

Abstract
The mechanisms involved in substrate-dependent regulation of a Magnaporthe oryzae gene encoding a cellulase which we designate MoCel7C (MGG_14954) were investigated. The levels of MoCel7C transcript were dramatically increased more than 1,000-fold, 16 to 24 h after transfer to a medium containing 2% carboxymethylcellulose (CMC), while levels were very low or undetectable in conventional rich medium. Green fluorescent protein reporter assays showed that the MoCel7C promoter was activated by cello-oligosaccharides larger than a pentamer. CMC-induced activation of the MoCel7C promoter was suppressed by glucose and cellobiose. Chromatin immunoprecipitation assays revealed that histone H3 methylation on lysine 4 (H3K4) at the MoCel7C locus was associated with activation of the gene by CMC. Consistently, CMC-induced MoCel7C gene activation was drastically diminished in a knockout (KO) mutant of the MoSET1 gene, which encodes a histone lysine methyltransferase that catalyzes H3K4 methylation in M. oryzae. Interestingly, however, MoCel7C transcript levels under noninducing conditions were significantly increased in the MoSET1 KO mutant, suggesting that MoSET1 directly or indirectly plays a role in both activation and suppression of the MoCel7C gene in response to environmental signals. In addition, gene expression and silencing vectors using the MoCel7C promoter were constructed.
INTRODUCTION
In eukaryotic cells, gene expression is often associated with covalent chemical modifications of histone proteins and DNA in the chromatin, which are referred to as epigenetic modifications. It is well-known that in mammals, changes in the patterns of epigenetic modification play important roles in stage- and/or tissue-specific gene expression as part of normal development. In addition to developmentally controlled gene expression, recent studies have indicated that epigenetic modifications can also be involved in transcriptional alterations induced by environmental signals. A variety of environmental and intrinsic signals can influence the activities of histone-modifying and -demodifying enzymes, which leads to specific combinations of histone modifications (1, 2). For example, methylation of histone H3 at lysine 4 (H3K4) and acetylation of histone H3 at lysine 9 (H3K9) are believed to be evolutionarily conserved marks of active chromatin, while di- and trimethylation of H3K9 are signatures of silent chromatin. Such modifications can occur in response to environmental signals (3, 4).
In filamentous fungi, epigenetic gene regulation has been studied in-depth in only a few model systems. Epigenetic modifications leading to gene suppression, such as cytosine methylation and trimethylation of histone H3 at lysine 9 (H3K9me3) have been intensively studied in Neurospora crassa and Ascobolus immersus, and their roles in heterochromatin formation and telomere silencing have been demonstrated in detail (5). In Neurospora and Aspergillus, histone acetylation was shown to be associated with transcriptional activation of genes involved in the light response and secondary metabolite (SM) production (6, 7). More recently, Bok et al. showed that CclA, a member of the H3K4 methylating COMPASS complex in Aspergillus nidulans, plays an important role in regulating SM gene clusters (8). The deletion of CclA resulted in a reduction in H3K4me2 and H3K4me3 as expected but, surprisingly, increased gene expression of cryptic SM gene clusters in A. nidulans and Aspergillus fumigatus (8, 9). LaeA is another regulator of SM gene clusters in Aspergillus, affecting the level of H3K9me3 (10, 11). Thus, in fungi, epigenetic regulation seems to be involved in various processes, including niche establishment through SM production and response to environmental signals.
Here we report that substrate-induced transcriptional activation of a cellulase gene in Magnaporthe oryzae is associated with H3K4 methylation catalyzed by the histone methyltransferase, MoSET1, an ortholog of Saccharomyces cerevisiae SET1. Rice blast caused by M. oryzae is one of the most devastating diseases of rice and is responsible for significant crop losses worldwide. M. oryzae has emerged as a model system for understanding fungus-plant interactions owing to its economic importance and genetic tractability, including fully sequenced genomes for both host and pathogen (12).
This fungus shows global changes in its transcription profile during the infection process (13). We previously showed that transcript levels of several cellulases belonging to glycoside hydrolase families 6 and 7 (GH6 and GH7) were upregulated more than 1,000-fold upon infection compared with the levels of cellulase transcripts in vegetative mycelia on rich medium (14). In this study, we show that one of these cellulases, designated MoCel7C (MGG_14954), is highly induced in the presence of cellulose, the major component of host cell walls. Thus, this fungus seems to sense the host cell wall upon infection and then strongly activates the MoCel7C gene.
Several cellulase promoters are known to be activated in the presence of a cellulose substrate (15–18). The best-characterized example is the cbh1 promoter in Trichoderma reesei. At least four transcription factors have been suggested to be involved in the regulation of the cbh1 promoter. ACEII and Xyr1 are positive regulators of the cbh1 promoter, while CRE1 and ACEI act as repressors (19–22). Interestingly, expression of all T. reesei cellulases was dependent on LAE1, which is a LaeA ortholog (23). However, LAE1 expression was not obviously correlated with any histone modifications examined (H3K4me2, H3K4me3, and H3K9me3) (23). Thus, to date, very little is known about the role of epigenetic modifications in the activation of cellulase genes in fungi.
In this work, we first characterized the transcriptional profile of the MoCel7C gene using a green fluorescent protein (GFP) reporter system. The mechanisms of substrate-dependent transcriptional regulation of the gene were also examined, especially in relation to H3K4 methylation. We also evaluated the gene expression and silencing vectors driven by the MoCel7C promoter, which should prove to be useful research tools for M. oryzae and closely related fungal species.
MATERIALS AND METHODS
Fungal strains, culture conditions, and transformation.
The M. oryzae strains used in this study were Br48 (wheat-infecting isolate) (24), and Br48-GFP, a transformant of Br48 with the GFP gene (25, 26). They were maintained and cultured as described previously (27). As a conventional rich medium, CM liquid (0.3% Casamino Acids, 0.3% yeast extract, 0.5% sucrose) or CM agar (CM liquid plus 1.5% agar) medium was used for GFP reporter assay, RNA extraction, and spheroplast preparation. As an induction medium, yeast nitrogen base without amino acids (Difco BD, Franklin Lakes, NJ) was used with carboxymethylcellulose (CMC) or another carbon source at appropriate concentrations. When grown in liquid medium, M. oryzae was cultured with gentle agitation on a rotary shaker set to 110 rpm. Fungal transformation was performed as described previously (27).
Construction of gene knockout mutants.
A knockout mutant of the MoSET1 gene (MGG_15053) was constructed using a split marker strategy (28). 5′ and 3′ flanking regions (2,119 bp and 2,129 bp) of the MoSET1 gene were inserted into upstream and downstream of the hygromycin resistance gene cassette in pSP72-HPH, respectively (29). Using the resulting construct as a template, upstream and downstream split marker amplicons were amplified by PCR with two pairs of primers (primers 5′-GCCGCAACCTCTACCAATGC-3′ and 5′-GGCTGCAGAACAGCGGGCAGTTCGG-3′ and primers 5′-GCCGTGCACAGGGTGTCACGTTGC-3′ and 5′-GTCCGTTCTCGGTGGTCTCT-3′). The split marker amplicons were introduced into M. oryzae by polyethylene glycol (PEG)-mediated transformation, and the resulting transformants were screened by PCR and subsequent genomic Southern blot analysis to obtain transformants with the expected homologous recombination event. To construct a complementary strain of the ΔMoSET1 mutant, a DNA fragment containing the wild-type MoSET1 gene with the putative promoter and terminator sequences was obtained by PCR amplified using the primers 5′-CAATTACCTCTTAACAACGACACC-3′ and 5′-TGACATGCGACTTCGGACT-3′. The PCR fragment was cloned at the EcoRV site in pBluescript SK-II (Stratagene, La Jolla, CA) and then introduced into the ΔMoSET1 mutant by the cotransformation method with pII99 carrying the Geneticin-resistant gene cassette (30).
Construction of gene expression and silencing vectors.
Two DNA fragments (1.1 kb and 0.2 kb) corresponded to promoter and terminator regions of the MoCel7C gene were first amplified by PCR using genomic DNA of Br48 as a template and two pairs of primers (primers 5′-CCTAGTGATGGTGGTATTGTTGAG-3′ and 5′-ATCGAATTCATGCATTTTCTTTGGCTGGCG-3′ and primers 5′-ACCGTCGACCTGGGAGGTTGTTGATGTGGA-3′ and 5′-CCGGGCCCACGTATAAACTTGGCGGTGCTT-3′). The resulting fragments were inserted into pBluescript SK-II (Stratagene) at the SmaI-EcoRI and SalI-ApaI sites, respectively. A NotI fragment containing the hygromycin resistance gene cassette was excised from pSilent-1 (31) and inserted at the NotI site in the resulting construct, establishing pEXEG2. To construct ipSilent, the KpnI site in the pEXEG2 was first disrupted by restriction digestion followed by filling in of the ends and self-ligation. Then, the multicloning site in pSilent-1 was PCR amplified with a pair of primers (5′-CTGACTTACCTATTCTACCCAAGCATC-3′ and 5′-ATGTCGACAAGTGGATCCGGGGCCCAG-3′) and inserted into the resulting construct at the HindIII-SalI sites, establishing ipSilent.
RNA isolation and quantitative RT-PCR (qRT-PCR).
RNA isolation and cDNA synthesis were performed as described previously with slight modifications (14). Total RNA was isolated from frozen mycelial powder using Sepasol RNA I Super (Nacalai Tesque, Kyoto, Japan). RNA quality and quantity were determined using a NanoDrop instrument (Thermo Scientific, Wilmington, DE) and denatured agarose gel electrophoresis. One microgram of total RNA was then subjected to cDNA synthesis using ReverTra Ace qPCR RT master mix with a genomic DNA (gDNA) remover kit (Toyobo, Osaka, Japan). qRT-PCR assay was carried out using FastStart SYBR green master mix (Roche Applied Science, Mannheim, Germany) according to the manufacturer's instructions with specific primers for targets and an internal control gene, actin; MGG_03982). The primer sequences are as follows; MGG_14954, 5′-ATGACCGTCCTCCTCACACT-3′ and 5′-AGCGGTAGGTGGTGATCTTG-3′; MGG_10712, 5′-GCAGTTCTGCACAGACCAGTT-3′ and 5′-AGGACCATTGGGATTTCGTAG-3′; MGG_07908, 5′-GTGAGGAGTGGGGAGAGTGG-3′ and 5′-CCTAAAGTACTCCTCGAACCAAGC-3′; MGG_05520, 5′-ATGGCTAGCAAGCTGTTCCTC-3′ and 5′-AGTCCTGTCCACCACATTGAC-3′; MGG_06834, 5′-TGATTCGCAAGATCACAACTCT-3′ and 5′-GCACTTTTGCCAGGTAAGAGAC-3′; GFP, 5′-AAGGACGACGGCAACTACAA-3′ and 5′-GTCCTCCTTGAAGTCGATGC-3′; and actin, 5′-GCGGTTACACCTCTCTACCAC-3′ and 5′-AGTCTGGATCTCCTGCTCAAAG-3′). Fluorescence from DNA-SYBR green complex was monitored with a Dice real-time thermal cycler system (TaKaRa Bio, Otsu, Japan) throughout the PCR. The level of target mRNA, relative to the mean of the actin gene was calculated by the comparative CT method.
Chromatin immunoprecipitation (ChIP) assay.
The ChIP assay was performed using a ChIP-IT Express kit (Active Motif, Carlsbad, CA) according to the manufacturer's protocol. Fungal mycelia were grown in CM liquid medium for 4 days. A portion of mycelia (100 mg) was harvested and incubated in 10 ml of phosphate-buffered saline (PBS) containing 1% formaldehyde for 15 min at room temperature. Chromatin was sheared on ice by sonication using a Bioruptor apparatus (Cosmo Bio Co., Ltd., Japan) for 3 cycles of 1 min on at high intensity (200 W) and 30 s off, followed by 4 cycles of 1 min on at medium intensity (160 W) and 30 s off. The size of the sheared chromatin was around 200 to 1,000 bp, as determined by agarose gel electrophoresis. Polyclonal antibodies against H3K4me2 were obtained from Active Motif. ChIP-enriched genomic DNA fragments were subjected to qPCR analysis using the primers above for the internal coding regions of the MoCel7A (MGG_14954) and actin genes. Input DNA was used to normalize the data.
Western blot analysis.
Fungal mycelia were homogenized in buffer composed of 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, and 1% Nonidet P-40. The homogenates were centrifuged (10,000 × g, 10 min), and the supernatants were collected. Equal amounts of homogenate protein (20 μg) were subjected to 15% SDS-polyacrylamide gel electrophoresis (PAGE), electroblotted onto a Immobilon-P polyvinylidene difluoride (PVDF) membrane (Millipore, Bedford, MA), and probed with the H3K4me2 antibody (Active Motif). Proteins reacting with the antibody were visualized by the ECL plus Western blotting detection reagents (GE Healthcare, Piscataway, NJ).
GFP reporter assay.
GFP fluorescence of the transformants was measured by the multilabel counter ARVO-SX (Perkin-Elmer, Waltham, MA) with 485-nm excitation and 535-nm emission wavelengths using 96-well microplates containing appropriate media. GFP fluorescence values of the transformants were calculated by subtracting an average fluorescence value of the wild-type Br48 strain.
Nucleotide sequence accession numbers.
The sequences for the inducible gene expression and silencing vectors, pEGEX2 and ipSilent, respectively, were deposited in GenBank under the respective accession numbers AB750392 and AB750393, and these vectors were deposited at the Fungal Genetics Stock Center.
RESULTS
Regulation of the MoCel7C promoter by carboxymethylcellulose (CMC).
We have previously shown that transcription of five (MGG_05520, MGG_10712, MGG_14954, MGG_07908, and MGG_06834) of the nine GH6 and GH7 cellulases in M. oryzae are highly upregulated during infection (14). Quantitative reverse transcription-PCR (qRT-PCR) analysis revealed that these five genes were all upregulated to different levels in a medium containing the substrate cellulose (2% carboxymethylcellulose [CMC]) as the sole carbon source (Fig. 1A). Among them, the transcript of MGG_14954, encoding the enzyme which we call here MoCel7C, was the most highly upregulated upon induction by CMC. The transcript abundance of the MoCel7C gene was approximately 30% relative to that of actin in the presence of CMC, while it was less than 0.1% in a conventional rich medium. MoCel7C expression was also highly induced by the crystalline celluloses Avicel and Sigmacell on agar medium but not in liquid medium (Fig. 1B). Thus, we used CMC as an inducer of the MoCel7C gene in further experiments. Previous phylogenetic analysis revealed that MoCel7C is a member of the GH7 family and is more closely related to endoglucanase than cellobiohydrolase (32).
Fig 1.
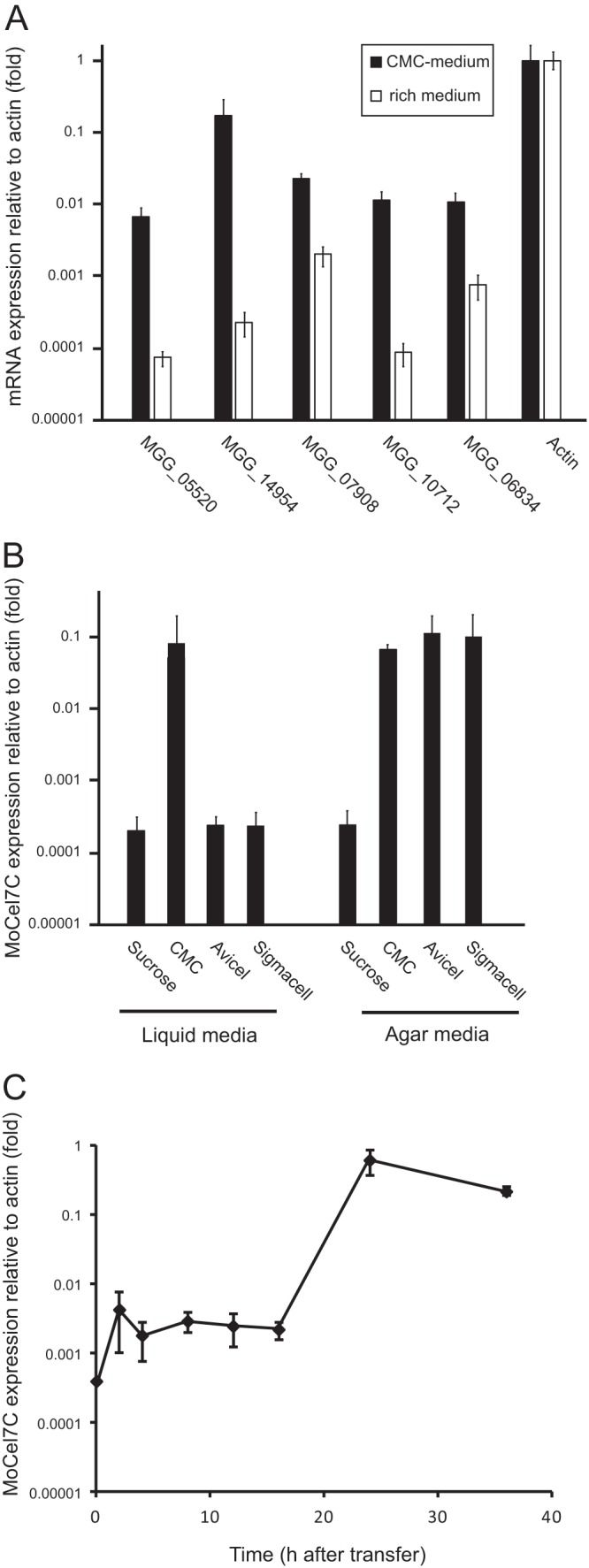
Expression profiles of Magnaporthe oryzae GH6 and GH7 cellulase genes in response to cellulose. (A) Quantitative RT-PCR (qRT-PCR) analysis of five GH6 and GH7 cellulase genes (MGG_05520, MGG_10712, MGG_14954, MGG_07908, and MGG_06834) in M. oryzae. The fungus was grown on medium containing 2% carboxymethylcellulose (CMC) as the sole carbon source (black bars) or on conventional rich medium (white bars). After 36 h of incubation at 25°C, total RNA was extracted from mycelia and subjected to the analysis. Values are expression levels of the cellulase genes relative to that of the internal control gene (actin; MGG_03982). This experiment was performed with two biological replicates and duplicate technical replicates. Error bars represent standard deviations. (B) Effects of different cellulose polymers on the expression of the MoCel7C gene (MGG_14954). M. oryzae was grown on agar or liquid medium containing each of the cellulose polymers shown in the graph or sucrose at a concentration 0.5% as the sole carbon source. After 36 h of incubation at 25°C, total RNA was extracted from mycelia and subjected to the analysis. Values are relative expression levels of the MoCel7C gene relative to that of the actin gene. Error bars represent standard deviations. This experiment was performed with three technical replicates. (C) Time course expression profile of MoCel7C after induction with CMC. Fungal mycelia were harvested at 2, 4, 8, 12, 16, 24, and 36 h after transfer to medium containing 2% CMC and subjected to qRT-PCR analysis. Expression changes were calculated with reference to levels of the actin gene (set at 1). This experiment was performed with two biological replicates and duplicate technical replicates. Error bars represent standard deviations.
Next we performed a time course experiment to monitor the expression kinetics of MoCel7C activated by 2% CMC. Total RNA was extracted at 2, 4, 8, 12, 16, 24, and 36 h after transfer to media containing CMC, and the transcript level of the MoCel7C gene was examined by qRT-PCR. As shown in Fig. 1C, a slight transcriptional upregulation was detected as early as 2 h after transfer, and a dramatic increase (more than 1,000-fold) in transcript abundance was observed between 16 and 24 h after transfer. Abundance was slightly decreased 36 h after transfer.
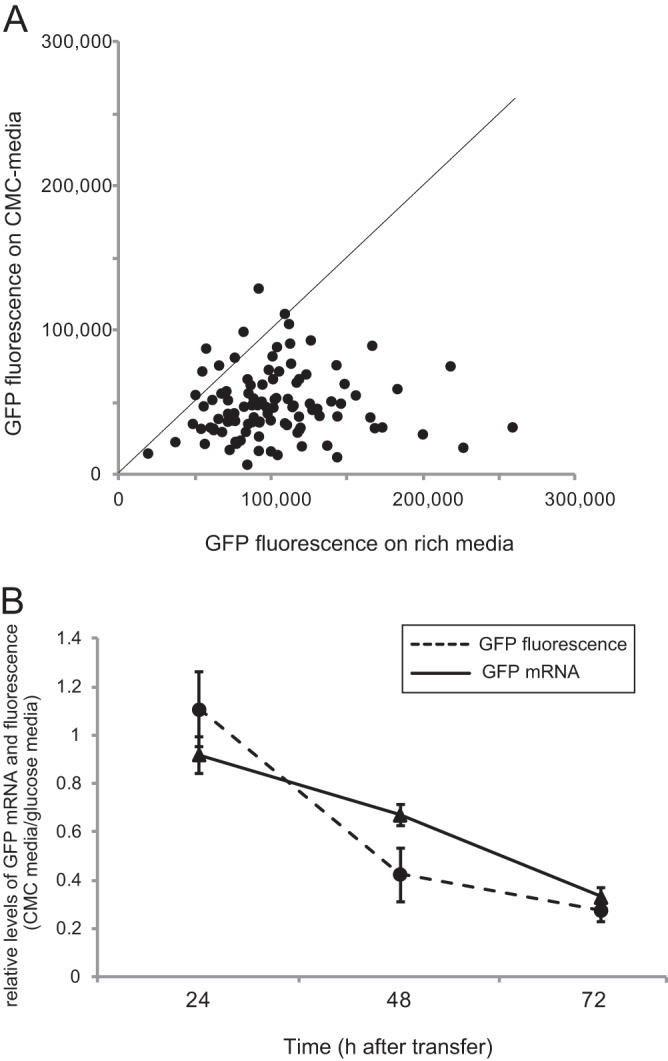
To further characterize the MoCel7c promoter, we performed a GFP reporter assay using pEXEG2-GFP, a GFP reporter vector carrying the full-length eGFP gene under the control of the MoCel7C promoter (see Materials and Methods). Fifty-six pEXEG2-GFP transformants were randomly picked, and their GFP fluorescence was measured 48 h after incubation on either rich or CMC-containing medium (Fig. 2A). GFP fluorescence was detectable in more than half of the transformants on the CMC-containing medium, clearly indicating that the MoCel7C promoter in the vector was responsive to CMC, similar to the endogenous copy. However, notably, approximately 20% of transformants on the rich medium also exhibited significant GFP fluorescence. This tendency was also observed by qRT-PCR validation of eight representative transformants (Fig. 2B). The relatively high level of GFP expression by the reporter construct in the transformants may be due to a proximal promoter around the integration site or to the lack of negative regulatory cis elements in the promoter fragment cloned in pEXEG2.
Fig 2.

Expression profiles of the GFP gene driven by the MoCel7C promoter in Magnaporthe oryzae. (A) Scatter plots of GFP fluorescence measurements of M. oryzae transformants grown on rich medium versus CMC medium. Fifty-six M. oryzae transformants with pEGEX2-GFP were randomly picked, and their GFP fluorescence was measured 48 h after incubation on each of the two media. (B) Quantitative RT-PCR (qRT-PCR) analysis of the GFP gene in eight representative M. oryzae transformants with pEGEX2-GFP (T1 through T8). The transformants were grown on medium containing 1% carboxymethylcellulose (CMC) (black bars) or conventional rich medium (white bars) for 48 h and subjected to the analysis. Values are expression levels of the GFP gene relative to the level of the actin gene (set at 1). As a control, the MoCel7C mRNA level in the wild-type strain (Br48) was also assessed under inducing and noninducing conditions. This experiment was performed with three technical replicates. Error bars represent standard deviations. (C) Effect of CMC concentration on the activation of the MoCel7C promoter. Two pEGEX2-GFP transformants (T3 and T4) were grown on medium containing CMC at the concentrations above for 48 h and subjected to GFP measurement. Error bars represent standard deviations. (D) Time course expression profiles of the GFP gene in two pEGEX2-GFP transformants (T3 and T4) after induction with 1% CMC. GFP fluorescence of fungal colonies on the CMC (▲) and rich (■) media was measured for 3 days at 12 h intervals. The experiment was performed with three biological replicates. Error bars represent standard deviations.
Using the GFP reporter system, we first assessed the optimal CMC concentration for activating the MoCel7C promoter. A small increase in GFP fluorescence was detectable in the presence of only 0.05% CMC, and the level of GFP fluorescence increased as the concentration of CMC increased, up to 2% (Fig. 2C). Thus, the MoCel7C promoter was activated by CMC in a concentration-dependent manner within the range tested. Next, the induction kinetics of the MoCel7C promoter was examined in two representative transformants. Consistent with the qRT-PCR analysis (Fig. 1C), GFP fluorescence in the two transformants increased dramatically 24 to 36 h after transfer to the CMC-containing medium, remained at high levels until 60 h after transfer, and decreased slightly at 72 h (Fig. 2D). In contrast, almost no increase in GFP fluorescence was detectable in transformants on the rich medium. These results were consistent with the qRT-PCR analysis of the endogenous MoCel7C gene (Fig. 1C).
The MoCel7C promoter is activated by cello-oligosaccharides larger than a pentamer but is suppressed by cellobiose and glucose.
The ability of various kinds of carbohydrates to induce the MoCel7C promoter was assessed using the GFP reporter system. In addition to CMC, glucose, sucrose, xylose, raffinose, galactose, and xylan were tested at a concentration of 0.5%. We also assessed how the degree of cello-oligosaccharide polymerization affects the induction of the MoCel7C promoter. Cello-oligosaccharide polymers, from dimers to hexamers (Seikagaku Kogyo, Tokyo, Japan), were used in the assay. GFP fluorescence of transformants was measured 60 h after transfer to media containing the different carbohydrates described above as the sole carbon source. As shown in Fig. 3A, high GFP fluorescence was detected only in the presence of CMC. However, a significant increase in GFP fluorescence was also detected in the presence of cello-oligosaccharides larger than pentamers. Since longer cello-oligosaccharides induced higher MoCel7C promoter activity, the induction appeared to occur in a polymerization-dependent manner. In addition, GFP fluorescence was slightly increased in the presence of xylan, suggesting that the MoCel7C promoter might respond, albeit at very low levels, to xylan.
Fig 3.

Effects of various carbohydrates on the activation of the MoCel7C promoter. (A) Two pEGEX2-GFP transformants (T3 and T4) were grown on medium containing each of the carbohydrates in the graph at a concentration of 0.5% for 60 h and subjected to GFP measurement. Average GFP values of the two transformants were plotted. This experiment was performed with three biological replicates. (B) Repression of CMC-induced activation of the MoCel7C promoter by glucose and cellobiose. The T3 and T4 transformants were grown on medium containing 1% CMC and cellobiose or glucose at different concentrations. GFP fluorescence of fungal colonies was measured 60 h after transfer to the medium. Average GFP values of the two transformants were plotted in the graph. This experiment was performed with three biological replicates. (C) qRT-PCR analysis of MoCel7C expression in response to various carbohydrates. M. oryzae was grown on medium containing each of the carbohydrates in the graph at a concentration of 0.5% as the sole carbon source. After 36 h of incubation at 25°C, total RNA was extracted from mycelia and subjected to the analysis. Values are expression levels of the MoCel7C gene relative to that of the actin gene. This experiment was performed with three technical replicates. Error bars represent standard deviations.
We next explored the possibility that cellobiose and glucose, catabolites of cellulase, could suppress CMC-triggered activation of the MoCel7C promoter. GFP fluorescence activated by 1% CMC was reduced by the addition of 0.2% glucose or 0.2% cellobiose to the medium, and the degree of reduction increased as their concentrations increased (Fig. 3B). MoCel7C promoter activation by 1% CMC was almost completely abrogated by the addition of 3% cellobiose or 3% glucose. Glucose and cellobiose suppressed the MoCel7C promoter at comparable levels (Fig. 3B). These results indicated that cellobiose and glucose may be useful to avoid unexpected gene expression driven by the MoCel7C promoter under noninducing conditions.
To verify the data from the GFP reporter assay, we performed qRT-PCR analysis of endogenous MoCel7C gene expression using glucose, sucrose, xylan, cellobiose, and cellohexaose as inducers. The suppressive effects of glucose and cellobiose on CMC-induced activation of the MoCel7C promoter were also tested with the endogenous gene. The results were mostly consistent with the GFP assay. However, the higher sensitivity and broader dynamic range of the qRT-PCR analysis allowed us to detect different levels of MoCel7C activation by the carbohydrates in more detail. Compared with the level of MoCel7C mRNA induced by CMC, the average levels induced by cellohexaose and xylan were 7.3% and 4.2%, respectively (Fig. 3C). The other carbohydrates tested showed much lower levels of MoCel7C promoter induction (0.01 to 0.1% relative to CMC). Among them, cellobiose and xylose induced the expression of MoCel7C at relatively high levels (Fig. 3C). However, cellobiose suppressed CMC-induced activation of the MoCel7C promoter to a level similar to that caused by glucose suppression (Fig. 3C).
Transcriptional upregulation of the MoCel7C gene is associated with histone H3 methylation on lysine 4.
To test the possibility that the activation of the MoCel7C promoter was modulated by chromatin modification, we performed a chromatin immunoprecipitation (ChIP)-qPCR assay using an antibody against H3K4me2. Studies on genome-wide histone methylation in yeast and mammals have revealed that H3K4me2 and H3K4me3 accumulate predominantly in transcriptionally active genes (33–37). In particular, H3K4me3 is associated with the promoter regions of active genes, and H3K4me2 is often detected in the coding regions. Thus, we performed ChIP-qPCR analysis using a pair of primers for the coding region of the MoCel7C gene. The results showed that the MoCel7C locus was significantly more associated with H3K4me2 following induction by CMC than under noninducing conditions (Fig. 4A), suggesting the involvement of H3K4me2 in the substrate-dependent activation of the MoCel7C gene. We next constructed a knockout (KO) M. oryzae mutant of the MoSET1 gene, an ortholog of Saccharomyces cerevisiae SET1, which is the histone methyltransferase catalyzing H3K4 methylation in S. cerevisiae. Western blot analysis revealed that H3K4me2 was reduced to an undetectable level in the MoSET1 KO mutant, indicating that MoSET1 catalyzes H3K4me methylation in M. oryzae (Fig. 4B). Upon induction with 2% CMC, the MoSET1 KO mutant showed only a 5.5-fold increase in MoCel7C mRNA abundance while the wild-type strain exhibited an approximately 1,000-fold increase (Fig. 4C), suggesting that H3K4 methylation is crucial for substrate-dependent activation of the MoCel7C gene. Consistently, no significant increase of GFP fluorescence was observed upon CMC induction in the MoSET1 KO transformant with pEXEG2-GFP (Fig. 4D). Notably, this resulted from two elements. First, the MoCel7C transcript level under inducing conditions was significantly lower in the MoSET1 KO strain than in the wild type, suggesting a positive effect of H3K4 methylation on gene expression. Meanwhile, interestingly, the abundance of the MoCel7 transcript was much higher in the MoSET1 KO strain than in the wild type under noninducing conditions (Fig. 4C). This tendency was also observed with all the CMC-responsive cellulase genes examined with the exception of MGG_05520 (Fig. 4E). These results suggest that MoSET1 can directly or indirectly play a role in gene repression under noninducing conditions as well as in gene activation under inducing conditions in M. oryzae.
Fig 4.

(A) Chromatin immunoprecipitation (ChIP) analysis of H3K4me2 levels at the MoCel7C and actin genes. The analysis was performed 36 h after transfer to medium containing 1% carboxymethylcellulose (CMC) (black bars) or conventional rich medium (white bars). Immunoprecipitated and input DNA were quantified by qPCR. ChIP signal was normalized to input DNA. Relative enrichment of H3K4me2 under inducing conditions was calculated by dividing normalized data by those under noninducing conditions. Error bars represent standard deviations. **, significant difference (two-tailed t test, P < 0.01). This experiment was performed with two biological replicates with duplicate technical replicates. (B) Western blot analysis of H3K4me2 in the MoSET1 KO strain. Total protein extracted from the wild type (WT), the MoSET1 KO mutant (Δset1), and the complementary strain (Δset1/MoSET1) was separated on a 15% polyacrylamide gel and probed with rabbit polyclonal antibodies against H3K4me2 (active motif). (C) qRT-PCR analysis of MoCel7C expression in response to CMC. The WT, Δset1, and Δset1/MoSET1 strains were grown on medium containing 1% CMC or on conventional rich medium for 36 h. Values are relative expression levels of the MoCel7C gene relative to that of the actin gene (set at 1). This experiment was performed with two biological replicates and duplicate technical replicates. Error bars represent standard deviations. (D) GFP reporter assay of the MoCel7C promoter in response to CMC. Two pEGEX2-GFP transformants each of the WT and Δset1 strains were grown on medium containing 1% CMC for 48 h and subjected to GFP measurement. Error bars represent standard deviations. This experiment was performed with three technical replicates. (E) qRT-PCR analysis of the CMC-responsive cellulase genes in the MoSET1 KO (Δset1) mutant. Experiments were performed as described for panel C.
The MoCel7C promoter can be used to regulate RNA silencing in M. oryzae.
We previously reported that pSilent-1, an RNA-silencing vector with the constitutive TrpC promoter, induced gene silencing efficiently in M. oryzae and other fungal species (31). To establish a regulatable gene silencing system, the gene silencing vector, ipSilent, was made by inserting the multiple cloning site of pSilent-1 into pEXEG2. To perform a reporter assay, ipSilent-GFP, an ipSilent-based gene silencing vector producing hairpin GFP RNA, was then constructed and introduced into a GFP-expressing strain of M. oryzae. Subsequently, 107 transformants were picked, and their GFP fluorescence was measured 72 h after incubation on either rich or CMC medium. Compared with the GFP-expressing parent strain, GFP fluorescence was reduced by an average of 12.5% and 58.2% in transformants grown on rich and CMC media, respectively (Fig. 5A), suggesting that gene silencing was induced by CMC. Approximately 30% of the transformants showed GFP silencing in a regulatable manner; they exhibited GFP fluorescence comparable to the parent strain (>90%) on the rich medium, which was reduced by more than 50% on the CMC medium. Among them, 14 transformants (13.1%) showed strong reduction (>80%) in GFP fluorescence on the CMC medium relative to that on the rich medium.
Fig 5.
Inducible gene silencing of the GFP gene using the MoCel7C promoter in Magnaporthe oryzae. (A) Scatter plots of GFP fluorescence measurements of M. oryzae transformants grown on rich medium versus CMC medium. The GFP silencing construct, ipSilent-GFP, which produces GFP hairpin RNA under the control of the MoCel7C promoter, was introduced into a GFP-expressing strain of M. oryzae. One hundred seven transformants were randomly picked, and their GFP fluorescence was measured 60 h after incubation on the two media. (B) Time course expression profile of the GFP gene in three transformants with ipSilent-GFP. GFP fluorescence of fungal colonies was measured at 24-h intervals for 3 days after transfer to medium containing 1% CMC. The experiment was performed with two biological replicates and two technical replicates. Error bars represent standard deviations.
Two representative ipSilent-GFP transformants were further subjected to time course experiments. Based on qRT-PCR analysis, GFP mRNA accumulation in the transformants on CMC medium was reduced by averages of 8.0%, 33.0%, and 67.0% compared with that in transformants grown on glucose-containing medium 24 h, 48 h, and 72 h after transfer, respectively (Fig. 5B). Consistently, GFP fluorescence of fungal colonies was repressed by an average of 57.4% and 72.3% at 48 h and 72 h after transfer to the CMC medium, respectively (Fig. 5B). These results clearly indicate that the MoCel7C promoter is a useful tool for the regulation of RNA silencing in M. oryzae.
DISCUSSION
From an ecological viewpoint, fungi are one of the principal decomposers in the ecosystem. Many fungi produce enzymes that hydrolyze cellulose, the most characteristic component of plant cell walls and the most abundant biomass on earth. Cellulose is a linear polymer consisting of β-1,4-linked glucose residues. Three types of cellulase, cellobiohydrolases (EC 3.2.1.91), endoglucanases (EC 3.2.1.4), and β-glucosidases (EC 3.2.1.21), synergistically catalyze different biochemical processes in cellulose degradation (38). Because they are decomposers, the proper regulation of cellulase production has been essential to fungi during evolution. In fact, previous studies have shown that fungal cellulase activity is often drastically increased in the presence of cellulose as a substrate (15–18).
Here, we examined if chromatin alteration was involved in such substrate-induced transcriptional activation of a cellulase gene in M. oryzae. It is generally believed that in eukaryotes, actively transcribed genes are associated with acetylation of histones H3 and H4 at various lysine residues and also with methylation of H3K4 (39). In the filamentous fungus N. crassa, acetylation on H3 lysine 14 (H3K14ac) is enriched at a blue-light-responsive promoter region as early as 20 min after a light pulse (6). In A. nidulans, H3K14ac and H3K9ac levels at nitrate-assimilating gene loci are increased after 120 min of nitrate starvation in a manner dependent on the nitrate status-sensing regulator AreA (40). However, a reduction in H3K4me2 and H3K4me3 caused by deletion of the CclA gene resulted in increased gene expression of cryptic SM gene clusters in A. nidulans and A. fumigatus (8, 9). Despite these observations, few experimental data are available on the role of H3K4 methylation in gene activation in filamentous fungi.
In this study, activation of the MoCel7C cellulase gene by CMC was associated with enrichment of H3K4 methylation at the locus (Fig. 4A). This substrate-dependent activation was drastically impaired in the KO mutant of MoSET1, the SET1 ortholog in M. oryzae. These results are consistent with the idea that H3K4 methylation is an epigenetic mark for gene activation in M. oryzae. Interestingly, however, the level of MoCel7C expression under noninducing conditions was increased in the MoSET1 KO mutant, suggesting a role for H3K4 methylation in gene suppression (Fig. 4C). Such apparent bilateral gene regulation by MoSET1 was also suggested for most but not all of the CMC-responsive cellulase genes in M. oryzae (Fig. 4E). Activation or loss of repression of M. oryzae cellulose genes in the MoSET1 KO mutant under noninducing conditions appeared to be consistent with upregulation of the SM cluster in Aspergillus CclA mutants (8, 9). In fact, SET1 in S. cerevisiae was originally characterized to be required for transcriptional silencing of silent mating-type loci in the subtelomeric region (41). Subsequently, involvement of SET1-mediated H3K4 methylation in silencing rDNA and the retrotransposon, Ty1, has also been demonstrated (42, 43). More recently, in N. crassa, SET1 was shown to be required for DNA methylation in the frequency gene (frq) promoter (44). A hypothesis to explain this apparent discrepancy in the role of SET1-like HMT is that H3K4 methylation would open the chromatin structure to enable promoter access for both gene activator and repressor. In this regard, Zeilinger et al. (45) showed that the carbon catabolite repressor Cre1 binds to a nucleosome-free region in the T. reesei cellulase gene (cbh2) promoter. Such binding may be affected by H3K4 methylation-mediated chromatin remodeling. Overall, our results indicated that MoSET1 is needed for proper gene regulation by modulating chromatin in M. oryzae, as suggested for SET1 in N. crassa (44).
An intriguing question is how fungi sense cellulose, a polymeric substrate that is, to a large extent, water insoluble. A hypothesis is that a low-level constitutive expression of cellulase might degrade cellulose into soluble cello-oligosaccharides that subsequently enter the fungal cells and cause induction (46). Consistent with the hypothesis, various types of cell wall-degrading enzymes, including cellulases, were coordinately induced in many fungal species grown in media containing cello-oligosaccharides or their derivatives. In our study, however, the MoCel7C promoter was repressed by cellobiose, which is known as an inducer of several fungal cellulase promoters. In some cellulolytic bacterial and fungal species, catabolite repression-like phenomena are likely to be provoked by cellobiose, especially at high concentrations (47–51). Therefore, cellobiose might also cause catabolite repression of the MoCel7C promoter in M. oryzae. Alternatively, as shown in N. crassa, the action of extracellular and intracellular β-glucosidases might readily process cellobiose to glucose and mask the activity of cellobiose for triggering the MoCel7C promoter (52). It should be noted that insoluble crystalline cellulose such as Avicel and Sigmacell activated the MoCel7C promoter on agar medium but not in liquid medium (Fig. 1B). This suggests that sensing cellulose by direct contact with fungal mycelia is essential for triggering activation of the MoCel7C promoter in M. oryzae.
An inducible promoter that can induce and silence gene expression offers an effective molecular tool and strategy for elucidating gene function. Inducible promoters facilitate genetic analysis, where a particular phenotype can be linked to expression or suppression of a specific gene. Inducible promoters also provide a means to study toxic proteins, whose presence at high levels causes lethality. The selective induction of gene expression is typically accomplished by the use of a specific inducer that directly or indirectly activates a corresponding promoter. Several inducible gene expression systems with various inducers, including copper-, thiamine-, alcohol-, doxycycline-, sodium acetate-, and cellulose-responsive promoters, have been developed in several model and industrially exploited fungi (53–59). However, some of them are prone to leaky expression in the absence of the inducer. The MoCel7C promoter, which is highly inducible by low-cost substrates and repressible by glucose, is highly suitable for developing an inducible gene expression system.
The promoters of glycosyl hydrolases are often used to develop inducible gene expression systems in filamentous fungi. For example, the cellobiohydrolase Cbh1 in T. reesei accounted for about 50% of all secreted proteins under inducing conditions (60), and its promoter has been successfully used to produce large amounts of homologous and heterologous proteins in an inducible manner (60). Based on the GFP reporter assay, the activation of the MoCel7C promoter was inducible by CMC in the “standard” rice-infecting strain, Guy11, as well as in a crabgrass-infecting isolate, which belongs to another Magnaporthe species, Magnaporthe grisea (data not shown). Thus, the molecular machineries required for CMC-triggered activation of the MoCel7C promoter might be conserved, at least partly, in the Magnaporthe complex, making MoCel7C promoter-based gene expression and silencing systems useful for genetic research.
ACKNOWLEDGMENTS
We thank Eric Selker, Shinji Honda (University of Oregon), and Michael Freitag (Oregon State University) for technical advice in ChIP analysis.
This work was supported in part by a Grant-in-Aid for Scientific Research and Special Coordination Funds for Promoting Science and Technology Japan.
Footnotes
Published ahead of print 30 August 2013
REFERENCES
- 1. Faulk C, Dolinoy DC. 2011. Timing is everything: the when and how of environmentally induced changes in the epigenome of animals. Epigenetics 6:791–797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Turner BM. 2011. Environmental sensing by chromatin: an epigenetic contribution to evolutionary change. FEBS Lett. 585:2032–2040 [DOI] [PubMed] [Google Scholar]
- 3. Chinnusamy V, Zhu JK. 2009. Epigenetic regulation of stress responses in plants. Curr. Opin. Plant Biol. 12:133–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cortessis VK, Thomas DC, Levine AJ, Breton CV, Mack TM, Siegmund KD, Haile RW, Laird PW. 2012. Environmental epigenetics: prospects for studying epigenetic mediation of exposure-response relationships. Hum. Genet. 131:1565–1589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Smith KM, Phatale PA, Bredeweg EL, Connolly LR, Pomraning KR, Freitag M. 2012. Epigenetics of filamentous fungi, p 1–43 In Meyers RA. (ed), Encyclopedia of molecular cell biology and molecular medicine. Wiley-VCH Verlag GmBH &Co., Weinheim, Germany [Google Scholar]
- 6. Grimaldi B, Coiro P, Filetici P, Berge E, Dobosy JR, Freitag M, Selker EU, Ballario P. 2006. The Neurospora crassa White Collar-1 dependent blue light response requires acetylation of histone H3 lysine 14 by NGF-1. Mol. Biol. Cell 17:4576–4583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shwab EK, Bok JW, Tribus M, Galehr J, Graessle S, Keller NP. 2007. Histone deacetylase activity regulates chemical diversity in Aspergillus. Eukaryot. Cell 6:1656–1664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bok JW, Chiang YM, Szewczyk E, Reyes-Dominguez Y, Davidson AD, Sanchez JF, Lo HC, Watanabe K, Strauss J, Oakley BR, Wang CC, Keller NP. 2009. Chromatin-level regulation of biosynthetic gene clusters. Nat. Chem. Biol. 5:462–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Palmer JM, Bok JW, Lee S, Dagenais TR, Andes DR, Kontoyiannis DP, Keller NP. 2013. Loss of CclA, required for histone 3 lysine 4 methylation, decreases growth but increases secondary metabolite production in Aspergillus fumigatus. PeerJ 1:e4. 10.7717/peerj.4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bok JW, Keller NP. 2004. LaeA, a regulator of secondary metabolism in Aspergillus spp. Eukaryot. Cell 3:527–535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Strauss J, Reyes-Dominguez Y. 2011. Regulation of secondary metabolism by chromatin structure and epigenetic codes. Fungal Genet. Biol. 48:62–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Xu J-R, Zhao X, Dean RA. 2007. From genes to genomes; a new paradigm for studying fungal pathogenesis in Magnaporthe oryzae. Adv. Genet. 57:175–218 [DOI] [PubMed] [Google Scholar]
- 13. Gowda M, Venu RC, Raghupathy MB, Nobuta K, Li H, Wing R, Stahlberg E, Couglan S, Haudenschild CD, Dean R, Nahm BH, Meyers BC, Wang GL. 2006. Deep and comparative analysis of the mycelium and appressorium transcriptomes of Magnaporthe grisea using MPSS, RL-SAGE, and oligoarray methods. BMC Genomics 7:310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Vu VB, Itoh K, Nguyen QB, Tosa Y, Nakayashiki H. 2012. Cellulases belonging to glycoside hydrolase families 6 and 7 contribute to the virulence of Magnaporthe oryzae. Mol. Plant Microbe Interact. 25:1135–1141 [DOI] [PubMed] [Google Scholar]
- 15. Mandels M, Reese ET. 1957. Induction of cellulase in Trichoderma viride as influenced by carbon sources and metals. J. Bacteriol. 73:269–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bisaria VS, Mishra S. 1989. Regulatory aspects of cellulase biosynthesis and secretion. CRC Crit. Rev. Biotechnol. 9:61–113 [DOI] [PubMed] [Google Scholar]
- 17. Schmoll M, Kubicek CP. 2003. Regulation of Trichoderma cellulase formation: lessons in molecular biology from an industrial fungus. A review. Acta Microbiol. Immunol. Hung. 50:125–145 [DOI] [PubMed] [Google Scholar]
- 18. Peterson R, Nevalainen H. 2012. Trichoderma reesei RUT-C30—thirty years of strain improvement. Microbiology 158:58–68 [DOI] [PubMed] [Google Scholar]
- 19. Aro N, Ilmen M, Saloheimo A, Penttila M. 2003. ACEI of Trichoderma reesei is a repressor of cellulase and xylanase expression. Appl. Environ. Microbiol. 69:56–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Aro N, Saloheimo A, Ilmen M, Penttila M. 2001. ACEII, a novel transcriptional activator involved in regulation of cellulase and xylanase genes of Trichoderma reesei. J. Biol. Chem. 276:24309–24314 [DOI] [PubMed] [Google Scholar]
- 21. Takashima S, Iikura H, Nakamura A, Masaki H, Uozumi T. 1996. Analysis of Cre1 binding sites in the Trichoderma reesei cbh1 upstream region. FEMS Microbiol. Lett. 145:361–366 [DOI] [PubMed] [Google Scholar]
- 22. Ling M, Qin Y, Li N, Liang Z. 2009. Binding of two transcriptional factors, Xyr1 and ACEI, in the promoter region of cellulase cbh1 gene. Biotechnol. Lett. 31:227–231 [DOI] [PubMed] [Google Scholar]
- 23. Seiboth B, Karimi RA, Phatale PA, Linke R, Hartl L, Sauer DG, Smith KM, Baker SE, Freitag M, Kubicek CP. 2012. The putative protein methyltransferase LAE1 controls cellulase gene expression in Trichoderma reesei. Mol. Microbiol. 84:1150–1164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Urashima AS, Hashimoto Y, Don LD, Kusaba M, Tosa Y, Nakayashiki H, Mayama S. 1999. Molecular analysis of the wheat blast population in Brazil with a homologue of retrotransposon MGR583. Ann. Phytopathol. Soc. Jpn. 65:429–436 [Google Scholar]
- 25. Kadotani N, Nakayashiki H, Tosa Y, Mayama S. 2003. RNA silencing in the phytopathogenic fungus Magnaporthe oryzae. Mol. Plant Microbe Interact. 16:769–776 [DOI] [PubMed] [Google Scholar]
- 26. Nguyen QB, Kadotani N, Kasahara S, Tosa Y, Mayama S, Nakayashiki H. 2008. Systematic functional analysis of calcium-signalling proteins in the genome of the rice-blast fungus, Magnaporthe oryzae, using a high-throughput RNA-silencing system. Mol. Microbiol. 68:1348–1365 [DOI] [PubMed] [Google Scholar]
- 27. Nakayashiki H, Kiyotomi K, Tosa Y, Mayama S. 1999. Transposition of the retrotransposon MAGGY in heterologous species of filamentous fungi. Genetics 153:693–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fairhead C, Llorente B, Denis F, Soler M, Dujon B. 1996. New vectors for combinatorial deletions in yeast chromosomes and for gap-repair cloning using ‘split-marker' recombination. Yeast 12:1439–1457 [DOI] [PubMed] [Google Scholar]
- 29. Morita Y, Hyon GS, Hosogi N, Miyata N, Nakayashiki H, Muranaka Y, Inada N, Park P, Ikeda K. 2013. Appressorium-localized NADPH oxidase B is essential for aggressiveness and pathogenicity in the host-specific, toxin-producing fungus Alternaria alternata Japanese pear pathotype. Mol. Plant Pathol. 14:365–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Namiki F, Matsunaga M, Okuda M, Inoue I, Nishi K, Fujita Y, Tsuge T. 2001. Mutation of an arginine biosynthesis gene causes reduced pathogenicity in Fusarium oxysporum f. sp. melonis. Mol. Plant Microbe Interact. 14:580–584 [DOI] [PubMed] [Google Scholar]
- 31. Nakayashiki H, Hanada S, Nguyen BQ, Kadotani N, Tosa Y, Mayama S. 2005. RNA silencing as a tool for exploring gene function in ascomycete fungi. Fungal Genet. Biol. 42:275–283 [DOI] [PubMed] [Google Scholar]
- 32. Zhou J, Zheng XZ, Lan L, Lin CZ, Wu YB, Lin XJ, Ebbole D, Lu GD, Wang ZH. 2008. Biochemical and molecular characterization of a putative endoglucanase in Magnaporthe grisea. Curr. Genet. 53:217–224 [DOI] [PubMed] [Google Scholar]
- 33. Bernstein BE, Humphrey EL, Erlich RL, Schneider R, Bouman P, Liu JS, Kouzarides T, Schreiber SL. 2002. Methylation of histone H3 Lys 4 in coding regions of active genes. Proc. Natl. Acad. Sci. U. S. A. 99:8695–8700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bernstein BE, Kamal M, Lindblad-Toh K, Bekiranov S, Bailey DK, Huebert DJ, McMahon S, Karlsson EK, Kulbokas EJ, III, Gingeras TR, Schreiber SL, Lander ES. 2005. Genomic maps and comparative analysis of histone modifications in human and mouse. Cell 120:169–181 [DOI] [PubMed] [Google Scholar]
- 35. Santos-Rosa H, Schneider R, Bannister AJ, Sherriff J, Bernstein BE, Emre NC, Schreiber SL, Mellor J, Kouzarides T. 2002. Active genes are tri-methylated at K4 of histone H3. Nature 419:407–411 [DOI] [PubMed] [Google Scholar]
- 36. Pokholok DK, Harbison CT, Levine S, Cole M, Hannett NM, Lee TI, Bell GW, Walker K, Rolfe PA, Herbolsheimer E, Zeitlinger J, Lewitter F, Gifford DK, Young RA. 2005. Genome-wide map of nucleosome acetylation and methylation in yeast. Cell 122:517–527 [DOI] [PubMed] [Google Scholar]
- 37. Zhang X, Clarenz O, Cokus S, Bernatavichute YV, Pellegrini M, Goodrich J, Jacobsen SE. 2007. Whole-genome analysis of histone H3 lysine 27 trimethylation in Arabidopsis. PLoS Biol. 5:e129. 10.1371/journal.pbio.0050129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Henrissat B, Bairoch A. 1996. Updating the sequence-based classification of glycosyl hydrolases. Biochem. J. 316:695–696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kouzarides T. 2007. Chromatin modifications and their function. Cell 128:693–705 [DOI] [PubMed] [Google Scholar]
- 40. Berger H, Pachlinger R, Morozov I, Goller S, Narendja F, Caddick M, Strauss J. 2006. The GATA factor AreA regulates localization and in vivo binding site occupancy of the nitrate activator NirA. Mol. Microbiol. 59:433–446 [DOI] [PubMed] [Google Scholar]
- 41. Nislow C, Ray E, Pillus L. 1997. SET1, a yeast member of the trithorax family, functions in transcriptional silencing and diverse cellular processes. Mol. Biol. Cell 8:2421–2436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Briggs SD, Bryk M, Strahl BD, Cheung WL, Davie JK, Dent SY, Winston F, Allis CD. 2001. Histone H3 lysine 4 methylation is mediated by Set1 and required for cell growth and rDNA silencing in Saccharomyces cerevisiae. Genes Dev. 15:3286–3295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Berretta J, Pinskaya M, Morillon A. 2008. A cryptic unstable transcript mediates transcriptional trans-silencing of the Ty1 retrotransposon in S. cerevisiae. Genes Dev. 22:615–626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Raduwan H, Isola AL, Belden WJ. 2013. Methylation of histone H3 on lysine 4 by the lysine methyltransferase SET1 protein is needed for normal clock gene expression. J. Biol. Chem. 288:8380–8390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zeilinger S, Schmoll M, Pail M, Mach RL, Kubicek CP. 2003. Nucleosome transactions on the Hypocrea jecorina (Trichoderma reesei) cellulase promoter cbh2 associated with cellulase induction. Mol. Genet. Genomics 270:46–55 [DOI] [PubMed] [Google Scholar]
- 46. el-Gogary S, Leite A, Crivellaro O, Eveleigh DE, El-Dorry H. 1989. Mechanism by which cellulose triggers cellobiohydrolase I gene expression in Trichoderma reesei. Proc. Natl. Acad. Sci. U. S. A. 86:6138–6141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Dekker RFH. 1981. Induction, localization and characterization of ß-d-glucosidase produced by a species of Monilia. J. Gen. Microbiol. 127:177–184 [Google Scholar]
- 48. Johnson EA, Bouchot F, Demain AL. 1985. Regulation of cellulase formation in Clostridium thermocellum. J. Gen. Microbiol. 131:2303–2308 [Google Scholar]
- 49. Murray WD. 1987. Effects of cellobiose and glucose on cellulose hydrolysis by both growing and resting cells of Bacteroides cellulosolvens. Biotechnol. Bioeng. 29:1151–1154 [DOI] [PubMed] [Google Scholar]
- 50. Mishra S, Beguin P, Aubert JP. 1991. Transcription of Clostridium thermocellum endoglucanase genes celF and celD. J. Bacteriol. 173:80–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kubicek CP, Penttila ME. 1998. Regulation of production of plant polysaccharide degrading enzymes by Trichoderma, p 49–72 In Harman GE, Kubicek CP. (ed), Trichoderma and Gliocladium, vol 2 Taylor & Francis Ltd., London, United Kingdom [Google Scholar]
- 52. Znameroski EA, Coradetti ST, Roche CM, Tsai JC, Iavarone AT, Cate JHD, Glass NL. 2012. Induction of lignocellulose-degrading enzymes in Neurospora crassa by cellodextrins. Proc. Natl. Acad. Sci. U. S. A. 109:6012–6017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bellemare DR, Sanschagrin M, Beaudoin J, Labbe S. 2001. A novel copper-regulated promoter system for expression of heterologous proteins in Schizosaccharomyces pombe. Gene 273:191–198 [DOI] [PubMed] [Google Scholar]
- 54. Tada S, Gomi K, Kitamoto K, Takahashi K, Tamura G, Hara S. 1991. Construction of a fusion gene comprising the Taka-amylase A promoter and the Escherichia coli beta-glucuronidase gene and analysis of its expression in Aspergillus oryzae. Mol. Gen. Genet. 229:301–306 [DOI] [PubMed] [Google Scholar]
- 55. Shoji JY, Maruyama J, Arioka M, Kitamoto K. 2005. Development of Aspergillus oryzae thiA promoter as a tool for molecular biological studies. FEMS Microbiol. Lett. 244:41–46 [DOI] [PubMed] [Google Scholar]
- 56. Gwynne DI, Buxton FP, Williams SA, Sills AM, Johnstone JA, Buch JK, Guo ZM, Drake D, Westphal M, Davies RW. 1989. Development of an expression system in Aspergillus nidulans. Biochem. Soc. Trans. 17:338–340 [DOI] [PubMed] [Google Scholar]
- 57. Vogt K, Bhabhra R, Rhodes JC, Askew DS. 2005. Doxycycline-regulated gene expression in the opportunistic fungal pathogen Aspergillus fumigatus. BMC Microbiol. 5:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wang ZY, Thornton CR, Kershaw MJ, Debao L, Talbot NJ. 2003. The glyoxylate cycle is required for temporal regulation of virulence by the plant pathogenic fungus Magnaporthe grisea. Mol. Microbiol. 47:1601–1612 [DOI] [PubMed] [Google Scholar]
- 59. Harkki A, Mantyla A, Penttila M, Muttilainen S, Buhler R, Suominen P, Knowles J, Nevalainen H. 1991. Genetic engineering of Trichoderma to produce strains with novel cellulase profiles. Enzyme Microb. Technol. 13:227–233 [DOI] [PubMed] [Google Scholar]
- 60. Keranen S, Penttila M. 1995. Production of recombinant proteins in the filamentous fungus Trichoderma reesei. Curr. Opin. Biotechnol. 6:534–537 [DOI] [PubMed] [Google Scholar]