

Abstract
The GNB/LNB (galacto-N-biose/lacto-N-biose) pathway plays a crucial role in bifidobacteria during growth on human milk or mucin from epithelial cells. It is thought to be the major route for galactose utilization in Bifidobacterium longum as it is an energy-saving variant of the Leloir pathway. Both pathways are present in B. bifidum, and galactose 1-phosphate (gal1P) is considered to play a key role. Due to its toxic nature, gal1P is further converted into its activated UDP-sugar through the action of poorly characterized uridylyltransferases. In this study, three uridylyltransferases (galT1, galT2, and ugpA) from Bifidobacterium bifidum were cloned in an Escherichia coli mutant and screened for activity on the key intermediate gal1P. GalT1 and GalT2 showed UDP-glucose-hexose-1-phosphate uridylyltransferase activity (EC 2.7.7.12), whereas UgpA showed promiscuous UTP-hexose-1-phosphate uridylyltransferase activity (EC 2.7.7.10). The activity of UgpA toward glucose 1-phosphate was about 33-fold higher than that toward gal1P. GalT1, as part of the bifidobacterial Leloir pathway, was about 357-fold more active than GalT2, the functional analog in the GNB/LNB pathway. These results suggest that GalT1 plays a more significant role than previously thought and predominates when B. bifidum grows on lactose and human milk oligosaccharides. GalT2 activity is required only during growth on substrates with a GNB core such as mucin glycans.
INTRODUCTION
In 1899, a bacterium was isolated by Henri Tissier from the feces of a breastfed infant (1). He introduced the name Bacillus bifidus (later reclassified as Bifidobacterium bifidum) after the Latin word bifidus, meaning forked, because of its Y-shaped morphology. Bifidobacteria are considered to benefit human health through inhibition of pathogens and regulation of intestinal microbial homeostasis (2). They are a predominant part of the gut microbiota of breastfed infants and occur naturally in the lower part of the human gastrointestinal tract, where common mono- and disaccharides are scarce (3). Therefore, bifidobacteria have developed alternative pathways that enable them to utilize various oligosaccharides such as mucin glycans and the 2 types of human milk oligosaccharides (HMO) based on their core sugars: lacto-N-biose I (LNB, type I) and N-acetyllactosamine (LacNAc, type II) (4–6). Growth of B. bifidum on these oligosaccharides by action of extracellular glycosidases has been investigated in great detail, demonstrating rapid release and subsequent uptake of LNB (7). LNB is hypothesized to be the bifidus factor in HMO and thus a key factor in intestinal colonization (8, 9). Degradation of this disaccharide and the related galacto-N-biose (GNB) occurs through the GNB/LNB pathway (4).
The GNB/LNB pathway was discovered in Bifidobacterium longum and is encoded by the lnpABCD operon. This operon codes for a galacto-N-biose/lacto-N-biose I phosphorylase (LNBP), an N-acetylhexosamine 1-kinase (NahK), a UDP-glucose-hexose-1-phosphate uridylyltransferase (GalT2), and a UDP-glucose 4-epimerase (GalE2) (4). Metabolic profiling and genome sequencing of various B. bifidum strains reveal that a similar GNB/LNB gene cluster is present in this species, and the corresponding locus tags are listed in Table 1 (10–13). However, this cluster is organized differently, as two sugar kinases lie between the coding sequences for LNBP and NahK (Fig. 1). LNBP catalyzes the first reaction of this pathway, the phosphorolytic cleavage of a galactosyl-beta-1,3-N-acetylhexosamine (GNB or LNB) into an N-acetylhexosamine (HexNAc) and galactose 1-phosphate (gal1P) (14). Due to its similarity to the Leloir pathway and the direct generation of gal1P without action of a galactokinase (GalK), the GNB/LNB pathway is considered an energy-saving variant of the Leloir pathway (Fig. 2). Therefore, it has been suggested that this is the main pathway for galactose metabolism in bifidobacteria and that it prevails over the more common Leloir pathway (4, 15, 16).
Table 1.
Locus tags of the genes investigated in this study based on the B. bifidum PRL2010 genome and their gene products (11)
| Gene | Locus tag | Gene product |
|---|---|---|
| ugpA | BBPR_0976 | UTP-glucose-1-phosphate uridylyltransferase |
| galE1 | BBPR_1456 | UDP-glucose 4-epimerase |
| galT1 | BBPR_0406 | UDP-glucose-hexose-1-phosphate uridylyltransferase |
| galK | BBPR_0407 | Galactokinase |
| lnbp | BBPR_1055 | Galacto-N-biose/lacto-N-biose I phosphorylase |
| nahK | BBPR_1052 | N-Acetylhexosamine kinase |
| galT2 | BBPR_1051 | UDP-glucose-hexose-1-phosphate uridylyltransferase |
| galE2 | BBPR_1050 | UDP-glucose 4-epimerase |
Fig 1.
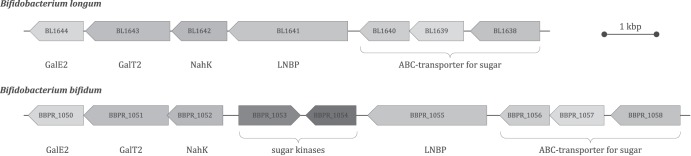
Schematic representation of the GNB/LNB gene cluster in B. longum NCC2705 (gene BL1638 to BL1644) and B. bifidum PRL2010 (gene BBPR_1050 to BBPR_1058). Two sugar kinases interrupt the GNB/LNB pathway of B. bifidum, and it is not organized as in the lnpABCD operon (BL1641 to BL1644) of B. longum.
Fig 2.

Overview of the Leloir pathway (A) and GNB/LNB pathway (B) in Bifidobacterium bifidum. LNBP, NahK, GalT2, and GalE2 are encoded in the same gene cluster (4). In contrast, the classic Leloir pathway (A) is encoded by scattered genes, and evidence is lacking for the existence of metabolic crossover between both pathways. Gene locus tags of the corresponding enzymes are listed in Table 1.
However, based on the presence of other uridylyltransferases and transcriptome profiling of B. bifidum, there are indications that the key intermediate gal1P may be metabolized via routes other than the GNB/LNB pathway. In this study, we investigate the function of three annotated and poorly characterized uridylyltransferases—GalT1, GalT2, and UgpA—of B. bifidum and their activity toward gal1P. GalT1 and GalT2 are both annotated as UDP-glucose-hexose-1-phosphate uridylyltransferases (EC 2.7.7.12) yet share little (12.1%) sequence identity. ugpA is annotated as a UTP-glucose-1-phosphate uridylyltransferase (EC 2.7.7.9), but activity toward other hexose 1-phosphates (H1P) was suggested in a previous study (17). In this paper, the enzymes were cloned and overexpressed in an engineered Escherichia coli strain in order to eliminate interference with the enzymes investigated and were screened for both activities, using a new chemoenzymatic assay. More detailed characterization of these uridylyltransferases provides new insights into gal1P degradation and its metabolic implications in B. bifidum.
MATERIALS AND METHODS
Bacterial strains and plasmids.
All strains and plasmids used in this study are listed in Table 2. E. coli DH5α (subcloning) was used for plasmid cloning and propagation, while E. coli MG1655 ΔgalETKM ΔgalU ΔushA Δugd Δagp (here named sMEMO_WT) was used for the expression and crude extract preparation of the uridylyltransferases. E. coli BL21(DE3) was used for the expression of the His6-tagged uridylyltransferases. The λ-red pKD46 plasmid, plasmid pKD4 for amplification of the kanamycin resistance marker, and plasmid pCP20 for removal of this marker were used for the one-step inactivation in E. coli MG1655 as described by Datsenko and Wanner (18).
Table 2.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Description | Source or reference |
|---|---|---|
| Strains | ||
| E. coli DH5α | General cloning host | Coli Genetic Stock Center |
| E. coli DH5α competent cells | Subcloning efficiency competent cells | Invitrogen |
| E. coli MG1655 | Escherichia coli λ− F− rph-1 | Coli Genetic Stock Center |
| E. coli BL21(DE3) | Escherichia coli F− λ(DE3 [lacI lacUV5-T7]) | Coli Genetic Stock Center |
| B. bifidum | Bifidobacterium bifidum (Tissier 1900) ATCC 29521 | BCCM/LMG |
| sMEMO_WT | E. coli MG1655 ΔgalETKM ΔgalU ΔushA Δugd Δagp | This study |
| sMEMO_UgpA | E. coli MG1655 ΔgalETKM ΔgalU ΔushA Δugd Δagp carrying pCX-Kan-P22-ugpA | This study |
| sMEMO_GalU | E. coli MG1655 ΔgalETKM ΔgalU ΔushA Δugd Δagp carrying pCX-Kan-P22-galU | This study |
| sMEMO_GalT1 | E. coli MG1655 ΔgalETKM ΔgalU ΔushA Δugd Δagp carrying p10-Trc-galT1 | This study |
| sMEMO_GalT2 | E. coli MG1655 ΔgalETKM ΔgalU ΔushA Δugd Δagp carrying p10-Trc-galT2 | This study |
| sMEMO_His_GalT1 | E. coli BL21(DE3) carrying p10-Trc-His-galT1 | This study |
| sMEMO_His_GalT2 | E. coli BL21(DE3) carrying p10-Trc-His-galT2 | This study |
| Plasmid vectors | ||
| pCX-Kan-P22 | Constitutive expression vector with P22 promoter, Ampr Kanr | 40 |
| pCX-Kan-P22-ugpA | pCX-Kan-P22 vector carrying ugpA from B. bifidum | This study |
| pCX-Kan-P22-galU | pCX-Kan-P22 vector carrying galU from E. coli | This study |
| p10-Trc | Inducible expression vector, Cmr | 41 |
| p10-Trc-galT1 | p10-Trc carrying galT1 from B. bifidum | This study |
| p10-Trc-galT2 | p10-Trc carrying galT2 from B. bifidum | This study |
| p10-Trc-His-galT1 | p10-Trc carrying galT1 from B. bifidum with His6 tag | This study |
| p10-Trc-His-galT2 | p10-Trc carrying galT2 from B. bifidum with His6 tag | This study |
| pKD46 | λ Red recombinase expression, Ampr | 18 |
| pCP20 | FLP recombinase expression, Ampr Cmr | 18 |
| pKD4 | Kan cassette template, Kanr Ampr | 18 |
Reagents.
T4 DNA ligase and all restriction enzymes were purchased from New England BioLabs (Ipswich, MA). PrimeSTAR polymerase was purchased from TaKaRa Bio (Japan). All chemicals used in crude extract preparation and assays were purchased from Sigma-Aldrich (Germany), except for gal1P and UTP, purchased from Merck (Darmstadt, Germany), and UDP-glucose and N-acetylglucosamine 1-phosphate from Carbosynth (Berkshire, United Kingdom).
Culture media.
E. coli cultures were grown on Luria-Bertani (LB) medium (Difco) with the necessary antibiotics (50 μg/ml kanamycin, 100 μg/ml ampicillin, 25 μg/ml chloramphenicol) for maintenance and selection of the plasmids. B. bifidum (ATCC 29521) was grown anaerobically at 37°C on LMG medium 144 (per liter of medium: 23 g special peptone [Difco], 1 g soluble starch, 5 g NaCl, 0.3 g cysteine hydrochloride, and 5 g glucose). The anaerobic environment, consisting of 5% H2 and 95% N2, was created by an anaerobic chamber (Concept 1000; Ruskinn Technology Ltd., United Kingdom).
DNA isolation, manipulation, and construction of uridylyltransferase vectors.
Genomic DNA (gDNA) was obtained by harvesting the cells of the culture by centrifugation at 22,000 × g (Heraeus Biofuge; Thermo). Cells were washed with saline and heated for 5 min at 100°C. The cell debris was removed by centrifugation at 22,000 × g for 10 min, and the supernatant was used as gDNA. All primers used in this study are listed in Table 3, and the gDNA of B. bifidum was used for the amplification of ugpA, galT1, and galT2 genes. gDNA of E. coli MG1655 was used for the amplification of galU. All the genes were amplified 2 times independently and sequenced to ensure that no mutations occurred in the constructs. The amplified galU and ugpA fragments were cut with EcoRI-HF and SacI-HF and ligated into the pCX-Kan-P22 vector, resulting in pCX-Kan-P22-galU and pCX-Kan-P22-ugpA, respectively. The galT1 and galT2 fragments were cut with NdeI-HF and PmeI and ligated into the p10-Trc vector, resulting in the p10-Trc-galT1 and p10-Trc-galT2 plasmids. In addition, both vectors were also redesigned so that an N-terminal His6 tag was added to allow purification, resulting in the p10-Trc-His-galT1 and p10-Trc-His-galT2 plasmids. These plasmids were created by using a 2-piece Gibson assembly method using cloning primers for the amplification of the p10-Trc backbone and His-galT1 and His-galT2 inserts listed in Table 3 (19). All plasmids were sequenced and showed no mutations.
Table 3.
List of primers used in this study
| Primer | Oligonucleotide sequence (5′–3′)a |
|---|---|
| Fw_galETKM_del | CTGGTGATTTGAACAATATGAGATAAAGCCCTCATGACGAGGGCGTAACAGTGTAGGCTGGAGCTGCTTC |
| Rv_galETKM_del | CTTTGTTATGCTATGGTTATTTCATACCATAAGCCTAATGGAGCGAATTATGCATATGAATATCCTCCTTAG |
| Fw_galU_del | CGCCTCCTTTTCAGAACTTAGCCCCTTCGGGGTGCTGATATACTGGGATGCGATACAGAAATATGAACACGTT |
| Rv_galU_del | GCAATCGACGCCGTTTTTTTATAGCTTATTCTTATTAAATTGTCTTAAACCGGACAATAAAAAATCCCGCCGC |
| Fw_ushA_del | TCGCGTCATACTATTTTTCAACACGTTGAAATCAGGTCAGGGAGAGAAGTGTGTAGGCTGGAGCTGCTTC |
| Rv_ushA_del | CCCGCCGCGATTAAGCATTGTGCCGGATGCAAACATCCGGCACTTTCGGACATATGAATATCCTCCTTAG |
| Fw_ugd_del | TGTAAGTAACAAAAGACAATCAGGGCGTAAATAGCCCTGATAACAAGATGGTGTAGGCTGGAGCTGCTTC |
| Rv_ugd_del | GATGCTAAAAACATCATGATTCACAGTTAAGTTAATTCTGAGAGCATGAACATATGAATATCCTCCTTAG |
| Fw_agp_del | CATATTTCTGTCACACTCTTTAGTGATTGATAACAAAAGAGGTGCCAGGAGTGTAGGCTGGAGCTGCTTC |
| Rv_agp_del | TAAAAACGTTTAACCAGCGACTCCCCCGCTTCTCGCGGGGGAGTTTTCTGCATATGAATATCCTCCTTAG |
| Fw_galU_EcoRI | CCGGCGAATTCGGAGGAAACAAAGATGGCTGCCATTAATACGAAAG |
| Rv_galU_SacI | CGCCGAGCTCTTACTTCTTAATGCCCATCTC |
| Fw_ugpA_EcoRI | CCGGCGAATTCGGAGGAAACAAAGATGTTTGCCGAAGATCTGAAACG |
| Rv_ugpA_SacI | CGCCGAGCTCTCACACCCAATCACCGGGCTCGATG |
| Fw_galT1_NdeI | AGGTCGCGCATATGGCAGAAATCACCAACTACAC |
| Rv_galT1_PmeI | TATGTTTAAACGCTAGCCTCGAGGCGGCCGCGAGCTCTAAAGGGCCGGCCTAAACGAATTCTCAGTCGGAGATGTCGATCTG |
| Fw_galT2_NdeI | AGGTCGCGCATATGACCACGGAAGAGAAGAAGG |
| Rv_galT2_PmeI | TATGTTTAAACGCTAGCCTCGAGGCGGCCGCGAGCTCTAAAGGGCCGGCCTAAACGAATTCCTAATGCTGAGTATGGAATCCGAGGCCTTCC |
| Fw_galT1_his | ATGGGCGGCTCACACCACCACCACCACCACGGTATGGCGTCTATGGCAGAAATCACCAACTAC |
| Fw_galT2_his | ATGGGCGGCTCACACCACCACCACCACCACGGTATGGCGTCTATGACCACGGAAGAGAAGAAG |
| Rv_galT1_his | CAATGATGATGATGATGATGGTCGACTCAGTCGGAGATGTCGATCTGTC |
| Rv_galT2_his | CAATGATGATGATGATGATGGTCGACCTAATGCTGAGTATGGAATC |
| Fw_backbone_p10 | AGACGCCATACCGTGGTGGTGGTGGTGGTGTGAGCCGCCCATATGTTATTCCTCCTTATTTAATC |
| Rv_backbone_p10 | GTCGACCATCATCATCATCATCATTG |
Restriction sites are underlined. His6 tags are underlined and in bold.
Construction of deletion mutants and expression strains.
To eliminate interference of the crude enzyme extract with the substrates used in the assay, an E. coli MG1655 knockout mutant was created by application of the one-step deletion system of Datsenko and Wanner (18). The linear DNA for the deletion of each target gene was amplified using deletion primers (Fw/Rv_gene_del) as mentioned in Table 3. The galETKM operon and the genes galU, ushA, ugd, and agp were deleted, resulting in the sMEMO_WT mutant (Table 2) as an expression host for the recombinant uridylyltransferases. Transformation of the constructed pCX-Kan-P22-galU, pCX-Kan-P22-ugpA, p10-Trc-galT1, and p10-Trc-galT2 plasmids in this sMEMO_WT mutant resulted in the sMEMO_GalU, sMEMO_UgpA, sMEMO_GalT1, and sMEMO_GalT2 strains, respectively. The p10-Trc-His-galT1 and p10-Trc-His-galT2 plasmids were transformed in BL21(DE3), resulting in sMEMO_His_GalT1 and sMEMO_His_GalT2 strains.
Preparation of crude enzyme extracts.
The sMEMO_GalU, sMEMO_UgpA, sMEMO_GalT1, and sMEMO_GalT2 strains were grown in tubes containing 5 ml LB (plus kanamycin or chloramphenicol when required) for 8 h at 37°C. The culture served as 2% inoculum for 250-ml Erlenmeyer flasks containing 50 ml LB medium with 1% glucose (and kanamycin or chloramphenicol when required). Strains sMEMO_GalT1 and sMEMO_GalT2 were induced with 0.2 mM IPTG (isopropyl-β-d-thiogalactopyranoside) after inoculation. Shake flasks were incubated at 37°C and 200 rpm for 16 h. Cells were harvested by centrifugation (Heraeus Biofuge; Thermo) for 10 min at 5,000 × g. The cell pellet was washed first with saline and then with 50 mM MOPS (morpholinepropanesulfonic acid) buffer (pH 6.5 or pH 7, depending on the assay). Finally, the pellet was dissolved in 5 ml of the above MOPS buffer and disrupted by sonication twice for 4 min each time (Branson sonifier, 50% duty cycle, output 3). The crude enzyme extract was obtained by centrifugation for 20 min at 22,000 × g. Protein concentration was determined via the Bradford assay (20).
Purification of GalT enzymes.
For the purification of both GalT1 and GalT2 enzymes, strains sMEMO_His_GalT1 and sMEMO_His_GalT2 were cultivated on 100 ml LB medium and chloramphenicol at 30°C and 200 rpm. When absorbance at 600 nm reached 0.6, IPTG was added at a final concentration of 0.4 mM. The crude extract was obtained in the same way as described above, and both enzymes were purified on a Ni-nitrilotriacetic acid agarose gel (Qiagen, Hilden, Germany) according to the manufacturer's instructions. After purification, a buffer exchange was performed to 50 mM MOPS buffer (pH 7) using an Amicon Ultra-4 centrifugal filter unit (30-kDa nominal molecular mass limit) from Merck (Darmstadt, Germany). Protein concentration was determined via the Bradford assay (20).
Assay for UTP-hexose-1-phosphate uridylyltransferase activity.
The assay for UTP-hexose-1-phosphate uridylyltransferase activity was based on the article by Wu et al. (21) but redesigned for pyrophosphate quantification. UTP-hexose-1-phosphate uridylyltransferase activity was measured in a 20-μl reaction mixture containing 50 mM MOPS buffer (pH 6.5), 2 mM UTP, 2 mM hexose 1-phosphate (glucose 1-phosphate [G1P] or gal1P), 2 mM MgCl2, 1 U of inorganic pyrophosphatase from E. coli, and various concentrations of crude enzyme extract. The reaction mixture was incubated at 37°C for 15 min, and the reaction was stopped by adding 180 μl of a 10 mM EDTA solution. Final activity was determined by quantifying the released phosphate during the reaction using a malachite green assay as described below. One unit of UTP-hexose-1-phosphate uridylyltransferase activity was defined as the amount of enzyme that formed 1 μmol of UDP-hexose per minute under these conditions.
Assay for UDP-glucose-hexose-1-phosphate uridylyltransferase.
UDP-glucose-hexose-1-phosphate uridylyltransferase activity was measured by using a continuous coupled assay for G1P quantification. The assay mixture consisted of 100 μl assay solution (50 mM MOPS buffer [pH 7], 2 mM NAD+, 10 mM MgCl2, 30 μM glucose-1,6-diphosphate, 1.2 U/ml phosphoglucomutase from rabbit muscle, and 1.2 U/ml glucose 6-phosphate dehydrogenase from Leuconostoc mesenteroides), 50 μl of substrate solution (50 mM MOPS buffer [pH 7], 4 mM gal1P, and 4 mM UDP-glucose), and a 50-μl dilution of crude enzyme extract or purified enzyme in 50 mM MOPS buffer (pH 7). The reaction was performed at 37°C in a microtiter plate, and NADH formation was monitored continuously by measuring the absorbance at 340 nm. One unit of UDP-glucose-hexose-1-phosphate uridylyltransferase activity was defined as the amount of enzyme that formed 1 μmol of G1P per minute under these conditions.
Kinetic analysis.
The kinetic constants were derived from initial rate analysis by varying the concentration of individual substrate. For the UTP-hexose-1-phosphate uridylyltransferase assay, UTP was varied from 0 to 2 mM in the presence of 2 mM hexose 1-phosphate (G1P or gal1P). Hexose 1-phosphate was subsequently varied from 0 to 2 mM at 2 mM saturation of UTP. For the UDP-glucose-hexose-1-phosphate uridylyltransferase assay, gal1P was varied from 1 to 0.02 mM in the presence of 1 mM UDP-glucose. Kinetic parameters were calculated from an S-V plot by nonlinear regression analysis using the Michaelis-Menten kinetic equations in R (“nlstools” package).
Malachite green assay.
Phosphate concentration of the samples in the micromolar range was determined using a malachite green assay: to 50 μl of sample, 30 μl reagent A, 100 μl Milli-Q water, and 30 μl reagent B was added (in this order). The mixture was incubated for 20 min in a microtiter plate at room temperature, and absorbance was measured at 630 nm. Reagent A consists of 50 mM ammonium heptamolybdate in 3 M sulfuric acid. Reagent B consists of 0.093% (wt/vol) malachite green and 0.93% (wt/vol) polyvinyl alcohol (Mw, 14,000). Concentrations were determined using a phosphate standard (serial dilutions ranging from 0 to 100 μM).
LC-MS.
The products—phosphorylated and nucleotide sugars—of the enzymatic reactions were also verified by liquid chromatography coupled to a mass spectrometer (LC-MS) using a Cosmosil Hilic (Nacalai USA, San Diego, CA) column (4.6 by 250 mm) with isocratic separation (0.1 M ammonium acetate [50%] and acetonitrile [50%] at a flow rate of 1 ml/min at 35°C for 30 min). The LC system was coupled to a Micromass Quattro LC (McKinley Scientific, USA). Detection was performed in a negative-mode electrospray ionization (ESI)-MS with a capillary voltage of 2.53 kV, a cone voltage of 20 V, cone and desolvation gas flows of 93 and 420 liters/h, and source and cone temperatures of 150 and 350°C, respectively.
Nucleotide sequence accession numbers.
The ugpA, galT1, and galT2 gene sequences have been deposited in GenBank under accession numbers KC261357, KC261358, and KC261359, respectively.
RESULTS
Cloning of the uridylyltransferase genes.
The nucleotide sequences of ugpA, galT1, and galT2 from B. bifidum ATCC 29521 were 99.4%, 99.6%, and 99.5% identical to the BBPR_0976, BBPR_0406, and BBPR_1051 genes of B. bifidum PRL2010, respectively (11). The predicted functions of these genes are UTP-glucose-1-phosphate uridylyltransferase (ugpA) and galactose-1-phosphate uridylyltransferase (galT1 and galT2), respectively. These genes were expressed in the E. coli MG1655 mutant, and an assay was performed to determine their function as described in Materials and Methods.
Assay validation.
A new UTP-hexose-1-phosphate uridylyltransferase assay was developed based on the principle of phosphate detection as described by Wu and coworkers (21). Crude extracts with this uridylyltransferase activity release pyrophosphate, which is converted to inorganic phosphate (Pi) when coupled with a pyrophosphatase. The subsequent reaction of phosphate with a malachite green reagent is spectrophotometrically detected in the micromolar range. Validation of the UTP-hexose-1-phosphate uridylyltransferase assay was performed using commercially available uridine-5′-diphosphoglucose pyrophosphorylase from baker's yeast (Sigma-Aldrich; 40 to 60% protein concentration, ≥50 U/mg protein). One unit was defined as forming 1 μmol glucose 1-phosphate (G1P) at 25°C at pH 7.6 in 1 min. Enzyme dilutions were incubated for 10 min with 2 mM UTP and G1P under the conditions described in Materials and Methods, and released phosphate was measured with the malachite green assay. A specific activity of 65.2 ± 1.7 U/mg protein was found. This assay coupled with subsequent malachite green phosphate detection proved to be a stable and precise method for determination of the specific activity. Validation of the UDP-glucose-hexose-1-phosphate uridylyltransferase assay was performed by adding 100 μl of 2 mM G1P to the assay solution.
Expression host validation.
An E. coli MG1655 mutant (sMEMO_WT) was created as an efficient screening host for the different uridylyltransferases from B. bifidum. The Leloir pathway (galETKM) and the gene coding for UTP-glucose-1-phosphate uridylyltransferase (galU) were deleted to prevent interference with the investigated enzymes. The degradation of hexose 1-phosphate or UDP-hexose substrates was also prevented by knocking out UDP-sugar hydrolase (ushA), UDP-glucose 6-dehydrogenase (ugd), and glucose-1-phosphatase (agp). The crude extract of sMEMO_WT was tested against the substrates of both assays to detect possible interference. No activity was observed for the UDP-glucose-hexose-1-phosphate uridylyltransferase assay. Testing of the crude extract against a hexose 1-phosphate and UTP showed no activity for the UTP-hexose-1-phosphate uridylyltransferase assay. However, minor activity was observed with UTP as sole substrate after extended incubation times (>30 min) and using undiluted crude extracts (data not shown). This is probably due to UTP hydrolysis by an unidentified hydrolase. Since incubation times were always shorter than 15 min and dilutions starting from 0.05 were used, sMEMO_WT was considered a suitable screening host.
UTP-hexose-1-phosphate uridylyltransferase activity.
Crude enzyme extract dilutions of UgpA, GalT1, and GalT2 were screened for UTP-hexose-1-phosphate uridylyltransferase activity against 2 mM hexose 1-phosphate (G1P or gal1P) and 2 mM UTP. Only UgpA showed activity against both substrates. Formation of the corresponding UDP-hexose (UDP-glucose or UDP-galactose, respectively) was confirmed by LC-MS. The specific activity of UgpA crude extract toward G1P was 13 U mg−1, which was about 33-fold higher than that toward gal1P. The apparent Km values were also calculated and seemed to be similar for the two substrates (Table 4). As a reference point for the substrate usage spectrum, the UgpA equivalent of E. coli (GalU) was also assayed against both substrates, showing activity only toward G1P. The specific activity of the GalU crude extract was about 6.9-fold lower than that of UgpA.
Table 4.
Kinetic parameters for UgpA and GalU crude protein extractsa
| Protein extract | Sp act (U mg−1) |
Km (mM) |
|
|---|---|---|---|
| H1P | UTP | ||
| UgpA (G1P) | 13.0 ± 0.08 | 0.098 ± 0.011 | 0.042 ± 0.006 |
| UgpA (gal1P) | 0.40 ± 0.01 | 0.148 ± 0.034 | 0.032 ± 0.010 |
| GalU (G1P) | 1.88 ± 0.03 | 0.010b | 0.070b |
| GalU (gal1P) | ND | ||
Reactions were carried out at pH 6.5 and 37°C. ND, not detected. Experiments were carried out in triplicate, and standard deviations are shown.
Data from the work of Weissborn et al. (27). Reaction carried out at pH 7.8 and 37°C.
Activity of GalT1 and GalT2.
The 3 recombinant uridylyltransferase crude extracts were screened for UDP-glucose-hexose-1-phosphate uridylyltransferase activity against 2 mM UDP-glucose and 2 mM gal1P at pH 7 and 37°C. GalT1 showed high activity; a 0.01 dilution of the crude extract converted all gal1P into G1P within a 5-min incubation. The same dilution of GalT2, however, displayed a very weak activity which was 417-fold lower than that of GalT1. The activity did not improve after adding cysteine, Fe2+, or Zn2+, as proposed in the literature (22, 23). Altering the expression conditions (0.1 mM IPTG induction), lowering the assay temperature to 25°C, or raising the buffer pH to 8.5 had no effect either. To confirm these results, the His6-tagged GalT1 and GalT2 were purified from the sMEMO_His_GalT1 and sMEMO_His_GalT2 strains and different dilutions were used to measure activity. Acceptors other than gal1P were also tested, including N-acetylgalactosamine 1-phosphate (galNAc1P) and N-acetylglucosamine 1-phosphate (glcNAc1P), in the presence of 2 mM UDP-glucose, and the specific activities are given in Table 5. GalT2 showed a 357-fold-lower activity than did GalT1, which is consistent with the results based on the crude extracts. The apparent Km of GalT1 for gal1P in the presence of 2 mM UDP-glucose was 0.29 ± 0.08 mM, which is in the range of previous findings (24). The apparent Km of GalT2 for gal1P was 0.61 ± 0.1 mM, while the Km of GalT2 toward galNAc1P was only 65.1 ± 15.9 μM.
Table 5.
Substrate acceptor specificities of the purified GalT1 and GalT2 enzymesa
| Enzyme | Sp act (U mg−1) |
|---|---|
| GalT1 (gal1P) | 93.1 ± 2.3 |
| GalT1 (galNAc1P) | ND |
| GalT1 (glcNAc1P) | ND |
| GalT2 (gal1P) | 0.261 ± 0.015 |
| GalT2 (galNAc1P) | 0.334 ± 0.028 |
| GalT2 (glcNAc1P) | 0.235 ± 0.013 |
Reactions were carried out at pH 7 and 37°C for various acceptors. Experiments were carried out in triplicate, and standard deviations are shown. ND, not detected.
DISCUSSION
Assay and expression host validation.
A new and reliable assay was developed that allows fast screening and characterization of enzymes with UTP-hexose-1-phosphate uridylyltransferase activity. The same assay can be used to investigate various other enzymes that liberate pyrophosphate, such as mannose-1-phosphate guanylyltransferase (EC 2.7.7.13) or isoprene synthase (EC 4.2.3.27). The development of the described expression host, lacking the Leloir pathway and relevant degradation reactions, bypasses the need to purify the enzymes investigated. These assays proved to be indispensable tools to characterize UgpA, GalT1, and GalT2 and gain new insights in the LNB/GNB metabolism of bifidobacteria.
Specificity of UgpA.
Our results indicate that UgpA is a promiscuous UTP-hexose-1-phosphate uridylyltransferase that catalyzes the formation of UDP-hexose from hexose 1-phosphate. Based on this activity and the size of the monomer (55 kDa), this enzyme is probably homologous to the enzyme that was purified from B. bifidum by Lee et al. but was never properly annotated (17, 25). They described the reverse (pyrophosphorylase) action of UgpA, which showed the same reactivity toward UDP-galactose as toward UDP-glucose. However, our findings indicate a 33-fold-lower activity in the synthesis direction of UDP-galactose. Because of this promiscuity, a new EC number (EC 2.7.7.10) was proposed to distinguish it from UTP-glucose-1-phosphate uridylyltransferase (EC 2.7.7.9; GalU), which predominantly acts on G1P in prokaryotes (26, 27). However, due to the activity on different sugar 1-phosphates, the absence of similarity with GalU-type enzymes, and a high level of identity with promiscuous eukaryotic uridylyltransferases, we suggest that UgpA should be classified as an UTP-monosaccharide-1-phosphate uridylyltransferase (EC 2.7.7.64, synonym of UDP-sugar pyrophosphorylase [USP]) (28–30). Moreover, a UDP-sugar pyrophosphorylase from B. longum (BLUSP) was recently cloned, showing 82% amino acid identity with UgpA, and was used for the efficient synthesis of a variety of UDP-sugars (31).
Specificity of GalT enzymes.
We observed that both GalT1 and GalT2 possessed UDP-glucose-hexose-1-phosphate uridylyltransferase activity (EC 2.7.7.12; GalT), yet the activity of GalT2 was 357-fold lower than that of GalT1. A similar result was reported in the large-scale preparation of LNB, where GalT1 of B. longum was used instead of GalT2 (4, 8). GalT2 shows a broad acceptor specificity, with a 9-fold-higher affinity toward galNAc1P than gal1P. The amino acid sequences of both GalT enzymes shared almost no identities (12.1%), which could be explained by convergent evolution of similar enzymatic function. However, it was shown that the majority of catalytic residues are under different evolutionary constraints in the two types of enzymes, and they likely have different functions (32). This is supported by the fact that purified GalT2 was 28% more active toward galNAc1P than gal1P as the substrate, providing a link between growth on GNB/LNB and aminosugar metabolism.
galT gene clusters.
GalT enzyme activity, which is essential for the transfer of the uridine nucleotide moiety from UDP-glucose to gal1P, is widespread in many organisms. However, based on the enzyme dissimilarities between GalT1 and GalT2, two classes seem to exist. Class I enzymes (GalT1 type) are found in various eukaryotic organisms and bacteria, while class II enzymes (GalT2 type) are almost exclusively present in Firmicutes and Actinobacteria (33, 34). Coding sequences for GalT1 or GalT2 do not co-occur in the same organism, with some Clostridiales and bifidobacteria as exceptions. Based on comparative bifidobacterial genome analysis, only B. bifidum, B. longum, and B. breve strains possess both galT1 and galT2, being part of a galTK gene cluster and a GNB/LNB degradation gene cluster, respectively (11, 12, 35). A schematic phylogenetic overview of these gene clusters is given in Fig. 3. Metabolic and genetic explanations for the unique coexistence of the two GalT enzymes in B. bifidum, B. longum, and B. breve strains remain unclear, but this coexistence is coupled to the occurrence of the GNB/LNB pathway, which is apparently absent in other bifidobacteria (7, 13).
Fig 3.

16S rRNA-based phylogenetic tree with gene cluster organization and prevalence of class I enzymes (GalT1 type) and class II enzymes (GalT2 type) in three representative bacterial phyla. The bacterial strain codes and bootstrapping values are included. Orthologues that match the galT1 or galT2 genes of B. bifidum are aligned vertically. GalT2-type enzymes are present only in Firmicutes and Actinobacteria and show a high degree of rearrangement with the neighboring genes.
Metabolic implications.
The occurrence of three uridylyltransferases in B. bifidum that act on gal1P can have metabolic implications that have been overlooked. While the Leloir pathway is in most organisms the major, if not the only, pathway for the degradation of galactose, an alternative route was discovered in B. longum under the form of the GNB/LNB pathway. The same gene cluster is also present in B. bifidum, and it is suggested that this energy-saving variant of the Leloir pathway is the main pathway for galactose metabolism (4). However, glycoprofiling of B. bifidum during growth on HMO shows the release of lactose and LNB as prominent disaccharides. While LNB enters the GNB/LNB pathway, galactose is released by action of a β-galactosidase from lactose (7). Our findings suggest that galactose is primarily metabolized via the Leloir pathway, together with gal1P originating from LNB or GNB.
Although GalT2 (as part of the GNB/LNB pathway) is put forward to play the key role in gal1P degradation (4), we suggest that GalT1 (as part of the Leloir pathway) predominates when grown on lactose, galactooligosaccharides (GOS), and HMO. In addition to the 357-fold-lower activity of GalT2, transcription data showed the upregulation of galT1 during growth on these substrates relative to glucose (36, 37). It is often stated that the genes of the GNB/LNB pathway are upregulated during growth on oligosaccharides or mucin, but whether a part or the whole cluster is upregulated is highly dependent on the substrate. Growth on LNB-containing oligosaccharides (HMO) requires only LNBP and NahK activity in principle, yielding gal1P and glcNAc1P. The latter is metabolized into fructose 6-phosphate (F6P) by action of a phosphoglucosamine mutase, an N-acetylglucosamine 6-phosphate deacetylase, and a glucosamine 6-phosphate deaminase, which then enters the bifid shunt (38). A significant upregulation of these genes was observed when grown on HMO (11, 39). Substrates with a GNB core (such as mucin glycans) require LNBP and NahK activity, yielding gal1P and galNAc1P and yet also require GalT2 and GalE2 activity to be converted to glcNAc1P in order to enter the bifid shunt. This hypothesis is supported by the substrate preference of GalT2 toward galNAc1P and by transcriptional data of bifidobacteria grown on lactose, HMO, GOS, or mucin-based media (11, 36, 37). A proposed metabolic route for lactose, LNB, and GNB degradation is depicted in Fig. 4. The metabolic implications of UgpA are probably limited to UDP-sugar generation, to provide the Leloir pathway with initial amounts of UDP-glucose (16, 26). This is supported by the fact that the Km for hexose 1-phosphate is more than 2-fold lower than that of GalT1.
Fig 4.

Proposed routes for the breakdown of galactose, lacto-N-biose I (LNB), and galacto-N-biose (GNB) in B. bifidum. All 3 substrates yield gal1P, which is metabolized solely by the Leloir pathway (left). Depending on the presence of LNB or GNB, N-acetylglucosamine (glcNAc) or N-acetylgalactosamine (galNAc), respectively, is formed through the action of LNBP. Utilization of glcNAc requires only N-acetylhexosamine kinase (NahK) activity to generate glcNAc1P. However, GNB-rich substrates such as mucin need the full set of enzymes encoded by the GNB/LNB gene cluster (right). galNAc is phosphorylated to galNAc1P by NahK, which is subsequently converted by GalT2 and GalE2 via a Leloir-like pathway to glcNAc1P, which can then enter the bifid shunt via fructose 6-phosphate (F6P) by consequent action of a glucosamine mutase, a deacetylase, and a deaminase. glcN6P, glucosamine 6-phosphate.
ACKNOWLEDGMENTS
We thank the Institute for the Promotion of Innovation through Science and Technology in Flanders (IWT-Vlaanderen) for financial support in the framework of the Ph.D. grant of F. De Bruyn. This research was also supported by the Multidisciplinary Research Partnership Ghent Bio-Economy.
We also thank Jan De Neve and Gaspard Lequeux for their critical view on the statistical methodology and Pieter Coussement for his remarks concerning the creation of a phylogenetic tree.
Footnotes
Published ahead of print 6 September 2013
REFERENCES
- 1.Tissier H. 1900. Recherches sur la flore intestinale des nourrissons (état normal et pathologique). Ph.D. thesis University of Paris, Paris, France [Google Scholar]
- 2.Guarner F, Malagelada JR. 2003. Gut flora in health and disease. Lancet 361:512–519 [DOI] [PubMed] [Google Scholar]
- 3.Turroni F, Foroni E, Pizzetti P, Giubellini V, Ribbera A, Merusi P, Cagnasso P, Bizzarri B, de'Angelis GL, Shanahan F, van Sinderen D, Ventura M. 2009. Exploring the diversity of the bifidobacterial population in the human intestinal tract. Appl. Environ. Microbiol. 75:1534–1545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nishimoto M, Kitaoka M. 2007. Identification of N-acetylhexosamine 1-kinase in the complete lacto-N-biose I/galacto-N-biose metabolic pathway in Bifidobacterium longum. Appl. Environ. Microbiol. 73:6444–6449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fushinobu S. 2010. Unique sugar metabolic pathways of bifidobacteria. Biosci. Biotechnol. Biochem. 74:2374–2384 [DOI] [PubMed] [Google Scholar]
- 6.Ruas-Madiedo P, Gueimonde M, Fernandez-Garcia M, de los Reyes-Gavilan CG, Margolles A. 2008. Mucin degradation by Bifidobacterium strains isolated from the human intestinal microbiota. Appl. Environ. Microbiol. 74:1936–1940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Asakuma S, Hatakeyama E, Urashima T, Yoshida E, Katayama T, Yamamoto K, Kumagai H, Ashida H, Hirose J, Kitaoka M. 2011. Physiology of consumption of human milk oligosaccharides by infant gut-associated bifidobacteria. J. Biol. Chem. 286:34583–34592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nishimoto M, Kitaoka M. 2007. Practical preparation of lacto-N-biose I, a candidate for the bifidus factor in human milk. Biosci. Biotechnol. Biochem. 71:2101–2104 [DOI] [PubMed] [Google Scholar]
- 9.Kitaoka M, Tian J, Nishimoto M. 2005. Novel putative galactose operon involving lacto-N-biose phosphorylase in Bifidobacterium longum. Appl. Environ. Microbiol. 71:3158–3162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhurina D, Zomer A, Gleinser M, Brancaccio VF, Auchter M, Waidmann MS, Westermann C, van Sinderen D, Riedel CU. 2011. Complete genome sequence of Bifidobacterium bifidum S17. J. Bacteriol. 193:301–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Turroni F, Bottacini F, Foroni E, Mulder I, Kim JH, Zomer A, Sanchez B, Bidossi A, Ferrarini A, Giubellini V, Delledonne M, Henrissat B, Coutinho P, Oggioni M, Fitzgerald GF, Mills D, Margolles A, Kelly D, van Sinderen D, Ventura M. 2010. Genome analysis of Bifidobacterium bifidum PRL2010 reveals metabolic pathways for host-derived glycan foraging. Proc. Natl. Acad. Sci. U. S. A. 107:19514–19519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yu DS, Jeong H, Lee DH, Kwon SK, Song JY, Kim BK, Park MS, Ji GE, Oh TK, Kim JF. 2012. Complete genome sequence of the probiotic bacterium Bifidobacterium bifidum strain BGN4. J. Bacteriol. 194:4757–4758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xiao JZ, Takahashi S, Nishimoto M, Odamaki T, Yaeshima T, Iwatsuki K, Kitaoka M. 2010. Distribution of in vitro fermentation ability of lacto-N-biose I, a major building block of human milk oligosaccharides, in bifidobacterial strains. Appl. Environ. Microbiol. 76:54–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hidaka M, Nishimoto M, Kitaoka M, Wakagi T, Shoun H, Fushinobu S. 2009. The crystal structure of galacto-N-biose/lacto-N-biose I phosphorylase: a large deformation of a TIM barrel scaffold. J. Biol. Chem. 284:7273–7283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kitaoka M. 2012. Bifidobacterial enzymes involved in the metabolism of human milk oligosaccharides. Adv. Nutr. 3:422S–429S [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frey PA. 1996. The Leloir pathway: a mechanistic imperative for three enzymes to change the stereochemical configuration of a single carbon in galactose. FASEB J. 10:461–470 [PubMed] [Google Scholar]
- 17.Lee LJ, Kimura A, Tochikura T. 1978. Presence of a single enzyme catalyzing the pyrophosphorolysis of UDP-glucose and UDP-galactose in Bifidobacterium bifidum. Biochim. Biophys. Acta 527:301–304 [DOI] [PubMed] [Google Scholar]
- 18.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U. S. A. 97:6640–6645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA, III, Smith HO. 2009. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6:343–345 [DOI] [PubMed] [Google Scholar]
- 20.Bradford MM. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248–254 [DOI] [PubMed] [Google Scholar]
- 21.Wu ZL, Ethen CM, Prather B, Machacek M, Jiang W. 2011. Universal phosphatase-coupled glycosyltransferase assay. Glycobiology 21:727–733 [DOI] [PubMed] [Google Scholar]
- 22.Geeganage S, Frey PA. 1999. Significance of metal ions in galactose-1-phosphate uridylyltransferase: an essential structural zinc and a nonessential structural iron. Biochemistry 38:13398–13406 [DOI] [PubMed] [Google Scholar]
- 23.Arabshahi A, Ruzicka FJ, Geeganage S, Frey PA. 1996. Standard free energies for uridylyl group transfer by hexose-1-P uridylyltransferase and UDP-hexose synthase and for the hydrolysis of uridine 5′-phosphoimidazolate. Biochemistry 35:3426–3428 [DOI] [PubMed] [Google Scholar]
- 24.Lee L, Kimura A, Tochikura T. 1978. Purification and properties of hexose 1-phosphate uridylyltransferase from Bifidobacterium bifidum. Agric. Biol. Chem. 42:723–730 [Google Scholar]
- 25.Lee L, Kimura A, Tochikura T. 1979. Purification and properties of UDP-glucose (UDP-galactose) pyrophosphorylase from Bifidobacterium bifidum. J. Biochem. 86:923–928 [DOI] [PubMed] [Google Scholar]
- 26.Thoden JB, Holden HM. 2007. The molecular architecture of glucose-1-phosphate uridylyltransferase. Protein Sci. 16:432–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Weissborn AC, Liu Q, Rumley MK, Kennedy EP. 1994. UTP: alpha-d-glucose-1-phosphate uridylyltransferase of Escherichia coli: isolation and DNA sequence of the galU gene and purification of the enzyme. J. Bacteriol. 176:2611–2618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kleczkowski LA, Decker D, Wilczynska M. 2011. UDP-sugar pyrophosphorylase: a new old mechanism for sugar activation. Plant Physiol. 156:3–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kotake T, Yamaguchi D, Ohzono H, Hojo S, Kaneko S, Ishida HK, Tsumuraya Y. 2004. UDP-sugar pyrophosphorylase with broad substrate specificity toward various monosaccharide 1-phosphates from pea sprouts. J. Biol. Chem. 279:45728–45736 [DOI] [PubMed] [Google Scholar]
- 30.Dai N, Petreikov M, Portnoy V, Katzir N, Pharr DM, Schaffer AA. 2006. Cloning and expression analysis of a UDP-galactose/glucose pyrophosphorylase from melon fruit provides evidence for the major metabolic pathway of galactose metabolism in raffinose oligosaccharide metabolizing plants. Plant Physiol. 142:294–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Muthana MM, Qu J, Li Y, Zhang L, Yu H, Ding L, Malekan H, Chen X. 2012. Efficient one-pot multienzyme synthesis of UDP-sugars using a promiscuous UDP-sugar pyrophosphorylase from Bifidobacterium longum (BLUSP). Chem. Commun. (Camb.) 48:2728–2730 [DOI] [PubMed] [Google Scholar]
- 32.Abhiman S, Sonnhammer EL. 2005. Large-scale prediction of function shift in protein families with a focus on enzymatic function. Proteins 60:758–768 [DOI] [PubMed] [Google Scholar]
- 33.Mollet B, Pilloud N. 1991. Galactose utilization in Lactobacillus helveticus: isolation and characterization of the galactokinase (galK) and galactose-1-phosphate uridyl transferase (galT) genes. J. Bacteriol. 173:4464–4473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jensen LJ, Kuhn M, Stark M, Chaffron S, Creevey C, Muller J, Doerks T, Julien P, Roth A, Simonovic M, Bork P, von Mering C. 2009. STRING 8—a global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res. 37:D412–D416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schell MA, Karmirantzou M, Snel B, Vilanova D, Berger B, Pessi G, Zwahlen MC, Desiere F, Bork P, Delley M, Pridmore RD, Arigoni F. 2002. The genome sequence of Bifidobacterium longum reflects its adaptation to the human gastrointestinal tract. Proc. Natl. Acad. Sci. U. S. A. 99:14422–14427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Klaassens ES, Ben-Amor K, Vriesema A, Vaughan EE, de Vos W. 2011. The fecal bifidobacterial transcriptome of adults: a microarray approach. Gut Microbes 2:217–226 [DOI] [PubMed] [Google Scholar]
- 37.Gonzalez R, Klaassens ES, Malinen E, de Vos WM, Vaughan EE. 2008. Differential transcriptional response of Bifidobacterium longum to human milk, formula milk, and galactooligosaccharide. Appl. Environ. Microbiol. 74:4686–4694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sela DA, Chapman J, Adeuya A, Kim JH, Chen F, Whitehead TR, Lapidus A, Rokhsar DS, Lebrilla CB, German JB, Price NP, Richardson PM, Mills DA. 2008. The genome sequence of Bifidobacterium longum subsp. infantis reveals adaptations for milk utilization within the infant microbiome. Proc. Natl. Acad. Sci. U. S. A. 105:18964–18969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Garrido D, Ruiz-Moyano S, Mills DA. 2012. Release and utilization of N-acetyl-d-glucosamine from human milk oligosaccharides by Bifidobacterium longum subsp. infantis. Anaerobe 18:430–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Aerts D, Verhaeghe T, De Mey M, Desmet T, Soetaert W. 2011. A constitutive expression system for high-throughput screening. Eng. Life Sci. 11:10–19 [Google Scholar]
- 41.Ajikumar PK, Xiao WH, Tyo KE, Wang Y, Simeon F, Leonard E, Mucha O, Phon TH, Pfeifer B, Stephanopoulos G. 2010. Isoprenoid pathway optimization for taxol precursor overproduction in Escherichia coli. Science 330:70–74 [DOI] [PMC free article] [PubMed] [Google Scholar]