

Abstract
Bacteria have the remarkable ability to communicate as a group in what has become known as quorum sensing (QS), and this trait has been associated with important bacterial phenotypes, such as virulence and biofilm formation. Bacteria also have an incredible ability to evolve resistance to all known antimicrobials. Hence, although inhibition of QS has been hailed as a means to reduce virulence in a manner that is impervious to bacterial resistance mechanisms, this approach is unlikely to be a panacea. Here we review the evidence that bacteria can evolve resistance to quorum-quenching compounds.
INTRODUCTION
Infectious diseases are the leading cause of death (1), and all antibiotics fail (2); therefore, it is imperative to develop novel ways to fight microbial infections. Here we review the use of chemicals that interfere with cell communication and investigate the likelihood that bacteria will evolve resistance to these compounds.
QS and QQ.
Bacteria use secreted chemicals as signals for a variety purposes, including virulence and biofilm formation. When the compounds build to a threshold concentration and trigger gene expression, the signals are known as quorum-sensing (QS) signals. Examples of well-studied QS signals include acylhomoserine lactones, autoinducer 2, and peptide signals, but many other signals, such as indole, exist (3). In addition to signals, signal synthases, signal receptors, signal response regulators, and regulated genes (QS regulon) are key components of any QS system (4). For example, LuxI-type enzymes are signal synthases which synthesize acylhomoserine lactones. In addition, LuxR-type regulators are receptor proteins for the autoinducer signals, and signal-receptor binding is responsible for the expression of QS regulons. Since numerous compounds that inhibit QS have been identified, since QS is linked to virulence, and since inhibition of QS does not usually affect growth (in rich medium), it has been reasoned that inhibition of QS may be an effective means of reducing pathogenicity that is not subject to the usual resistance mechanisms of bacteria (1, 5–8). Inhibition of QS is also known as quorum quenching (QQ) and is a form of antivirulence.
The well-known examples (9) of QQ compounds include lactonases/acylases that degrade the N-(3-oxoctanoyl)-homoserine lactone (HSL) autoinducers, synthase inhibitors, like analogues of anthranilic acid that block synthesis of quinolone signals (10), and receptor inhibitors, such as brominated furanones (11). In addition, low concentrations of azithromycin, ceftazidime, and ciprofloxacin (antibiotics) inhibit QS in Pseudomonas aeruginosa (12). Also, among the thousands of drugs approved for clinical use, the anthelmintic drug niclosamide is a QQ compound (13); this drug reduces surface motility, biofilm formation, and production of the secreted virulence factors elastase, pyocyanin, and rhamnolipids. The rhizosphere bacterium Stenotrophomonas maltophilia produces cis-9-octadecenoic acid, which is a QQ compound that reduces violacein production by Chromobacterium violaceum and biofilm formation by P. aeruginosa (14). The cyclic dipeptide 2,5-piperazinedione inhibits QS-dependent traits, such as protease activity, elastase activity, and the production of pyocyanin and extracellular polymeric substances (15). Further evidence that QQ compounds are readily found in nature include the finding that of 120 bacterial isolates from healthy coral species, up to 24% of the isolates showed anti-QS activity against three QS indicator strains (16); a possible explanation for this behavior is that the interaction of coral-associated bacteria is competitive, so QQ compounds are secreted from the dominant communities to diminish undesirable marine biofouling. From this group, a Favia sp. coral isolate inhibits the biofilm formation of P. aeruginosa and Acinetobacter baumannii through the secretion of a low-molecular-mass compound which is not inactivated by heat and protease K (16).
Mathematical modeling for resistance to QQ.
There are several manuscripts describing mathematical models of the QS systems of P. aeruginosa and other bacterial species, including planktonic cells (17) and biofilm cells (18) in closed and open spaces, such as microfluidic devices (19). Most of these models describe the effect of classical antibiotics and antivirulence compounds and suggest a narrow therapeutic concentration range for the QQ agent to be effective in biofilms. For example, Anguige et al. (20) found that there is a critical biofilm depth in which the QQ treatment is successful; hence, the biofilm penetrability of QQ drugs is a critical parameter to take into account for the design of antivirulence compounds. Therefore, perhaps resistance to QQ in biofilms may develop by restricting the permeability of the QQ drugs by overproducing extracellular matrix components that sequester the QQ agent, such as the ndvB-encoded glucans that sequester aminoglucosides (21). Other theoretical studies show that for biofilms, the time at which QQ treatment is initiated is critical for effective prevention of QS-mediated virulence (22).
Beckmann et al. (23) in 2012 developed what was purported to be the first in silico study that showed the possibility of QQ resistance; however, they did not evaluate resistance to QQ compounds, since a QQ compound was not introduced; instead, signal-blind or signal-deficient mutants were introduced into a wild-type culture, which is not the same as an organism developing resistance to QQ compounds. For QQ resistance, the original population would be inhibited from QS by the QQ compound, while resistant mutants would be unaffected and continue to QS in the presence of the QQ compound, which is a scenario different from that of Beckmann et al.'s simulations. The digital organisms were designed as a type of self-replicating computer program and were subject to the mutations and natural selection that exist in a computational environment. These digital organisms communicate with each other by sending messages, and QS is simulated by allowing each organism to receive one message and to send six messages to its neighbors, creating a positive “signal” feedback loop. In this digital setting, the authors found that the wild-type population became resistant to the deleterious effects of the QS mutants (the mutants increased the “energy consumption” in the system, making the system less efficient) by lowering the threshold of the signal necessary to trigger the QS-controlled phenotypes. Therefore, the model predicts that wild-type cells are resistant to takeover by the QS mutants. This interesting theoretical result remains to be tested experimentally.
Computational approaches and molecular-docking analysis have also been useful for understanding the binding of QQ compounds to receptor proteins to identify potential QQ compounds. Molecular alignment of receptor proteins (e.g., LuxR-type proteins) indicate that there are preserved motifs in the residues of Y53, Y71, W57, D70, and W85 of TraR and Y56, Y64, W60, D73, and W88 of LasR and that the amino acid residues D70, W57, and Y53 in TraR and D73, W60, Y56, and S129 in LasR are important for interacting with the autoinducer analogs (24). The autoinducer analogs rosmarinic acid, naringin, chlorogenic acid, morin, and mangiferin have been studied through in silico docking analysis, and the analyses demonstrated that these compounds can inhibit the production of protease, elastase, and hemolysin (25). In addition, five inducers and three inhibitors which are molecularly distant from the native autoinducer N-3-oxododecanoyl-l-homoserine lactone have been investigated as potential QQ compounds (26). As another example of these modeling approaches, competitive inhibitors of SdiA, a signal receptor of the QS signals of other bacteria in Escherichia coli, have been screened from Melia dubia seed extracts, and 27 compounds structurally unrelated to autoinducers show potential for attenuating QS in uropathogenic E. coli (27). Also, molecular docking was used to identify potential QQ compounds from bark extracts of the mangrove plant Rhizophora annamalayana (28). In addition, three compounds which can inhibit the activity of LuxS from Actinobacillus pleuropneumoniae (LuxS catalyzes S-ribosylhomocysteine into homocysteine and autoinducer 2) were identified computationally (29). In the same manner, possible QQ compounds which can inhibit growth and biofilm formation have been found in various extracts for cariogenic Streptococcus mutans isolates using ligand fit docking protocols (30).
Early studies on resistance to quorum quenching.
The first suggestion that cells may evolve resistance to QQ compounds was presented as an opinion piece by Defoirdt et al. (2) in 2010. The basis for this supposition was collected from several studies showing that the expression of core QS genes is highly varied between different strains of the same Vibrio species and other pathogenic bacteria, including P. aeruginosa. These core QS genes are involved in the production/detection of autoinducers as well as in QS signal transduction; since their variability is heritable, if this variation confers an advantage in fitness under QQ treatment, the authors concluded that natural selection would favor the spread of QQ resistance. Moreover, this group realized that previous arguments that concluded that resistance to QQ compounds was unlikely had been incorrectly predicated on the growth of pathogens in complex medium. Up to this point, QQ compounds were routinely tested in rich medium, where they were shown to not affect growth and therefore thought not subject to Darwinian selection pressure for resistance. Since pathogens are more likely to encounter conditions more closely resembling minimal media, where they are starved for nutrients and where QQ compounds affect growth, it was reasoned that cells may evolve resistance to QQ compounds. In addition, in mice infection models, the number of viable P. aeruginosa bacteria after QQ treatment in the lungs of infected mice decreases and the ability of this pathogen to disseminate in mice is inhibited; hence, even in the absence of a direct effect of the QS inhibitor, the fitness of bacteria during an infection clearly decreases under QS disruption (2). This is not surprising, since there are numerous studies that show that QS signals as well as QS-controlled virulence factors have a role in protecting bacteria against the immune system and that disruption of QS systems leads to the accelerated death of the bacterial pathogens (31–35).
There persists in the literature the misperception that some early QS work demonstrated resistance to QQ compounds. For example, there is an excellent paper by Koch et al. (36) based on a lock-and-key relationship between the receptor and autoinducers that identified substitutions in LuxR (L42A, a point mutation in the LuxR signal biding site) that altered both the binding of the natural ligand 3-oxo-C8-homoserine lactone and that of QQ compounds. However, resistance to QQ compounds was not investigated (36), as has been suggested (6). Koch et al. did not check the substituted LuxR protein in the original Vibrio host but instead did their work in E. coli, so there were no studies of resistance to a QS system (36). Also, far from conducting experiments on resistance and deducing that resistance is possible, the authors concluded the opposite, that resistance to QQ compounds was not likely, as they wrote the following:
Although there is no selective pressure imposed by the inhibitors per se, it is conceivable that pathogenic bacteria in the long run might develop resistance to quorum-sensing inhibitors that are based on agonist structure. In contrast, our furanone analysis suggests that through time inhibitors have been selected in nature where single amino acid changes in a separated receptor site leading to resistance are less likely to occur (36).
Similarly, Zhu et al. (37) studied the ability of AHL analogs to disrupt 3-oxo-C8-HSL signaling via TraR in Agrobacterium tumefaciens by investigating the ability of these compounds to activate expression of a TraR-regulated promoter. Although claimed otherwise (6), resistance to these compounds was not explored, since growth in the presence of these QQ compounds was not studied. Instead, the intent of the authors was to determine if differences in TraR levels affect the ability of A. tumefaciens to detect analogs of 3-oxo-C8-HSL, and “resistance” is not mentioned in the paper, nor was it explored.
Although distinct from demonstrating the development of resistance to QQ compounds, it has also been demonstrated that QQ compounds can select for a more virulent population by reducing the growth advantage of cells that are already deficient in QS relative to that of the wild-type strain. Kohler et al. (38) showed in a hospital setting and in the lab that the administration of azithromycin in cases with P. aeruginosa infection led to an enrichment of the more virulent wild-type strain relative to lasR strains.
Bacteriophages may also play a role in enhancing resistance to QQ compounds. For example, since QS in E. coli protects cells against λ phage attack (39), in the presence of bacteriophages and a QQ compound, QQ-resistant bacteria would have a competitive advantage relative to QQ-sensitive individuals, since the QQ-resistant bacteria would have an active QS system that would make them less susceptible to phage attack. Therefore, bacteriophages may select for QQ-resistant clones.
Resistance to QS inhibition.
The first demonstration that cells evolve resistance to QQ techniques was that of Maeda et al. (40) (published ahead of print in 2011). The opportunistic pathogen P. aeruginosa was used as the reference bacterium since it is notorious for causing severe infections and since it is one of the main QS bacterial model systems. A novel screen was developed to test if cells could evolve resistance to a QQ compound by using adenosine as the sole carbon source; growth on adenosine requires an active LasI/LasR N-3-oxododecanoyl homoserine lactone QS system, since the expression of the nucleoside hydrolase (nuh) gene is under its control. Hence, if QQ compounds inhibit the LasI/LasR system, the cells grow more slowly on adenosine (40), and if cells evolve resistance to the QQ compound, they will grow more rapidly on adenosine. In addition, the finding that adenosine inhibits the biofilm formation of P. aeruginosa (41) is theorized to be linked to QS to prevent cheating (42), and adenosine is produced from ATP at high levels in the human host (up to 5 mM) during surgical injury, ischemia, and inflammation, so it is a relevant carbon source for this pathogen and one that affects its physiology significantly. The gold standard of QQ compounds, the synthetic brominated furanone 4-bromo-5-(bromomethylene)-2(5H)-furanone, known as C-30 (43), which was derived from the natural brominated furanone (5Z)-4-bromo-5-(bromomethylene)-3-butyl-2(5H)-furanone of the algae Delisea pulchra, was used since it is by far the best-characterized QQ compound. For example, this family of compounds inhibits all three QS systems of Vibrio harveyi (11). Maeda et al. (40) used a concentration of brominated furanone (C-30) that did not affect growth in rich medium (so it did not inhibit growth as a toxin) and used both transposon mutagenesis and spontaneous mutants to identify resistant bacteria. The mechanism for this resistance in the transposon mutants was that the bacteria developed mexR and nalC mutations (40); these genes encode repressors of the MexAB-OprM multidrug resistance operon, so as a result of the mutations, the QQ compound was more readily effluxed (a result that was not anticipated). C-30 had a diminished ability to reduce several QS-controlled virulence factors and phenotypes in the mexR mutant, and the pathogenicity of the mexR mutant against the nematode Caenorhabditis elegans was not attenuated by the addition of C-30 (40), consistent with the resistance to C-30 of the mexR mutant during growth on adenosine. Critically, this group also used cells from cystic fibrosis patients (Liverpool epidemic strain 12142) with mexR and nalC mutations to show that even in the absence of the QS inhibitor, cells naturally evolve resistance to QQ compounds in the pathogenic state when confronted with the pressures of antibiotic treatment; hence, antibiotic treatment can lead to resistance to QQ compounds. In contrast to the transposon mutants, the spontaneous mutants isolated by Maeda et al. (40) had intact mexR and nalC genes, indicating that resistance can also rise by other uncharacterized mechanisms. Therefore, the authors showed that cells develop resistance to QQ compounds through different mechanisms and that these mutations actually occur in a clinical setting. The fact that the mutations arise in a clinical setting demonstrates that it does not matter whether growth depends on “public” or “private” goods; the crux is that cells were shown definitely to evolve resistance to QQ compounds even in the absence of previous exposure to them.
It may be argued that the Maeda et al. study (40) was predicated on using the QQ compound (C-30) under conditions in which it inhibited growth (growth on adenosine requires an active QS system). However, this situation of QQ affecting growth is common, since it has been shown that another well-publicized QQ compound, LED229, which inhibits QseC-based signaling in enterohemorrhagic E. coli (44), also affects growth (although claimed otherwise), since deletions in qseC results in numerous metabolic changes (9). Also, since QS often involves hundreds of genes (45, 46), it is reasonable to speculate that inhibiting QS outside laboratory conditions (i.e., growth in nonrich medium) may influence growth (9).
Additional clinical evidence of the ability of strains to evolve resistance to QQ compounds was provided by studying the resistance of Mexican clinical isolates from urine, blood, and catheter tip specimens from children to brominated furanone (C-30) and to 5-fluorouracil (5-FU) (47). From a screen of P. aeruginosa biofilm mutants, uracil was determined to act as a positive signal for biofilm formation, and 5-FU was shown to be effective in inhibiting this signaling, thereby repressing biofilm formation, significantly reducing QS phenotypes (10 μM 5-FU reduced elastase activity by 86%, eliminated pyocyanin production, reduced rhamnolipid production by 87%, eliminated swarming, and eliminated Pseudomonas quinolone signal production), and reducing pathogenicity (5-FU increased barley germination) (48). This reduction of P. aeruginosa pathogenicity by 5-FU was rediscovered by Imperi et al. (49) 4 years later, when they demonstrated that 5-fluorocytosine, which they showed is converted to 5-FU for its activity, also reduces pyoverdine, PrpL protease, and exotoxin in P. aeruginosa. 5-FU has also been used successfully in human trials as a coating for catheters (50), making it the first QQ compound to be used in medicine and the first QQ compound to have undergone large-scale human trials.
To identify strains resistant to 5-FU, García-Contreras et al. (47) assayed pyocyanin, elastase, and alkaline protease production of eight clinical strains and found two strains to be resistant to the brominated furanone C-30. One of the resistant strains was not sensitive to antibiotics, indicating that the C-30 resistance mechanism of this strain is likely not related to active efflux. Also, some clinical isolates showed resistance for at least one phenotype with 5-FU (47).
Subsequent to the first demonstration of resistance to QS compounds by Maeda et al. using both realistic lab constructs and clinical strains (40), Mellbye and Schuster (51) published a hypothesis/opinion report in which QS mimic approaches were used rather than realistic ones and in which no QS inhibitor was utilized. They utilized a P. aeruginosa lasR rhlR strain as a mimic of a QQ-sensitive strain and the wild-type strain as a QQ-resistant mimic. In this artificial system, they determined that cells resistant to QQ compounds should not have a growth advantage when public goods are utilized (i.e., when nutrients are processed extracellularly by QS-related enzymes) and that cells resistant to QQ compounds should have a growth advantage when nonpublic goods are utilized (i.e., when nutrients are processed intracellularly by QS enzymes) (51). Hence, their results using QS mimics corroborated the results of Maeda et al. (40) for their laboratory strains grown with adenosine as the intracellular nutrient. With regard to the more complex case of growth in the lungs of cystic fibrosis patients and the QQ-resistant mutants that were isolated from this real environment by Maeda et al. (40), the relevance of the Mellbye and Schuster study is not clear. Also, the result that the QQ-resistant mutations that were identified by Maeda et al. (40) had enhanced efflux rather than the predicted changes in QS receptors (6) shows that resistance may arise in ways not necessarily related to changes in QS receptors.
In addition, moderate resistance to the nonbiocidal antibiofilm group 2 capsule polysaccharide (G2cps), which works by a still-unknown mechanism in E. coli, can be achieved by creating mutations in several loci that affect the surface properties of the bacteria (52). This work confirms the idea that resistance to compounds that do not impair growth is possible, although multiple mutations were required in this case, and so it was reasoned that such resistance would be rare.
The above-discussed articles (2, 40, 47) are pioneering and open a whole new emergent research area, that of QQ resistance. In addition, the results shown (40, 47) may be significant for clinics since they indicate that the treatment of multiple-antibiotic-resistant strains with active efflux pumps with HSL analogues, such as C-30, may be futile and suggest that since there is a common resistance mechanism between antibiotics and QQ compounds, treatment with HSL analogues alone may select for multiple-antibiotic resistance as well. Also, it should be taken in to account that QS disruption renders bacteria more sensitive to some antimicrobials and antibiotics, like tobramycin, particularly in the biofilm mode of growth (43, 53). Therefore, for concomitant treatment of QQ and classical antibiotics, even if QQ compounds do not exert selective pressure by themselves, they will exert it indirectly by making cells more sensitive to antibiotics.
Perspectives on new QQ resistance mechanisms.
Ways of evolving resistance to QQ compounds other than active efflux should exist, as suggested previously (40). This is to be expected, since resistance to classical antibiotics can be achieved in many ways, such as by decreasing the permeability of the compounds, mutating the target, overexpressing antibiotic targets, and degrading/modifying the antibiotics. Along these lines, Maeda et al. found that C-30 can be degraded by PA14 (unpublished results), and they are currently investigating if this ability is enhanced in some C-30-resistant clinical isolates.
Further work is also required to determine if resistance to other kinds of quorum quenchers, such as signal-degrading enzymes, like lactonases or acylases for HSL autoinducers, is possible. Hence, it is important to distinguish those QQ compounds that must enter the cell to be effective (e.g., brominated furanones) from QQ compounds that work extracellularly (e.g., lactonases), since there may be less pressure to evolve resistance to extracellular compounds because greater efflux should not affect the use of these compounds (54). Although, to our knowledge, no experimental efforts have been devoted to explore this possibility, it can be anticipated that ways in which bacteria develop resistance to these agents may be to (i) increase autoinducer production, (ii) synthesize modified autoinducers (which are less susceptible to the attack of the degrading enzymes), or (iii) evolve mutations in the LuxR-like receptors that increase their affinity to the autoinducers (so that the necessary threshold of autoinducer concentration will decrease). Examples of the first two possibilities (an increase in autoinducer production and the presence of different variants of autoinducers) have already been reviewed (2), and for the third possibility, it has been demonstrated that some mutations in Vibrio fischeri LuxR, which normally recognizes the 3-oxo-C6-HSL signal, make it able to respond to different autoinducers, like octanoyl-HSL, pentanoyl-HSL, and tetradecanoyl-HSL, and moreover, some subset of these mutations also increases their sensitivity to the endogenous signal (55).
The choice to inhibit QS as a means of inhibiting pathogens (6) is also a questionable goal, since it violates one of the main postulates of preventing resistance, namely, that it is far better to make antivirulence drugs that are specific rather than to target general agents (56). Since QS often involves hundreds of gene targets (45, 46), bacteria may use multiple means of thwarting this approach. Additional complications for this approach are that since QS is used by many bacteria, beneficial microorganisms may also be affected by any general approach (9, 57), for example, in the gut, where hundreds of different species reside. Complicating matters further in mixed cultures is the fact that some pathogenic genes are activated by QS (e.g., P. aeruginosa) (46), while others are inactivated (e.g., Vibrio cholerae) (58); hence, QQ approaches may have unintended consequences in communities with many bacteria.
Conclusions.
As outlined here, bacteria have been shown to evolve resistance to QQ compounds both in lab studies and in clinics and to evolve resistance to QQ compounds even without their use (i.e., when bacteria are confronted with antibiotics and mutation in the efflux pump occurs); hence, we should be less sanguine about the possibilities that these novel QQ compounds are as robust as has been frequently indicated in the current literature (6). One actual mechanism of QQ resistance involving enhanced efflux (40) is shown in Fig. 1A, whereas Fig. 1B shows the predicted mechanism of QQ resistance of LasR receptor insensitivity based on the lock-and-key relationship through the amino acid change L42A, which led to an inability of the autoinducer to bind. Hopefully, even with resistance arising, QQ compounds may be used in combination with other antimicrobials. However, the exaggerated claims by many authors about the benefits of these compounds should be tempered.
Fig 1.
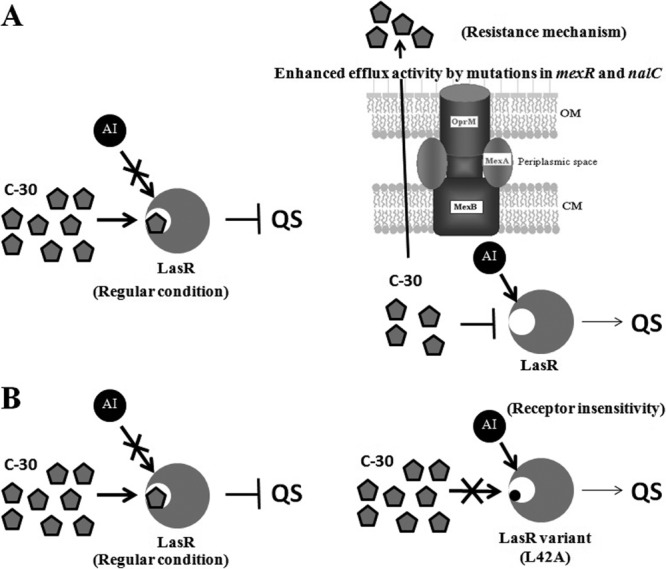
Mechanism of actual and predicted inhibition to QQ compounds. (A) Actual QQ resistance in P. aeruginosa based on enhanced efflux of the furanone C-30 due to mutations in the genes that encode the efflux repressors MexR and NalC (40); (B) predicted LasR receptor insensitivity to C-30 based on the lock-and-key concept (36). AI, autoinducer; OM, outer membrane; CM, cytosolic membrane.
ACKNOWLEDGMENTS
This work was supported by the NIH (grant R01 GM089999). T.K.W. is the Biotechnology Endowed Professor at the Pennsylvania State University. R.G.-C. was supported by SEP/CONACyT grant 152794 and by ICyTDF project PICSA 11-78. T.M. was supported by a Grant-in-Aid for Challenging Exploratory Research (25660062) of the Japan Society for the Promotion of Science.
Biographies

Rodolfo García-Contreras is an associate professor at the National Institute of Cardiology at Mexico City (since 2010). In 2005, he obtained his Ph.D. in Biomedical Sciences at the Faculty of Medicine UNAM in Mexico, where he studied the bioenergetics of photosynthetic bacteria. He completed two postdoctoral positions, the first at the Department of Chemical Engineering at Texas A&M University in the group of Prof. Thomas K. Wood, working on the genetic basis of biofilm formation in Escherichia coli, and the second at the Molecular Cell Physiology Department at the VU University of Amsterdam in the group of Fred Boogerd and Hans Westerhoff, working on E. coli central carbon and nitrogen metabolism from a systems biology perspective. Currently, his research is centered on the study of the resistance mechanisms of Pseudomonas aeruginosa against antivirulence compounds and novel antimicrobials using laboratory and clinical strains.

Toshinari Maeda is currently an associate professor of biological functions and engineering at the Kyushu Institute of Technology. He obtained his Ph.D. in biological engineering at the Graduate School of Life Science and Systems Engineering at the Kyushu Institute of Technology in March 2006 by studying the microbial degradation of 2,4,6-trinitrotoluene. He then worked mainly on bacterial hydrogen production in Escherichia coli as a postdoctoral researcher in the laboratory of Prof. Thomas K. Wood in the Department of Chemical Engineering at Texas A&M University (from April 2006 to September 2007). He obtained the position of assistant professor at his current institution in October 2007 and was promoted to associate professor in October 2011. His research focuses on microbial biotechnology with an emphasis on bioenergy production (hydrogen and methane), recycling and reduction of waste sludge, and the environmental adaptation of Pseudomonas aeruginosa and Bdellovibrio bacteriovorus.

Thomas K. Wood is the Biotechnology Endowed Chair and Professor of Chemical Engineering and Biochemistry and Molecular Biology at the Pennsylvania State University. He formerly held endowed chair positions at Texas A&M and at the University of Connecticut and started his academic career at the University of California, Irvine, in 1991. He obtained his Ph.D. in chemical engineering from North Carolina State University in 1991 by studying heterologous protein production and obtained his B.S. from the University of Kentucky in 1985. His current research pursuits include using systems biology approaches to understand cell persistence and determining the role of toxin-antitoxin systems in cells. He also explores the genetic basis of biofilm formation to prevent disease and to utilize biofilms for beneficial biotransformations including remediation, green chemistry, and energy production. His group has also utilized protein engineering to control biofilm formation and for bioremediation, green chemistry, and biofuels.
Footnotes
Published ahead of print 6 September 2013
REFERENCES
- 1.Rasko DA, Sperandio V. 2010. Anti-virulence strategies to combat bacteria-mediated disease. Nat. Rev. Drug Discov. 9:117–128 [DOI] [PubMed] [Google Scholar]
- 2.Defoirdt T, Boon N, Bossier P. 2010. Can bacteria evolve resistance to quorum sensing disruption? PLoS Pathog. 6:e1000989. 10.1371/journal.ppat.1000989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jayaraman A, Wood TK. 2008. Bacterial quorum sensing: signals, circuits, and implications for biofilms and disease. Annu. Rev. Biomed. Eng. 10:145–167 [DOI] [PubMed] [Google Scholar]
- 4.Nazzaro F, Fratianni F, Coppola R. 2013. Quorum sensing and phytochemicals. Int. J. Mol. Sci. 14:12607–12619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bjarnsholt T, Tolker-Nielsen T, Høiby N, Givskov M. 2010. Interference of Pseudomonas aeruginosa signalling and biofilm formation for infection control. Expert Rev. Mol. Med. 12:e11. 10.1017/S1462399410001420 [DOI] [PubMed] [Google Scholar]
- 6.Schuster M, Sexton DJ, Diggle SP, Greenberg EP. 2013. Acyl-homoserine lactone quorum sensing: from evolution to application. Annu. Rev. Microbiol. 67:43–63 [DOI] [PubMed] [Google Scholar]
- 7.Boyle KE, Heilmann S, van Ditmarsch D, Xavier JB. 2013. Exploiting social evolution in biofilms. Curr. Opin. Microbiol. 16:207–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ng WL, Perez L, Cong J, Semmelhack MF, Bassler BL. 2012. Broad spectrum pro-quorum-sensing molecules as inhibitors of virulence in vibrios. PLoS Pathog. 8:e1002767. 10.1371/journal.ppat.1002767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.LaSarre B, Federle MJ. 2013. Exploiting quorum sensing to confuse bacterial pathogens. Microbiol. Mol. Biol. Rev. 77:73–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lesic B, Lépine F, Déziel E, Zhang J, Zhang Q, Padfield K, Castonguay MH, Milot S, Stachel S, Tzika AA, Tompkins RG, Rahme LG. 2007. Inhibitors of pathogen intercellular signals as selective anti-infective compounds. PLoS Pathog. 3:e126. 10.1371/journal.ppat.0030126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Defoirdt T, Miyamoto CM, Wood TK, Meighen EA, Sorgeloos P, Verstraete W, Bossier P. 2007. The natural furanone (5Z)-4-bromo-5-(bromomethylene)-3-butyl-2(5H)-furanone disrupts quorum sensing-regulated gene expression in Vibrio harveyi by decreasing the DNA-binding activity of the transcriptional regulator protein luxR. Environ. Microbiol. 9:2486–2495 [DOI] [PubMed] [Google Scholar]
- 12.Skindersoe ME, Alhede M, Phipps R, Yang L, Jensen PØ, Rasmussen TB, Bjarnsholt T, Tolker-Nielsen T, Høiby N, Givskov M. 2008. Effects of antibiotics on quorum sensing in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 52:3648–3663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Imperi F, Massai F, Ramachandran Pillai C, Longo F, Zennaro E, Rampioni G, Visca P, Leoni L. 2013. New life for an old drug: the anthelmintic drug niclosamide inhibits Pseudomonas aeruginosa quorum sensing. Antimicrob. Agents Chemother. 57:996–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Singh VK, Kavita K, Prabhakaran R, Jha B. 2013. Cis-9-octadecenoic acid from the rhizospheric bacterium Stenotrophomonas maltophilia BJ01 shows quorum quenching and anti-biofilm activities. Biofouling 29:855–867 [DOI] [PubMed] [Google Scholar]
- 15.Musthafa KS, Balamurugan K, Pandian SK, Ravi AV. 2012. 2,5-Piperazinedione inhibits quorum sensing-dependent factor production in Pseudomonas aeruginosa PAO1. J. Basic Microbiol. 52:679–686 [DOI] [PubMed] [Google Scholar]
- 16.Golberg K, Pavlov V, Marks RS, Kushmaro A. 2013. Coral-associated bacteria, quorum sensing disrupters, and the regulation of biofouling. Biofouling 29:669–682 [DOI] [PubMed] [Google Scholar]
- 17.Dockery JD, Keener JP. 2001. A mathematical model for quorum sensing in Pseudomonas aeruginosa. Bull. Math. Biol. 63:95–116 [DOI] [PubMed] [Google Scholar]
- 18.Anguige K, King JR, Ward JP. 2006. A multi-phase mathematical model of quorum sensing in a maturing Pseudomonas aeruginosa biofilm. Math. Biosci. 203:240–276 [DOI] [PubMed] [Google Scholar]
- 19.Janakiraman V, Englert D, Jayaraman A, Baskaran H. 2009. Modeling growth and quorum sensing in biofilms grown in microfluidic chambers. Ann. Biomed. Eng. 37:1206–1216 [DOI] [PubMed] [Google Scholar]
- 20.Anguige K, King JR, Ward JP. 2005. Modelling antibiotic- and anti-quorum sensing treatment of a spatially-structured Pseudomonas aeruginosa population. J. Math. Biol. 51:557–594 [DOI] [PubMed] [Google Scholar]
- 21.Sadovskaya I, Vinogradov E, Li J, Hachani A, Kowalska K, Filloux A. 2010. High-level antibiotic resistance in Pseudomonas aeruginosa biofilm: the ndvB gene is involved in the production of highly glycerol-phosphorylated beta-(1→3)-glucans, which bind aminoglycosides. Glycobiology 20:895–904 [DOI] [PubMed] [Google Scholar]
- 22.Fozard JA, Lees M, King JR, Logan BS. 2012. Inhibition of quorum sensing in a computational biofilm simulation. Biosystems 109:105–114 [DOI] [PubMed] [Google Scholar]
- 23.Beckmann BE, Knoester DB, Connelly BD, Waters CM, McKinley PK. 2012. Evolution of resistance to quorum quenching in digital organisms. Artif. Life 18:291–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ahumedo M, Díaz A, Vivas-Reyes R. 2010. Theoretical and structural analysis of the active site of the transcriptional regulators LasR and TraR, using molecular docking methodology for identifying potential analogues of acyl homoserine lactones (AHLs) with anti-quorum sensing activity. Eur. J. Med. Chem. 45:608–615 [DOI] [PubMed] [Google Scholar]
- 25.Annapoorani A, Umamageswaran V, Parameswari R, Pandian SK, Ravi AV. 2012. Computational discovery of putative quorum sensing inhibitors against LasR and RhlR receptor proteins of Pseudomonas aeruginosa. J. Comput. Aided Mol. Des. 26:1067–1077 [DOI] [PubMed] [Google Scholar]
- 26.Skovstrup S, Le Quement ST, Hansen T, Jakobsen TH, Harmsen M, Tolker-Nielsen T, Nielsen TE, Givskov M, Taboureau O. 2013. Identification of LasR ligands through a virtual screening approach. ChemMedChem 8:157–163 [DOI] [PubMed] [Google Scholar]
- 27.Ravichandiran V, Shanmugam K, Solomon AP. 2013. Screening of SdiA inhibitors from Melia dubia seed extracts towards the hold back of uropathogenic E. coli quorum sensing-regulated factors. Med. Chem. 9:819–827 [DOI] [PubMed] [Google Scholar]
- 28.Musthafa KS, Sahu SK, Ravi AV, Kathiresan K. 17 April 2013. Anti-quorum sensing potential of the mangrove Rhizophora annamalayana. World J. Microbiol. Biotechnol. 10.1007/s11274-013-1347-8 [DOI] [PubMed] [Google Scholar]
- 29.Li L, Sun L, Song Y, Wu X, Zhou X, Liu Z, Zhou R. 17 June 2013. Screening of Actinobacillus pleuropneumoniae LuxS Inhibitors. Curr. Microbiol. 10.1007/s00284-013-0403-9 [DOI] [PubMed] [Google Scholar]
- 30.Al-Sohaibani S, Murugan K. 2012. Anti-biofilm activity of Salvadora persica on cariogenic isolates of Streptococcus mutans: in vitro and molecular docking studies. Biofouling 28:29–38 [DOI] [PubMed] [Google Scholar]
- 31.Telford G, Wheeler D, Williams P, Tomkins PT, Appleby P, Sewell H, Stewart GS, Bycroft BW, Pritchard DI. 1998. The Pseudomonas aeruginosa quorum-sensing signal molecule N-(3-oxododecanoyl)-l-homoserine lactone has immunomodulatory activity. Infect. Immun. 66:36–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bjarnsholt T, Jensen PØ, Burmølle M, Hentzer M, Haagensen JA, Hougen HP, Calum H, Madsen KG, Moser C, Molin S, Høiby N, Givskov M. 2005. Pseudomonas aeruginosa tolerance to tobramycin, hydrogen peroxide and polymorphonuclear leukocytes is quorum-sensing dependent. Microbiology 151:373–383 [DOI] [PubMed] [Google Scholar]
- 33.Kuang Z, Hao Y, Walling BE, Jeffries JL, Ohman DE, Lau GW. 2011. Pseudomonas aeruginosa elastase provides an escape from phagocytosis by degrading the pulmonary surfactant protein-A. PLoS One 6:e27091. 10.1371/journal.pone.0027091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dössel J, Meyer-Hoffert U, Schröder JM, Gerstel U. 2012. Pseudomonas aeruginosa-derived rhamnolipids subvert the host innate immune response through manipulation of the human beta-defensin-2 expression. Cell. Microbiol. 14:1364–1375 [DOI] [PubMed] [Google Scholar]
- 35.Leid JG, Willson CJ, Shirtliff ME, Hassett DJ, Parsek MR, Jeffers AK. 2005. The exopolysaccharide alginate protects Pseudomonas aeruginosa biofilm bacteria from IFN-gamma-mediated macrophage killing. J. Immunol. 175:7512–7518 [DOI] [PubMed] [Google Scholar]
- 36.Koch B, Liljefors T, Persson T, Nielsen J, Kjelleberg S, Givskov M. 2005. The LuxR receptor: the sites of interaction with quorum-sensing signals and inhibitors. Microbiology 151:3589–3602 [DOI] [PubMed] [Google Scholar]
- 37.Zhu J, Beaber JW, Moré MI, Fuqua C, Eberhard A, Winans SC. 1998. Analogs of the autoinducer 3-oxooctanoyl-homoserine lactone strongly inhibit activity of the TraR protein of Agrobacterium tumefaciens. J. Bacteriol. 180:5398–5405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kohler T, Perron GG, Buckling A, van Delden C. 2010. Quorum sensing inhibition selects for virulence and cooperation in Pseudomonas aeruginosa. PLoS Pathog. 6:e1000883. 10.1371/journal.ppat.1000883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Høyland-Kroghsbo NM, Maerkedahl RB, Svenningsen SL. 2013. A quorum-sensing-induced bacteriophage defense mechanism. mBio 4(1):e00362-12. 10.1128/mBio.00362-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Maeda T, García-Contreras R, Pu M, Sheng L, Garcia LR, Tomás M, Wood TK. 2012. Quorum quenching quandary: resistance to antivirulence compounds. ISME J. 6:493–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sheng L, Pu M, Hegde M, Zhang Y, Jayaraman A, Wood TK. 2012. Interkingdom adenosine signal reduces Pseudomonas aeruginosa pathogenicity. Microb. Biotechnol. 5:560–572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dandekar AA, Chugani S, Greenberg EP. 2012. Bacterial quorum sensing and metabolic incentives to cooperate. Science 338:264–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hentzer M, Wu H, Andersen JB, Riedel K, Rasmussen TB, Bagge N, Kumar N, Schembri MA, Song Z, Kristoffersen P, Manefield M, Costerton JW, Molin S, Eberl L, Steinberg P, Kjelleberg S, Høiby N, Givskov M. 2003. Attenuation of Pseudomonas aeruginosa virulence by quorum sensing inhibitors. EMBO J. 22:3803–3815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rasko DA, Moreira CG, Li DR, Reading NC, Ritchie JM, Waldor MK, Williams N, Taussig R, Wei S, Roth M, Hughes DT, Huntley JF, Fina MW, Falck JR, Sperandio V. 2008. Targeting QseC signaling and virulence for antibiotic development. Science 321:1078–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schuster M, Lostroh CP, Ogi T, Greenberg EP. 2003. Identification, timing, and signal specificity of Pseudomonas aeruginosa quorum-controlled genes: a transcriptome analysis. J. Bacteriol. 185:2066–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wagner VE, Bushnell D, Passador L, Brooks AI, Iglewski BH. 2003. Microarray analysis of Pseudomonas aeruginosa quorum-sensing regulons: effects of growth phase and environment. J. Bacteriol. 185:2080–2095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.García-Contreras R, Martínez-Vázquez M, Velázquez Guadarrama N, Villegas Paneda AG, Hashimoto T, Maeda T, Quezada H, Wood TK. 2013. Resistance to the quorum-quenching compounds brominated furanone C-30 and 5-fluorouracil in Pseudomonas aeruginosa clinical isolates. Pathog. Dis. 68:8–11 [DOI] [PubMed] [Google Scholar]
- 48.Ueda A, Attila C, Whiteley M, Wood TK. 2009. Uracil influences quorum sensing and biofilm formation in Pseudomonas aeruginosa and fluorouracil is an antagonist. Microb. Biotechnol. 2:62–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Imperi F, Massai F, Facchini M, Frangipani E, Visaggio D, Leoni L, Bragonzi A, Visca P. 2013. Repurposing the antimycotic drug flucytosine for suppression of Pseudomonas aeruginosa pathogenicity. Proc. Natl. Acad. Sci. U. S. A. 110:7458–7463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Walz JM, Avelar RL, Longtine KJ, Carter KL, Mermel LA, Heard SO. 2010. Anti-infective external coating of central venous catheters: a randomized, noninferiority trial comparing 5-fluorouracil with chlorhexidine/silver sulfadiazine in preventing catheter colonization. Crit. Care Med. 38:2095–2102 [DOI] [PubMed] [Google Scholar]
- 51.Mellbye B, Schuster M. 2011. The sociomicrobiology of antivirulence drug resistance: a proof of concept. mBio 2(5):e00131-11. 10.1128/mBio.00131-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Travier L, Rendueles O, Ferrières L, Herry J-M, Ghigo J-M. 2013. Escherichia coli resistance to nonbiocidal antibiofilm polysaccharides is rare and mediated by multiple mutations leading to surface physicochemical modifications. Antimicrob. Agents Chemother. 57:3960–3968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Davies DG, Parsek MR, Pearson JP, Iglewski BH, Costerton JW, Greenberg EP. 1998. The involvement of cell-to-cell signals in the development of a bacterial biofilm. Science 280:295–298 [DOI] [PubMed] [Google Scholar]
- 54.Defoirdt T. 2013. Antivirulence therapy for animal production: filling an arsenal with novel weapons for sustainable disease control. PLoS Pathog. 10.1371/journal.ppat.1003603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Collins CH, Arnold FH, Leadbetter JR. 2005. Directed evolution of Vibrio fischeri LuxR for increased sensitivity to a broad spectrum of acyl-homoserine lactones. Mol. Microbiol. 55:712–723 [DOI] [PubMed] [Google Scholar]
- 56.Greene SE, Reid A. 2013. Moving targets: fighting resistance in infections, cancers, pests. Microbe 8:279–285 [Google Scholar]
- 57.Hong K-W, Koh C-L, Sam C-K, Yin W-F, Chan K-G. 2012. Quorum quenching revisited—from signal decays to signalling confusion. Sensors 12:4661–4696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ng W-L, Perez L, Cong J, Semmelhack MF, Bassler BL. 2012. Broad spectrum pro-quorum-sensing molecules as inhibitors of virulence in vibrios. PLoS Pathog. 8:e1002767. 10.1371/journal.ppat.1002767 [DOI] [PMC free article] [PubMed] [Google Scholar]