

Abstract
Eukaryotic parasites of the genus Plasmodium cause malaria by invading and developing within host erythrocytes. Here, we demonstrate that PfShelph2, a gene product of Plasmodium falciparum that belongs to the Shewanella-like phosphatase (Shelph) subfamily, selectively hydrolyzes phosphotyrosine, as shown for other previously studied Shelph family members. In the extracellular merozoite stage, PfShelph2 localizes to vesicles that appear to be distinct from those of rhoptry, dense granule, or microneme organelles. During invasion, PfShelph2 is released from these vesicles and exported to the host erythrocyte. In vitro, PfShelph2 shows tyrosine phosphatase activity against the host erythrocyte protein Band 3, which is the most abundant tyrosine-phosphorylated species of the erythrocyte. During P. falciparum invasion, Band 3 undergoes dynamic and rapid clearance from the invasion junction within 1 to 2 s of parasite attachment to the erythrocyte. Release of Pfshelph2 occurs after clearance of Band 3 from the parasite-host cell interface and when the parasite is nearly or completely enclosed in the nascent vacuole. We propose a model in which the phosphatase modifies Band 3 in time to restore its interaction with the cytoskeleton and thus reestablishes the erythrocyte cytoskeletal network at the end of the invasion process.
INTRODUCTION
Parasites of the genus Plasmodium cause a great deal of morbidity and mortality worldwide, largely in regions with limited access to the tools necessary to control mosquito populations and to treat human infections (1, 2). Five species of this class of eukaryotic pathogens cause human disease, with Plasmodium falciparum alone infecting approximately 500 million people per year and resulting in approximately 1 million deaths (3). All symptoms and disease pathologies of malaria occur during the asexual blood stage, in which the invasive merozoites infect host erythrocytes, establish residence within these host cells, and divide into daughter merozoites that then rupture out of the host cell and subsequently reinvade new erythrocytes.
Previous analyses of the Plasmodium sp. genomes identified 27 genes that are predicted to be protein phosphatases, including several prokaryote-like phosphatases (4–6). Interestingly, members of this genus of eukaryotic pathogens possess several phosphatases that cluster by sequence homology with prokaryote-like phosphatases. Two prokaryote-like Plasmodium phosphatases were found to cluster with a family of bacterial phosphatases termed the Shewanella-like phosphatases (Shelphs) after the first and thus far only characterized enzyme of the family (7, 8). Despite clustering as a subgroup of the serine/threonine phosphatase family, the Shewanella phosphotyrosine phosphatase (PTPase) has phosphotyrosine phosphatase activity in vitro (9–11). In contrast to typical PTPases of eukaryotes, which utilize a catalytic-site cysteine residue, the Shelph PTPase utilizes a catalytic mechanism involving divalent metal cations, which is the mechanism used by other members of the serine/threonine phosphatase family to which it belongs (9–11). In addition to being present in bacteria and Plasmodium spp., orthologues of pfshelph2 (PlasmoDB ID PF3D7_1206000; Uniprot ID Q8I5Y5) exist in other lower eukaryotes, including Schizosaccharomyces pombe, Leishmania major, Trypanosoma brucei, and Cryptosporidium parvum (5, 8). Currently, it remains to be determined whether Shelphs from other organisms possess the same substrate specificity as the original Shewanella PTPase.
The two genes encoding the Shewanella-like phosphatases of P. falciparum, here termed PfShelph1 and PfShelph2, are conserved among members of the genus Plasmodium. Both PfShelphs possess all of the residues that define the catalytic site of phosphoprotein phosphatase family members, suggesting that they are catalytically active (4, 5, 7, 8). Additionally, both PfShelphs possess predicted N-terminal signal sequences, with PfShelph1 predicted to have an additional apicoplast-targeting motif (4–6). A wide-scale study of transcripts in P. falciparum showed that PfShelph2 is likely expressed in the invasive merozoite stage that invades host erythrocytes (12). Consistent with their prediction, the investigators showed that an exogenously expressed PfShelph2-green fluorescent protein (GFP) transgene traffics to a punctate organelle in schizonts, a pattern seen with other proteins involved in invasion (12). However, whether PfShelph2 expressed under the control of its endogenous promoter is also localized to merozoites and whether the protein is an active phosphoprotein phosphatase involved in host-pathogen interactions during invasion remains unknown.
Despite their unique phylogenetic classification and potentially paradoxical substrate specificity, much work remains to be done in terms of validating the predicted activity and function of the PfShelph proteins. To advance knowledge of this interesting class of enzymes, we set out to characterize PfShelph2, specifically with the aims of determining its (i) substrate specificity, (ii) subcellular location, and (iii) dynamics and possible contribution to changes in erythrocyte membrane protein dynamics during the invasion process. We show that recombinant PfShelph2 expressed and purified from either Escherichia coli or P. falciparum possesses substrate specificities similar to those of its Shewanella counterpart, hydrolyzing both tyrosine-phosphorylated peptides and the tyrosine-phosphorylated cytoplasmic domain of Band 3. In addition, we tagged the endogenous pfshelph2 gene with GFP and found that the fusion protein is detectable in vesicular structures in late-stage parasites and at the apical end of the invasive merozoite stage. We show that the vesicles are discharged at a late stage in the invasion process. Based on immunofluorescence and immunoelectron microscopy, we also show that the phosphatase concentrates in vesicles that do not colocalize with markers of rhoptry, dense granule, or microneme organelles. Our data further demonstrate that PfShelph2 is released into to the host cell at a time when Band 3, the major tyrosine-phosphorylated protein of the erythrocyte membrane, begins to return to the site of invasion from which it is cleared early in the invasion process. These data suggest a possible model in which PfShelph2 is a parasite-derived signaling molecule that plays a role in regulating interactions between the host erythrocyte membrane protein Band 3 and possibly other proteins of the erythrocyte cytoskeleton during the genesis of the parasitophorous vacuole.
MATERIALS AND METHODS
Parasite culturing.
The 3D7 strain of P. falciparum was cultured and synchronized as previously described (13, 14). For invasion experiments, parasite cultures were synchronized by sorbitol lysis and Percoll gradient purification of schizonts. Uninfected erythrocytes at 2 to 5% hematocrit were incubated in complete RPMI 1640 medium (cRPMI) with synchronized schizonts at a parasitemia of 0.5 to 3% in 96-well plates under standard culture conditions. Giemsa-stained thin blood smears were made at 0 and 12 to 18 h postinvasion to determine parasitemia. Fold invasion was calculated as the ratio of percent rings at 12 to 18 h to percent schizont parasitemia at 0 h.
MBP-PfShelph2 purification.
The pfshelph2 gene was codon optimized by Genscript for E. coli expression, amplified by PCR (primers Forward, 5′-CCGCCGGAATTCAACGAAAGCTACTCTAACATCAAATGGGAACATG, and Reverse, 5′-CGACGCGTCGACTTAAATATCGCTGTTGATGTAGTTCACTTTGTAACTACC), and cloned into the pMAL-p4E (New England BioLabs) vector for periplasmic expression. The clones were confirmed by restriction digestion and sequencing and subsequently used to transform E. coli BL21(DE3). For protein production, 3 ml of overnight culture (in LB plus 100 μg/ml ampicillin and 34 μg/ml chloramphenicol) was inoculated into 1 liter LB with 100 μg/ml ampicillin and 34 μg/ml chloramphenicol. The culture was grown to log phase (optical density at 600 nm [OD600], ∼0.4 to 0.6), and protein expression was induced by addition of 1 mM IPTG (isopropyl-β-d-thiogalactopyranoside) and overnight growth at 18°C. Bacteria were collected by centrifugation at 6,000 × g for 20 min, resuspended in 15 ml column buffer (50 mM Tris-HCl, pH 7.4, 200 mM NaCl, 1 mM dithiothreitol [DTT], 400 μM MnCl2), and lysed by treatment with lysozyme (250 μg/ml; 1 h; 4°C), followed by sonication. The resulting lysate was cleared of debris by centrifugation at 90,000 × g for 90 min at 4°C and loaded onto preequilibrated amylose resin (New England BioLabs). The resin was washed with column buffer, and maltose binding protein (MBP)-PfShelph2 was eluted with column buffer containing 10 mM maltose. The eluted protein was concentrated in a centrifugal filter device (Millipore) with a 3.5-kDa cutoff.
PfShelph2-Strep purification.
Full-length pfshelp2 was amplified from P. falciparum strain 3D7 genomic DNA using the following primers: primer A, 5′-GCACGCGTCGACATGAATATATCATATTTAAGGAATTTTTCTTGTATAT; primer B, 5′-GCACGCAGATCTTATATCGGAATTTATATAATTTACTTTATATGATCCATC. The PCR product was cloned downstream of the calmodulin promoter into the SalI and BglII sites of pA171, a modified version of vector pA2 containing two tandem Strep-tag II tags 3′ of the multiple-cloning site (15). The parasites were transfected with 100 μg of the pHTH vector containing the piggyBac transposase and 100 μg of pA171 with pfshelph2. Transfected parasites were drug selected with 2.5 nM WR99210 and maintained under continuous drug selection. The parasites were periodically synchronized by treatment with sorbitol and purification with 65% Percoll. Late-stage parasites were collected every 2 days, washed 3 times in PBS plus 11 mM glucose, and stored at −80°C. Frozen parasites were lysed by resuspending them in lysis buffer (50 mM Tris-HCl, pH 8.0, 500 mM NaCl, 1 mM MnCl2, 1 mM DTT) and sonicating them on ice. The resulting lysate was cleared by centrifugation at 90,000 × g for 60 min and then loaded onto an Akta 10 fast protein liquid chromatograph (FPLC) with a preequilibrated 1-ml Qiagen Streptactin Superflow Plus column. The loaded column was washed with lysis buffer (∼100 ml), and protein was eluted with lysis buffer plus 2.5 mM desthiobiotin (Sigma). The eluted fractions were pooled and concentrated in a 3.5-kDa-cutoff concentrator.
para-Nitrophenyl phosphate assays.
Purified enzyme was incubated in a total volume of 100 μl of buffer (20 mM Tris-HCl, pH 8.0, 400 mM NaCl, 200 μM MnCl2, 1 mM DTT unless otherwise indicated) with 5 mM para-nitrophenyl phosphate (pNPP) (Anaspec) in a 96-well plate. The OD405 was measured using a Spectramax M2 plate reader at the indicated times. Blank samples were wells without enzyme or wells with a catalytically inactive mutant as indicated in the figures.
FDP assays.
Purified enzyme was incubated in 100 μl of buffer (20 mM Tris-HCl, pH 7.4, 400 mM NaCl, 200 μM MnCl2, 1 mM DTT unless otherwise indicated) in a 96-well plate. Fluorescein diphosphate (FDP) (Anaspec) hydrolysis was monitored by following fluorescence (excitation [Ex] and emission [Em] wavelengths, 490 nm and 514 nm) at the indicated times. Samples to which no enzyme was added were used as blanks.
Malachite green assays.
Purified enzyme was incubated with 200 μM of the indicated substrate in 50 μl buffer (20 mM Tris-HCl, pH 8.0, 400 mM NaCl, 200 μM MnCl2, 1 mM DTT) for the indicated times at 37°C. Following incubation, 100 µl of Biomol Green (Enzo) reagent was added, and after incubation for 30 min at room temperature, absorbance was measured at 620 nm using a Spectramax M2 plate reader. Samples were blanked with samples to which no enzyme was added. The phosphopeptides used were as follows: EGFR2 substrate (Anaspec), DADEYLIPQQG; PTP1B substrate (Anaspec), ELEFYMDYE-NH2; PP1/2 substrate (Anaspec), GRPRTSSFAEG; histone H1 substrate (H1) (Anaspec), GGGPATPKKAKKL; insulin receptor substrate (IR) (Sigma), TRDIYETDYYRK. All peptides were assayed for the presence of hydrolyzable phosphate by incubation with alkaline phosphatase (New England BioLabs) and measuring free-phosphate release with Biomol Green.
Production of PfShelph2-GFP parasites.
The pPM2GT vector (obtained from MR4; deposited by D. E. Goldberg [16]) was digested with XhoI and AvrII to remove the plasmepsin II fragment, and a fragment of pfShelph2 (nucleotides 301 to 912) was amplified by PCR (5′ primer, CCGCCGCTCGAGGCAAAACCATTAAATTCGAAAATACAATTAATATTAGG; 3′ primer, AATCAACCTAGGTATATCGGAATTTATATAATTTACTTTATATGATCCATC) and cloned into the digested pPM2GT vector to yield pPFLGT. Positive clones were identified by PCR and sequenced. pPFLGT was purified with the Sigma Maxiprep kit, and 100 μg of plasmid was transfected as previously described (17). Drug selection with 10 nM WR99210 was initiated 48 h posttransfection and maintained until parasitemia reached ∼1%, at which point the parasites were grown without drug for 21 days to allow loss of unintegrated episomes. After 21 days, drug selection (10 nM WR99210) was resumed for another period until parasitemia reached ∼1%. The parasites were then grown for another 21-day cycle in the absence of drug, followed by resumption of drug selection and then cloning by limiting dilution. The clones were analyzed for integration by PCR. Genomic DNA was extracted using a Qiagen Blood and Cell Culture DNA minikit.
Immunofluorescence assays.
Immunofluorescence assays were performed by washing parasites in phenol red-free RPMI medium (Gibco) and then allowing them to settle onto poly-l-lysine-coated coverslips. The cells were washed 3 times in PBS and then fixed in 9:1 acetone-methanol for 10 min at −20°C, washed 3 times in PBS, blocked in 1% fish skin gelatin in PBS (FSG-PBS) for 10 min, and then probed with antibodies diluted in 1% FSG-PBS. The primary antibodies used were chicken anti-GFP (1:500) (Abcam), mouse anti-RhopH2 (1:500), anti-PfRON3, rabbit anti-PTRAMP (1:500), and rabbit anti-RESA (1:500). The secondary antibodies used at 1:500 dilution were donkey anti-chicken DyLight 488 (Jackson ImmunoResearch) and rhodamine goat anti-mouse (MP Biomedicals). Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI) (1 μg/ml) or Hoechst 33342 (1 μg/ml) as indicated. The lack of colocalization between PfShelph2-GFP and apical-organelle markers in merozoites was measured using the Softworx 2.0 colocalization tool. A total of 100 to 150 optical sections were examined for quantitative analyses.
Real-time live fluorescence imaging.
For real-time imaging of merozoite invasion, we adapted the method described by Gilson and Crabb (18). Briefly, 20 μl of synchronized culture at 1 to 2% hematocrit and 10 to 15% parasitemia containing rupturing late-stage schizonts (∼1 to 5% parasitemia) was added to 4 ml of prewarmed cRPMI 1640 medium. 300 μl of this suspension was then added to a well of a Lab-Tek II 8-well coverglass chamber (Nunc). The cells were then allowed to settle for 30 min at 37°C. Images were captured using an inverted Olympus IX fluorescence microscope and a CoolSnap HQ2 charge-coupled-device (CCD) camera controlled by Deltavision software (Applied Precision, Seattle, WA). The frame capture rates for the CCD camera per optical section were 1 image/1.2 s (see Fig. 8A and Fig. S7 in the supplemental material) and 1 image/1.9 s (see Fig. 8B). The images were deconvolved with Softworx.
Fig 8.

Real-time imaging of PfShelph2-GFP discharge during invasion. (A) Merozoite invasion was imaged as described in Materials and Methods. The arrowhead indicates the invading merozoite. Complete discharge of apical green fluorescence occurs by 15 s. At the end of invasion, the cells were imaged in 10 optical sections with 0.2 μm between sections. PfShelph2-GFP signal was not detected in other focal planes, as shown in the cumulative z stack. The asterisks indicate an extracellular merozoite where PfShelph2-GFP fluorescence remains detectable at 26 s (although it is not seen in a few earlier sections because it moves out of the plane of focus). Scale bars, 5 μm. (B) Merozoite invasion was imaged as in panel A using erythrocytes treated to label Band 3 with CY5 as described in Materials and Methods. (Bottom) Area of Band 3 clearance and subsequent return at the site of parasite invasion (arrows). Green, PfShelph2-GFP: red, Band 3. Scale bars, 5 μm.
Erythrocyte Band 3 was labeled by suspending 3 μl of packed red blood cells (RBCs) in 200 μl of cRPMI and adding 0.5 μl of a CY5-coupled anti-Band 3 antibody that recognizes the extracellular region of Band 3 (19). The suspension of RBCs was incubated with the antibody at room temperature on a rotating mixer for 2 h. After the incubation, the RBCs were washed 3 times with 5 ml of cRPMI and resuspended in 8 ml of cRPMI. This suspension of labeled cells was incubated at 37°C overnight. On the day of the invasion experiment, Percoll-purified schizonts were washed twice with cRPMI and resuspended in a 1-ml volume. One to 10 μl of the schizont suspension was added to the 8-ml suspension of labeled RBCs; 300 μl of this schizont plus CY5-Band 3 RBC suspension was then added to a well of a Lab-Tek II 8-well coverglass chamber previously coated with anti-CD59 antibody.
Band 3 phosphorylation and dephosphorylation assays. (i) CDB3 phosphorylation.
A total of 2.25 μg of glutathione S-transferase (GST)-tagged N-terminal cytoplasmic domain of Band 3 (CDB3) was phosphorylated by incubation with 28 ng recombinant His-Syk (Millipore) in kinase buffer (50 mM Tris, pH 7.5, 150 mM NaCl, 5 mM MgCl2, 5 mM MnCl2, 400 μM ATP, 0.1 mg/ml bovine serum albumin [BSA]) at 37°C for 60 min. The Syk inhibitors Syk IV and R406 were added to 5 μM final concentration to stop Syk activity.
(ii) Phospho-CDB3 dephosphorylation.
Fifty nanograms of phosphorylated CDB3 was incubated with 50 ng of PfShelph2-Strep in phosphatase assay buffer (20 mM Tris, pH 7.4, 400 mM NaCl, 200 μM MnCl2, 1 mM DTT, 0.01% Triton X-100). Calf alkaline phosphatase, 5 units (New England BioLabs), was added to control reaction mixtures.
Immunoblotting.
The primary antibodies used were mouse anti-GST (Santa Cruz; B-14) and rabbit anti-Band 3 phospho-Y8. The secondary antibodies used were anti-rabbit Dylight 680 and anti-mouse Dylight 800 (Cell Signaling). The secondary antibodies were detected using a LiCor Odyssey imager.
Immunoelectron microscopy.
Transgenic parasites expressing PfShelph2-GFP comprised of rupturing late-stage schizonts were mixed with red blood cells for 30 min at 37°C, fixed in glutaraldehyde, and embedded in LR White (Electron Microscopy Sciences). Thin sections (70 nm) were probed with rabbit anti-GFP, followed by secondary-antibody–gold conjugate (10 nm), and contrasted with aqueous uranyl acetate. Images were obtained using a Titan 80-300 electron microscope at 80 kV. For double immunolabeling, sections were probed with antibodies to GFP (to label PfShelph2-GFP) and 10-nm gold-conjugated secondary antibody.
RESULTS
Recombinant PfShelph2 shows in vitro phosphotyrosine phosphatase activity.
As discussed above, prior bioinformatic analyses predicted that PfShelph2 is an active phosphatase (5, 7, 8). To determine if PfShelph2 possesses phosphatase activity, we expressed and purified wild-type (WT) PfShelph2 as an MBP fusion protein from E. coli. As a control, we purified a modified protein with a substitution in a residue predicted to be essential for catalytic activity (D79N). Analysis of the purified products by SDS-PAGE and anti-MBP immunoblotting revealed that both WT and D79N fusion proteins consist of a major MBP fusion band of the expected molecular weight, as well as lower-abundance cleaved fusion products (Fig. 1A). In vitro phosphatase assays using the purified MBP-PfShelph2 showed that the WT, but not the D79N mutant, preparations hydrolyzed the artificial substrate pNPP (Fig. 1B). The D79N mutant did not inhibit the activity of the WT MBP-PfShelph2, demonstrating that lack of D79N activity is not due to the presence of an inhibitor of phosphatase activity in the D79N preparation. The data shown are representative of data obtained from three independent purifications. In addition, WT MBP-PfShelph2 hydrolyzed FDP, another artificial phosphatase substrate (see Fig. S2 in the supplemental material). Consistent with its classification as an Mn2+/Mg2+-dependent phosphatase, MBP-PfShelph2 activity required Mn2+ for activity, with Co2+ also supporting a high level of activity (Fig. 1C). Further, MBP-PfShelph2 exhibited a pH optimum of approximately pH 8.0 to 8.2 (Fig. 1D), indicating that the recombinant MBP-PfShelph2 is most active under slightly alkaline conditions.
Fig 1.

Purification and in vitro activity of MBP-PfShelph2. (A) Coomassie blue-stained SDS-PAGE gels (left; 10 μg protein per lane) and anti-MBP immunoblot (right; 1 μg of protein per lane) of affinity-purified MBP-PfShelph2. The arrowhead indicates full-length MBP-PfShelph2. Molecular masses are indicated on the left in kDa. (B) Activity of purified MBP-PfShelph2. Purified MBP-PfShelph2 WT or D79N mutant enzymes were incubated at a concentration of 10 μg/ml (100-μl total volume) with pNPP at 37°C for 30 min as described in Materials and Methods. The data are representative of three independent experiments, each performed in duplicate. The error bars indicate standard deviations. (C) Divalent metal dependency of MBP-PfShelph2. WT PfShelph2 activity was measured as for panel B with the indicated divalent metals at a concentration of 100 μM. (D) Determination of pH maximum for MBP-PfShelph2. WT MBP-PfShelph2 activity was measured as for panel B in buffer of the indicated pHs.
Having established that MBP-PfShelph2 hydrolyzes two artificial substrates, we examined whether PfShelph2 exhibits specificity for phosphotyrosine residues, as seen for the Shewanella PTPase. We incubated MBP-PfShelph2 with different phosphorylated substrates and used malachite green to measure release of free phosphate (Fig. 2A). Only phosphotyrosine-containing species, both free phosphotyrosine residues (data not shown) and phosphopeptides, served as efficient substrates for the WT MBP-PfShelph2 (Fig. 2A). In contrast, phosphoserine- and phosphothreonine-containing species were not efficiently hydrolyzed. Consistent with its substrate specificity, MBP-PfShelph2 activity was inhibited by sodium orthovanadate (Fig. 2B), a PTPase inhibitor, but not by two S/T phosphatase inhibitors, microcystin and calyculin (Fig. 2C). Figure 2D confirms that the microcystin and calyculin were indeed active in inhibiting protein phosphatase 1 (PP1), an S/T phosphatase that is sensitive to these two inhibitors (20). Together, these data provide strong evidence that, like its Shewanella sp. counterpart enzyme, PfShelph2 is a phosphotyrosine phosphatase.
Fig 2.

MBP-PfShelph2 hydrolyzes tyrosine-phosphorylated peptides. (A) Purified WT MBP-PfShelph2 (20 μg/ml) was incubated with the indicated phosphorylated substrates for 2 h at 37°C. After addition of malachite green reagent, the OD620 was measured and converted to pmol of phosphate. The data are representative of three independent experiments, each performed in duplicate. The error bars indicate standard deviations. (B) MBP-PfShelph2 activity was measured using the pNPP assay in the presence of sodium orthovanadate at the indicated concentrations. (C) MBP-PfShelph2 activity was measured as for panel B in the presence of S/T phosphatase inhibitors at the indicated concentrations. (D) The S/T phosphatase inhibitors were tested using PP1 (Sigma) as in panel A.
PfShelph2-Strep purified from P. falciparum has phosphotyrosine phosphatase activity.
To investigate whether PfShelph2 expressed in P. falciparum also functions as a phosphotyrosine phosphatase, we generated transgenic P. falciparum lines expressing a WT or D79N version of full-length Strep-tag II-tagged PfShelph2. Both fusion proteins were purified from parasite lysate as described in Materials and Methods, resulting in protein preparations shown by silver stain analysis (Fig. 3A).
Fig 3.

PfShelph2-Strep purified from P. falciparum is a tyrosine phosphatase. (A) Silver-stained SDS-PAGE gels (left; 2 μg protein per well) and anti-Strep immunoblot (right; 100 ng protein per well) of affinity-purified WT and D79N PfShelph2-Strep. The arrowhead indicates PfShelph2-Strep. (B) FDP assay of WT-PfShelph2-Strep and D79N-PfShelph2-Strep with 1 μg/ml of enzyme performed as described in Materials and Methods. AFU, arbitrary fluorescence units. (C) Substrate specificity of PfShelph2-Strep determined by malachite green assay as described in Materials and Methods. Gray bars, WT PfShelph2-Strep; white bars, D79N PfShelph2-Strep. The data are representative of three independent experiments, each performed in duplicate. The error bars indicate standard deviations. (D) Immunoblot evaluation of PfShelph2-GFP fusion. Uninfected human RBCs (2 × 106), 3D7 schizonts, and two clones (F11 and F12) of PfShelph2-GFP were analyzed by SDS-PAGE and immunoblotting with anti-GFP antibody. The arrowhead indicates PfShelph2-GFP fusion protein.
In phosphatase assays using FDP as a substrate, we found the D79N-Strep protein possessed no phosphatase activity, while the WT-Strep protein showed a high level of activity relative to the mutant (Fig. 3B). We found similar results using pNPP as a substrate (data not shown). When we incubated the PfShelph2-Strep proteins with phosphorylated peptides, we found that the WT, but not the D79N mutant, hydrolyzed phosphotyrosine, but not phosphothreonine or phosphoserine residues (Fig. 3C). Compared with the MBP-PfShelph2 enzyme, the enzyme purified from parasites had a slightly lower pH optimum, ranging from pH 7.4 to 8.0 (see Fig. S1 in the supplemental material). These results provide further confirmation that, like the previously studied Shelph from Shewanella, PfShelph2 is an active phosphotyrosine phosphatase and suggest that the enzyme functions optimally under neutral to slightly basic conditions.
Endogenously tagged PfShelph2-GFP localizes to a perinuclear region in live schizonts and merozoites.
To further characterize PfShelph2, we tagged the endogenous pfshelph2 gene with GFP using a single-crossover homologous-recombination strategy developed by Klemba et al. (16). The presence of full-length pfshelph2-gfp was confirmed by PCR (see Fig. S3 in the supplemental material), and expression of full-length PfShelph-GFP was established by immunoblotting (Fig. 3D). After validating the PfShelph2-GFP clones for correct modification of the pfshelph2 locus, we synchronized PfShelph2-GFP transgenic parasites to within approximately 6 h and imaged live cells to examine the timing of PfShelph2-GFP expression and its location. Examination of trophozoite stage parasites (∼26 h postinvasion) revealed no GFP fluorescence over background levels (Fig. 4A). In contrast, in schizonts (∼44 h postinvasion), PfShelph2-GFP was clearly detectable in well-defined punctae (Fig. 4A). The youngest parasites in which fluorescence was clearly observed were those with clearly separated individual daughter nuclei. In late schizonts, each spot of fluorescence appeared to be paired with a daughter nucleus. Free merozoites also showed PfShelph2-GFP fluorescence localized to punctae in a position opposite the pole containing the nucleus (Fig. 4A). Close examination of fluorescence in free merozoites revealed that PfShelph2-GFP fluorescence may be composed of one intense point of fluorescence contiguous with a less intense “tail.” Alternatively, the fluorescence pattern may be due to the presence of two or more separate GFP-positive vesicles (Fig. 4A). The PfShelph2-GFP-containing organelle appears to be stable in extracellular merozoites, as greater than 95% of merozoites imaged possess this apical signal (95.6% ± 9.4% [standard deviation {SD}]; n = 153 from 14 independent fields). In young rings (0 to 6 h), we observed no punctate GFP signal. Weak, diffuse signal was seen, suggestive of dissipation of PfShelph2 during either invasion or the first 6 h of intraerythrocytic growth. By the late ring/early trophozoite stage (26 h), PfShelph2 was no longer detectable in the parasite.
Fig 4.

Location of PfShelph2-GFP. (A) PfShelph2-GFP parasites synchronized to within approximately 6 h were Percoll purified and incubated with uninfected RBCs. The parasites were imaged at 44 h (schizont and merozoites), 6 h (ring), and 26 h (late ring/early trophozoite) after the start of the experiment. Blue, Hoechst (nuclei); green, GFP. Scale bars, 2 μm. The images represent projections of 12 optical sections. (B) Percoll-purified PfShelph2-GFP schizonts and merozoites were matured, fixed, and stained for rhoptry (RhopH2 and RON3) markers. (C and D) Merozoites were fixed, permeabilized, and stained for microneme (PTRAMP) (C) and dense granule (RESA) (D) markers as described in Materials and Methods. (B to D) Blue, DAPI (nuclei); red, organelle marker; green, anti-GFP. Scale bars, 1 µM (A and B) and 5 µM (C and D). The images represent single optical sections.
Immunolocalization studies indicate that the distribution of PfShelph2-GFP in merozoites is distinct from markers of the rhoptry, dense granules, and micronemes.
To determine the specific compartment where PfShelph2-GFP is located in merozoites, we performed coimmunolocalization assays with known apical-organelle markers. We first examined whether PfShelph2-GFP colocalizes with the rhoptry markers RhopH2 and PfRON3. We found no colocalization between RhopH2, a rhoptry bulb marker (21), and PfShelph2-GFP in indirect immunofluorescence assays (Fig. 4B). Similarly, PfRON3, a rhoptry neck marker (22), showed no colocalization with PfShelph2 (Fig. 4B). In addition, RESA, a dense granule marker (23), showed no colocalization with PfShelph2 and the distribution of PfShelph2 was also distinct from that of PTRAMP, a marker for the micronemes (24) (Fig. 4C and D). Occasionally, a low level of overlap was seen between the distributions of PfShelph2 and a known marker in a single optical section, but it was not sustained in a consecutive section, and frequently, the major site of PfShelph2 concentration was in a plane distinct from that of the bulk of microneme and dense-granule structures.
To confirm that PfShelph2 resides in membrane structures, we examined its distribution by immunoelectron microscopy. We found that PfShelph2-labeled vesicular structures reside primarily in a perinuclear region (Fig. 5A to C; see Fig. S4 in the supplemental material). PfShelph2 was detected behind the nucleus, away from the rhoptries (Fig. 5A and C), as well as closer to the rhoptries (Fig. 5B). Although PfShelph2 does not colocalize with known markers of the rhoptries, dense granules, and micronemes, vesicles containing PfShelph2 may be a subdomain of granules or micronemes. Further work is required to identify and establish the nature of PfShelph2-labeled structures.
Fig 5.
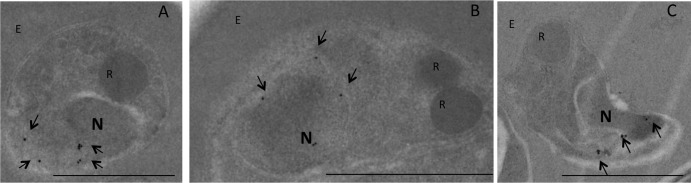
Localization of PfShelph2 in merozoites by immunoelectron microscopy. The immunoelectron micrographs show merozoites probed for PfShelph2-GFP (10-nm gold particles). R, rhoptry; N, nucleus; E, erythrocyte. Bars, 1.0 μm. The arrows in panels A and C show PfShelph2 with a perinuclear pattern on the side of the nucleus opposite that of the rhoptries. The arrows in panel B show an example of PfShelph2 located in a perinuclear pattern on the same side as the rhoptries.
PfShelph2 can access the erythrocyte in newly formed rings and dephosphorylates the major tyrosine-phosphorylated erythrocyte protein, Band 3, in vitro.
We performed immunoelectron microscopy with newly formed rings to investigate the localization of PfShelph2 during and after invasion. As shown in Fig. 6A and B (see Fig. S5 in the supplemental material), in rings aged 0 to 30 min, we detected PfShelph2 within the parasite (shown by dark arrows; sometimes in concentrated foci), as well as in the erythrocyte (light arrows). Further, PfShelph2 is detected within the erythrocyte, just as the parasite is completing invasion (Fig. 6A). Secretory organelle proteins are known to be exported from the merozoite to the erythrocyte in young rings (25, 26). Our results suggest that PfShelph2 may be released either by the invading merozoite into the erythrocyte at terminal stages of entry or by very young rings.
Fig 6.
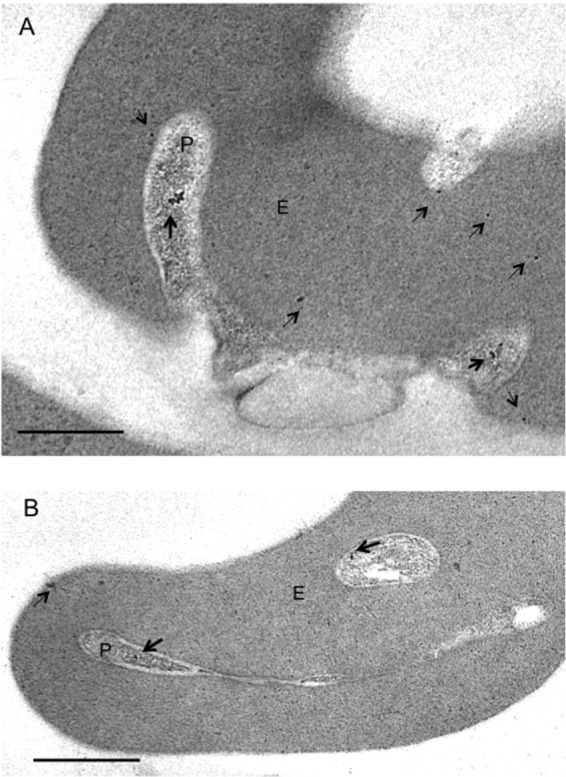
Immunoelectron microscopy indicates the location of PfShelph2 in early-ring-infected erythrocytes. Invasion events and early rings were captured, and thin sections were probed for the PfShelph2 location as described in Materials and Methods. Gold particles (10 nm) indicate the location of PfShelph2. Dark and light arrows, respectively, mark PfShelph in the parasite and erythrocyte. (A) Parasite (P) during entry into the erythrocyte (E) and in the process of ring formation. (B) Newly formed intracellular ring stage parasite (P) in erythrocyte (E). Scale bars, 0.5 um. Control sections probed with secondary antibodies alone showed no gold particles (see Fig. S6 in the supplemental material).
As PfShelph2 appears to gain access to the erythrocyte cytosol, we hypothesized that PfShelph2 may target a substrate within the host cell. Band 3 is a major tyrosine-phosphorylated protein in the erythrocyte, and tyrosine phosphorylation alters several measures of its interaction with the erythrocyte cytoskeleton (27). In an in vitro phosphatase assay, we found that purified PfShelph2, but not the inactive variant, dephosphorylates the tyrosine-phosphorylated N-terminal cytoplasmic domain of Band 3 (Fig. 7). Our data suggest that PfShelph2 is translocated into the erythrocyte cytoplasm during the invasion process and that the phosphatase may act to dephosphorylate Band 3.
Fig 7.
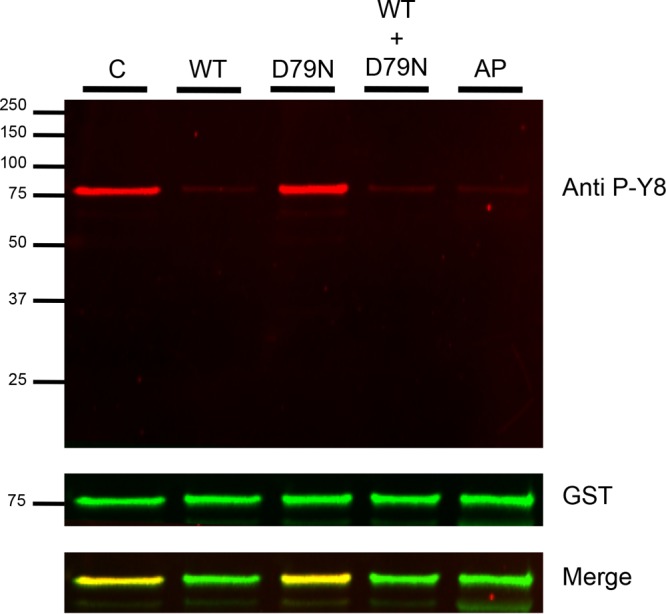
PfShelph2 dephosphorylation of the phospho-Band 3 N-terminal cytoplasmic domain. Fifty nanograms of phosphorylated CDB3 was incubated in the absence of any phosphatase (C) or with 50 ng of PfShelph2 (WT), the D79N inactive mutant (D79N), both WT and D79N, or alkaline phosphatase (AP) for 30 min. The reaction was stopped by the addition of 5× SDS buffer containing 1 M DTT. Samples were incubated at 55°C for 15 min and then analyzed by SDS-PAGE (12% gel), followed by immunoblotting with both anti-Band 3 phosphotyrosine 8 (red) and anti-GST (green). The CDB3 sequence is MEELQDDYEDMMEENLEQEEYEDPDIPESQMEEPAAHDTEATATDYHTTS (tyrosine 8 and tyrosine 21 are in boldface).
Band 3 dynamics and phosphorylation in malaria invasion.
Phosphorylation of Band 3 is known to occur during malaria infection (28). It reduces the affinity of Band 3 for the cytoskeleton and decreases its attachment to the submembrane cytoskeleton (27). While Band 3 is the most abundant protein in the erythrocyte membrane, it is excluded from the malarial parasitophorous vacuole membrane (PVM) (29, 30). Thus, by some unknown mechanism, Band 3 must be excluded from the PVM, while other host erythrocyte membrane components are incorporated into the PVM during invasion. The timing and mechanism of Band 3 exclusion from the PVM during invasion and the potential role of Band 3 phosphorylation in these events are largely unknown.
To better understand Band 3 dynamics, we labeled Band 3 on erythrocytes with a fluorescently labeled antibody directed to the extracellular region of Band 3 and observed malarial invasion in real time. Malarial invasion proceeds through a series of steps, starting with an initial binding event that is reversible. After the initial binding to the erythrocyte surface, the extracellular merozoite must orient itself so that its apical end faces the host erythrocyte. Prior to that, the merozoite can interact with the erythrocyte and deform it without stable attachment. Once attachment is observed, entry is a rapid process, accomplished in ∼15 to 60 s. As shown in Fig. 8, and in Fig. S6 and Movie S1 in the supplemental material, within 1 to 2 s of parasite attachment to the erythrocyte, Band 3 is cleared from the merozoite-erythrocyte junction. As the parasite enters, Band 3 is excluded from the newly forming parasitophorous vacuole. However, when the parasite is entirely intracellular (23 s), Band 3 is restored to the junction between the newly formed vacuole and the erythrocyte membrane. The data in Fig. 8 suggest that the parasite induces clearance of Band 3 within the first 1 to 2 s after attachment. Later, when the new vacuole closes around the entering parasite, Band 3, possibly via a parasite-induced process, returns to the site of invasion.
Given these data, one might reasonably hypothesize that the clearance of Band 3 at the invasion junction involves an event or events that lead to its disassociation from the underlying skeleton. Phosphorylation has been shown to influence the interaction between Band 3 and the cytoskeleton (27). Band 3 tyrosine phosphorylation affects its association with the submembrane cytoskeleton (27) and may play a role in malaria parasite invasion. We hypothesized that dephosphorylation of Band 3 may be important to restore erythrocyte stability and that PfShelph2 may play a role in dephosphorylation of Band 3. To determine if PfShelph2 might contribute to the return of Band 3 to the site of invasion, we examined the dynamics of its discharge in relation to Band 3 dynamics during invasion. While Band 3 clears from the contact site within 1 to 2 s of parasite attachment, PfShelph2 remains concentrated in the merozoite (Fig. 8B; see Fig. S7 in the supplemental material). Thus, Band 3 clearance from the invasion junction precedes any apparent dynamic change in the distribution of PfShelph2.
The dynamics of PfShelph2 release suggest that it is released after parasite attachment and during biogenesis of the new vacuole.
Having observed that PfShelph2 is redistributed after Band 3 clearance and before the return of Band 3 to the site of invasion, we proceeded to further study the timing of PfShelph2 redistribution by examining more invasion events. As shown in Fig. 8A, we observed that PfShelph2-GFP retains its apical punctate pattern during initial parasite-host cell interaction, apical orientation, attachment, and initial movement into the host cell (Fig. 8A; see Fig. S6 and Movie S1 in the supplemental material). Analysis of 15 successful invasion events (Table 1) suggests that the time taken for a merozoite to complete invasion ranges from ∼10 to 30 s. PfShelph2 discharge occurs approximately halfway through the invasion process or later, with an average time of 13.6 (±5.3; n = 15) seconds from initial movement of the merozoite into the host cell (Table 1). This also corresponds to a position where approximately half of the merozoite is engulfed by the newly forming parasitophorous vacuole. In the same frames where we saw invading merozoites, we saw no loss of apical GFP fluorescence in extracellular merozoites, arguing against photobleaching as the reason for loss of apical GFP signal in invading merozoites (Fig. 8A; see Fig. S6 in the supplemental material). Moreover, in the presence of cytochalasin, which allows parasites to attach to the erythrocyte but blocks the parasite's actin-myosin motor and thus subsequent steps of invasion (31), PfShelph2 was not released or photobleached in attached parasites despite continuous imaging over 100 s (Fig. 9). These experiments suggest that parasite attachment to the erythrocyte is not sufficient to release PfShelph2 and that PfShelph2 release occurs after activation of the parasite actin-myosin motor.
Table 1.
Dynamics of PfShelph1-GFP discharge during merozoite invasion of erythrocytes
| Event no.a | Time until loss of GFP signal (s)b | Time taken for completion of invasion (s)c |
|---|---|---|
| 1 | 18.60 | 21.24 |
| 2 | 6.57 | 14.47 |
| 3 | 7.91 | 17.10 |
| 4 | 7.29 | 10.93 |
| 5 | 20.04 | 23.68 |
| 6 | 10.93 | 18.21 |
| 7 | 9.67 | 15.19 |
| 8 | 16.58 | 17.96 |
| 9 | 15.20 | 27.63 |
| 10 | 9.46 | 21.28 |
| 11 | 24.83 | 28.38 |
| 12 | 11.82 | 18.92 |
| 13 | 11.82 | 21.28 |
| 14 | 16.55 | 20.10 |
| 15 | 16.56 | 21.29 |
Data are shown for 15 merozoite entry events, measured as described in Materials and Methods.
Average, 13.59 ± 5.27 s.
Average, 19.84 ± 4.64 s.
Fig 9.
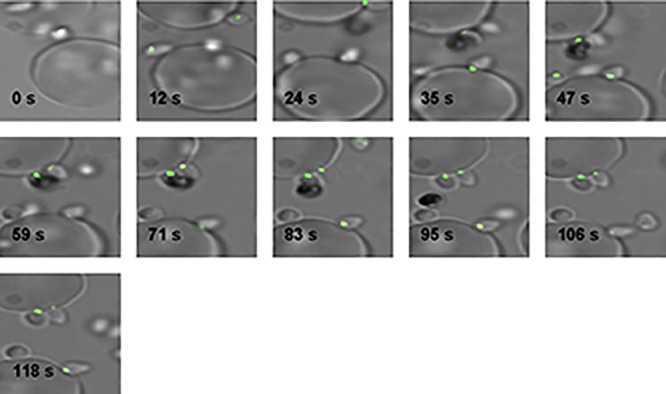
PfShelph2-GFP is not discharged from merozoites in cytochalasin D-treated cultures. Merozoite invasion was imaged as described in Materials and Methods in the presence of 1 μM cytochalasin D (Sigma).
Together, these data strongly support the idea that PfShelph2 dissipation needs active secretion by the parasite and occurs well after parasite attachment and Band 3 clearance. This is consistent with PfShelph2 location in vesicles distinct from micronemes, whose contents are observed to be released early during the invasion process (31–33). PfShelph2 vesicles may be more closely akin to the specialized granules whose contents are transferred to the erythrocyte well after attachment, in the second half of the entry process, and in early-ring-stage parasites.
DISCUSSION
Host cell invasion is an essential step in the ability of Plasmodium spp. to cause malaria and thus represents an attractive step to target with drugs and vaccines. A detailed understanding of invasion requires extensive characterization of the parasite and host factors involved, including evaluating the activity and subcellular localization of the putative kinases and phosphatases identified in the genomes of these organisms. While a number of malaria parasite-derived kinases (34, 35) and phosphatases (4, 6, 36, 37) have been identified, the activities and functions of most of the kinases and phosphatases of Plasmodium sp. parasites remain to be fully delineated. In the present study, we characterize PfShelph2. Specifically we elucidate several of its in vitro biochemical properties and investigate its subcellular localization, release, and export to the erythrocyte during invasion. In the process, we also define the timing of its redistribution relative to host erythrocyte Band 3 dynamics, further contributing to the body of knowledge regarding host cell membrane protein dynamics during the invasion process.
In vitro, both MBP-PfShelph2 purified from E. coli and PfShelph2-Strep purified from P. falciparum hydrolyze two artificial substrates, as well as the more biologically relevant phosphotyrosine in peptides and free phosphotyrosine. Mutation of a predicted active-site residue abolishes activity, demonstrating that the activity we detect in our purified preparations depends on the presence of WT PfShelph2. As predicted by bioinformatic analysis, PfShelph2 activity requires the presence of divalent metal ions, specifically Mn2+ or Co2+. The pH optimum of 7.4 to 8.0 for PfShelph2 suggests that PfShelph2 acts in a subcellular location with an alkaline pH. Also consistent with PfShelph2's phosphotyrosine phosphatase activity is the observation that two serine/threonine phosphatase inhibitors had no effect on PfShelph2 activity while sodium orthovanadate at a commonly used concentration inhibits PfShelph2 activity. These findings suggest that, with respect to inhibitors, the active site of PfShelph2 more closely resembles that of PTPases than that of S/T phosphatases. Most studied PTPases utilize a conserved catalytic-site cysteine residue (38), while S/T phosphatases utilize a divalent metal ion-based mechanism of catalysis (39). The lack of a catalytic-site cysteine residue in PfShelph2, in addition to its Mn2+ dependence, suggests that PfShelph2 hydrolyzes phosphotyrosine via a mechanism more similar to that of S/T phosphatases than to that of PTPases. This is in accordance with the mechanism proposed for the previously studied Shewanella phosphotyrosine phosphatase (11).
Since the first submission of our study, Patzewitz et al. have reported that the Plasmodium berghei PfShelph1 homologue SHLP1 localizes to the endoplasmic reticulum (ER) throughout all asexual and sexual stages, is important in ookinete development in mosquito stages, and is thus needed for parasite transmission (40). Our work delineates a Shelph involved in blood-stage infection. Like the original bacterial Shewanella PTPase previously studied, PfShelph2 hydrolyzes phosphotyrosine residues while having little or no activity against phosphoserine or phosphothreonine (9, 10). Including our current study, two Shelphs have now been shown to possess PTPase activity, as opposed to phosphoserine/phosphothreonine activity. This gives further support to the idea that members of this class of enzymes possess conserved residues that alter the substrate specificity of their parent phosphatase family and thus make them potentially unique targets for pharmacologic intervention.
In addition to its interesting substrate specificity, PfShelph2 possesses a predicted ER-targeting signal sequence, suggesting that the protein is recruited to the secretory pathway. We examined the localization of endogenously tagged PfShelph2-GFP using live-cell microscopy and observed that that PfShelph2-GFP traffics to vesicles in schizonts and merozoites. This is consistent with previous work demonstrating that an exogenous pfshelph2-gfp transgene under the control of the AMA-1 promoter shows punctate fluorescence in schizonts (12). These authors also suggested that PfShelph2 plays a role in invasion, but neither its function, subcellular location, dynamics, or timing of action was studied.
Consistent with the proposed role in invasion, we found that PfShelph2 is distributed in a punctate pattern in late schizonts and merozoites. Immunofluorescence studies showed that these vesicles did not colocalize with the two rhoptry markers RhopH2 and RON3, the dense granule marker RESA, or the microneme marker PTRAMP. It is possible that PfShelph2 resides in a subcompartment of an apical organelle whose contents are released late in the invasion process. As our studies relied on following the localization of a GFP-tagged PfShelph2, it is possible that the GFP affected the targeting of PfShelph2. However, we think that it is unlikely that the C-terminal GFP tag promotes release into the erythrocyte. The tight folding of GFP might in fact retard export, a process that usually requires protein unfolding. Furthermore, tagging the gene on the C terminus with Strep-tag II does not abrogate biochemical activity, and the GFP-labeled protein is dynamic and is rapidly released during invasion, dependent on the parasite's actin-myosin motor. While we could not follow the localization of PfShelph2-GFP after its release in live cells, immunoelectron microscopy suggested that PfShelph2-GFP translocates to the host cell cytosol. The translocation of merozoite organellar components to the erythrocyte cytosol has been observed before, as in the cases of Rhoph2 (41) and RESA (26), and thus, PfShelph2 may represent another parasite antigen that gains access to the erythrocyte cytosol and, once there, may play a role in phosphotyrosine-based signaling events.
To further characterize the nature of the PfShelph2-containing organelle, we examined PfShelph2-GFP movement with real-time microscopy. Real-time observation of invasion of PfShelph2-GFP-expressing merozoites demonstrated that the punctate organelle in which the enzyme resides is likely discharged or redistributed at a late stage of invasion, after the point at which the merozoite is halfway inside the host cell and before the point at which the posterior pole of the merozoite is fully enclosed by the parasitophorous vacuole. One study defines this as a “second phase” of invasion (18) in which the merozoite moves inward into its nascent vacuole.
While our studies with PfShelph2-GFP made us strongly suspect that the PfShelph2-containing organelle redistributes in the second phase of invasion, we sought to more precisely resolve the timing of PfShelph2-GFP redistribution, as well as its redistribution relative to host erythrocyte membrane protein dynamics. To do this, we fluorescently labeled Band 3 on erythrocytes and thus were able to visualize the clearance of a major erythrocyte membrane protein and its subsequent return to the site of invasion. These studies convincingly show that the punctate distribution of PfShelph2-GFP is lost after the initial clearance of Band 3 and before its return to the site of invasion. To our knowledge, clearance of Band 3 from the site of invasion had not been visualized in real time prior to our study, and this may provide a useful tool to define the temporal relationship of other steps in the invasion process relative to host erythrocyte Band 3 dynamics.
The timing of PfShelph2-GFP redistribution and our identification of the phosphatase in the host erythrocyte cytosol lead us to propose a model in which phosphorylation of Band 3 plays a role in regulating the integrity of the erythrocyte cytoskeleton during invasion (Fig. 10). We suggest that at the start of the invasion process a parasite or host tyrosine kinase phosphorylates Band 3, promoting its dissociation from ankyrin and the spectrin-based cytoskeleton. This results in a defect in the cytoskeleton through which the merozoite can move into the erythrocyte as it drives the formation of the parasitophorous vacuole. Later, as the merozoite is nearly or entirely within the erythrocyte and enveloped in the parasitophorous vacuole membrane, it releases PfShelph2 into the erythrocyte cytosol, where its phosphatase activity acts on phosphorylated Band 3 and restores Band 3's affinity for ankyrin, thus promoting the return of erythrocyte cytoskeletal components to the site of invasion.
Fig 10.

Model of erythrocyte cytoskeleton dynamics during merozoite invasion and a possible role for PfShelph2 in regulating Band 3 dynamics. The merozoite attaches to the erythrocyte via adhesion ligands on its surface and in micronemes. An unknown parasite or erythrocyte kinase (blue diamonds) phosphorylates Band 3, reducing its affinity for the erythrocyte cytoskeleton through ankyrin. This allows the merozoite to create a defect in the erythrocyte cytoskeleton through which it can enter the erythrocyte as it drives the formation of the parasitophorous vacuole, which is derived from host and parasite components, including components discharged from the rhoptries. As the merozoite enters the erythrocyte, it discharges PfShelph2 into the erythrocyte, where it can dephosphorylate Band 3 and thus repair the defect in the erythrocyte cytoskeleton created by the invading merozoite.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge Andrew R. Osborne Institute of Structural and Molecular Biology, Department of Biological Sciences, Birkbeck, University of London, London, United Kingdom, for help in designing experiments and in editing the manuscript and for development of the pA171 vector.
This work was supported by NRSA F30 HL094042-01 to S.F.-P.; MRC (U117532067) to A.A.H; and NIH grants R01 AI039071 and 5 R03 MH 96573 to K.H. and P01 HL078826 to K.H., N.M., and A.A.H.
Footnotes
Published ahead of print 3 July 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/EC.00027-13.
REFERENCES
- 1.Breman JG. 2001. The ears of the hippopotamus: manifestations, determinants, and estimates of the malaria burden. Am. J. Trop. Med. Hyg. 64:1–11 [DOI] [PubMed] [Google Scholar]
- 2.Sachs J, Malaney P. 2002. The economic and social burden of malaria. Nature 415:680–685 [DOI] [PubMed] [Google Scholar]
- 3.WHO 2008. World malaria report. WHO, Geneva, Switzerland [Google Scholar]
- 4.Kutuzov MA, Andreeva AV. 2008. Protein Ser/Thr phosphatases of parasitic protozoa. Mol. Biochem. Parasitol. 161:81–90 [DOI] [PubMed] [Google Scholar]
- 5.Andreeva AV, Kutuzov MA. 2008. Protozoan protein tyrosine phosphatases. Int. J. Parasitol. 38:1279–1295 [DOI] [PubMed] [Google Scholar]
- 6.Bajsa J, Duke SO, Tekwani BL. 2008. Plasmodium falciparum serine/threonine phosphoprotein phosphatases (PPP): from housekeeper to the ‘holy grail'. Curr. Drug Targets 9:997–1012 [DOI] [PubMed] [Google Scholar]
- 7.Wilkes JM, Doerig C. 2008. The protein-phosphatome of the human malaria parasite Plasmodium falciparum. BMC Genomics 9:412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Andreeva AV, Kutuzov MA. 2004. Widespread presence of “bacterial-like” PPP phosphatases in eukaryotes. BMC Evol. Biol. 4:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tsuruta H, Aizono Y. 1999. Enzymatical properties of psychrophilic phosphatase I. J. Biochem. 125:690–695 [DOI] [PubMed] [Google Scholar]
- 10.Tsuruta H, Aizono Y. 2003. Catalytic efficiency and some structural properties of cold-active protein-tyrosine-phosphatase. J. Biochem. 133:225–230 [DOI] [PubMed] [Google Scholar]
- 11.Tsuruta H, Mikami B, Aizono Y. 2005. Crystal structure of cold-active protein-tyrosine phosphatase from a psychrophile, Shewanella sp. J. Biochem. 137:69–77 [DOI] [PubMed] [Google Scholar]
- 12.Hu G, Cabrera A, Kono M, Mok S, Chaal BK, Haase S, Engelberg K, Cheemadan S, Spielmann T, Preiser PR, Gilberger TW, Bozdech Z. 2010. Transcriptional profiling of growth perturbations of the human malaria parasite Plasmodium falciparum. Nat. Biotechnol. 28:91–98 [DOI] [PubMed] [Google Scholar]
- 13.Trager W, Jensen JB. 1976. Human malaria parasites in continuous culture. Science 193:673–675 [DOI] [PubMed] [Google Scholar]
- 14.Haldar K, Elmendorf HG, Das A, Li WL, Ferguson DJ, Elford BC. 1994. In vitro secretory assays with erythrocyte-free malaria parasites. Methods Cell Biol. 45:221–246 [DOI] [PubMed] [Google Scholar]
- 15.van Ooij C, Tamez P, Bhattacharjee S, Hiller NL, Harrison T, Liolios K, Kooij T, Ramesar J, Balu B, Adams J, Waters AP, Janse CJ, Haldar K. 2008. The malaria secretome: from algorithms to essential function in blood stage infection. PLoS Pathog. 4:e1000084. 10.1371/journal.ppat.1000084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Klemba M, Beatty W, Gluzman I, Goldberg DE. 2004. Trafficking of plasmepsin II to the food vacuole of the malaria parasite Plasmodium falciparum. J. Cell Biol. 164:47–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fidock DA, Wellems TE. 1997. Transformation with human dihydrofolate reductase renders malaria parasites insensitive to WR99210 but does not affect the intrinsic activity of proguanil. Proc. Natl. Acad. Sci. U. S. A. 94:10931–10936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gilson PR, Crabb BS. 2009. Morphology and kinetics of the three distinct phases of red blood cell invasion by Plasmodium falciparum merozoites. Int. J. Parasitol. 39:91–96 [DOI] [PubMed] [Google Scholar]
- 19.Salomao M, Zhang X, Yang Y, Lee S, Hartwig JH, Chasis JA, Mohandas N, An X. 2008. Protein 4.1R-dependent multiprotein complex: new insights into the structural organization of the red blood cell membrane. Proc. Natl. Acad. Sci. U. S. A. 105:8026–8031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Swingle M, Ni L, Honkanen RE. 2007. Small-molecule inhibitors of Ser/Thr protein phosphatases: specificity, use and common forms of abuse. Methods Mol. Biol. 365:23–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ling IT, Florens L, Dluzewski AR, Kaneko O, Grainger M, Yim Lim BY, Tsuboi T, Hopkins JM, Johnson JR, Torii M, Bannister LH, Yates JR, III, Holder AA, Mattei D. 2004. The Plasmodium falciparum clag9 gene encodes a rhoptry protein that is transferred to the host erythrocyte upon invasion. Mol. Microbiol. 52:107–118 [DOI] [PubMed] [Google Scholar]
- 22.Ito D, Han ET, Takeo S, Thongkukiatkul A, Otsuki H, Torii M, Tsuboi T. 2011. Plasmodial ortholog of Toxoplasma gondii rhoptry neck protein 3 is localized to the rhoptry body. Parasitol. Int. 60:132–138 [DOI] [PubMed] [Google Scholar]
- 23.Pei X, Guo X, Coppel R, Bhattacharjee S, Haldar K, Gratzer W, Mohandas N, An X. 2007. The ring-infected erythrocyte surface antigen (RESA) of Plasmodium falciparum stabilizes spectrin tetramers and suppresses further invasion. Blood 110:1036–1042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thompson J, Cooke RE, Moore S, Anderson LF, Janse CJ, Waters AP. 2004. PTRAMP; a conserved Plasmodium thrombospondin-related apical merozoite protein. Mol. Biochem. Parasitol. 134:225–232 [DOI] [PubMed] [Google Scholar]
- 25.Culvenor JG, Day KP, Anders RF. 1991. Plasmodium falciparum ring-infected erythrocyte surface antigen is released from merozoite dense granules after erythrocyte invasion. Infect. Immun. 59:1183–1187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Foley M, Tilley L, Sawyer WH, Anders RF. 1991. The ring-infected erythrocyte surface antigen of Plasmodium falciparum associates with spectrin in the erythrocyte membrane. Mol. Biochem. Parasitol. 46:137–147 [DOI] [PubMed] [Google Scholar]
- 27.Ferru E, Giger K, Pantaleo A, Campanella E, Grey J, Ritchie K, Vono R, Turrini F, Low PS. 2011. Regulation of membrane-cytoskeletal interactions by tyrosine phosphorylation of erythrocyte band 3. Blood 117:5998–6006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pantaleo A, Ferru E, Carta F, Mannu F, Giribaldi G, Vono R, Lepedda AJ, Pippia P, Turrini F. 2010. Analysis of changes in tyrosine and serine phosphorylation of red cell membrane proteins induced by P. falciparum growth. Proteomics 10:3469–3479 [DOI] [PubMed] [Google Scholar]
- 29.Dluzewski AR, Fryer PR, Griffiths S, Wilson RJ, Gratzer WB. 1989. Red cell membrane protein distribution during malarial invasion. J. Cell Sci. 92:691–699 [DOI] [PubMed] [Google Scholar]
- 30.Lauer S, VanWye J, Harrison T, McManus H, Samuel BU, Hiller NL, Mohandas N, Haldar K. 2000. Vacuolar uptake of host components, and a role for cholesterol and sphingomyelin in malarial infection. EMBO J. 19:3556–3564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miller LH, Aikawa M, Johnson JG, Shiroishi T. 1979. Interaction between cytochalasin B-treated malarial parasites and erythrocytes. Attachment and junction formation. J. Exp. Med. 149:172–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aikawa M, Miller LH, Rabbege JR, Epstein N. 1981. Freeze-fracture study on the erythrocyte membrane during malarial parasite invasion. J. Cell Biol. 91:55–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Riglar DT, Richard D, Wilson DW, Boyle MJ, Dekiwadia C, Turnbull L, Angrisano F, Marapana DS, Rogers KL, Whitchurch CB, Beeson JG, Cowman AF, Ralph SA, Baum J. 2011. Super-resolution dissection of coordinated events during malaria parasite invasion of the human erythrocyte. Cell Host Microbe 9:9–20 [DOI] [PubMed] [Google Scholar]
- 34.Lim DC, Cooke BM, Doerig C, Saeij JP. 2012. Toxoplasma and Plasmodium protein kinases: roles in invasion and host cell remodelling. Int. J. Parasitol. 42:21–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Doerig C, Billker O, Haystead T, Sharma P, Tobin AB, Waters NC. 2008. Protein kinases of malaria parasites: an update. Trends Parasitol. 24:570–577 [DOI] [PubMed] [Google Scholar]
- 36.Kumar R, Musiyenko A, Cioffi E, Oldenburg A, Adams B, Bitko V, Krishna SS, Barik S. 2004. A zinc-binding dual-specificity YVH1 phosphatase in the malaria parasite, Plasmodium falciparum, and its interaction with the nuclear protein, pescadillo. Mol. Biochem. Parasitol. 133:297–310 [DOI] [PubMed] [Google Scholar]
- 37.Pendyala PR, Ayong L, Eatrides J, Schreiber M, Pham C, Chakrabarti R, Fidock DA, Allen CM, Chakrabarti D. 2008. Characterization of a PRL protein tyrosine phosphatase from Plasmodium falciparum. Mol. Biochem. Parasitol. 158:1–10 [DOI] [PubMed] [Google Scholar]
- 38.Tonks NK. 2005. Redox redux: revisiting PTPs and the control of cell signaling. Cell 121:667–670 [DOI] [PubMed] [Google Scholar]
- 39.Barford D. 1996. Molecular mechanisms of the protein serine/threonine phosphatases. Trends Biochem. Sci. 21:407–412 [DOI] [PubMed] [Google Scholar]
- 40.Patzewitz EM, Guttery DS, Poulin B, Ramakrishnan C, Ferguson DJ, Wall RJ, Brady D, Holder AA, Szoor B, Tewari R. 2013. An ancient protein phosphatase, SHLP1, is critical to microneme development in Plasmodium ookinetes and parasite transmission. Cell Rep. 3:622–629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hiller NL, Akompong T, Morrow JS, Holder AA, Haldar K. 2003. Identification of a stomatin orthologue in vacuoles induced in human erythrocytes by malaria parasites. A role for microbial raft proteins in apicomplexan vacuole biogenesis. J. Biol. Chem. 278:48413–48421 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.