

Abstract
A fundamental aspect of most infectious diseases is the need for the invading microbe to proliferate in the host. However, little is known about the metabolic pathways required for pathogenic microbes to colonize and persist in their hosts. In this study, we used RNA sequencing (RNA-seq) to generate a high-resolution transcriptome of the opportunistic pathogen Aggregatibacter actinomycetemcomitans in vivo. We identified 691 A. actinomycetemcomitans transcriptional start sites and 210 noncoding RNAs during growth in vivo and as a biofilm in vitro. Compared to in vitro biofilm growth on a defined medium, ∼14% of the A. actinomycetemcomitans genes were differentially regulated in vivo. A disproportionate number of genes coding for proteins involved in metabolic pathways were differentially regulated in vivo, suggesting that A. actinomycetemcomitans in vivo metabolism is distinct from in vitro growth. Mutational analyses of differentially regulated genes revealed that formate dehydrogenase H and fumarate reductase are important A. actinomycetemcomitans fitness determinants in vivo. These results not only provide a high-resolution genomic analysis of a bacterial pathogen during in vivo growth but also provide new insight into metabolic pathways required for A. actinomycetemcomitans in vivo fitness.
INTRODUCTION
Over 100 years ago, Louis Pasteur et al. emphasized the importance of understanding the metabolic processes involved in bacterial colonization and persistence during infection (1). However, progress in this area has proven challenging, in part because of the difficulty in characterizing the nutritional content of infection sites. One approach that has been successful in probing the growth environment of the infection site is transcriptomics using microarrays and RNA sequencing (RNA-seq). Microarrays have yielded insights into the carbon sources and metabolic pathways used by pathogens during infection (2–5), although these studies are technically challenging because of the large amounts of host RNA often present in disease samples. In contrast, high-throughput RNA-seq provides a robust tool for global gene expression analyses in vivo since it can be performed with small amounts of RNA and bacterial and host gene expression can be separated computationally (6). However, to date, very few in vivo RNA-seq studies of mammalian pathogens have been performed. One study revealed significant insight into the in vivo physiology of Vibrio cholerae during infection (7); however, the undefined medium used as an in vitro control limited the conclusions that could be drawn about the metabolic pathways active in vivo.
Our laboratory has studied nutrition in numerous pathogenic bacteria, including the opportunistic human pathogen Aggregatibacter actinomycetemcomitans (8–14). A. actinomycetemcomitans is a Gram-negative, facultative anaerobe that resides in the oral cavities of humans and old-world primates. Specifically, A. actinomycetemcomitans colonizes the subgingival crevice, defined as the area around the tooth bounded by the gingival epithelium on one side and the tooth surface on the other. A. actinomycetemcomitans is the proposed causative agent of localized aggressive periodontitis (15, 16), an acute disease characterized by massive tissue destruction and tooth loss. A. actinomycetemcomitans also causes extraoral infections, including abscess infections (17–19), and our laboratory has used a murine abscess infection model to characterize A. actinomycetemcomitans genes required for growth in vivo (14). In this study, we used high-resolution transcriptomics to examine the physiology of A. actinomycetemcomitans during growth in the murine abscess. These analyses revealed significant insights into the transcriptional organization of the A. actinomycetemcomitans genome, including transcription start sites (TSS) and noncoding RNAs (ncRNAs). In addition, numerous metabolic genes displaying enhanced transcription during in vivo growth were identified. Targeted mutagenesis of these differentially regulated genes revealed roles for fermentative metabolism and anaerobic respiration for in vivo persistence.
MATERIALS AND METHODS
Bacterial growth conditions.
A. actinomycetemcomitans strain 624, a rough serotype A clinical isolate, was used in this study. A. actinomycetemcomitans 624 was routinely cultured in brain heart infusion (BHI) broth or tryptic soy broth supplemented with 0.5% yeast extract (TSBYE) while shaking at 150 rpm in a 5% CO2 atmosphere at 37°C. For mutant A. actinomycetemcomitans, BHI and TSBYE were supplemented with 50 μg/ml spectinomycin. For RNA-seq, A. actinomycetemcomitans was grown in chemically defined, modified Socransky medium supplemented with 20 mM glucose and 50 mM morpholinepropanesulfonic acid (MOPS), pH 7.2 (CDM) (8, 20), while shaking at 150 rpm in a 5% CO2 atmosphere at 37°C. Prior to biofilm growth, overnight liquid cultures of A. actinomycetemcomitans were diluted 1:2 with fresh CDM, grown for 2 h, centrifuged at >16,000 × g, resuspended in CDM, and inoculated onto CDM agar plates as colony biofilms (21). Colony biofilms were grown for 4 h on CDM agar, transferred to fresh CDM agar for 4 more h of growth, and collected in RNAlater solution.
Determination of generation times.
Wild-type A. actinomycetemcomitans 624 and the ΔfdhF1F2, ΔfrdABCD, and Δpfl mutants were grown while shaking at 150 rpm in liquid CDM aerobically at 37°C in a 5% CO2 atmosphere and statically in an anaerobic chamber (Coy). Because A. actinomycetemcomitans 624 grows in large aggregates in vitro that cannot be adequately dispersed, cellular protein concentrations were determined with a Bradford assay (Bio-Rad) as a proxy for cellular growth and for calculation of generation times. To prepare lysates for a Bradford assay, cells were removed throughout the exponential growth phase, pelleted in a microcentrifuge, resuspended in 6 M urea, and boiled for 30 min at 100°C to lyse the cells. Generation times were calculated by plotting cellular protein concentrations over time.
A. actinomycetemcomitans murine abscess infections.
Three-day murine abscess infections were established with wild-type and mutant A. actinomycetemcomitans strains as described previously (14). Severity of infections was determined by plate counting on BHI agar and on BHI agar supplemented with 50 μg/ml spectinomycin for the wild-type and mutant A. actinomycetemcomitans strains, respectively. The experiments described here were conducted according to the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The Texas Tech University Health Sciences Center Institutional Animal Care and Use Committee approved the protocol (09039).
RNA isolation and high-throughput sequencing library preparation.
A. actinomycetemcomitans biofilm cells stored in RNAlater solution were centrifuged at 5,000 × g, resuspended in 100 μl of 10 mg/ml lysozyme in TE buffer, and incubated for 10 min at 25°C. After lysozyme treatment, 200 μl of phosphate-buffered saline (PBS) was added to each lysed cell solution and further lysis was carried out by mechanical disruption of each sample four times for 30 s at maximum speed in a Mini-Beadbeater (Biospec Products) and lysed cell solutions were stored in an ice bath for 2 min between bead beatings. Total RNA was isolated from the resulting lysed cells with 1 ml RNA Bee solution (Tel-Test) according to the manufacturer's protocol. Murine abscesses were resuspended immediately in 1 ml RNA Bee and bead beaten four times as described above, and RNA was purified from pooled abscesses (see Table S1 in the supplemental material). DNA contamination was removed from RNA samples by treating 5 μg of total RNA with 2.5 U of RQ1 DNase (Promega) for 30 min at 37°C, and RNA was purified with 500 μl of RNA Bee solution. DNA removal was verified by PCR amplification of the A. actinomycetemcomitans clpX protease gene from DNase-treated RNA (see Table S7). Bacterial and host rRNAs were depleted by commercially available capture methods (Ambion MICROBExpress and MICROBEnrich kits) or enzymatic degradation (Epicentre Terminator 5′-monophosphate-dependent RNase) according to the manufacturers' protocols. To produce RNA between 20 and 500 nucleotides (nt) that is optimal for sequencing, 1 μg of an rRNA-depleted RNA sample was treated with NEBNext RNA fragmentation buffer (NEB). Strand-specific cDNA libraries were prepared with commercially available T4 RNA ligase-based kits and sequenced on the SOLiD V4 and Illumina HiSeq2000 platforms. Both SOLiD and Illumina cDNA libraries were subjected to polyacrylamide gel extraction to purify cDNA between 130 and 500 nt, removing library adapters and primers from the samples. For a summary of the sequencing library preparation kits and sequencing outputs used, see Table S1 in the supplemental material.
Computational methods.
The A. actinomycetemcomitans D7S-1 reference genome (GenBank accession no. ADCF01000001.1) was used for RNA-seq read alignment (22). The 50-bp single-end SOLiD sequence reads were aligned in colorspace to the reference genome by using SHRiMP version 2.2.1 to produce sam format read alignment files (23). The longer ∼100-bp Illumina sequence reads were prefiltered with FLEXBAR version 2.0 (24) to remove library adapter sequences from the sequence reads (Illumina index sequencing primer sequence, 5′-AGATCGGAAGAGCACACGTCTGAACTCCAGTCAC-3′; Illumina 3′ adapter sequence, 5′-TCGTATGCCGTCTTCTGCTTG-3′) and improve alignment with the reference genome. Filtered Illumina reads were aligned with the reference using Bowtie version 2.0.0 to produce read alignment files in sam format (25). The read alignment files were converted from sam to bam format, sorted, and indexed with Samtools (26). Individual read alignments in bam file format from each condition were visualized with the Integrative Genomics Viewer (IGV) (27, 28). By using the read alignments visualized with the IGV, TSS were manually recorded in GFF format by identifying the position where reads aligned upstream of genes (see Table S2 in the supplemental material). By an analogous approach, ncRNAs visualized in the IGV were manually annotated by the identification of reads mapping to intergenic regions and antisense to coding sequences (see Table S3). Whole-genome annotations for TSS and ncRNAs are provided in GFF and GenBank formats with instructions for opening the files on our laboratory's website at http://web.biosci.utexas.edu/whiteley_lab/pages/resources.html (DataSetS1.gff and DataSetS2.gbk). Prior to the determination of differential gene expression, read alignment files produced by SHRiMP were reformatted for HTSeq read counting with a custom Perl script. The number of reads aligned to each gene and ncRNA in the newly annotated reference genome was calculated with HTSeq (http://www.huber.embl.de/users/anders/HTSeq), not including tRNAs and rRNAs. Prior to the calculation of differential gene expression, read counts per gene were summed for technical replicates in accordance with the DESeq recommendation. Differences in gene and ncRNA expression between A. actinomycetemcomitans biofilms and abscesses were determined on the basis of a negative binomial distribution with R package DESeq version 1.6.1 (29). Clusters of orthologous groups (COG) gene enrichment analysis was conducted with COGs determined previously (30). Enrichment of differentially regulated genes in a given COG category was determined by comparing the prevalence of up- or downregulated genes assigned to a specific COG category to the prevalence of genes in the entire genome assigned to that COG category. Enrichment of a COG category in either the up- or downregulated gene set relative to the genome was calculated with a Fisher's exact test custom macro in Microsoft Excel. Whole-genome diagrams were produced with Circos version 0.55 (31).
Construction of A. actinomycetemcomitans deletion mutants.
A. actinomycetemcomitans mutants were constructed for the formate dehydrogenase (FDH) H operon (fdhF1F2; D7S_2219-D7S_2220), the fumarate reductase operon (frdABCD; D7S_1533-D7S_1536), and pyruvate formate lyase (pfl; D7S_2028) by double homologous recombination. Wild-type genes of interest were replaced with aad9 (encodes spectinomycin resistance) from pVT1461 by overlap extension PCR and natural transformation of A. actinomycetemcomitans 624 (32, 33). The aad9 spectinomycin resistance gene was amplified from pVT1461 with Spec-F and Spec-R to generate Specr. The PCR overlap extension constructs were amplified for the fdhF1F2 operon with primers fdhKO-P1F-USS and fdhKO-P1R-Sp to generate the fdhKO-P1 PCR product and with primers fdhKO-P2F-Sp and fdhKO-P2R-USS to generate the fdhKO-P2 PCR product. Overlap extension PCR was done by mixing 500 ng each of fdhKO-P1, Specr, and fdhKO-P2 with primers fdhKO-P1F-USS and fdhKO-P2R-USS, making the fdhKO PCR product containing the ∼1-kb region upstream of the fdhF1F2 operon, aad9, and the ∼1-kb region downstream of the fdhF1F2 operon and A. actinomycetemcomitans-specific DNA uptake signal sequences on each end to enhance natural transformation. Prior to natural transformation, the ∼3-kb fdhKO overlap extension PCR product was gel extracted with a Fermentas gel extraction kit. The fdhF1F2 operon was replaced by natural transformation on tryptic soy agar supplemented with 0.5% yeast extract (TSAYE) with 5% heat-inactivated horse serum and 1 mM cyclic AMP in an anaerobic chamber via double homologous recombination with 1 μg of the fdhKO overlap extension PCR product. The resulting ΔfdhF1F2 mutants were selected on TSAYE with 50 μg/ml spectinomycin. The ΔfdhF1F2 mutant was verified by growing the mutant in TSBYE with 50 μg/ml spectinomycin, purifying genomic DNA with a Qiagen DNeasy kit, and PCR amplifying the aad9 gene with the Spec-R primer specific to the aad9 gene and the fdhKO-verify primer, which anneals upstream of the fdhKO-P1F-USS primer. The verification product was observed for ΔfdhF1F2 mutant genomic DNA but not wild-type genomic DNA. Analogous methods were used to generate the ΔfrdABCD mutant with primers frdKO-P1F-USS, frdKO-P1R-Sp, frdKO-P2F-Sp, frdKO-P2R-USS, and frdKO-verify, as well as the Δpfl mutant with primers pflKO-P1F-USS, pflKO-P1R-Sp, pflKO-P2F-Sp, pflKO-P2R-USS, and pflKO-verify. For the sequences of the primers used, see Table S6 in the supplemental material.
Sequence read accession number.
Sequence reads from this study have been deposited in the NCBI Sequence Read Archive (http://www.ncbi.nlm.nih.gov/sra) under accession number SRP022893.
RESULTS
In vitro and in vivo RNA-seq.
The goal of this study was to use RNA-seq to provide a high-resolution analysis of the A. actinomycetemcomitans genetic elements expressed during in vivo growth, with a specific interest in metabolic genes. While A. actinomycetemcomitans is most noted for its ability to cause oral infections, our laboratory has used an extraoral abscess model to study A. actinomycetemcomitans virulence (14). We prefer this model for the following reasons. (i) A. actinomycetemcomitans causes extraoral infections, including abscess infections; thus, the model has clinical relevance (17–19). (ii) The infectious dose is easily controlled and results in a contained infection that has been used to study the pathogenesis of oral bacteria (14, 34, 35). (iii) Unlike periodontal models of infection, infected tissue can be easily removed to assess disease severity (14) or for RNA isolation. In addition to performing transcriptome analysis in a relevant animal model of infection, it is also critical to examine A. actinomycetemcomitans gene expression during in vitro growth under defined conditions. Growth under known nutritional conditions is essential to provide a well-defined control transcriptome for comparison with the undefined in vivo transcriptome. Therefore, we grew A. actinomycetemcomitans as a colony biofilm on a solid defined medium with glucose as the sole energy source. Since glucose metabolism is well characterized and A. actinomycetemcomitans forms robust biofilms, this growth condition provided a well-defined, relevant control for the in vivo experiments.
Total RNA was harvested from biofilms and abscesses, and strand-specific cDNA libraries were subjected to RNA-seq analysis. Because murine abscesses contain small populations of bacteria (approximately 106 bacteria per abscess), it was essential to develop a method by which to efficiently purify RNA from infected tissue, deplete the abundant murine rRNA, and enrich for bacterial transcripts in order to obtain sufficient bacterial sequence reads for analysis. Since sequencing technologies are constantly evolving, we used two sequencing platforms to quantify transcript levels. However, it is important to note that all sequencing libraries were prepared by using similar RNA ligation-based protocols and each technology produces reads of sufficient length to map specifically to reference genomes. For both technologies, gene expression data were normalized among all replicates for differential expression analyses. The resulting sequence reads were processed and aligned to the A. actinomycetemcomitans genome to determine the A. actinomycetemcomitans transcriptome during in vitro and in vivo growth. In total, 7.1 million biofilm and 63.3 million in vivo sequence reads were aligned to the A. actinomycetemcomitans genome (see Table S1 in the supplemental material), resulting in averages of ∼190 and 870 reads/gene for in vitro- and in vivo-grown A. actinomycetemcomitans, respectively. By using these data, we manually annotated A. actinomycetemcomitans TSS and ncRNAs. Differential expression of protein-coding genes and ncRNAs was determined after normalization for read number to identify in vivo-regulated genes.
Transcription start site mapping.
To begin characterizing the primary transcriptome of A. actinomycetemcomitans, TSS were manually annotated by using the aligned RNA-seq data (as detailed in the “computational methods” portion of Materials and Methods). TSS were identified by manually recording the position where reads aligned upstream of annotated genes with the IGV (see Table S2 in the supplemental material) (27, 28). In total, 691 mRNA TSS were identified in A. actinomycetemcomitans (Fig. 1). Among these TSS, several previously characterized TSS, including katA, apiA, and lysT were verified by our RNA-seq analyses, thus validating our approach (21, 36). Because of the methods used to generate the RNA-seq libraries, it is important to note that the TSS identified represent authentic TSS, as well as the 5′ ends of processed RNAs. However, most bacterial transcripts are not specifically processed at a defined nucleotide but are processively degraded (37). Moreover, since several of the TSS identified here correspond to TSS identified by other approaches (21, 36), many of these TSS likely represent true start sites.
Fig 1.

A. actinomycetemcomitans mRNA 5′ ends and 5′-end switching in vivo. (A) A. actinomycetemcomitans 5′ ends mapped along the genome to the positive strand (outer circle, black) and negative strand (inner circle, red). The putative origin of replication is indicated (dnaA). Locus tags for genes with different start sites in vivo and in vitro are marked along the outermost circle and colored according to the read strand. (B) Genes with different start sites in vivo. Locus tags for genes are shown, significant differences in mRNA expression (fold change) during in vivo growth are shown below the locus tags, and the location of the in vivo TSS relative to the in vitro TSS (Diff.) and the gene function follow. When no fold change is indicated, genes were not differentially expressed in vivo.
Secondary TSS have been observed among genes in Helicobacter pylori and in a cyanobacterium, but the roles of alternate TSS are relatively obscure (38, 39). When TSS for genes differ from one growth condition to another, it suggests that the gene is transcribed from multiple promoters or the mRNA is differentially processed. We observed 11 genes with different TSS in vivo and in vitro (Fig. 1). One potential hypothesis is that alternative promoters are used to differentially regulate genes during in vivo and in vitro growth; however, 9 of the 11 genes were not differentially expressed, suggesting that the use of alternate promoters does not correlate with differential regulation.
A. actinomycetemcomitans ncRNA discovery.
RNA-seq provides tremendous insight into the identification and potential functions of ncRNAs transcribed from intergenic regions and antisense to coding genes. Like TSS and operons, ncRNAs were identified on the basis of contiguous reads aligning with intergenic regions and antisense to protein-coding genes observed in the IGV (Fig. 2; see Table S3 in the supplemental material). The ncRNA sequences were collected and compared to the Rfam database to determine homology to ncRNAs characterized in other bacteria (40, 41). In biofilms and in vivo, A. actinomycetemcomitans expressed a number of ncRNAs, including riboswitches, cis-antisense RNAs (asRNAs), small ncRNAs (sRNAs), and clustered regularly interspaced short palindromic repeat RNAs (crRNAs) (Fig. 2). In total, A. actinomycetemcomitans expressed 210 ncRNAs, including 127 asRNAs, 3 pre-crRNAs, 3 riboswitches, 75 sRNAs, and 1 unidentified tRNA. Among these ncRNAs were housekeeping ncRNAs involved in transcription, translation, and protein secretion (Table 1) and several previously discovered and predicted ncRNAs (21, 42–47). In addition, many ncRNAs were expressed differently in vivo than during in vitro biofilm growth. Indeed, 80 out of 210 ncRNAs were differentially expressed in vivo, including 39 upregulated and 41 downregulated ncRNAs (Fig. 2). This result is significant but perhaps expected, since the roles of ncRNAs in in vivo growth and persistence have been described for other bacteria (48–50).
Fig 2.

A. actinomycetemcomitans ncRNAs are differentially regulated in vivo. A. actinomycetemcomitans ncRNAs mapped along the genome to the positive strand (outer circle) and negative strand (inner circle). ncRNAs are colored according to type as follows: sRNA, red; asRNA, blue; riboswitch, green; pre-crRNA, light blue. The circular histogram shows log2 fold changes in ncRNA expression in vivo (−10 to 10, inner line to outer line). Bars are colored relative to the changes in ncRNA expression as follows: >2-fold upregulated, yellow; >5-fold upregulated, red; >2-fold downregulated, light blue; >5-fold downregulated, purple; no change, gray.
Table 1.
Rfam predictions for A. actinomycetemcomitans ncRNAsa
| Locus tag | Start | Stop | Strand | RNA family(ies) | Rfam prediction |
||
|---|---|---|---|---|---|---|---|
| Start | End | E value | |||||
| D7S_0043.1 | 41437 | 41836 | + | sRNA, RNase P class A | 2 | 390 | 4.91E-40 |
| D7S_0235.1 | 240380 | 240648 | − | sRNA, gcvB | 18 | 174 | 8.90E-23 |
| D7S_0343.1 | 340697 | 340887 | + | sRNA, tfoR | 91 | 181 | 5.10E-03 |
| D7S_0661.1 | 640001 | 640182 | + | sRNA, His leader | 42 | 171 | 4.28E-17 |
| D7S_0743.1 | 704845 | 704936 | − | tRNA | 2 | 74 | 5.29E-14 |
| D7S_0849.1 | 794824 | 795016 | − | sRNA, 6S RNA | 1 | 184 | 1.52E-23 |
| D7S_1442.2 | 1323184 | 1323273 | − | sRNA, C4 | 1 | 74 | 4.90E-12 |
| D7S_1454.1 | 1330343 | 1330779 | − | sRNA, tmRNA | 1 | 366 | 7.92E-92 |
| D7S_1607.1 | 1477807 | 1477998 | − | Riboswitch, FMNb riboswitch | 1 | 182 | 5.17E-29 |
| D7S_1716.1 | 1572027 | 1572259 | − | Riboswitch, glycine riboswitch | 87 | 231 | 1.00E-10 |
| D7S_2270.1 | 2096164 | 2096331 | + | sRNA bacterial SRP | 47 | 145 | 2.08E-18 |
ncRNA nucleotide sequences were collected and used to search Rfam database families (RNA family). Locus tags are in Table S3 in the supplemental material. Start and stop refer to the location of the ncRNA on the A. actinomycetemcomitans reference genome. The portions of the A. actinomycetemcomitans ncRNA sequences that possessed sequence homology to RNAs in the Rfam database are indicated (Start and End), as are the expectation (E) values.
FMN, flavin mononucleotide.
Fermentative metabolism and anaerobic respiration promote in vivo survival.
Our main goal was to use RNA-seq to identify metabolic pathways that impact A. actinomycetemcomitans fitness in vivo. The hypothesis was that genes differentially regulated in vivo will be enriched for those important for growth in the murine abscess. To identify differentially regulated genes, in vitro and in vivo transcriptomes were compared. This analysis yielded 337 genes (∼14% of all predicted genes) that were differentially regulated 2-fold or more (Fig. 3A; see Tables S4 and S5 in the supplemental material) in the abscess infection than in the in vitro biofilm. Of these genes, 107 were differentially regulated 5-fold or more. Notably, genes that encode the A. actinomycetemcomitans virulence factors leukotoxin (locus tag D7S_0615) and cytolethal distending toxin (locus tag D7S_2348) (51–55) were upregulated in vivo. While leukotoxin is upregulated under low-oxygen conditions (51), prior to this study, neither toxin was known to be upregulated during infection.
Fig 3.
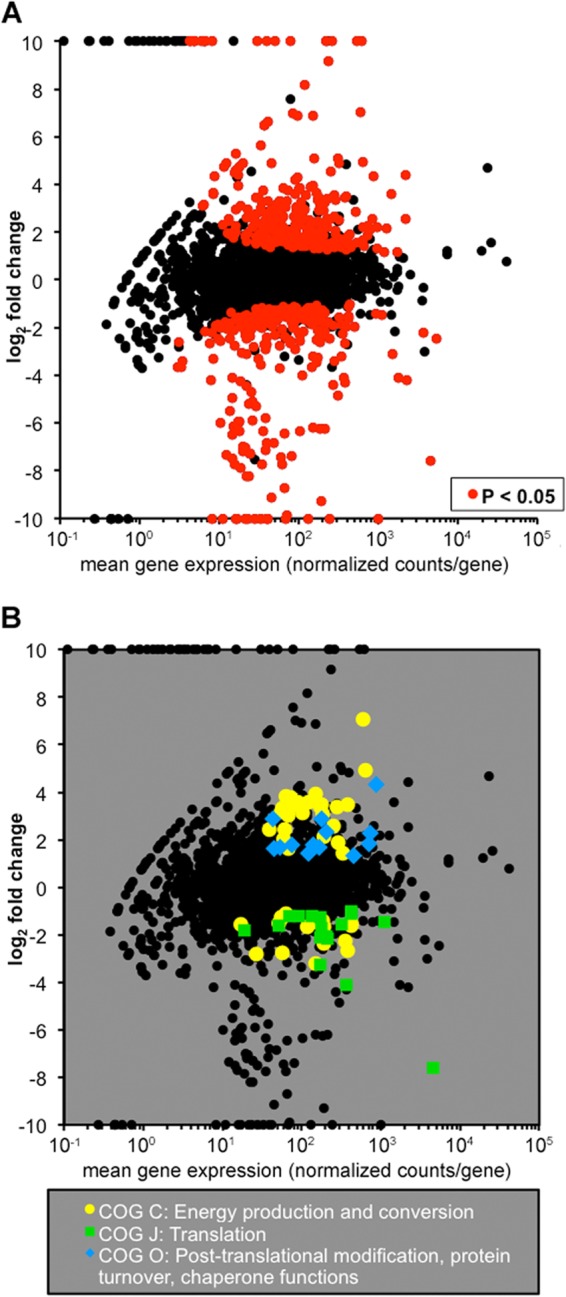
Differential RNA-seq and COG enrichment analyses reveal the importance of energy metabolism in vivo. (A) In vivo differential gene expression plot. The fold change in the expression of each gene in vivo is plotted against mean gene expression. Red points represent the 337 genes with significant differential expression in vivo (P < 0.05, DESeq). (B) In vivo COG enrichment analysis. The enrichment of COGs among differentially regulated genes compared to the abundance of the COGs in the genome was determined with Fisher's exact test (P < 0.05).
To gain a broader perspective on genes differentially regulated in vivo, we performed a COG enrichment analysis to define (56, 57) differentially regulated groups of genes. This analysis first clusters genes on the basis of their putative function and then examines whether a particular category is enriched for differentially regulated genes compared to what would be expected by chance. Using the COGs defined by Kittichotirat et al. (30), only one category, COG C, was significantly enriched in both up- and downregulated genes. COG C includes genes involved in energy production and conversion (Fig. 3B), suggesting that A. actinomycetemcomitans undergoes substantial changes in metabolism during in vivo growth.
Included within the differentially regulated metabolic genes were operons that encode FDH H (fdhF1F2) and fumarate reductase (frdABCD), which are involved in fermentative metabolism and anaerobic respiration, respectively (Fig. 4). Thus, we hypothesized that these operons would be important for A. actinomycetemcomitans growth in the murine abscess. To test this hypothesis, individual mutants containing deletions of fdhF1F2 or frdABCD were constructed via allelic exchange. In addition, a strain containing a deletion of the gene that encodes pyruvate formate lyase (pfl) was also constructed. Since pfl was not differentially regulated in vivo, we hypothesized that it would not be critical for A. actinomycetemcomitans growth in the abscess; thus, this mutant served as a control for subsequent in vivo experiments.
Fig 4.

Metabolic genes and pathways upregulated in vivo. Genes that putatively encode fructose, glucose, mannose, and ribose transporters were upregulated in a murine abscess along with multiple fermentative and anaerobic respiratory pathways. Red, >5-fold upregulation; yellow, >2-fold upregulation; gray, no change. Dashed lines indicate genes/pathways selected for mutagenesis. TMAO, trimethylamine-N-oxide; TMA, trimethylamine; DMSO, dimethyl sulfoxide; DMS, dimethyl sulfide; ETC, electron transport chain; TCA, tricarboxylic acid.
Abscesses produced by both the ΔfdhF1F2 and ΔfrdABCD mutants contained ∼10-fold less bacteria than wild-type infections (Fig. 5), indicating that these pathways are critical for A. actinomycetemcomitans fitness in the murine abscess. In contrast, the Δpfl mutant showed bacterial numbers similar to those of wild-type A. actinomycetemcomitans (Fig. 5). Importantly, deletion of the fdhF1F2 and frdABCD operons did not impact aerobic or anaerobic in vitro growth in a glucose-defined medium (data not shown). Collectively, these data suggest that the fdhF1F2 and frdABCD operons are not only highly upregulated during in vivo growth but also impact A. actinomycetemcomitans fitness in the murine abscess.
Fig 5.

Metabolic mutants are attenuated in vivo. Monoculture abscess infections with the wild-type and ΔfdhF1F2, ΔfrdABCD, and Δpfl mutant strains were processed 3 days postinfection, and CFU counts per abscess were determined by plate counting (*, P < 0.05 compared to the wild type by a Mann-Whitney U test). Each symbol represents the infection of an individual mouse.
DISCUSSION
In this study, a high-resolution transcriptome analysis of in vitro- and in vivo-grown A. actinomycetemcomitans was performed by RNA-seq. Protocols were developed to isolate total RNA from abscess infections and enrich for bacterial RNA before RNA-seq analysis. From these data, the terminal 5′ sequences of ∼700 RNAs were mapped, most of which likely represent authentic transcriptional start sites. In addition, over 300 A. actinomycetemcomitans genes were shown to be differentially regulated during abscess infection in comparison with in vitro-grown bacteria. This study provides the first high-resolution transcriptome analysis of a bacterium during growth in an abscess and establishes a method for performing RNA-seq with samples containing predominantly host RNA.
The primary goal of this work was to identify metabolic pathways used by A. actinomycetemcomitans in vivo by focusing on metabolic genes differentially regulated during growth in an abscess. The use of defined in vitro growth conditions provided a robust control to probe A. actinomycetemcomitans metabolism during in vivo growth, allowing the identification of both fermentative and respiratory pathways important for A. actinomycetemcomitans growth in the murine abscess (Fig. 4). The enrichment of pathways including fdhF1F2 and frdABCD, which are involved primarily in the regeneration of NAD+ during anaerobic growth, suggests that A. actinomycetemcomitans experiences low oxygen levels in an abscess. This observation is curious in regard to a previous study by our group that demonstrated that during coculture infection with the peroxigenic oral bacterium Streptococcus gordonii, A. actinomycetemcomitans grows aerobically in an abscess (14). Why the A. actinomycetemcomitans-S. gordonii coculture abscess infection is aerobic is unknown, but these results emphasize that a pathogen may require distinct metabolic pathways to persist in an infection site dependent on whether it is alone or in the presence of other microbes. Of course, the infection site may also impact gene expression, and it will be interesting to examine A. actinomycetemcomitans gene expression in the oral cavity during mono- and coculture.
One of the primary advantages of RNA-seq is that it provides unparalleled insight into ncRNAs expressed under a given growth condition. Here we reported the differential regulation of 80 A. actinomycetemcomitans ncRNAs and increased expression of hfq in vivo. The upregulation of hfq is particularly striking, since it encodes an sRNA chaperone that promotes ncRNA interactions with mRNA targets (58, 59). Similar to other studies, this suggests that A. actinomycetemcomitans ncRNAs may play important regulatory roles in vivo (48, 49, 60). Differential regulation of ncRNAs also provided insight into the nutritional content of the infection site. For example, the A. actinomycetemcomitans lysine riboswitch and the sRNA GcvB (21, 61, 62) showed reduced abundance during infection. These ncRNAs regulate the expression of lysine and glycine transport proteins, respectively, and their decreased levels suggest that these amino acids are found at reduced levels in an abscess.
Transcriptome analysis by RNA-seq is a powerful and sensitive tool for studying infectious disease processes in vivo. The development of methods for performing RNA-seq with in vivo samples dominated by host RNA allowed tremendous insight into the physiology and metabolism of A. actinomycetemcomitans during in vivo growth. As evidenced by the abscess CFU counts, the method used here can be used to perform gene expression analyses of as few as 105 in vivo bacteria. Ultimately, the application of this technology to defined polymicrobial infections, as well as undefined complex human infections, will provide a window into the physiology of bacterial pathogens in human infections.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants from the NIH (1R01DE020100 to M.W. and 5F31DE021633-02 to P.J. and M.W.). M.W. is a Burroughs Wellcome Investigator in the Pathogenesis of Infectious Disease.
We thank the Whiteley lab members for critical discussions of the manuscript and the University of Texas Genome Sequencing and Analysis Facility for discussions about computational analyses.
Footnotes
Published ahead of print 23 August 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00875-13.
REFERENCES
- 1.Pasteur L, Joubert J, Chamberland C. 1878. La théorie des germes et ses applications à la médecine et à la chirurgie. G. Masson, Paris, France [Google Scholar]
- 2.Mashburn LM, Jett AM, Akins DR, Whiteley M. 2005. Staphylococcus aureus serves as an iron source for Pseudomonas aeruginosa during in vivo coculture. J. Bacteriol. 187:554–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Orihuela CJ, Radin JN, Sublett JE, Gao G, Kaushal D, Tuomanen EI. 2004. Microarray analysis of pneumococcal gene expression during invasive disease. Infect. Immun. 72:5582–5596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Snyder JA, Haugen BJ, Buckles EL, Lockatell CV, Johnson DE, Donnenberg MS, Welch RA, Mobley HL. 2004. Transcriptome of uropathogenic Escherichia coli during urinary tract infection. Infect. Immun. 72:6373–6381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xu Q, Dziejman M, Mekalanos JJ. 2003. Determination of the transcriptome of Vibrio cholerae during intraintestinal growth and midexponential phase in vitro. Proc. Natl. Acad. Sci. U. S. A. 100:1286–1291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Westermann AJ, Gorski SA, Vogel J. 2012. Dual RNA-seq of pathogen and host. Nat. Rev. Microbiol. 10:618–630 [DOI] [PubMed] [Google Scholar]
- 7.Mandlik A, Livny J, Robins WP, Ritchie JM, Mekalanos JJ, Waldor MK. 2011. RNA-Seq-based monitoring of infection-linked changes in Vibrio cholerae gene expression. Cell Host Microbe 10:165–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brown SA, Whiteley M. 2007. A novel exclusion mechanism for carbon resource partitioning in Aggregatibacter actinomycetemcomitans. J. Bacteriol. 189:6407–6414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brown SA, Whiteley M. 2009. Characterization of the l-lactate dehydrogenase from Aggregatibacter actinomycetemcomitans. PLoS One 4:e7864. 10.1371/journal.pone.0007864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Palmer GC, Palmer KL, Jorth PA, Whiteley M. 2010. Characterization of the Pseudomonas aeruginosa transcriptional response to phenylalanine and tyrosine. J. Bacteriol. 192:2722–2728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Palmer KL, Aye LM, Whiteley M. 2007. Nutritional cues control Pseudomonas aeruginosa multicellular behavior in cystic fibrosis sputum. J. Bacteriol. 189:8079–8087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Palmer KL, Brown SA, Whiteley M. 2007. Membrane-bound nitrate reductase is required for anaerobic growth in cystic fibrosis sputum. J. Bacteriol. 189:4449–4455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Palmer KL, Mashburn LM, Singh PK, Whiteley M. 2005. Cystic fibrosis sputum supports growth and cues key aspects of Pseudomonas aeruginosa physiology. J. Bacteriol. 187:5267–5277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ramsey MM, Rumbaugh KP, Whiteley M. 2011. Metabolite cross-feeding enhances virulence in a model polymicrobial infection. PLoS Pathog. 7:e1002012. 10.1371/journal.ppat.1002012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Slots J, Reynolds HS, Genco RJ. 1980. Actinobacillus actinomycetemcomitans in human periodontal disease: a cross-sectional microbiological investigation. Infect. Immun. 29:1013–1020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zambon JJ. 1985. Actinobacillus actinomycetemcomitans in human periodontal disease. J. Clin. Periodontol. 12:1–20 [DOI] [PubMed] [Google Scholar]
- 17.Kozarov EV, Dorn BR, Shelburne CE, Dunn WA, Jr, Progulske-Fox A. 2005. Human atherosclerotic plaque contains viable invasive Actinobacillus actinomycetemcomitans and Porphyromonas gingivalis. Arterioscler. Thromb. Vasc. Biol. 25:e17–18 [DOI] [PubMed] [Google Scholar]
- 18.Stepanović S, Tosic T, Savic B, Jovanovic M, K'Ouas G, Carlier JP. 2005. Brain abscess due to Actinobacillus actinomycetemcomitans. APMIS 113:225–228 [DOI] [PubMed] [Google Scholar]
- 19.Yuan A, Yang PC, Lee LN, Chang DB, Kuo SH, Luh KT. 1992. Actinobacillus actinomycetemcomitans pneumonia with chest wall involvement and rib destruction. Chest 101:1450–1452 [DOI] [PubMed] [Google Scholar]
- 20.Socransky SS, Dzink JL, Smith CM. 1985. Chemically defined medium for oral microorganisms. J. Clin. Microbiol. 22:303–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jorth P, Whiteley M. 2010. Characterization of a novel riboswitch-regulated lysine transporter in Aggregatibacter actinomycetemcomitans. J. Bacteriol. 192:6240–6250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.David M, Dzamba M, Lister D, Ilie L, Brudno M. 2011. SHRiMP2: sensitive yet practical SHort Read Mapping. Bioinformatics 27:1011–1012 [DOI] [PubMed] [Google Scholar]
- 23.Chen C, Kittichotirat W, Chen W, Downey JS, Si Y, Bumgarner R. 2010. Genome sequence of naturally competent Aggregatibacter actinomycetemcomitans serotype a strain D7S-1. J. Bacteriol. 192:2643–2644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dodt M, Roehr J, Ahmed R, Dieterich C. 2012. FLEXBAR—flexible barcode and adapter processing for next-generation sequencing platforms. Biology 1:895–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9:357–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25:2078–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. 2011. Integrative genomics viewer. Nat. Biotechnol. 29:24–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thorvaldsdóttir H, Robinson JT, Mesirov JP. 2013. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief. Bioinform. 14:178–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Anders S, Huber W. 2010. Differential expression analysis for sequence count data. Genome Biol. 11:R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kittichotirat W, Bumgarner RE, Asikainen S, Chen C. 2011. Identification of the pangenome and its components in 14 distinct Aggregatibacter actinomycetemcomitans strains by comparative genomic analysis. PLoS One 6:e22420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA. 2009. Circos: an information aesthetic for comparative genomics. Genome Res. 19:1639–1645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mintz KP, Brissette C, Fives-Taylor PM. 2002. A recombinase A-deficient strain of Actinobacillus actinomycetemcomitans constructed by insertional mutagenesis using a mobilizable plasmid. FEMS Microbiol. Lett. 206:87–92 [DOI] [PubMed] [Google Scholar]
- 33.Wang Y, Goodman SD, Redfield RJ, Chen C. 2002. Natural transformation and DNA uptake signal sequences in Actinobacillus actinomycetemcomitans. J. Bacteriol. 184:3442–3449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ebersole JL, Kesavalu L, Schneider SL, Machen RL, Holt SC. 1995. Comparative virulence of periodontopathogens in a mouse abscess model. Oral Dis. 1:115–128 [DOI] [PubMed] [Google Scholar]
- 35.Kesavalu L, Holt SC, Ebersole JL. 1998. Virulence of a polymicrobic complex, Treponema denticola and Porphyromonas gingivalis, in a murine model. Oral Microbiol. Immunol. 13:373–377 [DOI] [PubMed] [Google Scholar]
- 36.Ramsey MM, Whiteley M. 2009. Polymicrobial interactions stimulate resistance to host innate immunity through metabolite perception. Proc. Natl. Acad. Sci. U. S. A. 106:1578–1583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Evguenieva-Hackenberg E, Klug G. 2011. New aspects of RNA processing in prokaryotes. Curr. Opin. Microbiol. 14:587–592 [DOI] [PubMed] [Google Scholar]
- 38.Mitschke J, Vioque A, Haas F, Hess WR, Muro-Pastor AM. 2011. Dynamics of transcriptional start site selection during nitrogen stress-induced cell differentiation in Anabaena sp PCC7120. Proc. Natl. Acad. Sci. U. S. A. 108:20130–20135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sharma CM, Hoffmann S, Darfeuille F, Reignier J, Findeiss S, Sittka A, Chabas S, Reiche K, Hackermuller J, Reinhardt R, Stadler PF, Vogel J. 2010. The primary transcriptome of the major human pathogen Helicobacter pylori. Nature 464:250–255 [DOI] [PubMed] [Google Scholar]
- 40.Gardner PP, Daub J, Tate J, Moore BL, Osuch IH, Griffiths-Jones S, Finn RD, Nawrocki EP, Kolbe DL, Eddy SR, Bateman A. 2011. Rfam: Wikipedia, clans and the “decimal” release. Nucleic Acids Res. 39:D141–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Griffiths-Jones S, Bateman A, Marshall M, Khanna A, Eddy SR. 2003. Rfam: an RNA family database. Nucleic Acids Res. 31:439–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guerrier-Takada C, Gardiner K, Marsh T, Pace N, Altman S. 1983. The RNA moiety of ribonuclease P is the catalytic subunit of the enzyme. Cell 35:849–857 [DOI] [PubMed] [Google Scholar]
- 43.Jorth P, Whiteley M. 2012. An evolutionary link between natural transformation and CRISPR adaptive immunity. mBio 3(5):e00309–12. 10.1128/mBio.00309-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Keiler KC. 2008. Biology of trans-translation. Annu. Rev. Microbiol. 62:133–151 [DOI] [PubMed] [Google Scholar]
- 45.Ray BK, Apirion D. 1979. Characterization of 10S RNA: a new stable RNA molecule from Escherichia coli. Mol. Gen. Genet. 174:25–32 [DOI] [PubMed] [Google Scholar]
- 46.Ulbrandt ND, Newitt JA, Bernstein HD. 1997. The E. coli signal recognition particle is required for the insertion of a subset of inner membrane proteins. Cell 88:187–196 [DOI] [PubMed] [Google Scholar]
- 47.Wassarman KM, Storz G. 2000. 6S RNA regulates E. coli RNA polymerase activity. Cell 101:613–623 [DOI] [PubMed] [Google Scholar]
- 48.Mann B, van Opijnen T, Wang J, Obert C, Wang YD, Carter R, McGoldrick DJ, Ridout G, Camilli A, Tuomanen EI, Rosch JW. 2012. Control of virulence by small RNAs in Streptococcus pneumoniae. PLoS Pathog. 8:e1002788. 10.1371/journal.ppat.1002788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Meibom KL, Forslund AL, Kuoppa K, Alkhuder K, Dubail I, Dupuis M, Forsberg A, Charbit A. 2009. Hfq, a novel pleiotropic regulator of virulence-associated genes in Francisella tularensis. Infect. Immun. 77:1866–1880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sampson TR, Saroj SD, Llewellyn AC, Tzeng YL, Weiss DS. 2013. A CRISPR/Cas system mediates bacterial innate immune evasion and virulence. Nature 497:254–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Childress C, Feuerbacher LA, Phillips L, Burgum A, Kolodrubetz D. 2013. Mlc is a transcriptional activator with a key role in integrating cyclic AMP receptor protein and integration host factor regulation of leukotoxin RNA synthesis in Aggregatibacter actinomycetemcomitans. J. Bacteriol. 195:2284–2297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Damek-Poprawa M, Jang JY, Volgina A, Korostoff J, DiRienzo JM. 2012. Localization of Aggregatibacter actinomycetemcomitans cytolethal distending toxin subunits during intoxication of live cells. Infect. Immun. 80:2761–2770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fong KP, Tang HY, Brown AC, Kieba IR, Speicher DW, Boesze-Battaglia K, Lally ET. 2011. Aggregatibacter actinomycetemcomitans leukotoxin is post-translationally modified by addition of either saturated or hydroxylated fatty acyl chains. Mol. Oral Microbiol. 26:262–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Matangkasombut O, Wattanawaraporn R, Tsuruda K, Ohara M, Sugai M, Mongkolsuk S. 2010. Cytolethal distending toxin from Aggregatibacter actinomycetemcomitans induces DNA damage, S/G2 cell cycle arrest, and caspase-independent death in a Saccharomyces cerevisiae model. Infect. Immun. 78:783–792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rompikuntal PK, Thay B, Khan MK, Alanko J, Penttinen AM, Asikainen S, Wai SN, Oscarsson J. 2012. Perinuclear localization of internalized outer membrane vesicles carrying active cytolethal distending toxin from Aggregatibacter actinomycetemcomitans. Infect. Immun. 80:31–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tatusov RL, Koonin EV, Lipman DJ. 1997. A genomic perspective on protein families. Science 278:631–637 [DOI] [PubMed] [Google Scholar]
- 57.Tatusov RL, Federova ND, Jackson JD, Jacobs AR, Kiryutin B, Koonin EV, Krylov DM, Mazumder R, Mekhedov SL, Nikolskaya AN, Rao BS, Smirnov S, Sverdlov AV, Vasudevan S, Wolf YI, Yin JJ, Natale DA. 2003. The COG database: an updated version includes eukaryotes. BMC Bioinformatics 4:41. 10.1186/1471-2105-4-41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fender A, Elf J, Hampel K, Zimmermann B, Wagner EG. 2010. RNAs actively cycle on the Sm-like protein Hfq. Genes Dev. 24:2621–2626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Link TM, Valentin-Hansen P, Brennan RG. 2009. Structure of Escherichia coli Hfq bound to polyriboadenylate RNA. Proc. Natl. Acad. Sci. U. S. A. 106:19292–19297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Koo JT, Alleyne TM, Schiano CA, Jafari N, Lathem WW. 2011. Global discovery of small RNAs in Yersinia pseudotuberculosis identifies Yersinia-specific small, noncoding RNAs required for virulence. Proc. Natl. Acad. Sci. U. S. A. 108:E709–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sharma CM, Darfeuille F, Plantinga TH, Vogel J. 2007. A small RNA regulates multiple ABC transporter mRNAs by targeting C/A-rich elements inside and upstream of ribosome-binding sites. Genes Dev. 21:2804–2817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Urbanowski ML, Stauffer LT, Stauffer GV. 2000. The gcvB gene encodes a small untranslated RNA involved in expression of the dipeptide and oligopeptide transport systems in Escherichia coli. Mol. Microbiol. 37:856–868 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.