

Abstract
Streptomyces species dedicate a large portion of their genomes to secondary metabolite biosynthesis. A diverse and largely marine-derived lineage within this genus has been designated MAR4 and identified as a prolific source of hybrid isoprenoid (HI) secondary metabolites. These terpenoid-containing compounds are common in nature but rarely observed as bacterial secondary metabolites. To assess the phylogenetic diversity of the MAR4 lineage, complementary culture-based and culture-independent techniques were applied to marine sediment samples collected off the Channel Islands, CA. The results, including those from an analysis of publically available sequence data and strains isolated as part of prior studies, placed 40 new strains in the MAR4 clade, of which 32 originated from marine sources. When combined with sequences cloned from environmental DNA, 28 MAR4 operational taxonomic units (0.01% genetic distance) were identified. Of these, 82% consisted exclusively of either cloned sequences or cultured strains, supporting the complementarity of these two approaches. Chemical analyses of diverse MAR4 strains revealed the production of five different HI structure classes. All 21 MAR4 strains tested produced at least one HI class, with most strains producing from two to four classes. The two major clades within the MAR4 lineage displayed distinct patterns in the structural classes and the number and amount of HIs produced, suggesting a relationship between taxonomy and secondary metabolite production. The production of HI secondary metabolites appears to be a phenotypic trait of the MAR4 lineage, which represents an emerging model with which to study the ecology and evolution of HI biosynthesis.
INTRODUCTION
The genus Streptomyces is extraordinarily diverse, with nearly 600 formally described species (http://www.ezbiocloud.net). These filamentous, Gram-positive bacteria have been heavily exploited for their ability to produce structurally diverse secondary metabolites, many of which have become useful pharmaceutical agents (1, 2). Streptomyces spp. are generally saprophytic and are known soil inhabitants, where they play important ecological roles in the breakdown of recalcitrant organic materials (3) and the suppression of plant pathogens (4). They are also widely distributed in marine sediments (5, 6), form symbiotic relationships with plants (7) and insects (8), and are observed in association with marine sponges (8). Some streptomycete lineages have been observed broadly both in soils and in marine sediments (9), suggesting a high degree of environmental flexibility.
The ecological functions of most streptomycete secondary metabolites have not been defined, and thus, potential links between secondary metabolite production and environmental adaptation remain largely unknown. One example where such links have been reported is the production of thaxtomins by the phytopathogen Streptomyces scabies. These phytotoxins are the causal agent of potato scab disease, and the transfer of the associated biosynthetic genes among related streptomycetes allows strains to emerge as new pathogens (10). Another example is the production of the antifungal agent candicidin by Streptomyces spp. involved in the tripartite mutualism with attine ants and their fungal gardens (11). Although streptomycetes have been observed broadly in ocean sediments, we are aware of no evidence linking the production of a specific class of secondary metabolites to the survival of Streptomyces spp. in the marine environment.
The MAR4 streptomycete lineage was first recognized as part of a survey of cultured marine actinobacteria (12). Prior to the present study, the S. aculeolatus and S. synnematoformans type strains were the only nonmarine strains recognized within this clade (13). These strains were derived from a terrestrial soil sample and a sand dune sample, respectively, and are the only formally described species within the clade.
The MAR4 lineage has been linked to the production of hybrid isoprenoid (HI) secondary metabolites (13), which are of mixed biosynthetic origin and include an isoprene-derived moiety. The attachment of isoprene, the building block of terpenes, to a secondary metabolite can occur on a variety of biosynthetically distinct chemical scaffolds, including phenazines, polyketides, peptides, aminocoumarins, phenylpropanoids, and alkaloids (14). While terpenoids are relatively common plant secondary metabolites (15), they are considered a rare part of actinomycete secondary metabolism (16). Nonetheless, actinomycete-derived HIs, such as the commercially important aminocoumarin antibiotics (17), can display potent biological activities and thus are relevant targets for drug discovery efforts.
The major goals of this study are to expand our understanding of the diversity and distributions of actinomycetes in the MAR4 lineage and to test for correlations between MAR4 phylogeny and HI production. To assess the diversity within this group, complementary culture-based and culture-independent methods were applied to marine sediment samples collected off the Channel Islands, CA. In addition, strains previously identified as MAR4 candidates on the basis of prior research or derived from publically available sequence data were also analyzed.
MATERIALS AND METHODS
Sample collection.
Two hundred thirty-two near-shore sediment samples were collected from five sites (34°02.67′N, 119°32.67′W; 33°59.65′N, 119°33.39′W; 34°03.08′N, 119°34.51′W; 34°01.99′N, 119°42.14′W; 34°03.29′N, 119°49.16′W) surrounding Santa Cruz Island, CA, from 20 to 22 September 2010. Surface sediment samples from water depths of 8 to 12 m were collected in sterile 4-oz Whirl-Pak bags (Nasco, Modesto, CA) by divers. Two cores were collected at one site (34°01.99′N, 119°42.14′W) using a handheld coring device. These were cut into 1- to 3-cm sections immediately after collection and stored frozen. A subset of the samples from each site was processed for actinomycete cultivation immediately after collection. All remaining samples were stored at −20°C before further processing.
Sixty-eight additional surface sediment samples were collected from 53 sites along the coast between Ventura and Long Beach, CA, and surrounding Santa Cruz Island. These samples were collected at depths from 5 to 833 m using a modified surface-deployed sampler (model 214WA110; Kahlsico, El Cajon, CA) on 4 to 5 December 2010. Samples were stored on ice for at most 3 days before processing and storage at −20°C.
Strain isolation.
All samples were processed using previously described methods, including treatments that select for spore-forming actinomycetes (18–20). The 232 samples collected in September 2010 were processed using up to four methods. These included serial dilution, heat shock, drying, and enrichment culturing (20), followed by plating on agar media. The 68 samples collected in December 2010 were processed by serial dilution and plating. All samples were inoculated onto the following media prepared with 18 g agar: A1 (10 g soluble starch, 4 g yeast extract, 2 g peptone, 1 liter seawater), 50% A1, 20% A1, B1 (2.5 g peptone, 1.5 g yeast extract, 1.5 ml glycerol, 1 liter seawater), Difco marine broth 2215 (Becton, Dickinson and Company, Sparks, MD), and SWA (1 liter seawater, 1 ml trace mineral solution [21]). All plates contained 100 μg/ml cycloheximide to reduce fungal growth.
Serial dilutions (1:50, 1:1,000, and 1:10,000) were made in sterile seawater. Heat shock was performed on samples diluted 1:4 in sterile seawater, followed by heating at 55°C in a water bath for 6 to 9 min. Twenty-five microliters of each dilution was inoculated onto agar medium and spread with a sterile glass rod. For the drying treatment, sediment samples were air dried in a laminar flow hood for at least 24 h and stamped onto agar medium using a sterile foam plug. The enrichment cultures consisted of 1.5 ml of sediment in a 15-ml test tube containing 10 ml of either sterile seawater and 1% chitin or 20% A1 supplemented with either polymyxin B sulfate (5 μg/ml), novobiocin (25 μg/ml), kanamycin (20 μg/ml), or no antibiotic. The tubes were incubated without shaking at room temperature for 100 days. The supernatant was then serially diluted as described above, and 25 μl was plated prior to treatment and following heat shock treatment. The underlying sediment was dried and stamped as described above.
All plates were incubated for up to 90 days at room temperature following inoculation. Actinomycete colonies were identified by colony morphology, texture, and spore formation and transferred with sterile toothpicks onto medium A1. This process was repeated until pure cultures were obtained, as evidenced by uniform morphology. Pure strains were cultured in liquid A1 and stored at −80°C with 10% glycerol added as a cryoprotectant.
DNA extraction, PCR amplification, and sequencing.
Genomic DNA was isolated using the DNeasy protocol (Qiagen Inc., Valencia, CA) with previously described modifications (19) and stored at −20°C. The 16S rRNA gene was PCR amplified using primers FC27 and RC1492 (see Table S2 in the supplemental material). Each 20-μl PCR mixture contained 20 to 50 ng genomic DNA, 200 μM (each) the forward and reverse primers, 2.5 mM MgCl2, PCR buffer (Applied Biosystems), 2 U AmpliTaq Gold DNA polymerase (Applied Biosystems), 400 μM deoxynucleoside triphosphate mixture, and 7% dimethyl sulfoxide. The PCR protocol consisted of 12.5 min at 94°C and 35 cycles of 1 min at 94°C, 1 min at 63°C, and 1 min at 72°C, followed by 7 min at 72°C. The PCR products were purified using a Zymo Clean and Concentrator kit (Zymo Research) according to the manufacturer's protocol and sequenced by Sanger sequencing (SeqXcel, Inc., San Diego, CA).
eDNA extraction and PCR.
Environmental DNA (eDNA) was extracted and purified from frozen sediment samples according to the PowerSoil DNA isolation protocol (MoBio, Carlsbad, CA), with the bead-beating step extended to 1 h. Purified DNA was stored at −20°C. Two seminested PCR protocols were used to specifically amplify MAR4 16S rRNA genes from eDNA (see Tables S2 and S3 in the supplemental material). The primer sets were designed on the basis of a 16S rRNA alignment of previously identified MAR4 strains and several non-MAR4 Streptomyces spp. Primer specificity was confirmed using the Probe Match program (22) (http://rdp.cme.msu.edu/probematch/search.jsp). PCRs were carried out as described above with various annealing temperatures (see Table S3 in the supplemental material). Due to the low yields of eDNA, only 5 to 10 ng was added to each 20-μl PCR mixture during the first round of amplification. PCR products from the first round were purified with the Zymo Clean and Concentrator kit (Zymo Research) and immediately used in the second round of amplification with an annealing temperature of 60°C.
Clone libraries.
Purified PCR products derived from eDNA were cloned using a TOPO TA cloning kit (Invitrogen, Carlsbad, CA) according to the manufacturer's protocol with the following modifications. The 6-μl cloning reaction mixtures contained 4 μl PCR product, 1 μl salt solution, and 1 μl TOPO vector. The entire volume was used in the transformation of competent TOP10 cells. Plasmids were purified and sequenced by Sanger sequencing using primer M13 (SeqXcel, San Diego, CA, and Beckman Coulter Genomics, Danvers, MA). Forward and reverse reads were obtained for each clone, yielding approximately 950 bp per clone. Chimeric clones were identified using the Bellerophon program (23) and removed from the analysis.
Identification of MAR4 sequences.
Phylogenetic analyses included previously identified MAR4 16S rRNA gene sequences (13), as well as strains cultured as part of the present study and strains cultured as part of prior studies (9, 12, 19) that were subsequently considered MAR4 candidates on the basis of sequence data or the production of HI secondary metabolites. Diverse MAR4 sequence representatives (accession numbers KC261629, KC261602, EF121313, KC261627, KC261626, X87316, NR041166, EF538742, KC261604, JF346436, EF581384, KC261611, and KC261615) were additionally used as BLASTn queries of the NCBI database. Redundancies were removed from the top 100 hits, and the remaining sequences were subjected to phylogenetic analysis along with the cloned sequences generated as part of the present study. 16S rRNA gene sequences were aligned using the MUSCLE program (24). Maximum likelihood phylogenies were built using raxmlGUI (25) and the GTR+G substitution model with 100 thorough bootstraps. Maximum parsimony trees were created using PAUP* (26). The tree topology and position of the previously identified MAR4 strains within the least inclusive node were used to identify the MAR4 lineage. The strains within this lineage were clustered into operational taxonomic units (OTUs) on the basis of a 16S rRNA sequence distance of 0.01 using the mothur program (27, 28) with no terminal gap penalty and an average neighbor algorithm with precision set at 100.
Secondary metabolite analysis.
Twenty-one MAR4 strains, up to five strains from each available OTU, were chosen for secondary metabolite analysis. Nine strains were cultured in duplicate to assess the reproducibility of the analyses. Each strain was cultured in 25 ml medium A1 at 27°C with shaking at 230 rpm for 4 to 7 days, after which 10 ml was inoculated into 1 liter of medium A1 in a 2.8-liter Fernbach flask. Following 3, 5, and 7 days of growth, 45 ml was extracted with an equal volume of ethyl acetate. The ethyl acetate extract was separated and dried under vacuum. On day 9, the remainder of the culture was extracted and dried.
The extracts were analyzed by liquid chromatography (LC)-mass spectrometry (MS) (Hewlett-Packard series 1100) using UV detection, a reversed-phase C18 column (4.6 by 100 mm; pore size, 5 μm; Phenomenex Luna), and a solvent gradient from 10% to 100% CH3CN in water. Low-resolution mass spectra were obtained in the positive mode (electrospray ionization [ESI] voltage, 6.0 kV; capillary temperature, 200°C; auxiliary and sheath gas pressure, 5 units and 70 lb/in2, respectively). LC traces were generated at 210 and 254 nm, and the UV absorbance spectra associated with each peak were evaluated by comparison to an in-house spectral library (see Fig. S2 in the supplemental material). Peaks were assigned to a particular compound class if they had a UV matching score of 900 or greater, as reported using the Agilent Technologies (Santa Clara, CA) ChemStation software, and a mass within the range previously reported for compounds in that class. Positive scores ranged from 903 to 999 (mean, 969.2; median, 973.5). Some peaks assigned to the napyradiomycin and marinone classes eluted with 100% CH3CN and were assigned solely on the basis of highly diagnostic UV spectra. Peaks with absorption spectra characteristic for phenazines but with masses that could not be assigned to compounds in the lavanducyanin class were analyzed by high-resolution LC-tandem MS (MS/MS; 6530 Accurate Mass quadrupole time-of-flight [Q-TOF] system; Agilent Technologies) using the column and conditions described above. High-resolution MS/MS data were obtained in the positive mode (ESI voltage, 4.5 kV; ion source and drying gas temperatures, 350°C; drying gas pressure, 11.0 liters/min; collision energy, 20 V). The total number of peaks associated with each compound class was recorded for each strain. The area under each peak was measured at 254 nm for the napyradiomycin and phenazine classes and 210 nm for the marinone, lavanducyanin, and nitropyrrolin classes. In cases where multiple peaks were detected within one compound class, the area under the peaks (mass absorbance units multiplied by time) was summed and divided by the total number of peaks, resulting in an average area per peak for each compound class.
Nucleotide sequence accession numbers.
The GenBank accession numbers generated as part of this study are KC261602 to KC261629 (cultured strains) and KC477442 to KC477620 (cloned sequences).
RESULTS
MAR4 diversity.
The 16S rRNA gene sequence diversity of the MAR4 clade was evaluated using cultured strains and cloned sequences generated from marine sediment samples collected off the Channel Islands, CA. In addition, strains cultured as part of prior studies and sequence data from public databases were also evaluated. Cultivation efforts applied to 300 sediment samples yielded 155 strains with morphological features typical of the genus Streptomyces, including the production of aerial hyphae, tough, leathery colonies, and spores. NCBI BLAST analyses of 16S rRNA gene sequences indicated that 125 of these strains were most closely related to Streptomyces spp. A maximum likelihood phylogeny placed five of these strains (CNY-714, CNY-715, CNY-716, CNY-717, and CNY-718) within a consistent and well-supported node that contained the 17 previously identified MAR4 strains (13) (see Fig. S1 in the supplemental material). These strains were obtained using a variety of media and processing methods (see Table S1 in the supplemental material). Phylogenetic placement within this node was used to assign these and other strains and cloned sequences to the MAR4 lineage.
Culture-independent analyses performed on four of the Channel Island sediment samples using MAR4-specific 16S rRNA primers yielded 196 nonchimeric sequences, of which 179 fell within the MAR4 clade (GenBank accession numbers KC477442 to KC477620). An additional 15 strains, isolated as part of prior studies (9, 12, 19) and considered candidates for the MAR4 clade on the basis of preliminary sequence data or secondary metabolite analysis, were also confirmed to be members of this lineage. To further expand the search, 13 diverse MAR4 sequences were used as queries of the NCBI database. The top 100 BLAST matches yielded 465 nonredundant sequences, of which 21 fell within the MAR4 lineage. These consisted of 20 cultured strains and one cloned sequence (Table 1). Slightly more that 50% of these were of marine origin.
Table 1.
Sources of MAR4 strains and sequences identified in this study
In total, 801 candidate MAR4 sequences were analyzed as part of this study. Phylogenetic analyses placed 220 of these sequences in the MAR4 clade (Table 1). The inclusion of the 17 previously identified MAR4 strains (13) with the 40 strains identified here raises the total number of cultured strains that can be placed in this lineage to 57. A comprehensive phylogeny of the 57 cultured strains and representative cloned sequences defines the extant diversity of the MAR4 lineage (Fig. 1), which encompasses 4.1% 16S rRNA sequence divergence within the genus Streptomyces. The MAR4 lineage includes the S. synnematoformans and S. aculeolatus type strains, which are the only named species within the clade. It consistently bifurcates into two distinct yet poorly supported subclades, each of which contains one of the type strains. Only one strain within the MAR4 node (the strain with accession number X87316) does not fall within these two subclades.
Fig 1.

16S rRNA gene phylogeny of the MAR4 clade. The maximum likelihood (ML) phylogeny includes all cultured MAR4 strains and a representative from each OTU (Fig. 2) that contains only cloned sequences. Sequences are identified by NCBI accession number followed by the strain number and source, when available. Sequences added to the NCBI database as a result of this study are indicated in bold. * and ** indicate that the clades represented by the S. aculeolatus and S. synnematoformans type strains, respectively. Maximum likelihood and maximum parsimony (MP) trees had no conflicting nodes. Bootstrap (maximum likelihood) and jackknife (maximum parsimony) values higher than 50% are indicated.
The MAR4 lineage as currently described is largely of marine origin, with 46 of 57 cultured strains and 179 of 180 cloned sequences originating from the marine environment (see Table S4 in the supplemental material). The 12 sequences that are not linked to the marine environment include 1 sequence cloned from a uranium-contaminated subsurface sediment (GenBank accession number HM186499), three strains isolated from terrestrial sources (accession numbers NR041166, EF121313, and AB373966), six strains isolated from Lechuguilla Cave, NM (accession numbers JQ014364, JQ014418, JQ014483, JQ014508, JQ014513, and JQ014571), and two sequence deposits that include no environmental source information (accession numbers X87316 and HQ992743).
Cultured and culture-independent MAR4 diversity.
The 16S rRNA sequences within the MAR4 clade were clustered at a genetic distance of 0.01 (29), a value commonly used to delineate actinomycete taxonomic units (30, 31). This resulted in 28 OTUs (Fig. 2), of which 14 were comprised entirely of cloned sequences, 8 were comprised entirely of cultured sequences, and 6 were comprised of both cultured and cloned sequences. Thus, most OTUs (82%) were comprised entirely of either cultured strains or cloned sequences, with little overlap between the two. OTUs 13 and 17 contained 105 and 37 clones, respectively, which accounted for 71% of the cloned sequences. Each of these OTUs also contained 3 to 5 cultured strains. Approximately one-half of the cultured strains fell within OTU 14, which did not include any cloned sequences. Overall, 88% of the MAR4 strains and cloned sequences were assigned to 3 of the 28 OTUs. The five MAR4 strains cultured as part of this study all fell within OTUs containing previously isolated strains, while the 179 cloned sequences fell within 20 OTUs, of which 14 were composed entirely of Channel Islands clone sequences.
Fig 2.

Distribution of MAR4 sequences among OTUs with an average neighbor clustering distance of 0.01.
A rarefaction curve generated from the combined MAR4 OTU data reveals a steep slope, indicating that considerable diversity has yet to be sampled (Fig. 3) (29). This result is supported by the Chao1 and abundance coverage estimator (ACE) richness estimators, which predict that continued sequencing will yield from 58 to 86 OTUs. Individual rarefaction curves of cultured strains and cloned sequences similarly revealed that more sampling will yield additional MAR4 diversity.
Fig 3.

Rarefaction curve for MAR4 cultured, cloned, and combined sequences. Chao1 and ACE richness estimates are indicated.
HI production by MAR4 strains.
A phenotypic trait previously associated with the MAR4 clade is the production of HI secondary metabolites (13). To further explore the relationships between HI production and MAR4 phylogeny, we analyzed the culture extracts of 21 diverse MAR4 strains over a 9-day cultivation period. These strains belonged to 9 OTUs that were broadly distributed throughout the MAR4 clade and included representatives from both the S. synnematoformans and S. aculeolatus clades (see Table S4 in the supplemental material). Based on UV absorbance and mass spectral data, we observed the production of compounds in four previously reported HI classes. These classes were the napyradiomycins (32), the marinones (33, 34), the lavanducyanins (35), and the nitropyrrolins (36) (Fig. 4). In addition, a compound with a UV spectrum that was diagnostic for phenazines but that did not match any of these four classes was also detected. Further investigation of this compound by high-resolution MS/MS revealed a parent ion, [M + H]+, with an exact mass of 377.2236 and a fragmentation pattern consistent with phenazine-1-carboxylic acid (37, 38) with methyl and monoterpene substituents (see Fig. S3 in the supplemental material). These results provide preliminary evidence that the unknown compound is a prenylated phenazine, which we have tentatively assigned to a fifth HI class while further structural characterization is ongoing.
Fig 4.
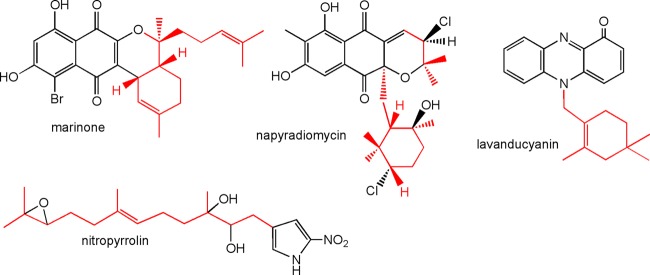
Previously reported hybrid isoprenoid structure classes detected from MAR4 strains. The terpene-derived portion of each molecule is highlighted in red.
All 21 of the MAR4 strains investigated produced at least one HI and, in most cases, produced more than one (Fig. 5). In many cases, multiple derivatives within the same structural class were observed from a single strain. The S. aculeolatus and S. synnematoformans lineages showed differences in the numbers and types of HI secondary metabolites produced, with the former including the only four strains observed to produce nitropyrrolins. In addition, the majority of the strains tested in the S. aculeolatus clade (10 of 14) produced napyradiomycins, whereas only 1 strain in the S. synnematoformans clade produced a napyradiomycin. Eight of the S. aculeolatus strains also produced more than one napyradiomycin derivative; in comparison, the S. synnematoformans strain produced only one. The opposite pattern was observed with the marinones, with all of the S. synnematoformans strains producing multiple derivatives but the S. aculeolatus strains producing only one. Interestingly, no strain produced multiple compounds in both the napyradiomycin and marinone classes.
Fig 5.

Lineage-specific HI production by MAR4 strains. The 16S rRNA gene phylogeny shown in Fig. 1 is displayed as a cladogram. Strains analyzed for HI production are in bold. For each strain analyzed, the number of compounds detected within each class is indicated by number and color intensity according to the number detected. * and ** indicate the clades represented by the S. aculeolatus and S. synnematoformans type strains, respectively.
In cases where strains produced multiple derivatives within a compound class, they generally produced more of each derivative than strains that produced only a single compound. This is best observed with napyradiomycin production, in which case the S. aculeolatus clade produced more peaks per strain (Fig. 5) and the average area under each peak was 25.3 times larger than that in the S. synnematoformans clade (Table 2). Similarly, the average area under each marinone peak in the S. synnematoformans clade, within which all strains produced multiple derivatives, was 4.3 times larger than that in the S. aculeolatus clade, where only one derivative was produced. The relative amounts and number of derivatives of the lavanducyanins and the uncharacterized prenylated phenazine produced by both clades were similar; however, both compound classes were observed more consistently from members of the S. synnematoformans clade. No major differences were observed between duplicate extracts except for strain CNQ-181, where a lavanducyanin peak was observed in only one of the replicates (data not shown).
Table 2.
Relative compound production for members of the S. aculeolatus and S. synnematoformans clades
| Compound | Avg area under the peak (no. of peaks) for: |
Ratio | |
|---|---|---|---|
| S. aculeolatus clade | S. synnematoformans clade | ||
| Napyradiomycin | 1,451 ± 312 (41) | 57 (1) | 25.6:1 |
| Marinone | 380 ± 234 (8) | 1,638 ± 470 (28) | 1:4.3 |
| Lavanducyanin | 2,216 ± 1,120 (5) | 1,347 ± 474 (7) | 1.6:1 |
| Nitropyrrolin | 447 ± 102 (20) | ND | |
| Prenylated phenazine | 661 ± 364 (2) | 878 ± 364 (7) | 1:1.3 |
Values represent the average area under the peaks associated with each compound class ± standard error of the mean. ND, not detected.
DISCUSSION
The MAR4 lineage of streptomycetes has been reported to be largely of marine origin and recognized as a source of the rare HI class of bacterial secondary metabolites (13, 39). The aim of the present study was to further explore the diversity and distribution of this lineage through the application of culture-dependent and culture-independent methods and through the mining of publically available sequence data. Secondary aims were to characterize the numbers, types, and relative amounts of HI secondary metabolites produced by these bacteria and to search for correlations between MAR4 phylogeny and HI production.
The MAR4 lineage is defined on the basis of a well-supported node observed in the 16S rRNA gene phylogeny (Fig. 1). The present study expands the number of cultured strains within this lineage from 17 to 57. Seventy-nine percent of these strains, including strains reported from eight independent research groups and strains from a range of sample types (see Table S4 in the supplemental material), originated from marine sources, suggesting that the lineage may preferentially occur in marine habitats. At the level of resolution obtained in this study, there is no evidence for specific marine or terrestrial lineages within the MAR4 clade, suggesting that strains are broadly distributed across these environments. Interestingly, the six strains isolated from Lechuguilla Cave, NM, do not cluster together, suggesting that the cave system has been colonized multiple times. However, a more highly resolved phylogeny generated using less conserved loci will be needed before conclusions can be drawn about intraclade relationships and biogeographic patterns within the MAR4 lineage.
Of the 125 Streptomyces spp. cultured as part of this study, only 5 fell within the MAR4 clade, suggesting that these bacteria are rare members of the streptomycete community. The 20 additional MAR4 strains identified on the basis of NCBI sequence deposits further support the rarity of this group, given the nearly 30,000 NCBI sequence deposits labeled “Streptomyces 16S.” While it remains possible that the selective cultivation methods typically employed for Streptomyces spp. are not particularly effective for this group, only one MAR4 clone was detected among the 589 environmental Streptomyces 16S sequence deposits in the NCBI database. Despite this apparent rarity, the results of a culture-independent analysis performed on a subset of the Channel Island sediment samples revealed that considerable MAR4 OTU diversity has yet to be obtained in culture. Surprisingly, 82% of the OTUs were comprised entirely of either cloned or cultured sequences. These included several OTUs from the Channel Island sites with which both techniques were performed yet only cultured OTUs were observed. Similar observations have been made for soil samples (40) and could reflect the presence of readily cultured members of the rare biosphere that remain undetected in clone libraries due to low abundance. These observations support the application of both culture and culture-independent methods in the assessment of MAR4 diversity.
The results presented here further establish the production of HI secondary metabolites as a phenotypic trait of the MAR4 lineage, with all 21 of the strains tested producing HI secondary metabolites. These compounds could be delineated into five structural classes, including a prenylated phenazine that could not be assigned to a known chemical class. Surprisingly, many strains produced multiple HI classes, suggesting that they possess more than one pathway associated with HI biosynthesis.
Despite the lack of statistical support for the intraclade relationships in the MAR4 phylogeny, distinct patterns were observed in the production of HIs by the two clades defined by the S. aculeolatus and the S. synnematoformans type strains. These patterns suggest that an investment in the production of either the napyradiomycin or marinone class appears to be a phenotypic trait that differentiates the two clades. The relationship between napyradiomycin production and the S. aculeolatus clade is further supported by an independent study of the MAR4 strains CNH-070 and CNQ-329, which yielded six novel and three known napyradiomycin derivatives (43). In addition, the S. synnematoformans clade preferentially produces the prenylated phenazine, while the only strains observed to produce the nitropyrrolin class were in the S. aculeolatus clade. Prenylated pyrroles are rare in nature; to the best of our knowledge, the only other streptomycete-produced compounds of this type are the glaciapyrroles isolated from a marine sediment-derived Streptomyces sp. (41) and a pyrrolostatin produced by a Brazilian soil streptomycete (42). Future comparative genomic studies will provide better resolution of the lineage specificity of the biosynthetic pathways associated with these compound classes as well as the full HI biosynthetic potential of MAR4 strains.
The enrichment of HI secondary metabolites in the MAR4 lineage provides evidence that positive selection has favored their production. While the natural functions of MAR4 HIs have yet to be defined, it will be interesting to evaluate nonmarine MAR4 strains to determine if this enrichment represents a marine adaptation. Ultimately, identifying the ecological functions of HI secondary metabolites and how they may enhance survival in specific habitats remain important goals for future studies of MAR4 bacteria.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by NOAA California Sea Grant College Program project R/NMP-100 (grant NA100AR4170060 to P.R.J.) through NOAA's National Sea Grant College Program, U.S. Department of Commerce. P.R.J. acknowledges NIH for support to acquire high-resolution LC-MS instrumentation under grant S10 OD010640-01. K.A.G. acknowledges support from an NIH Training Grant in Marine Biotechnology (GM067550). L.P.I. thanks CAPES-FIPSE an FAPESP (2011/08064-2) for undergraduate investigator scholarships.
The statements, findings, conclusions, and recommendations are those of the authors and do not necessarily reflect the views of California Sea Grant or the U.S. Department of Commerce.
C. Hughes and W. Fenical are gratefully acknowledged for guidance on analytical chemistry. L.P.I. thanks Roberto G. S. Berlinck (Universidade de Sao Paulo) and Fabiano L. Thompson (Universidade Federal do Rio de Janeiro) for providing continuous support during the development of this project.
Footnotes
Published ahead of print 30 August 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.01814-13.
REFERENCES
- 1.Berdy J. 2005. Bioactive microbial metabolites. A personal view. J. Antibiot. 58:1–26 [DOI] [PubMed] [Google Scholar]
- 2.Watve MG, Tickoo R, Jog MM, Bhole BD. 2001. How many antibiotics are produced by the genus Streptomyces? Arch. Microbiol. 176:386–390 [DOI] [PubMed] [Google Scholar]
- 3.Zimmermann W. 1990. Degradation of lignin by bacteria. J. Biotechnol. 13:119–130 [Google Scholar]
- 4.Weller DM, Raaijmakers JM, Gardener BB, Thomashow LS. 2002. Microbial populations responsible for specific soil suppressiveness to plant pathogens. Annu. Rev. Phytopathol. 40:309–348 [DOI] [PubMed] [Google Scholar]
- 5.Maldonado LA, Stach JEM, Pathom-aree W, Ward AC, Bull AT, Goodfellow M. 2005. Diversity of cultivable actinobacteria in geographically widespread marine sediments. Antonie Van Leeuwenhoek Int. J. Gen. Mol. Microbiol. 87:11–18 [DOI] [PubMed] [Google Scholar]
- 6.Moran MA, Rutherford LT, Hodson RE. 1995. Evidence for indigenous Streptomyces populations in a marine environment determined with a 16S rRNA probe. Appl. Environ. Microbiol. 61:3695–3700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Strobel G, Daisy B. 2003. Bioprospecting for microbial endophytes and their natural products. Microbiol. Mol. Biol. Rev. 67:491–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Seipke RF, Kaltenpoth M, Hutchings MI. 2012. Streptomyces as symbionts: an emerging and widespread theme? FEMS Microbiol. Rev. 36:862–876 [DOI] [PubMed] [Google Scholar]
- 9.Prieto-Davo A, Fenical W, Jensen PR. 2008. Comparative actinomycete diversity in marine sediments. Aquat. Microb. Ecol. 52:1–11 [Google Scholar]
- 10.Lerat S, Simao-Beaunoir A-M, Beaulieu C. 2009. Genetic and physiological determinants of Streptomyces scabies pathogenicity. Mol. Plant Pathol. 10:579–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barke J, Seipke R, Gruschow S, Heavens D, Drou N, Bibb M, Goss R, Yu D, Hutchings M. 2010. A mixed community of actinomycetes produce multiple antibiotics for the fungus farming ant Acromyrmex octospinosus. BMC Biol. 8:109. 10.1186/1741-7007-8-109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jensen PR, Gontang E, Mafnas C, Mincer TJ, Fenical W. 2005. Culturable marine actinomycete diversity from tropical Pacific Ocean sediments. Environ. Microbiol. 7:1039–1048 [DOI] [PubMed] [Google Scholar]
- 13.Gallagher KA, Fenical W, Jensen PR. 2010. Hybrid isoprenoid secondary metabolite production in terrestrial and marine actinomycetes. Curr. Opin. Biotechnol. 21:794–800 [DOI] [PubMed] [Google Scholar]
- 14.Tello M, Kuzuyama T, Heide L, Noel JP, Richard SB. 2008. The ABBA family of aromatic prenyltransferases: broadening natural product diversity. Cell. Mol. Life Sci. 65:1459–1463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gershenzon J, Dudareva N. 2007. The function of terpene natural products in the natural world. Nat. Chem. Biol. 3:408–414 [DOI] [PubMed] [Google Scholar]
- 16.Izumikawa M, Khan ST, Takagi M, Shin-ya K. 2010. Sponge-derived Streptomyces producing isoprenoids via the mevalonate pathway. J. Nat. Prod. 73:208–212 [DOI] [PubMed] [Google Scholar]
- 17.Heide L. 2009. Genetic engineering of antibiotic biosynthesis for the generation of new aminocoumarins. Biotechnol. Adv. 27:1006–1014 [DOI] [PubMed] [Google Scholar]
- 18.Freel KC, Edlund A, Jensen PR. 2012. Microdiversity and evidence for high dispersal rates in the marine actinomycete ‘Salinispora pacifica.’ Environ. Microbiol. 14:480–493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gontang EA, Fenical W, Jensen PR. 2007. Phylogenetic diversity of gram-positive bacteria cultured from marine sediments. Appl. Environ. Microbiol. 73:3272–3282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mincer TJ, Fenical W, Jensen PR. 2005. Culture-dependent and culture-independent diversity within the obligate marine actinomycete genus Salinispora. Appl. Environ. Microbiol. 71:7019–7028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee SD, Kang SO, Hah YC. 2000. Catellatospora koreensis sp. nov., a novel actinomycete isolated from a gold-mine cave. Int. J. Syst. Evol. Microbiol. 50:1103–1111 [DOI] [PubMed] [Google Scholar]
- 22.Cole JR, Chai B, Farris RJ, Wang Q, Kulam SA, McGarrell DM, Garrity GM, Tiedje JM. 2005. The Ribosomal Database Project (RDP-II): sequences and tools for high-throughput rRNA analysis. Nucleic Acids Res. 33:D294–D296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huber T, Faulkner G, Hugenholtz P. 2004. Bellerophon: a program to detect chimeric sequences in multiple sequence alignments. Bioinformatics 20:2317–2319 [DOI] [PubMed] [Google Scholar]
- 24.Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32:1792–1797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Silvestro D, Michalak I. 2012. raxmlGUI: a graphical front-end for RAxML. Org. Divers. Evol. 12:335–337 [Google Scholar]
- 26.Swofford D. 2003. PAUP*: phylogenetic analysis using parsimony, version 4.0b10. Illinois Natural History Survey, University of Illinois, Champaign, IL [Google Scholar]
- 27.Schloss PD, Westcott SL. 2011. Assessing and improving methods used in operational taxonomic unit-based approaches for 16S rRNA gene sequence analysis. Appl. Environ. Microbiol. 77:3219–3226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF. 2009. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75:7537–7541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hughes JB, Hellmann JJ, Ricketts TH, Bohannan BJM. 2001. Counting the uncountable: statistical approaches to estimating microbial diversity. Appl. Environ. Microbiol. 67:4399–4406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Prieto-Davó A, Villarreal-Gómez LJ, Forschner-Dancause S, Bull AT, Stach JEM, Smith DC, Rowley DC, Jensen PR. 2013. Targeted search for actinomycetes from nearshore and deep-sea marine sediments. FEMS Microbiol. Ecol. 84:510–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stach JE, Maldonado LA, Masson DG, Ward AC, Goodfellow M, Bull AT. 2003. Statistical approaches for estimating actinobacterial diversity in marine sediments. Appl. Environ. Microbiol. 69:6189–6200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shiomi K, Iinuma H, Hamada M, Naganawa H, Manabe M, Matsuki C, Takeuchi T, Umezawa H. 1986. Novel antibiotics napyradiomycins—production, isolation, physicochemical properties and biological activity. J. Antibiot. 39:487–493 [DOI] [PubMed] [Google Scholar]
- 33.Hardt IH, Jensen PR, Fenical W. 2000. Neomarinone, and new cytotoxic marinone derivatives, produced by a marine filamentous bacterium (actinomycetales). Tetrahedron Lett. 41:2073–2076 [Google Scholar]
- 34.Pathirana C, Jensen PR, Fenical W. 1992. Marinone and debromomarinone—antibiotic sesquiterpenoid naphthoquinones of a new structure class from a marine bacterium. Tetrahedron Lett. 33:7663–7666 [Google Scholar]
- 35.Imai S, Furihata K, Hayakawa Y, Noguchi T, Seto H. 1989. Lavanducyanin, a new antitumor substance produced by Streptomyces sp. J. Antibiot. 42:1196–1198 [DOI] [PubMed] [Google Scholar]
- 36.Kwon HC, Espindola PDM, Park J-S, Prieto-Davo A, Rose M, Jensen PR, Fenical W. 2010. Nitropyrrolins A-E, cytotoxic farnesyl-alpha-nitropyrroles from a marine-derived bacterium within the actinomycete family streptomycetaceae. J. Nat. Prod. 73:2047–2052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moree WJ, Phelan VV, Wu C-H, Bandeira N, Cornett DS, Duggan BM, Dorrestein PC. 2012. Interkingdom metabolic transformations captured by microbial imaging mass spectrometry. Proc. Natl. Acad. Sci. U. S. A. 109:13811–13816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Watson D, Taylor GW, Wilson R, Cole PJ, Rowe C. 1988. Thermospray mass spectrometric analysis of phenazines. Biol. Mass Spectrom. 17:251–255 [Google Scholar]
- 39.Fenical W, Jensen PR. 2006. Developing a new resource for drug discovery: marine actinomycete bacteria. Nat. Chem. Biol. 2:666–673 [DOI] [PubMed] [Google Scholar]
- 40.Shade A, Hogan CS, Klimowicz AK, Linske M, McManus PS, Handelsman J. 2012. Culturing captures members of the soil rare biosphere. Environ. Microbiol. 14:2247–2252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Macherla VR, Liu J, Bellows C, Teisan S, Nicholson B, Lam KS, Potts BCM. 2005. Glaciapyrroles A, B, and C, pyrrolosesquiterpenes from a Streptomyces sp. isolated from an Alaskan marine sediment. J. Nat. Prod. 68:780–783 [DOI] [PubMed] [Google Scholar]
- 42.Kato S, Shindo K, Kawai H, Odagawa A, Matsuoka M, Mochizuki J. 1993. Pyrrolostatin, a novel lipid peroxidation inhibitor from Streptomyces chrestomyceticus—taxonomy, fermentation, isolation, structure elucidation and biological properties. J. Antibiot. 46:892–899 [DOI] [PubMed] [Google Scholar]
- 43.Cheng Y-B, Jensen PR, Fenical W. 2013. Cytotoxic and antimicrobial napyradiomycins from two marine-derived Streptomyces strains. Eur. J. Org. Chem. 18:3751–3757 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.