted according to the protocol of Zhou et al. (36).
Genome sequencing and assembly.
The genome of F. agariphila KMM 3901T was sequenced at the Max Planck Genome Centre in Cologne, Germany. At first, a draft genome was generated using the 454 FLX and GS Junior sequencers (454 Life Sciences/Roche, Branford, CT, USA). Reads (686,070) with an estimated error rate of 1.08% were aligned using the software program Newbler v.2.6 (Roche), resulting in 99 contigs of 4.48 Mbp total (average size, 45,232 bp; N50, 104,744 bp; largest contig, 318,947 bp). These contigs were afterwards linked to 40 scaffolds by means of 1,378 paired Sanger reads from an end-sequenced fosmid library (average gap size, 37,970 bp ± 9,492 bp), using an ABI 3730XL analyzer (Applied Biosystems, Foster City, CA, USA). Gaps were subsequently filled by targeted fosmid sequencing and editing in the software program consed v.23 (37). A single scaffold was obtained that comprised nine contigs with a total of 4,205,536 sequenced bases. This scaffold is estimated to represent >99% of the F. agariphila KMM 3901T genome and has an average coverage of >53× and an estimated error of less than 1:10,000.
Genome annotation.
Prediction of genes and functional annotation were carried out through the Rapid Annotation using Subsystem Technology (RAST) server (38). The RAST annotations were subsequently imported into a local installation of the GenDB (39) annotation system (version 2.2) for data mining, using similarity searches against the NCBI nonredundant protein (40), InterPro (41), PFAM (42), KEGG (43), COG (44), and CAZy (32) databases, as well as predictions of signal peptides with SignalP v3.0 (45) and transmembrane regions with TMHMM v2.0c (46). The annotations of selected genes were manually curated using the JCOAST software tool (47). All CAZymes were subjected to a detailed manual expert annotation involving phylogenetic reconstructions in order to elucidate their substrate specificities.
Substrate tests.
Growth assays were carried out twice and in replicates in sterile 17-ml polystyrene vials with 10 ml marine mineral medium with trace elements but without vitamins (48) containing 0.6 mg/liter glucose, cellobiose, yeast extract, peptone, and Casamino Acids, 300 μM ammonium, and 16 μM phosphate.
Stock solutions (10 g/liter) of soluble sugars (l-arabinose, d-arabinose, dl-xylose, d-galactose, d-glucose, d-fructose, d-mannitol, d-mannose, l-rhamnose, trehalose, d-sucrose, maltose, N-acetyl-d-glucosamine, and raffinose) were prepared in ultrapure water (Aquintus system; membraPure, Berlin, Germany), adjusted to pH 7.0 with 1 M NaOH or 1 M HCl and sterile filtered through a 0.2-μm-pore-size filter.
Stock solutions of polysaccharides (glycogen, cellulose, xylan, κ- and ι-carrageenan, and laminarin) were prepared as follows: Glycogen (G-8751; Sigma-Aldrich) was dissolved in sterile artificial seawater (ASW) and sterile filtrated. For cellulose, small pieces of filter paper (grade 595 1/2, Whatman; GE Healthcare, Freiburg, Germany) were washed three times with ultrapure water followed by 70% ethanol and subsequently autoclaved in ultrapure water at 121°C for 21 min. Xylan (4414.1; Carl Roth, Karlsruhe, Germany) and κ- or ι-carrageenan (22048 and 22045; Sigma-Aldrich, Hamburg, Germany) were prepared according to the protocol of Widdel and Bak (48) as 4% (wt/vol) solutions and autoclaved. Afterwards, the substrate solution was mixed 1:1 (vol/vol) with sterile doubly concentrated ASW while stirring at 80°C. The pH was adjusted to 7.0, and the mixtures were poured into sterile polypropylene petri dishes. Pieces of the gels served as a carbon source. Laminarin from Laminaria saccharina (L-1760; Sigma-Aldrich) was washed and then three times pasteurized in ultrapure water at 70°C for 1 h and washed with autoclaved ultrapure water to remove d-mannitol impurities.
Media were supplemented with saccharide stock solutions to a final saccharide content of 0.1 g/liter. Inocula were 100 μl of a F. agariphila KMM 3901T stock culture. Growth at 21°C was observed for up to 2 weeks (mono- and disaccharides) or 8 weeks (polysaccharides), respectively, and detected as formation of biomass against control cultures with either inoculated but nonsupplemented medium or noninoculated but supplemented medium. Growth of bacteria in medium was not inhibited by the presence of applied saccharides or polysaccharides.
Nucleotide sequence accession number.
The annotated genome sequence of F. agariphila KMM 3901T has been submitted to the European Nucleotide Archive of the EMBL (HG315671).
RESULTS
Genome properties.
The F. agariphila KMM 3901T genome comprises a single chromosome (GC content, 33.5%) and no extrachromosomal elements. A single scaffold of 4,229,450 bp was obtained, consisting of nine contigs (Fig. 2). Three thousand six hundred forty-two genes were predicted, 3,582 of which were protein-coding genes. Two thousand six hundred twenty-two of the protein-coding genes were assigned putative functions, with the remaining annotated as hypothetical proteins. The genome comprises four rRNA operons, two of which could be only partially resolved since they constitute repeats on the DNA level. In total, 45 tRNAs for all 20 standard amino acids were found.
Fig 2.

Circular representation of the F. agariphila KMM 3901T genome. From outward to inward: genes (circles 1 and 2), CAZymes (circle 3), RNAs (circle 4), GC content (circle 5), GC skew (circle 6), DNA curvature (circle 7), DNA bending (circle 8), and tetranucleotide skew (circle 9) are shown. The features of the inner five circles were computed for a 5-kbp sliding window with a self-written PERL script (circles 5 and 6), the program banana from the EMBOSS package (circles 7 and 8), and the program TETRA (circle 9) (81). The red boxes on the outside indicate the remaining gaps in the genome.
Energy metabolism.
F. agariphila KMM 3901T has the complete Embden-Meyerhof-Parnas (EMP) pathway. It is missing the genes for the oxidative branch of the pentose 5-phosphate (PP) pathway but has the genes for the nonoxidative pentose interconversions. For pyruvate oxidation to acetyl-coenzyme A (CoA), F. agariphila KMM 3901T contains both a three-component pyruvate dehydrogenase complex and a pyruvate formate lyase. F. agariphila KMM 3901T has a complete Krebs cycle without the glyoxylate shunt and a redox chain for oxygen respiration, including a sodium-transporting NAD(H):quinone oxidoreductase (complex I), succinate dehydrogenase (complex II), cytochrome d and c type (complex IV) terminal oxidases, and a F0F1-type ATPase. The complex III (cytochrome bc1) is absent.
Under anoxic conditions, F. agariphila KMM 3901T has the potential for a mixed acid fermentation. Pyruvate may be reduced to l- or d-lactate, converted via acetyl phosphate to acetate, or converted to acetyl-CoA and subsequently to butanoyl-CoA, from which butanol might be produced, as indicated by presence of a butanol dehydrogenase. Formate from the pyruvate formate lyase is likely excreted, since no indications of hydrogen production were found. In addition, F. agariphila KMM 3901T has genes for the denitrification of nitrate via nitrite to dinitrogen oxide.
F. agariphila KMM 3901T likely stores energy and phosphorus in the form of polyphosphate, since the genome encodes an exopolyphosphatase and a polyphosphate kinase. In addition, F. agariphila KMM 3901T might store energy and carbon in the form of glycogen. The latter was not clear, since the genome features a glycogen synthase and a branching enzyme for glycogen biosynthesis but seems to lack known genes for glycogen breakdown, such as glycogen phosphorylase and a glycogen debranching enzyme.
Mono- and disaccharide utilization.
F. agariphila KMM 3901T converts pentoses such as ribose, xylose, and possibly arabinose via the nonoxidative part of the PP pathway to α-d-glucose 6-phosphate, which enters the EMP pathway. Pentose-specific transporters could be annotated only for xylose.
F. agariphila KMM 3901T has genes that indicate usage of the hexoses fucose, rhamnose, galactose, mannose, N-acetylglucosamine, galacturonate/glucuronate, and the sugar alcohol mannitol. Fucose and rhamnose are likely degraded via l-lactaldehyde to pyruvate. Galactose is likely metabolized via the complete Leloir pathway. Mannose may be converted to fructose-6-phosphate and funneled into the EMP pathway. Likewise, N-acetylglucosamine is deacetylated (nagA) and deaminated (nagB) to fructose-6-phosphate and funneled into the EMP pathway. Glucuronate can potentially be released from glucuronides by a GH88 (49) and further converted to 2-dehydro-3-deoxy-d-gluconate, which subsequently can enter the Entner-Doudoroff (ED) pathway. Mannuronate is likely released from alginates by PL6 alginate lyases. Mannitol can be converted to d-fructose-6-phosphate, which can enter the EMP pathway. Hexose-specific transporters could be annotated for fucose, galactose, hexuronides, N-acetyl-d-glucosamine, N-acetylneuraminate, mannuronate, and mannitol.
F. agariphila KMM 3901T has genes for the usage of the disaccharides lactose and maltose. Lactose can be potentially cleaved to d-glucose and d-galactose, and maltose can potentially be cleaved to d-glucose and β-d-glucose-1-phosphate. Likewise, the nonreducing disaccharide (and osmolyte) trehalose can be cleaved to d-glucose and β-d-glucose-1-phosphate via a trehalose phosphorylase. Disaccharide-specific transporters could be annotated only for maltose/maltodextrins.
In agreement with genome analysis, growth of F. agariphila KMM 3901T was positive for the monosaccharides l-arabinose, d-arabinose, dl-xylose, d-galactose, d-glucose, d-fructose, d-mannitol, d-mannose, and l-rhamnose, the disaccharides trehalose, d-sucrose, and maltose, and the trisaccharide raffinose, as well as N-acetyl-d-glucosamine.
Polysaccharide utilization.
We identified 193 CAZymes in the F. agariphila KMM 3901T genome (Table 1), comprising 88 GHs, 70 GTs, 13 PLs, 13 CEs, and 9 CBMs. The GHs belong to 27 and the PLs to 4 known families and are indicative of the degradation of the algal polysaccharides agar/agarose, alginate, arabinan, fucosides/fucoidan, α-glucans (e.g., starch), laminarin, mannan, polygalacturonans, porphyran, and xylan. In addition, genes for the degradation of algal chondroitin sulfate-like mucopolysaccharides, glycoproteins, and d-glucosyl-N-acylsphingosine were annotated, whereas annotated β-N-acetylhexosaminidases are likely involved in bacterial cell wall metabolism or detachment from biofilms (50) and thus may play a role in algal colonization. Likewise, we found genes for the synthesis of glycogen and the turnover of peptidoglycan.
Table 1.
CAZyme profile of F. agariphila KMM 3901T
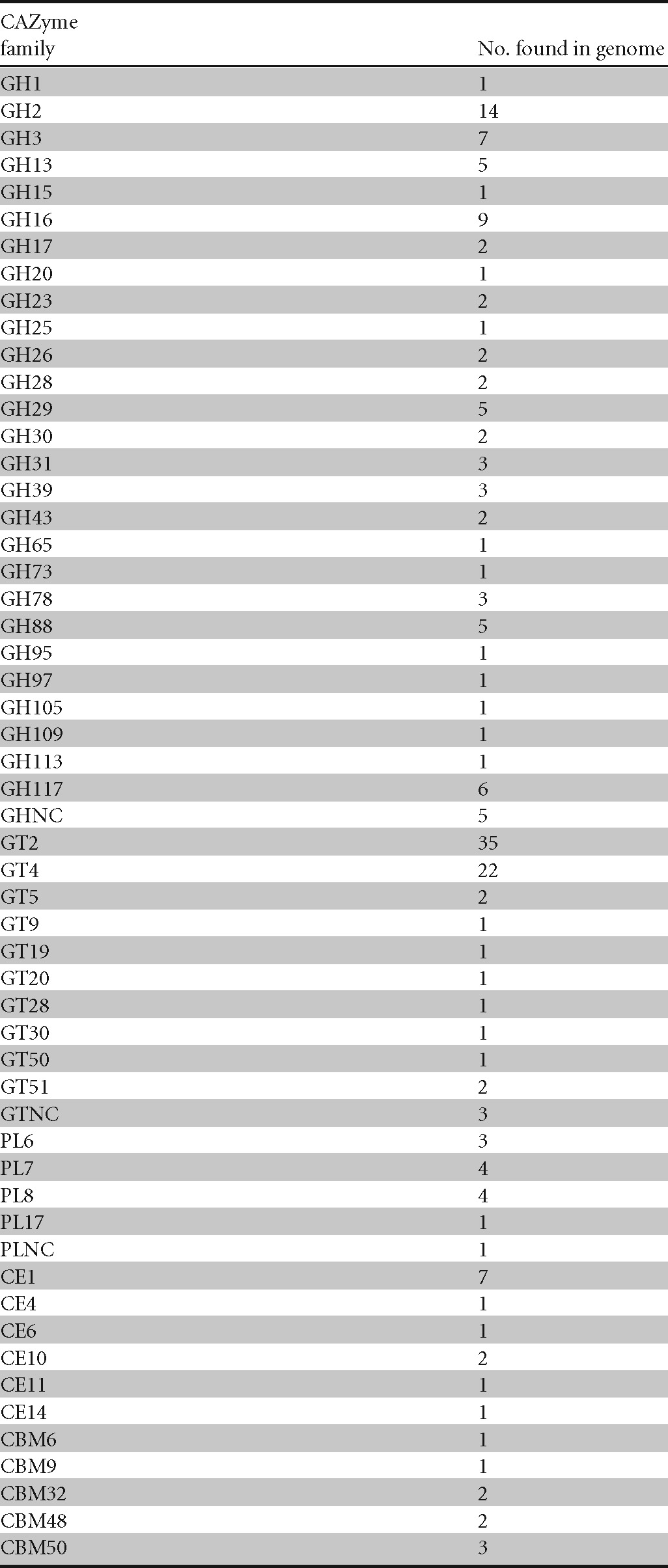
As implied by its name, F. agariphila KMM 3901T can degrade agarose by a β-agarase (GH16) and the resulting agarobiose dimers by a 3,6-anhydro-α-l-galactosidase (GH117) (51). The presence of eight alginate lyases of the CAZy families PL6, PL7, and PL17 furthermore indicates the potential for alginate degradation. These alginate lyases comprise the genes alyA2, alyA3, alyA4, and alyA6, as described for the genome of the alginate-degrading flavobacterium Zobellia galactanivorans DsijT (52). Arabinan degradation potential is provided by an α-l-arabinofuranosidase (GH3). Fucosides (found in N-linked glycans, e.g., on plant surfaces) and the sulfated polysaccharide fucoidan can likely be degraded by α-l-fucosidases (GH29 and GH95). An α-amylase (GH13) and three α-glucosidases (GH31) indicate the capacity for α-1,4-glucan degradation. Degradation of the β-1,3-glucan laminarin is indicated by an endo-β-1,3-glucanase (GH16) (53). Mannan can likely be degraded via an endo-β-1,4-mannosidase (GH26) and β-glucosidases (GH3) (54). Two polygalacturonases (GH28) for the degradation of polygalacturonans and four β-porphyranases (GH16) for the degradation of porphyran were also found in the genome. β-d-Xylans can likely be degraded to β-d-xylopyronase by an endo-β-1,4-xylanase and a β-1,4-xylosidase (GH39). A bifunctional β-xylosidase/α-l-arabinofuranosidase of the family GH3, which is known to degrade arabinoxylan hemicelluloses (55), was also found.
These annotations were corroborated by substrate tests with selected polysaccharides. Growth was positive with laminarin and xylan, weak with glycogen and κ-carrageenan, and negative with ι-carrageenan and cellulose.
Polysaccharide utilization loci.
PULs constitute coregulated transcriptional units that can encompass one or several operons. The boundaries of these units are often difficult to determine based only on a genome sequence, i.e., without expression data. For this study, we used as a delimiting criterion the presence of at least one TBDR/SusD-like protein gene pair with adjacent polysaccharide utilization genes (30). F. agariphila KMM 3901T has at least 13 of such gene clusters (Fig. 3). The colocation of TBDRs and SusD-like outer membrane proteins indicates an ancient functional coupling for polysaccharide recognition and direction to the TBDT pore. SusD-like proteins are known to play a role in starch binding (56) and have been previously shown to be colocated with TBDRs (2, 33, 57).
Fig 3.

Tentative polysaccharide utilization loci of F. agariphila KMM 3901T. CAZy families are indicated by numbers. The numbers to the PUL's left and right indicate positions in the genome.
F. agariphila KMM 3901T has 45 sulfatases, half of which were found in six of the PULs (Fig. 3B, D, F, G, H, and L). They were found to be always colocated with GH genes, indicating that the sulfatases are essential for the degradation of sulfated marine algal polysaccharides. The respective GHs include β-xylosidases (B, G, and H), α-glucosidases (B), β-glucosidases (B), α-fucosidases (B, D, and F), polygalacturonase (F), β-galactosidases (F, G, and H), α-rhamnosidases (H), and β-mannosidases (L). Some GHs in these PULs are not colocated with sulfatases, such as α-glucosidases (K), β-glucosidases (C), and β-galactosidases (I). GHs in other PULs include β-N-acetylhexosaminidases (E and M), mannosidase (L), α-amylases (K), maltose phosphorylase (K), and β-porphyranases (F).
In-depth CAZyme analysis allowed deduction of the most probable substrates of most PULs. PULs E, I, and M are likely specific for (decorated) N-acetyl-d-glucosamines, PUL B for complex sulfated O-linked glucans, PUL D for fucoidan, PUL F for porphyran, PUL G for agar/agaroid, PUL H for sulfated (decorated) rhamnogalacturonans, PUL J for alginate, PUL K for starch, and PUL L for sulfated mannan.
Transporters.
Besides TBDTs, the genome of F. agariphila KMM 3901T features a wide range of other transporters. A total of 31 genes could be assigned to ABC transporters, whereas only two tripartite ATP-independent periplasmic (TRAP) transport systems were found. Ion transporters were annotated for ammonium, iodide, iron, magnesium/cobalt, manganese, nickel, nitrate/nitrite, phosphate, potassium, sulfate, sulfur, and lead/cadmium/zinc/mercury. Besides the already-mentioned mono- and disaccharide importers, transport systems were found for organic compounds such as dipeptides, formate, glycine betaine, methionine, oligopeptides, and vitamin B12. Four sodium/proton transporters, one sodium/calcium transporter, and one arginine/ornithine transporter were annotated.
Proteases.
F. agariphila KMM 3901T encodes 129 peptidases, the majority of which are serine peptidases and metalloproteinases (see Table S1 in the supplemental material). Among serine peptidases, members of the families S9 and S15, both of which cleave mainly prolyl bonds (http://merops.sanger.ac.uk [58]), are most prevalent in F. agariphila KMM 3901T. S9 members act mostly on oligopeptides, probably due to the confined space in the N terminus of their β-propeller tunnel (59, 60), and S15 members release Xaa-Pro dipeptides (59). So far, S9 and S15 peptidases have been connected to the degradation of proline-rich proteins from animals (61–64) and are not known for a role in the biodegradation of algal biomass.
Among the present metalloproteinases, members of the families M24A and M23 belong to the most frequent ones. M24A family proteinases are known to remove the initial methionine found in many eukaryotic proteins (65), whereas M23 family members have been shown to take part in the extracellular degradation of bacterial peptidoglycan, either as a defense or as a feeding mechanism (66). The complete extracellular decomposition of peptides to amino acids requires M20 and M28 family exopeptidases (http://merops.sanger.ac.uk [58]), both of which can be found abundantly in the F. agariphila KMM 3901T genome as well.
Assimilation of nitrogen.
Besides a dissimilatory nitrate reductase, F. agariphila KMM 3901T has an assimilatory nitrate reductase and a cytochrome-dependent nitrite reductase for subsequent assimilatory reduction to ammonium. However, it has been observed that Formosa strains, including F. algariphila KMM 3901T, do not consistently exhibit nitrate reductase activity in physiological tests (24).
Motility.
F. agariphila KMM 3901T has genes for gliding motility (gldBCDFGHIJN). Gliding movement is an important feature for the colonization of surfaces and formation of biofilms.
Phages and transposases.
The F. agariphila KMM 3901T genome harbors 18 identifiable phage genes, most of which are concentrated in a single locus of around 50 genes (3.40 to 3.44 Mbp), with many hypothetical genes that most likely constitute a temperate phage. F. agariphila KMM 3901T also has 17 transposases, which mostly belong to the transposase mutator and IS116/IS110/IS902 Pfam families.
DISCUSSION
Some pelagic marine members of the Bacteroidetes, such as Polaribacter sp. MED152 (7) and Dokdonia sp. MED134 (9, 67), feature relatively small genomes of around 3 Mbp, are unable to ferment, and contain the light-dependent proton-pumping membrane protein proteorhodopsin. For Dokdonia sp. MED134, it has been shown that it can harness its proteorhodopsin for enhanced growth during light exposure, probably by using the additional ATP for the fixation of carbon dioxide via anaplerotic reactions (9). This mechanism likely aids pelagic members of the Bacteroidetes to cope with times of low nutrient availability in the ocean (7, 9). In contrast, F. agariphila KMM 3901T has a substantially larger genome, exceeding 4 Mbp, has the capacity to ferment and denitrify, and is devoid of proteorhodopsin. This indicates a notably different purely heterotrophic lifestyle in a nutrient-rich environment in line with algal colonization, where macromolecules, such as proteins and polysaccharides, are aplenty.
While proteases are important for the ecology of marine Bacteroidetes (10), it is difficult to attribute individual protease genes in a bacteroidetal genome to specific external proteinaceous substrates (e.g., an algal cell wall protein) based on annotations. Analysis of the F. agariphila KMM 3901T peptidases using the MEROPS classification system (56) at least did not allow the deduction of a particular lifestyle. However, the CAZyme profile of F. agariphila KMM 3901T is strongly indicative for its alga-associated lifestyle since it features CAZymes for the breakdown of many known polysaccharides specific to marine algae.
Polysaccharides constitute the most diverse class of macromolecules in nature in terms of primary structure, since they feature a wide variety of sugar monomers (including anhydro sugars) that can be linked in various ways in a linear or branched fashion and decorated with a large number of moieties (e.g., sulfate, methyl, or acetyl groups). There is no bacterium known that can degrade all naturally occurring polysaccharides. Instead, known polysaccharide-degrading bacteria specialize in dedicated polysaccharides subsets and their monomer constituents and thus harbor a restricted set of enzymes for polysaccharide decomposition. CAZymes typically account for not more than about 2% of the genes in most bacterial genomes (68, 69) and seldom exceed 5% even in bacteria that are specialized on carbohydrate degradation. This entails that multiple such niche-adapted microbial specialists are typically required for the concerted breakdown of complex polysaccharide mixtures in nature. A recent example is the study by Martens et al. (34), who showed this type of resource and niche partitioning in two human gut members of the Bacteroidetes.
The genome of F. agariphila KMM 3901T features ∼2% proteases and ∼5% CAZymes, which reflects the strong specialization of this species on macromolecule degradation. Within the so-far-sequenced members of the Flavobacteria, F. agariphila KMM 3901T exhibits one of the highest CAZymes densities (Table 2). More than 70% of the GHs in the F. agariphila KMM 3901T genome are located within PULs, where they are colocated with genes of TBDTs. These mediate the uptake of polysaccharides across the outer membrane into the periplasmic space, typically at the level of short oligomers (34). Periplasmic GHs cleave these oligomers into dimers and monomers that are finally taken up by ABC transporters or permeases over the cytoplasmic membrane into the cytoplasm. The TBDT system consists of an outer membrane TBDR and the TonB, ExbD, and ExbB proteins that form a complex in the cytoplasmic membrane (70). The TonB protein interlinks the components of the inner and outer membrane by β-strand paring with the TBDR N terminus. This connection is believed to translate the proton motive force across the cytoplasmic membrane into a movement that opens or closes a pore formed by the TBDR via its plug domain (71, 72). Most of the so-far-investigated TBDT systems are involved in the uptake of bulky iron-siderophore complexes. TBDTs are also known for the uptake of vitamin B12 and nickel. However, the importance of TBDTs for the uptake of carbohydrate oligomers is increasingly being recognized (28, 70, 72, 73). The number of TBDR genes in bacterial genomes differs widely. While the majority host only about 14 TBDRs, single genomes can harbor up to 120 TBDRs (23). Organisms with high numbers of TBDR genes belong mostly to the Alphaproteobacteria, the Gammaproteobacteria, and the Bacteroidetes (23). F. agariphila KMM 3901T harbors 66 TBDR genes, and a third of these TBDRs are located in PULs, which underpins the relevance of TBDT systems for polysaccharide degradation and uptake in F. agariphila KMM 3901T.
Table 2.
The 10 sequenced Flavobacteria strains with the as-yet-highest CAZyme numbersa
| Strain | No. of genes | No. of CAZymes | Proportion of CAZymes (%) |
|---|---|---|---|
| Formosa agariphila KMM 3901T | 3,642 | 193 | 5.30 |
| Zobellia galactanivorans DsiJT | 4,784 | 247 | 5.16 |
| Flavobacterium johnsoniae UW101 | 5,138 | 258 | 5.02 |
| Zunongwangia profunda SM-A87 | 4,714 | 208 | 4.41 |
| Capnocytophaga ochracea DSM 7271 | 2,256 | 95 | 4.21 |
| Flavobacteriaceae bacterium 3519-10 | 2,583 | 102 | 3.95 |
| Flavobacteriales bacterium HTCC2170 | 3,460 | 135 | 3.90 |
| Ornithobacterium rhinotracheale DSM 15997 | 2,371 | 86 | 3.63 |
| Flavobacterium branchiophilum FL-15 | 3,083 | 108 | 3.50 |
| Cellulophaga algicola DSM 14237 | 4,345 | 151 | 3.48 |
Thirty-two Flavobacteria genomes were analyzed in total. CAZyme annotations apart from those of F. agariphila KMM 3901T were obtained from the CAZy database.
Even though F. agariphila KMM 3901T has been isolated from a specimen of the green alga Acrosiphonia sonderi, its CAZmye spectrum covers polysaccharides such as the following: (i) sulfated mannan, which is not restricted to green algae (74) but is also found in red algae (75), (ii) typical red alga polysaccharides, such as agar and porphyran, and (iii) typical brown alga polysaccharides, such as alginate, fucoidan, and laminarin. The genetic potential for mannitol degradation is probably an adaption to brown algae that synthesize d-mannitol as a storage compound (76) and have laminarin, whose reducing ends are masked by mannitol residues (in contrast to chrysolaminarin in diatoms). The CAZyme repertoire of F. agariphila KMM 3901T includes typical algal cell wall polysaccharides, such as agar, alginate, arabinan, mannan, and polygalacturonans, as well as storage polysaccharides, such as laminarin and mannan. This indicates that F. agariphila KMM 3901T is unlikely to be restricted to algal surface colonization but probably is also able to degrade various algal cells. Both a substrate spectrum that is not restricted to green algae and the capacity for algal degradation are supported by the fact that its close relative, F. algae KMM 3553T, has been isolated from the thallus of a decaying brown alga, Fucus evanescens. F. agariphila KMM 3901T can use xylan, a typical tissue interconnecting plant polysaccharide. Therefore, it is possible that F. agariphila KMM 3901T is potentially harmful and can cause algal necrosis when entering lesions in algal tissues. The capability to thrive under anoxic conditions might be used during such algal infections. F. agariphila KMM 3309T might be also successful at beach rift lines, where algae of various types are washed up and oxic and anoxic conditions change on a small scale.
Some PULs in F. agariphila KMM 3901T show a strong synteny with PULs of other Flavobacteria, for example, the alginate PUL J (Fig. 4) with PULs in the marine flavobacterium Zobellia galactanivorans DsiJT (52) and “Gramella forsetii” KT0803 (6). Alginolytic activity of the respective PUL in Zobellia galactanivorans DsijT has recently been experimentally verified (52). Such PUL syntenies indicate that a set of well-conserved PULs exist, for which a relationship between gene arrangement and polysaccharide substrate can be established. These PULs could become useful as bioindicators in future metagenomic studies of phytoplankton-associated bacteria.
Fig 4.

Synteny between the alginate degradation PUL in F. agariphila KMM 3901T and PULs in other Flavobacteria. Genes include the following: 1, acetoin reductase; 2, mannunorate transporter; 3, transcriptional regulator, GntR family; 4, alginate lyase (PL7); 5, PKD domain protein; 6, PKD domain protein; 7, SusD-like protein; 8, TBDR; 9, alginate lyase (PL17); 10, alginate lyase (PL6); 11,; 4-hydroxy-2-oxoglutarate aldolase; 12, 2-dehydro-3-deoxygluconate kinase; 13, pectin degradation protein KdgF; 14, alginate lyase; 15, hypothetical protein.
The annotation of CAZymes and their specificities is prone to false positives when carried out in silico, since some former CAZymes have evolved to act on noncarbohydrate substrates while still displaying typical CAZyme characteristics (77). We therefore conducted a manual fine annotation of all CAZymes of the F. agariphila KMM 3901T genome. Of course, we cannot rule out false annotations that might lead to an overestimation of the polysaccharide degradation spectrum of F. agariphila KMM 3901T. These limitations notwithstanding, our genome analysis clearly shows how profoundly the alga-associated lifestyle has shaped the CAZyme inventory of F. agariphila KMM 3901T over the course of its evolution.
Polysaccharides in nature are very diverse, and polysaccharide-degrading bacteria have adapted to this by grouping genes for the degradation of specific polysaccharides into distinct PULs. It has been suggested that PUL specificities go as far as even the branching patterns of a polysaccharide playing a role (e.g., see references 30 and 31 and references therein). From other members of the Bacteroidetes, it is known that PULs are coregulated. For example, for Bacteroides thetaiotaomicron VPI-5482, it has been shown that binding of a monosaccharide to a hybrid two-component system can induce one specific PUL while simultaneously repressing others, which indicates “hierarchical PUL expression and prioritization of polysaccharide use” (78). Thus, PUL expression in F. agariphila KMM 3901T likely also constitutes a highly regulated network that reacts in a controlled and specific manner when a polysaccharide or naturally cooccurring polysaccharides become available.
We are just beginning to uncover the intricacies of these mechanisms, and we know substantially less about Bacteroidetes from marine algae than about those from the human gut. The almost-complete genome of F. agariphila KMM 3901T will enable future studies of the mechanisms and regulation of its polysaccharide degradation capabilities and will thus allow deeper insights into the characteristics of marine alga-associated Flavobacteria.
Supplementary Material
ACKNOWLEDGMENTS
We thank Clara Martinéz Peréz for her initial work on CAZymes, Lennart Kappelmann for helping with annotation, Bernhard M. Fuchs for critical reading of the manuscript, Tanja Hilleker from the Max Planck Genome Centre in Cologne for her work on gap closure, and Gurvan Michel and Mirjam Czjzek from the Station Biologique de Roscoff for fruitful discussions.
This study was supported by the Max Planck Society.
ADDENDUM IN PROOF
The remaining gaps in the F. agariphila genome were resolved after the manuscript's acceptance. The genome sequence in the public databases will be updated accordingly.
Footnotes
Published ahead of print 30 August 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.01937-13.
REFERENCES
- 1. Eilers H, Pernthaler J, Glöckner FO, Amann R. 2000. Culturability and in situ abundance of pelagic bacteria from the North Sea. Appl. Environ. Microbiol. 66:3044–3051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gómez-Pereira PR, Schüler M, Fuchs BM, Bennke C, Teeling H, Waldmann J, Richter M, Barbe V, Bataille E, Glöckner FO, Amann R. 2012. Genomic content of uncultured Bacteroidetes from contrasting oceanic provinces in the North Atlantic Ocean. Environ. Microbiol. 14:52–66 [DOI] [PubMed] [Google Scholar]
- 3. Gómez-Pereira PR, Fuchs BM, Alonso C, Oliver MJ, van Beusekom JEE, Amann R. 2010. Distinct flavobacterial communities in contrasting water masses of the North Atlantic Ocean. ISME J. 4:472–487 [DOI] [PubMed] [Google Scholar]
- 4. Llobet-Brossa E, Rosselló-Mora R, Amann R. 1998. Microbial community composition of Wadden Sea sediments as revealed by fluorescence in situ hybridization. Appl. Environ. Microbiol. 64:2691–2696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kirchman DL. 2002. The ecology of Cytophaga-Flavobacteria in aquatic environments. FEMS Microbiol. Ecol. 39:91–100 [DOI] [PubMed] [Google Scholar]
- 6. Bauer M, Kube M, Teeling H, Richter M, Lombardot T, Allers E, Würdemann CA, Quast C, Kuhl H, Knaust F, Woebken D, Bischof K, Mussmann M, Choudhuri JV, Meyer F, Reinhardt R, Amann RI, Glöckner FO. 2006. Whole genome analysis of the marine Bacteroidetes ‘Gramella forsetii' reveals adaptations to degradation of polymeric organic matter. Environ. Microbiol. 8:2201–2213 [DOI] [PubMed] [Google Scholar]
- 7. González JM, Fernández-Gómez B, Fernàndez-Guerra A, Gómez-Consarnau L, Sánchez O, Coll-Lladó M, del Campo J, Escuerdo L, Rodríguez-Martínez R, Alonso-Sáez L, Latasa M, Paulsen I, Nedashkovskaya O, Lekunberri I, Pinhassi J, Pedrós-Alió C. 2008. Genome analysis of the proteorhodopsin-containing marine bacterium Polaribacter sp. MED152 (Flavobacteria). Proc. Natl. Acad. Sci. U. S. A. 105:8724–8729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Thomas F, Hehemann J-H, Rebuffet E, Czjzek M, Michel G. 2011. Environmental and gut Bacteroidetes: the food connection. Front. Microbiol. 2:93. 10.3389/fmicb.2011.00093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. González JM, Pinhassi J, Fernández-Gómez B, Coll-Lladó M, González-Velázquez M, Puigbò P, Jaenicke S, Gómez-Consarnau L, Fernàndez-Guerra A, Goesmann A, Pedrós-Alió C. 2011. Genomics of the proteorhodopsin-containing marine flavobacterium Dokdonia sp. strain MED134. Appl. Environ. Microbiol. 77:8676–8686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fernández-Gómez B, Richter M, Schüler M, Pinhassi J, Acinas SG, González JM, Pedrós-Alió C. 2013. Ecology of marine Bacteroidetes: a comparative genomics approach. ISME J. 7:1026–1037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Riedel T, Gómez-Consarnau L, Tomasch J, Martin M, Jarek M, González JM, Spring S, Rohlfs M, Brinkhoff T, Cypionka H, Göker M, Fiebig A, Klein J, Goesmann A, Fuhrman JA, Wagner-Döbler I. 2013. Genomics and physiology of a marine flavobacterium encoding a proteorhodopsin and a xanthorhodopsin-like protein. PLoS One 8:e57487. 10.1371/journal.pone.0057487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Frias-Lopez J, Zerkle AL, Bonheyo GT, Fouke BW. 2002. Partitioning of bacterial communities between seawater and healthy, black band diseased, and dead coral surfaces. Appl. Environ. Microbiol. 68:2214–2228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rohwer F, Seguritan V, Azam F, Knowlton N. 2002. Diversity and distribution of coral-associated bacteria. Mar. Ecol. Prog. Ser. 243:1–10 [Google Scholar]
- 14. Barbeyron T, L'Haridon S, Corre E, Kloareg B, Potin P. 2001. Zobellia galactanovorans gen. nov., sp. nov., a marine species of Flavobacteriaceae isolated from a red alga, and classification of [Cytophaga] uliginosa (ZoBell and Upham 1944) Reichenbach 1989 as Zobellia uliginosa gen. nov., comb. nov. Int. J. Syst. Evol. Microbiol. 51:985–997 [DOI] [PubMed] [Google Scholar]
- 15. Nedashkovskaya OI, Kim SB, Han SK, Lysenko AM, Rohde M, Zhukova NV, Falsen E, Frolova GM, Mikhailov VV, Bae KS. 2003. Mesonia algae gen. nov., sp. nov., a novel marine bacterium of the family Flavobacteriaceae isolated from the green alga Acrosiphonia sonderi (Kutz) Kornm. Int. J. Syst. Evol. Microbiol. 53:1967–1971 [DOI] [PubMed] [Google Scholar]
- 16. Nedashkovskaya OI, Kim SB, Han SK, Rhee M-S, Lysenko AM, Rohde M, Zhukova NV, Frolova GM, Mikhailov VV, Bae KS. 2004. Algibacter lectus gen. nov., sp. nov., a novel member of the family Flavobacteriaceae isolated from green algae. Int. J. Syst. Evol. Microbiol. 54:1257–1261 [DOI] [PubMed] [Google Scholar]
- 17. Ivanova EP, Alexeeva YV, Flavier S, Wright JP, Zhukova NV, Gorshkova NM, Mikhailov VV, Nicolau DV, Christen R. 2004. Formosa algae gen. nov., sp. nov., a novel member of the family Flavobacteriaceae. Int. J. Syst. Evol. Microbiol. 54:705–711 [DOI] [PubMed] [Google Scholar]
- 18. Nedashkovskaya OI, Kim SB, Lee KH, Bae KS, Frolova GM, Mikhailov VV, Kim IS. 2005. Pibocella ponti gen. nov., sp. nov., a novel marine bacterium of the family Flavobacteriaceae isolated from the green alga Acrosiphonia sonderi. Int. J. Syst. Evol. Microbiol. 55:177–181 [DOI] [PubMed] [Google Scholar]
- 19. Beleneva IA, Zhukova NV. 2006. Bacterial communities of some brown and red algae from Peter the Great Bay, the Sea of Japan. Microbiology 75:348–357 [PubMed] [Google Scholar]
- 20. Staufenberger T, Thiel V, Wiese J, Imhoff JF. 2008. Phylogenetic analysis of bacteria associated with Laminaria saccharina. FEMS Microbiol. Ecol. 64:65–77 [DOI] [PubMed] [Google Scholar]
- 21. Salaün S, Kervarec N, Potin P, Haras D, Piotto M, La Barre S. 2010. Whole-cell spectroscopy is a convenient tool to assist molecular identification of cultivatable marine bacteria and to investigate their adaptive metabolism. Talanta 80:1758–1770 [DOI] [PubMed] [Google Scholar]
- 22. Crump BC, Koch EW. 2008. Attached bacterial populations shared by four species of aquatic angiosperms. Appl. Environ. Microbiol. 74:5948–5957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Blanvillain S, Meyer D, Boulanger A, Lautier M, Guynet C, Denancé N, Vasse J, Lauber E, Arlat M. 2007. Plant carbohydrate scavenging through TonB-dependent receptors: a feature shared by phytopathogenic and aquatic bacteria. PLoS One 2:e224. 10.1371/journal.pone.0000224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nedashkovskaya OI, Kim SB, Vancanneyt M, Snauwaert C, Lysenko AM, Rohde M, Frolova GM, Zhukova NV, Mikhailov VV, Bae KS, Oh HW, Swings J. 2006. Formosa agariphila sp. nov., a budding bacterium of the family Flavobacteriaceae isolated from marine environments, and emended description of the genus Formosa. Int. J. Syst. Evol. Microbiol. 56:161–167 [DOI] [PubMed] [Google Scholar]
- 25. Yoon B-J, Oh D-C. 2011. Formosa spongicola sp. nov., isolated from the marine sponge Hymeniacidon flavia. Int. J. Syst. Evol. Microbiol. 61:330–333 [DOI] [PubMed] [Google Scholar]
- 26. Li Q, Zhang Y, Juck D, Fortin N, Greer CW. 2011. Impact of intensive land-based fish culture in Qingdao, China, on the bacterial communities in surrounding marine waters and sediments. Evid. Based Complement. Alternat. Med. 2011:487543. 10.1155/2011/487543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Prabagaran SR, Manorama R, Delille D, Shivaji S. 2007. Predominance of Roseobacter, Sulfitobacter, Glaciecola and Psychrobacter in seawater collected off Ushuaia, Argentina, Sub-Antarctica. FEMS Microbiol. Ecol. 59:342–355 [DOI] [PubMed] [Google Scholar]
- 28. Teeling H, Fuchs BM, Becher D, Klockow C, Gardebrecht A, Bennke CM, Kassabgy M, Huang S, Mann AJ, Waldmann J, Weber M, Klindworth A, Otto A, Lange J, Bernhardt J, Reinsch C, Hecker M, Peplies J, Bockelmann FD, Callies U, Gerdts G, Wichels A, Wiltshire KH, Glöckner FO, Schweder T, Amann R. 2012. Substrate-controlled succession of marine bacterioplankton populations induced by a phytoplankton bloom. Science 336:608–611 [DOI] [PubMed] [Google Scholar]
- 29. Kraan S. 2012. Algal polysaccharides, novel applications and outlook. In Chang C-F. (ed), Carbohydrates—comprehensive studies on glycobiology and glycotechnology. InTech, New York, NY: http://www.intechopen.com/books/carbohydrates-comprehensive-studies-on-glycobiology-and-glycotechnology/algal-polysaccharides-novel-applications-and-outlook [Google Scholar]
- 30. Sonnenburg ED, Zheng H, Joglekar P, Higginbottom SK, Firbank SJ, Bolam DN, Sonnenburg JL. 2010. Specificity of polysaccharide use in intestinal Bacteroides species determines diet-induced microbiota alterations. Cell 141:1241–1252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sonnenburg ED, Sonnenburg JL, Manchester JK, Hansen EE, Chiang HC, Gordon JI. 2006. A hybrid two-component system protein of a prominent human gut symbiont couples glycan sensing in vivo to carbohydrate metabolism. Proc. Natl. Acad. Sci. U. S. A. 103:8834–8839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cantarel BL, Coutinho PM, Rancurel C, Bernard T, Lombard V, Henrissat B. 2009. The Carbohydrate-Active EnZymes database (CAZy): an expert resource for glycogenomics. Nucleic Acids Res. 37:D233–D238. 10.1093/nar/gkn663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Martens EC, Koropatkin NM, Smith TJ, Gordon JI. 2009. Complex glycan catabolism by the human gut microbiota: the Bacteroidetes Sus-like paradigm. J. Biol. Chem. 284:24673–24677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Martens EC, Lowe EC, Chiang H, Pudlo N, Wu M, McNulty NP, Abbott DW, Henrissat B, Gilbert HJ, Bolam DN, Gordon JI. 2011. Recognition and degradation of plant cell wall polysaccharides by two human gut symbionts. PLoS Biol. 9:e1001221. 10.1371/journal.pbio.1001221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hehemann J-H, Kelly AG, Pudlo N, Martens EC, Boraston AB. 2012. Bacteria of the human gut microbiome catabolize red seaweed glycans with carbohydrate-active enzyme updates from extrinsic microbes. Proc. Natl. Acad. Sci. U. S. A. 109:19786–19791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhou J, Bruns MA, Tiedje JM. 1996. DNA recovery from soils of diverse composition. Appl. Environ. Microbiol. 62:316–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gordon D. 2004. Viewing and editing assembled sequences using Consed. Curr. Protoc. Bioinformatics 2:11.2.1–11.2.43. 10.1002/0471250953.bi1102s02 [DOI] [PubMed] [Google Scholar]
- 38. Aziz RK, Bartels D, Best A, DeJongh M, Disz T, Edwards R, Formsma K, Gerdes S, Glass EM, Kubal M, Meyer F, Olsen GJ, Olson R, Osterman AL, Overbeek R, McNeil LK, Paarmann D, Paczian T, Parrello B, Pusch GD, Reich C, Stevens R, Vassieva O, Vonstein V, Wilke A, Zagnitko O. 2008. The RAST server: rapid annotations using subsystems technology. BMC Genomics 9:75. 10.1186/1471-2164-9-75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Meyer F, Goesmann A, McHardy A, Bartels D, Bekel T, Clausen J, Kalinowski J, Linke B, Rupp O, Giegerich R, Puehler A. 2003. GenDB—an open source genome annotation system for prokaryote genomes. Nucleic Acids Res. 31:2187–2195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pruitt KD, Tatusova T, Brown GR, Maglott DR. 2012. NCBI Reference Sequences (RefSeq): current status, new features and genome annotation policy. Nucleic Acids Res. 40:D130–D135. 10.1093/nar/gkr1079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hunter S, Jones P, Mitchell A, Apweiler R, Attwood TK, Bateman A, Bernard T, Binns D, Bork P, Burge S, de Castro E, Coggill P, Corbett M, Das U, Daugherty L, Duquenne L, Finn RD, Fraser M, Gough J, Haft D, Hulo N, Kahn D, Kelly E, Letunic I, Lonsdale D, Lopez R, Madera M, Maslen J, McAnulla C, McDowall J, McMenamin C, Mi H, Mutowo-Muellenet P, Mulder N, Natale D, Orengo C, Pesseat S, Punta M, Quinn AF, Rivoire C, Sangrador-Vegas A, Selengut JD, Sigrist CJ, Scheremetjew M, Tate J, Thimmajanarthanan M, Thomas PD, Wu CH, Yeats C, Yong S-Y. 2012. InterPro in 2011: new developments in the family and domain prediction database. Nucleic Acids Res. 40:D306–D312. 10.1093/nar/gkr948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sonnhammer EL, Eddy SR, Durbin R. 1997. Pfam: a comprehensive database of protein domain families based on seed alignments. Proteins 28:405–420 [DOI] [PubMed] [Google Scholar]
- 43. Ogata H, Goto S, Sato K, Fujibuchi W, Bono H, Kanehisa M. 1999. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 27:29–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tatusov RL. 1997. A genomic perspective on protein families. Science 278:631–637 [DOI] [PubMed] [Google Scholar]
- 45. Nielsen H, Engelbrecht J, Brunak S, von Heijne G. 1997. Identification of prokaryotic and eukaryotic signal peptides and prediction of their cleavage sites. Protein Eng. 10:1–6 [DOI] [PubMed] [Google Scholar]
- 46. Krogh A, Larsson B, von Heijne G, Sonnhammer EL. 2001. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 305:567–580 [DOI] [PubMed] [Google Scholar]
- 47. Richter M, Lombardot T, Kostadinov I, Kottmann R, Duhaime MB, Peplies J, Glöckner FO. 2008. JCoast—a biologist-centric software tool for data mining and comparison of prokaryotic (meta)genomes. BMC Bioinformatics 9:177. 10.1186/1471-2105-9-177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Widdel F, Bak F. 1992. Gram-negative mesophilic sulfate-reducing bacteria, p 3352–3378 In Balows A, Tr̈uper HG, Dworkin M, Harder W, Schleifer KH. (ed), The Prokaryotes, 2nd ed. Springer Verlag, Berlin, Germany [Google Scholar]
- 49. Jongkees SAK, Withers SG. 2011. Glycoside cleavage by a new mechanism in unsaturated glucoronyl hydrolases. J. Am. Chem. Soc. 133:19334–19337 [DOI] [PubMed] [Google Scholar]
- 50. Itoh Y, Wang X, Hinnebusch BJ, James F, Iii P, Romeo T, Iii JFP. 2005. Depolymerization of β-1,6-N-acetyl-d-glucosamine disrupts the integrity of diverse bacterial biofilms. J. Bacteriol. 187:382–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hehemann J-H, Correc G, Barbeyron T, Helbert W, Czjzek M, Michel G. 2010. Transfer of carbohydrate-active enzymes from marine bacteria to Japanese gut microbiota. Nature 464:908–912 [DOI] [PubMed] [Google Scholar]
- 52. Thomas F, Barbeyron T, Tonon T, Génicot S, Czjzek M, Michel G. 2012. Characterization of the first alginolytic operons in a marine bacterium: from their emergence in marine Flavobacteriia to their independent transfers to marine Proteobacteria and human gut Bacteroides. Environ. Microbiol. 14:2379–2394 [DOI] [PubMed] [Google Scholar]
- 53. Kumagai Y, Ojima T. 2010. Isolation and characterization of two types of beta-1,3-glucanases from the common sea hare Aplysia kurodai. Comp. Biochem. Physiol. 155:138–144 [DOI] [PubMed] [Google Scholar]
- 54. Moreira LRS, Filho EXF. 2008. An overview of mannan structure and mannan-degrading enzyme systems. Appl. Microbiol. Biotechnol. 79:165–178 [DOI] [PubMed] [Google Scholar]
- 55. Lee RC, Hrmova M, Burton RA, Lahnstein J, Fincher GB. 2003. Bifunctional family 3 glycoside hydrolases from barley with alpha-L-arabinofuranosidase and beta-D-xylosidase activity. Characterization, primary structures, and COOH-terminal processing. J. Biol. Chem. 278:5377–5387 [DOI] [PubMed] [Google Scholar]
- 56. Cho KY, Salyers AA. 2001. Biochemical analysis of interactions between outer membrane proteins that contribute to starch utilization by Bacteroides thetaiotaomicron. J. Bacteriol. 183:7224–7230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Xu J, Bjursell MK, Himrod J, Deng S, Carmichael LK, Chiang HC, Hooper LV, Gordon JI. 2003. A genomic view of the human-Bacteroides thetaiotaomicron symbiosis. Science 299:2074–2076 [DOI] [PubMed] [Google Scholar]
- 58. Rawlings ND, Barrett AJ, Bateman A. 2012. MEROPS: the database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res. 40:D343–D350. 10.1093/nar/gkr987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Fülöp V. 1998. Prolyl oligopeptidase: an unusual beta-propeller domain regulates proteolysis. Cell 94:161–170 [DOI] [PubMed] [Google Scholar]
- 60. Polgár L. 2002. The prolyl oligopeptidase family. Cell. Mol. Life Sci. 59:349–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Matos J, Nardi M, Kumura H, Monnet V. 1998. Genetic characterization of pepP, which encodes an aminopeptidase P whose deficiency does not affect Lactococcus lactis growth in milk, unlike deficiency of the X-prolyl dipeptidyl aminopeptidase. Appl. Environ. Microbiol. 64:4591–4595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Rawlings N, Polgar L, Barrett A. 1991. A new family of serine-type peptidases related to prolyl oligopeptidase. Biochem. J. 279:907–908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Holst JJ, Deacon CF. 1998. Inhibition of the activity of dipeptidyl-peptidase IV as a treatment for type 2 diabetes. Diabetes. 47:1663–1670 [DOI] [PubMed] [Google Scholar]
- 64. Rigolet P, Mechin I, Delage M-M, Chich J-F. 2002. The structural basis for catalysis and specificity of the X-prolyl dipeptidyl aminopeptidase from Lactococcus lactis. Structure (London, England) 10:1383–1394 [DOI] [PubMed] [Google Scholar]
- 65. Bradshaw RA, Brickey WW, Walker KW. 1998. N-terminal processing: the methionine aminopeptidase and N alpha-acetyl transferase families. Trends Biochem. Sci. 23:263–267 [DOI] [PubMed] [Google Scholar]
- 66. Baba T, Schneewind O. 1996. Target cell specificity of a bacteriocin molecule: a C-terminal signal directs lysostaphin to the cell wall of Staphylococcus aureus. EMBO J. 15:4789–4797 [PMC free article] [PubMed] [Google Scholar]
- 67. Gómez-Consarnau L, González JM, Coll-Lladó M, Gourdon P, Pascher T, Neutze R, Pedrós-Alió C, Pinhassi J. 2007. Light stimulates growth of proteorhodopsin-containing marine Flavobacteria. Nature 445:210–213 [DOI] [PubMed] [Google Scholar]
- 68. Coutinho PM, Deleury E, Davies GJ, Henrissat B. 2003. An evolving hierarchical family classification for glycosyltransferases. J. Mol. Biol. 328:307–317 [DOI] [PubMed] [Google Scholar]
- 69. Lairson LL, Henrissat B, Davies GJ, Withers SG. 2008. Glycosyltransferases: structures, functions, and mechanisms. Annu. Rev. Biochem. 77:521–555 [DOI] [PubMed] [Google Scholar]
- 70. Schauer K, Rodionov DA, de Reuse H. 2008. New substrates for TonB-dependent transport: do we only see the “tip of the iceberg”? Trends Biochem. Sci. 33:330–338 [DOI] [PubMed] [Google Scholar]
- 71. Noinaj N, Guillier M, Barnard T, Buchanan S. 2010. TonB-dependent transporters: regulation, structure, and function. Annu. Rev. Microbiol. 64:43–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Krewulak KD, Vogel HJ. 2011. TonB or not TonB: is that the question? Biochem. Cell Biol. 89:87–97 [DOI] [PubMed] [Google Scholar]
- 73. Neugebauer H, Herrmann C, Kammer W, Schwarz G, Nordheim A, Braun V. 2005. ExbBD-dependent transport of maltodextrins through the novel MalA protein across the outer membrane of Caulobacter crescentus. J. Bacteriol. 187:8300–8311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Fernández PV, Estevez JM, Cerezo AS, Ciancia M. 2012. Sulfated β-d-mannan from green seaweed Codium vermilara. Carbohydr. Polym. 87:916–919 [DOI] [PubMed] [Google Scholar]
- 75. Pérez Recalde M, Noseda MD, Pujol CA, Carlucci MJ, Matulewicz MC. 2009. Sulfated mannans from the red seaweed Nemalion helminthoides of the South Atlantic. Phytochemistry 70:1062–1068 [DOI] [PubMed] [Google Scholar]
- 76. Michel G, Tonon T, Scornet D, Cock JM, Kloareg B. 2010. Central and storage carbon metabolism of the brown alga Ectocarpus siliculosus: insights into the origin and evolution of storage carbohydrates in eukaryotes. New Phytol. 188:67–81 [DOI] [PubMed] [Google Scholar]
- 77. Coutinho PM, Stam M, Blanc E, Henrissat B. 2003. Why are there so many carbohydrate-active enzyme-related genes in plants? Trends Plant Sci. 8:563–565 [DOI] [PubMed] [Google Scholar]
- 78. Lynch JB, Sonnenburg JL. 2012. Prioritization of a plant polysaccharide over a mucus carbohydrate is enforced by a Bacteroides hybrid two-component system. Mol. Microbiol. 85:478–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Stamatakis A. 2006. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22:2688–2690 [DOI] [PubMed] [Google Scholar]
- 80. Ludwig W, Strunk O, Westram R, Richter L, Meier H, Yadhukumar Buchner A, Lai T, Steppi S, Jobb G, Förster W, Brettske I, Gerber S, Ginhart AW, Gross O, Grumann S, Hermann S, Jost R, König A, Liss T, Lüssmann R, May M, Nonhoff B, Reichel B, Strehlow R, Stamatakis A, Stuckmann N, Vilbig A, Lenke M, Ludwig T, Bode A, Schleifer K-H. 2004. ARB: a software environment for sequence data. Nucleic Acids Res. 32:1363–1371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Teeling H, Meyerdierks A, Bauer M, Amann R, Glöckner FO. 2004. Application of tetranucleotide frequencies for the assignment of genomic fragments. Environ. Microbiol. 6:938–947 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.