

Abstract
Accumulation of d-leucine, d-allo-isoleucine, and d-valine was observed in the growth medium of a lactic acid bacterium, Lactobacillus otakiensis JCM 15040, and the racemase responsible was purified from the cells and identified. The N-terminal amino acid sequence of the purified enzyme was GKLDKASKLI, which is consistent with that of a putative γ-aminobutyrate aminotransferase from Lactobacillus buchneri. The putative γ-aminobutyrate aminotransferase gene from L. buchneri JCM 1115 was expressed in recombinant Escherichia coli and then purified to homogeneity. The enzyme catalyzed the racemization of a broad spectrum of nonpolar amino acids. In particular, it catalyzed at high rates the epimerization of l-isoleucine to d-allo-isoleucine and d-allo-isoleucine to l-isoleucine. In contrast, the enzyme showed no γ-aminobutyrate aminotransferase activity. The relative molecular masses of the subunit and native enzyme were estimated to be about 49 kDa and 200 kDa, respectively, indicating that the enzyme was composed of four subunits of equal molecular masses. The Km and Vmax values of the enzyme for l-isoleucine were 5.00 mM and 153 μmol·min−1·mg−1, respectively, and those for d-allo-isoleucine were 13.2 mM and 286 μmol·min−1·mg−1, respectively. Hydroxylamine and other inhibitors of pyridoxal 5′-phosphate-dependent enzymes completely blocked the enzyme activity, indicating the enzyme requires pyridoxal 5′-phosphate as a coenzyme. This is the first evidence of an amino acid racemase that specifically catalyzes racemization of nonpolar amino acids at the C-2 position.
INTRODUCTION
d-Amino acids, enantiomers of l-amino acids, play key roles as components of the peptidoglycan cell wall of bacteria. Within bacterial cell walls, d-alanine (d-Ala) and d-glutamate (d-Glu) are often the most common d-amino acids (1, 2), although significant levels of d-aspartate (d-Asp) are found in the peptidoglycans of Lactococcus lactis (3) and Enterococcus faecium (4), and d-serine (d-Ser) is found in the peptidoglycan of E. gallinarum (5) and vancomycin-resistant Staphylococcus aureus (6–8). These d-amino acids are primarily produced from the corresponding l-enantiomers by enzymes such as alanine racemase (EC 5.1.1.1), glutamate racemase (EC 5.1.1.3), aspartate racemase (EC 5.1.1.13), and serine racemase (EC 5.1.1.18). On the other hand, various bacteria have been known to produce free d-branched amino acids (d-BCAAs), such as d-isoleucine (d-Ile), d-leucine (d-Leu), and d-valine (d-Val) (9), and d-BCAAs are now thought to be specific molecular signals in some bacterial species. For example, d-Leu, d-Ile, and d-Val are reportedly involved in regulation of cell wall remodeling in Vibrio cholerae (9), while d-Leu is involved in controlling biofilm dispersal in Bacillus subtilis (10). It has been suggested that d-BCAAs are produced by a few pyridoxal 5′-phosphate (PLP)-dependent amino acid racemases that have broad substrate specificity. At present, however, little is known about the characteristics of these racemases. Although Pseudomonas graveolens arginine racemase (EC 5.1.1.9) and Pseudomonas putida amino acid racemase (EC 5.1.1.10) have been reported (11, 12), these two racemases show much less activity toward Leu than the main substrate, lysine (Lys), and none toward Ile and Val. Thus, the details of the production mechanisms of d-BCAAs in bacteria have been unclear.
We recently analyzed several strains of Lactobacillus and found that growth of Lactobacillus otakiensis JCM 15040 cultures was accompanied by secretion of d-Leu, d-allo-Ile, and d-Val into the culture medium. In the present study, we report a novel racemase probably responsible for production of these d-BCAAs.
MATERIALS AND METHODS
Materials.
Toyopearl phenyl-650M, butyl-650M, and SuperQ-650M were purchased from Tosoh (Tokyo, Japan). Red Sepharose CL-4B (reactive red 120 dye; Sigma, St. Louis, MO) was prepared as described previously (13). Ni-nitrilotriacetic acid (NTA) agarose was obtained from Qiagen (Tokyo, Japan). N-Acetyl-l-cysteine (NAC), N-tert-butyloxycarbonyl-l-cysteine (NBC), and o-phthaldialdehyde (OPA) were purchased from Wako Pure Chemical Industries (Tokyo, Japan), Nova Biochemical Corp. (Waltham, MA), and Nacalai Tesque (Kyoto, Japan), respectively. Escherichia coli BL21(DE3) and the plasmid pCold ProS2 were from Stratagene (La Jolla, CA) and TaKaRa Bio, Inc. (Otsu, Japan), respectively.
Microorganisms and growth conditions.
L. otakiensis JCM 15040 and L. buchneri JCM 1115 were obtained from the Japan Collection of Microorganisms (JCM; Tsukuba, Japan). L. otakiensis JCM 15040 was used as the enzyme source for isoleucine 2-epimerase. The L. buchneri JCM 1115 genome, which contains a putative γ-aminobutyrate aminotransferase (GABA-AT) gene, was used as a gene source. These strains were cultured in MRS medium (Becton, Dickinson and Co., Franklin Lakes, NJ) at 30°C (L. otakiensis JCM 15040) or 37°C (L. buchneri JCM 1115).
Preparation of culture media for d-amino acid analyses.
For sequential analysis of the d-amino acid content of the culture medium conditioned by L. otakiensis JCM 15040 and L. buchneri JCM 1115, sample solutions were prepared as follows. Aliquots (1 ml) of the culture medium were centrifuged (10,000 × g for 1 min at 4°C), after which the supernatants were filtered through an Amicon Ultra 0.5-ml centrifugal filter 3K device (Merck Millipore, Darmstadt, Germany). The prepared samples were stored at −20°C until use.
Analysis of l- and d-amino acids using UPLC.
d, l-Amino acids in sample solutions were derivatized with OPA and NAC (14) or NBC (15) using two methanolic solutions (A and B) containing the derivatizing reagents. Methanolic solution A was prepared by dissolving 8 mg of OPA and 10 mg of NAC in 1 ml of methanol, while methanolic solution B was prepared by dissolving 10 mg of OPA and 10 mg of NBC in 1 ml of methanol. The reaction mixture (250 μl) for the derivatization contained 25 μl of amino acid sample, 50 μl of methanolic solution (A or B), and 175 μl of 0.4 M borate sodium hydroxide buffer (pH 10.4). After derivatization for 2 min at room temperature in the dark, an aliquot (1 μl) of the reaction mixture was introduced into an ultraperformance liquid chromatography (UPLC) system (Waters, Milford, MA).
The diastereoisomeric derivatives of amino acids formed with OPA-NAC or OPA-NBC were applied to a Waters AccQ-Tag Ultra 2.1- by 100-mm column (Waters) in a ultraperformance liquid chromatograph (the Acquity UPLC tunable UV system consisted of a Waters binary solvent manager, a Waters sample manager, and a Waters fluorescence detector). The excitation and emission wavelengths for fluorescent detection of the diastereoisomeric amino acid derivatives were 350 nm and 450 nm, respectively, and the data were processed using Empower 2 (Waters). The system was operated at a flow rate of 0.25 ml/min at 30°C. The UPLC gradient system for analysis of OPA-NAC derivatives (A = 50 mM sodium acetate [pH 5.9], and B = methanol) was 10 to 20% B for 3.2 min, 20% B for 1 min, 20 to 40% B for 3.6 min, 40% B for 1.2 min, 40 to 60% B for 3.8 min, 60% B for 1 min, and 60 to 10% B for 0.01 min. The gradient system for analysis of OPA-NBC derivatives (A = 50 mM sodium acetate [pH 5.9], and B = acetonitrile) was 15 to 21% B for 7 min, 21 to 27.5% B for 1.5 min, 27.5% B for 2 min, 27.5 to 30% B for 1 min, 30 to 40% B for 2 min, 40% B for 0.5 min, and 40 to 15% B for 0.01 min. The peak heights and retention times were used for amino acid quantification and identification, respectively. Using OPA-NAC derivatization, 28 kinds of amino acids were analyzed at one time: the d- and l-forms of Asp, Ser, Ala, Val, Leu, methionine (Met), tryptophan (Trp), tyrosine (Tyr), phenylalanine (Phe), arginine (Arg), histidine (His), threonine (Thr), glutamine (Gln), Ile, and allo-Ile. Using OPA-NBC derivatization, 4 kinds of amino acids were analyzed at one time: the d- and l-forms of Glu and asparagine (Asn). With respect to the substrate specificity, the d- and l-forms of norleucine (Nle), ornithine (Orn), and Lys were derivatized to diastereomers using the OPA-NAC system, and the d- and l-forms of norvaline (Nva), 2-aminobutanoic acid (Abu), and tert-leucine (tert-Leu) were derivatized to diastereomers using the OPA-NBC system.
Protein determination.
Protein concentrations were measured using the method of Bradford (16); bovine serum albumin was used as the standard.
Enzyme assay.
Two different conditions were used to assay isoleucine 2-epimerase activity during purification and characterization of the enzyme. During enzyme purification, the reaction mixture (500 μl) consisted of 100 mM Na-phosphate buffer (pH 8.0), 10 mM l-Ile, 0.1 mM PLP, and the proper amount of enzyme. This reaction mixture was incubated for 6 h at 30°C (during purification of the enzyme from the crude extract of L. otakiensis JCM 15040) or at 37°C for 5 min (during purification of the recombinant enzyme from L. buchneri JCM 1115). During characterization of the recombinant enzyme from L. buchneri JCM 1115, the reaction mixture (500 μl), consisting of 200 mM Na-citrate buffer (pH 5.5), 50 mM l-Ile or d-allo-Ile, 0.1 mM PLP, and the proper amount of the recombinant enzyme, was incubated at 37°C for 5 min. The latter reaction condition was used as the standard assay condition for characterizations of the recombinant enzyme.
Under both sets of assay conditions, the reaction was stopped by addition of 125 μl of 50% trichloroacetic acid (TCA) to 500 μl of the reaction mixture. After incubation for 5 min at room temperature, the mixture was centrifuged (10,000 × g for 15 min at 4°C), and an aliquot (500 μl) of the supernatant was neutralized by addition of 300 μl of 1 M NaOH. The amount of product, d-allo-Ile or l-Ile, in the solution was then measured using UPLC as described above.
Purification of a protein exhibiting isoleucine 2-epimerase activity from L. otakiensis JCM 15040.
L. otakiensis JCM 15040 was aerobically cultivated at 30°C for 30 h in MRS medium (10 liters), after which the cells were collected by centrifugation (8,000 × g for 30 min at 4°C). The harvested cells (ca. 32.2 g, wet weight) were used as the starting material for purification of protein exhibiting isoleucine 2-epimerase activity. Unless otherwise indicated, 50 mM Na-phosphate buffer (pH 7.2) containing 1 mM EDTA and 1 mM dithiothreitol was used as the standard buffer throughout the purification procedures, and all purification procedures were carried out at room temperature.
Step 1.
To prepare a crude extract, the cells were washed twice with the standard buffer, suspended in the same buffer, disrupted using a multibead shocker (Yasui Kikai, Osaka, Japan), and centrifuged (10,000 × g for 30 min at 4°C). The resultant supernatant was used as the crude extract.
Step 2.
The crude extract was mixed with two volumes of 3.6 M (NH4)2SO4 dissolved in the standard buffer. After incubation for 4 h at 4°C, the mixture was centrifuged (10,000 × g for 30 min at 4°C), and the supernatant was retrieved.
Step 3.
The collected supernatant was applied to a Toyopearl phenyl-650M column (diameter, 2.5 cm; length, 10 cm) equilibrated with standard buffer containing 2.4 M (NH4)2SO4. The column was then washed with the same buffer and eluted with a linear gradient of 2.4 to 0.4 M (NH4)2SO4 in buffer. The active fractions were pooled and then dialyzed against the standard buffer at 4°C.
Step 4.
The first dialysate was mixed with 2 volumes of 3.6 M (NH4)2SO4 dissolved in the standard buffer, and the mixture was applied to a Toyopearl butyl-650M column (diameter, 2.5 cm; length, 10 cm) equilibrated with the standard buffer containing 2.4 M (NH4)2SO4. The column was then washed with the same buffer and eluted with a linear gradient of 2.4 to 0.4 M (NH4)2SO4 in the buffer. The active fractions were pooled and dialyzed against the standard buffer at 4°C.
Step 5.
The second dialysate was applied to a Toyopearl SuperQ-650M column (diameter, 2.5 cm; length, 10 cm) equilibrated with the standard buffer. The column was then washed with the buffer and eluted with a linear gradient of 0 to 250 mM NaCl in the buffer. The active fractions were pooled and dialyzed against the standard buffer at 4°C.
Step 6.
The third dialysate was applied to a Red Sepharose CL-4B column (diameter, 1.5 cm; length, 5.75 cm) equilibrated with the standard buffer. The column was then washed with the standard buffer, and the through and wash fractions were mixed as the active fractions. The resultant enzyme solution was then concentrated using an Amicon Ultra 15-ml centrifugal filter 3K device.
Step 7.
Native PAGE of the concentrated enzyme solution was run on a 7.5% polyacrylamide slab gel for further enzyme purification. The electrophoresis was performed using the method of Laemmli (17) with some modifications; buffers without SDS were used, and the protein sample was not heated during the pretreatment procedures. After electrophoresis using a constant current of 20 mA for 90 min, the gel was cut into 16 pieces. The gel pieces were individually crushed in the standard buffer, and the resultant solutions were centrifuged (17,000 × g for 15 min at 4°C). The enzyme activity of the supernatants was then assayed, and the active enzyme solution was used as the final purified enzyme solution from L. otakiensis JCM 15040.
Analysis of N-terminal amino acid sequence.
Approximately 3 μg of the purified enzyme from L. otakiensis JCM 15040 was subjected to SDS-PAGE as described below (17). The resolved proteins were then electroblotted onto a polyvinylidene difluoride membrane, after which the membrane was stained with Ponceau S and destained. To determine of the N-terminal amino acid sequence, each protein band was then subjected to automated Edman degradation using a PPSQ-10 protein sequencer (Shimadzu, Kyoto, Japan). Then with the N-terminal amino acid sequences obtained from the analysis, homology searches were conducted using a protein-protein BLAST (18).
Cloning a putative GABA-AT gene from L. buchneri JCM 1115.
A putative GABA-AT gene fragment from L. buchneri JCM 1115 was amplified by PCR using the oligonucleotide primers 5′-TATAGAGCTCATGGGTAAATTAGACAAAGCGTCTAAATTAATTGATGAAG-3′, which contained a unique SacI restriction site (underlined), and 5′-TATACTCGAGTTATTACCAGCCAATTTTACCTGTATCCTTGGG-3′, which contained a unique XhoI restriction site proximal to the 5′ end of the termination codon. These primers were designed based on the nucleotide sequence of the Lbuc_2316 gene of L. buchneri NRRL B-30929. Chromosomal DNA isolated from L. buchneri JCM 1115 with Isoplant II (Nippon Gene, Tokyo, Japan) was used as the template. The amplified 1.35-kb fragment was digested with SacI and XhoI and ligated into the expression vector pCold ProS2 linearized with SacI and XhoI sites.
Expression and purification of the recombinant L. buchneri JCM 1115 enzyme.
E. coli strain BL21(DE3) was transformed with the expression vector harboring the putative GABA-AT gene from L. buchneri JCM 1115, and the transformants were cultivated at 37°C in 1 liter of Luria-Bertani medium (Becton, Dickinson and Co.) containing 100 μg·ml−1 ampicillin until the optical density at 600 nm (OD600) reached 0.6. Thereafter, 0.1 mM isopropyl-β-d-thiogalactopyranoside was added to the culture medium, and the cultivation was continued for an additional 10 h at 15°C, after which the cells were collected by centrifugation (6,500 × g for 10 min at 4°C). The harvested cells (ca. 2.23 g, wet weight) were used as the starting material for purification of the recombinant enzyme. Unless indicated otherwise, the following purification procedures were carried out at room temperature.
Step 1.
To prepare the crude extract, the cells were washed twice with buffer A consisting of 50 mM Na-phosphate buffer (pH 7.9), 1 mM 2-mercaptoethanol (2-ME), 0.5 M NaCl, and 5 mM imidazole, suspended in the same buffer, lysed using a 500-W ultrasonic processor (Sonics Materials, Newtown, CT), and centrifuged at 10,000 × g for 30 min at 4°C. The resultant supernatant was used as the crude extract.
Step 2.
The crude extract was applied to a Ni-NTA agarose column (diameter, 1.5 cm; length, 5.75 cm) previously equilibrated with buffer A. After the column had been washed with buffer B consisting of 50 mM Na-phosphate buffer (pH 7.9), 1 mM 2-ME, 0.5 M NaCl, and 60 mM imidazole, the enzyme with fused ProS2 and His tags was eluted using a linear gradient of 60 to 600 mM imidazole in buffer B. The active fractions were pooled and dialyzed against 50 mM Na-phosphate buffer (pH 7.2) containing 0.1 mM 2-ME at 4°C.
Step 3.
The first dialysate was incubated for 10 min at 25°C with 0.2-mg/ml thrombin (Sigma) to cleave the ProS2 and His tags. The solution was then applied to a Toyopearl SuperQ-650 M column (diameter, 1.5 cm; length, 5.75 cm) equilibrated with 50 mM Na-phosphate buffer (pH 7.2) containing 1 mM 2-ME. The column was then washed with the same buffer and eluted with a linear gradient of 0 to 250 mM NaCl in the same buffer. The active fractions were pooled and dialyzed against 50 mM Na-phosphate buffer (pH 7.2) containing 0.1 mM 2-ME, 0.5 M NaCl, and 5 mM imidazole at 4°C.
Step 4.
The second dialysate was applied to a Ni-NTA agarose column (diameter, 1.5 cm; length, 5.75 cm) previously equilibrated with buffer A. After the column was washed with buffer B, the wash fractions were mixed as the active fraction and dialyzed against 10 mM Na-phosphate buffer (pH 7.2) containing 0.1 mM 2-ME. The third dialysate was used as the final recombinant enzyme preparation.
SDS-PAGE and molecular mass determination.
SDS-PAGE (10% acrylamide slab gel, 1-mm thick) was carried out using the procedure of Laemmli (17), after which the protein band was stained with Coomassie brilliant blue R-250 or fluorescent gel stain (Bio-Rad, Hercules, CA). The molecular mass of the purified recombinant enzyme was determined using a HiLoad 26/60 Superdex 200 prep-grade column (GE Healthcare, Fairfield, CT) previously equilibrated with 20 mM Tris-HCl buffer (pH 7.5) containing 0.2 M NaCl. The enzyme was eluted with the same buffer at a flow rate of 3 ml/min using an ÄKTA Explorer system (GE Healthcare). The apparent molecular mass was estimated by comparing the retention time of the enzyme with those of a set of molecular mass markers (MWGF1000-1KT; Sigma). The subunit molecular mass of the enzyme was estimated from the amino acid sequence of the enzyme.
Kinetic analyses.
Enzyme assays were carried out under the standard assay conditions using the recombinant enzyme (1.3 μg/ml) and l-Ile (1.25, 2.5, 5, 10, 20, 40, and 60 mM) or d-allo-Ile (5, 10, 20, 40, 60, 80, and 100 mM). The reaction was stopped after 5 min, when less than 10% of the substrate was consumed. The Km and Vmax values for the isoleucine 2-epimerase reaction were determined from Lineweaver-Burk plots of the data (the double-reciprocal plots of the specific activities versus the concentrations of substrates for each direction of reaction). In addition, kcat (molecular activity) was defined as molecules of product formed per min per molecule of enzyme and was calculated from the Vmax (μmol of product formed per minute per mg of enzyme) and molecular weight (49,422.4) of the enzyme.
Effects of PLP and divalent metal ions on the enzyme activity.
The effects of specific PLP inhibitors (0.1 and 1.0 mM hydroxylamine, aminooxyacetate, and phenylhydrazine) on the isoleucine 2-epimerase (6.50 μg/ml) activity were examined under the standard assay conditions using l-Ile as the substrate. In addition, we investigated the effect of PLP on the epimerase activity using dialysis experiments for the recombinant enzyme. The enzyme (65.0 μg/ml) was dialyzed against 1,800 volumes of 100 mM sodium phosphate buffer (pH 7.2) containing 0.1 mM 2-ME and either 50 mM or no hydroxylamine for 18 h at 4°C (first dialysis). This was followed by dialysis against 1,800 volumes of 10 mM sodium phosphate buffer (pH 7.2) containing 0.1 mM 2-ME and either 1 mM or no PLP for 18 h at 4°C (second dialysis). After that, the enzyme activity was measured under the standard assay conditions with 6.50 μg/ml of the enzyme and l-Ile as the substrate without PLP. The effect of divalent metal ion on the enzyme activity was also examined. The isoleucine 2-epimerase activity was determined under the standard assay conditions with 6.50 μg/ml of the enzyme, l-Ile as the substrate, and 10 mM CaCl2, MgCl2, SrCl2, MnCl2, CuCl2, NiCl2, CoCl2, FeCl2, or ZnCl2.
Effects of pH and temperature on enzyme activity and stability.
To investigate the pH dependency of the enzyme activity, isoleucine 2-epimerase (6.50 μg/ml) activity was assayed at various pHs (3.5 to 10.0) under the standard assay conditions. In addition, to determine the effect of temperature on the enzyme activity, the isoleucine 2-epimerase (6.50 μg/ml) activity was assayed at various temperatures (15 to 75°C) under the standard assay conditions. Furthermore, we also investigated the effects of pH and temperature on the enzyme stability. The recombinant enzyme (65.0 μg/ml) was incubated in 100 mM buffer at various pHs (3.0 to 12.0) at 30°C for 30 min or in 10 mM sodium phosphate buffer (pH 7.2) at various temperatures (30 to 90°C) for 30 min, after which the residual isoleucine 2-epimerase activity was measured under the standard assay conditions with 6.50 μg/ml of the enzyme and l-Ile as the substrate.
Homology search of the isoleucine 2-epimerase in lactic acid bacteria.
To examine a distribution of the isoleucine 2-epimerase in lactic acid bacteria, we performed the homology search based on the amino acid sequence of L. buchneri isoleucine 2-epimerase using BLAST. The database of reference proteins was used in the BLAST search.
RESULTS
Determination of BCAA racemase activity in L. otakiensis JCM 15040.
We observed that by the time cultivation of L. otakiensis JCM 15040 reached the stationary phase, there had been marked increases in the concentrations of d-Leu, d-allo-Ile, and d-Val in the medium; that is, growth of L. otakiensis JCM 15040 was accompanied by increases in the levels of these d-BCAAs in the culture medium (Fig. 1A). This suggests that the d-BCAAs detected in the culture medium were produced by L. otakiensis JCM 15040. We therefore tested for the activities of racemases and epimerases, which are well known as d-amino acid synthetases. Crude extract prepared from L. otakiensis JCM 15040 cells was incubated for 6 h at 30°C in a reaction mixture containing 100 mM Na-phosphate buffer (pH 8.0), 0.1 mM PLP, and 10 mM l- or d-amino acid, after which the produced enantiomeric amino acids were assayed using UPLC. We found that the enzyme reaction produced d-allo-Ile, d-Leu, and d-Val from substrates l-Ile, l-Leu, and l-Val, respectively, and that, conversely, l-Ile, l-Leu, and l-Val were produced from d-allo-Ile, d-Leu, and d-Val, respectively. This shows the presence of both BCAA racemase and epimerase activities in L. otakiensis JCM 15040. Moreover, the specific activity of the apparent isoleucine 2-epimerase was higher than those of the Leu and Val racemases (Table 1). In addition, the isoleucine 2-epimerase activity was higher than the Ala racemase activity broadly observed in Lactobacillus species.
Fig 1.
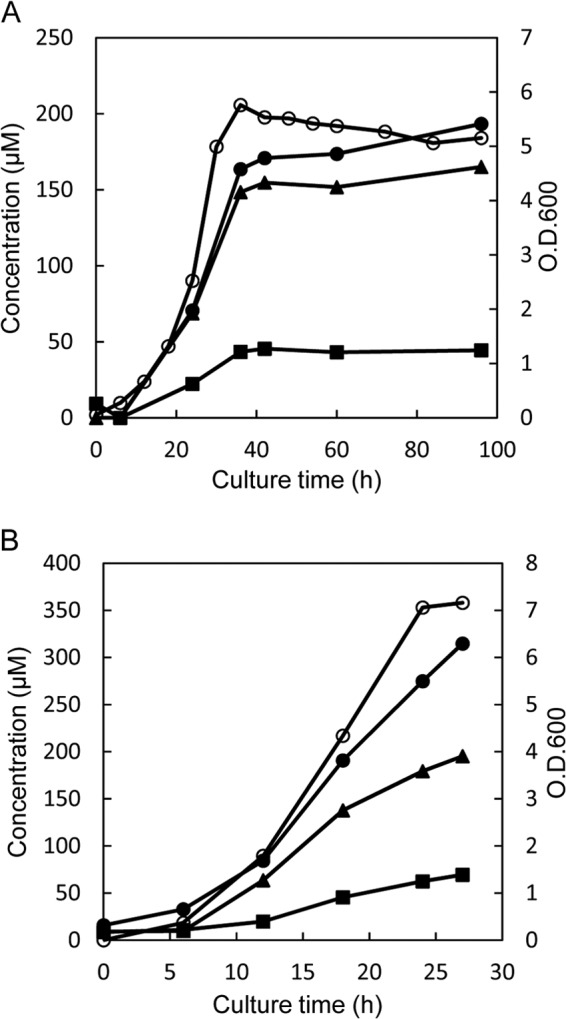
Time course of D-BCAA accumulation in culture media of L. otakiensis JCM 15040 (A) and L. buchneri JCM 1115 (B). Each of the Lactobacillus strains was cultured in MRS medium, and the time-dependent accumulation of d-Leu (solid circles), d-allo-Ile (solid triangles), and d-Val (solid squares) in the culture medium was monitored using a UPLC system. The cell concentration was assayed at OD600 (open circles).
Table 1.
Racemase and epimerase activities in the crude extract of L. otakiensis JCM 15040
| Substrate | Sp act (nmol·mg−1·min−1) |
|
|---|---|---|
| l→d | d→l | |
| l- or d-form | ||
| Ala | 7.03 | 6.16 |
| Val | 2.41 | 1.65 |
| Leu | 2.86 | 3.04 |
| l-Ile or d-allo-Ile | 8.26 | 7.69 |
Purification of a protein exhibiting isoleucine 2-epimerase activity from L. otakiensis JCM 15040 and N-terminal amino acid sequence analysis.
The isoleucine 2-epimerase activity was purified from a crude extract of L. otakiensis JCM 15040 using an ammonium sulfate fractionation, four successive chromatography steps, and native PAGE. Ultimately, the enzyme was purified about 4,270 times, with an overall yield of about 1.27% (Table 2). When the purified enzyme was subjected to SDS-PAGE, two main bands were observed (see the data at https://www.dropbox.com/s/h4084ry9b3406ji/Supplemental%20materials%20for%20JB00709-13.pdf), and the corresponding molecular masses were calculated to be about 90 and 50 kDa. In addition, their N-terminal amino acid sequences were determined to be MKINQFAYVPTD and GKLDKASKLI, respectively.
Table 2.
Purification of the isoleucine 2-epimerase activity from L. otakiensis JCM 15040
| Purification step | Total protein (mg) | Total activity (nmol·min−1) | Sp act (nmol·mg−1·min−1)a | Yield (%)a | Purification (fold)a |
|---|---|---|---|---|---|
| Crude extract | 1,009 | 8,132 | 8.04 | 100 | 1.00 |
| (NH4)2SO4 (supernatant, (60%) | 475 | 5,178 | 10.9 | 63.7 | 1.35 |
| Toyopearl phenyl-650M | 149 | 5,036 | 33.8 | 61.9 | 4.20 |
| Toyopearl butyl-650M | 45.0 | 3,528 | 78.4 | 43.4 | 9.75 |
| Toyopearl SuperQ-650M | 1.18 | 2,541 | 2,154 | 31.2 | 268 |
| Red Sepharose | 0.81 | 2,252 | 2,781 | 27.6 | 346 |
| Native PAGE | 0.003 | 103 | 34,332 | 1.27 | 4,270 |
Cell weight, 32.2 g.
Homology search and d-amino acid detection in the culture medium of L. buchneri JCM 1115.
Based on the N-terminal amino acid sequences of the two proteins detected on SDS-PAGE of the final purified enzyme from L. otakiensis JCM 15040, we conducted a homology search of the GenBank database using a protein-protein BLAST. The search revealed the N-terminal amino acid sequence of the 90-kDa protein to be completely identical to those deduced from the Lbuc_1851 gene of L. buchneri NRRL B-30929 and the LBUCD034_1936 gene of L. buchneri CD034, while the sequence of the 50-kDa protein was also completely identical to those deduced from the Lbuc_2316 gene of L. buchneri NRRL B-30929 and the LBUCD034_2418 gene of L. buchneri CD034. The products of the Lbuc_1851 and LBUCD034_1936 genes are annotated as X-prolyl-dipeptidyl aminopeptidases, while the product of LBUCD034_2418 is annotated as a GABA-AT. On the other hand, the product of Lbuc_2316 is annotated as an acetylornithine transaminase (AO-TA), although the amino acid sequence deduced from Lbuc_2316 is more homologous to that of bacterial GABA-AT than AO-TA, and the Kyoto Encyclopedia of Gene and Genome Orthology database lists the product of Lbuc_2316 as a GABA-AT (http://www.genome.jp/dbget-bin/www_bget?lbh:Lbuc_2316). Between the two putative enzymes, we would predict that BCAA racemase is encoded by a putative GABA-AT gene rather than a putative X-prolyl-dipeptidyl aminopeptidase gene, because racemases are generally PLP-dependent enzymes, similar to transaminases, including GABA-AT, and the amino acid sequences deduced from both Lbuc_2316 and LBUCD034_2418 include PLP-binding sites.
Cloning and sequence of the putative GABA-AT gene from L. buchneri JCM 1115.
When the concentrations of d-amino acids in the culture medium of L. buchneri JCM 1115 were sequentially measured, we found that the levels of d-Leu, d-allo-Ile, and d-Val in the culture medium increased accompanied by the increase in bacterial growth (Fig. 1B). A DNA fragment (1.35 kb) containing the putative GABA-AT gene from L. buchneri JCM 1115 was amplified by PCR and cloned into expression vector pCold ProS2, after which analysis of the nucleotide sequence was entrusted to Hokkaido System Science (Sapporo, Japan). The sequence of the analyzed fragment (1.35 kb) contained one open reading frame encoding the enzyme, which was composed of 450 amino acid residues (GenBank accession no. KC413940). The amino acid sequence deduced from the L. buchneri JCM 1115 gene showed 99.8% and 99.6% identity to those deduced from Lbuc_2316 of L. buchneri NRRL B-30929 and LBUCD034_2418 of L. buchneri CD 034, respectively (data not shown).
Purification and molecular mass determination of the recombinant enzyme.
E. coli BL21(DE3) cells were transformed by the expression vector pCold ProS2 containing the putative GABA-AT gene from L. buchneri JCM 1115. The crude extract from the recombinant cells showed high isoleucine 2-epimerase activity (see the data at https://www.dropbox.com/s/h4084ry9b3406ji/Supplemental%20materials%20for%20JB00709-13.pdf), and three chromatography steps, including excision of the ProS2- and His-tags, yielded 11.1 mg of the purified enzyme from the extract prepared from 1 liter of culture. SDS-PAGE of the purified enzyme gave a single band corresponding to a molecular mass of about 49 kDa (see the URL mentioned above), which is consistent with the molecular weight (49,422.4) calculated from the amino acid sequence obtained as described above. In addition, the molecular mass of the recombinant enzyme was estimated by gel filtration and the amino acid sequence of the enzyme. With gel filtration on high-performance liquid chromatography (HPLC) using a Superdex 200 column, the purified recombinant enzyme eluted as a single peak with a retention time corresponding to 200 kDa. Thus, the native enzyme appears to exist as a tetramer.
Identification of the enzyme as isoleucine 2-epimerase.
We next identified the product of the enzymatic reaction catalyzed by the recombinant enzyme (6.5 μg/ml) under the standard assay conditions using 10 mM l-Ile or d-allo-Ile as the substrate. After the enzymatic reaction using l-Ile as the substrate, UPLC showed only a d-allo-Ile peak beside an l-Ile peak in the reaction mixture. The appearance of d-allo-Ile was accompanied by a reduction in l-Ile (Fig. 2A), suggesting that the enzyme catalyzes 2-epimerization of l-Ile to d-allo-Ile. The epimerization nearly reached equilibrium after incubation for 20 min, and the final concentrations of l-Ile and d-allo-Ile were 4.33 mM and 5.67 mM, respectively. A similar reaction time course was observed with d-allo-Ile as the substrate, and the concentrations of d-allo-Ile and l-Ile at equilibrium were also 5.67 mM and 4.33 mM, respectively (Fig. 2B).
Fig 2.
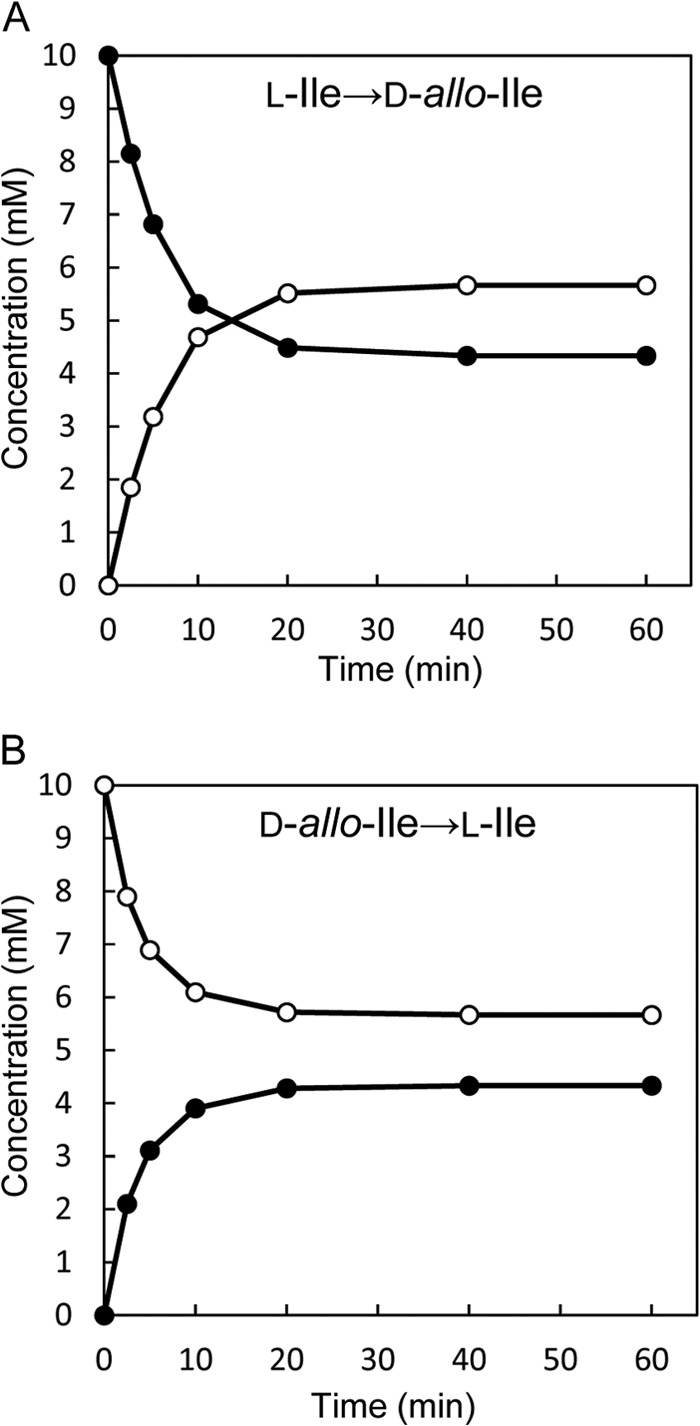
Time courses of the isoleucine 2-epimerase reaction with l-Ile (A) or d-allo-Ile (B) as the substrate. The data for l-Ile and d-allo-Ile are depicted as solid circles and open circles, respectively.
In addition, because a gene homologous to the recombinant enzyme was annotated as GABA-AT in the GenBank database, we also assessed the GABA-AT activity of the recombinant enzyme. The purified recombinant enzyme (32.5 μg/ml) was incubated in a reaction mixture (500 μl) containing 200 mM Na-phosphate buffer (pH 8.0), 100 mM α-ketoglutarate, 100 mM GABA, and 0.1 mM PLP for 60 min at 37°C. The optimum pH of bacterial GABA-AT is reportedly around 8.0 (19–23). After running the reaction, we used UPLC to assess the formation of l-Glu as the reaction product, but no l-Glu peak was detected.
Substrate specificity.
The substrate specificities of the racemase and epimerase activities of the recombinant enzyme were investigated using the d- and l-forms of Asp, Glu, Lys, Arg, His, Orn, Thr, Asn, Gln, Trp, Ile, Leu, Val, Phe, Met, Ser, Ala, Nva, Nle, Abu, allo-Ile, and tert-Leu. Each substrate (50 mM) was incubated with the enzyme (6.50 μg/ml) under the standard assay conditions, and the enantiomer produced was assayed by UPLC. The enzyme preferentially catalyzed epimerization of l-Ile and d-allo-Ile, as well as racemization of many nonpolar amino acids, including Leu and Val (Table 3). In contrast, the enzyme had no activity toward the l- and d-forms of Asp, Glu, Lys, Arg, His, Orn, Thr, Asn, Gln, Trp, and tert-Leu.
Table 3.
Substrate specificity for racemase or epimerase activity of the recombinant enzymea
|
l→d reaction |
d→l reaction |
||||
|---|---|---|---|---|---|
| l-Form substrate | Sp act (μmol·mg−1·min−1) | Relative activity (%) | d-Form substrate | Sp act (μmol·mg−1·min−1) | Relative activity (%) |
| Ile | 149 ± 4.01 | 100 | allo-Ile | 221 ± 0.400 | 100 |
| Nva | 83.4 ± 3.30 | 56 | Nva | 183 ± 4.26 | 83 |
| Nle | 74.1 ± 2.82 | 50 | Val | 131 ± 7.14 | 59 |
| Val | 71.7 ± 4.14 | 48 | Nle | 116 ± 3.42 | 52 |
| Abu | 47.7 ± 2.34 | 32 | Met | 93.7 ± 2.59 | 42 |
| Leu | 44.2 ± 1.00 | 30 | Abu | 66.3 ± 1.94 | 30 |
| Phe | 35.7 ± 0.444 | 24 | Leu | 54.8 ± 2.07 | 25 |
| Met | 32.0 ± 0.487 | 21 | Ile | 44.5 ± 2.03 | 20 |
| allo-Ile | 27.8 ± 1.42 | 19 | Phe | 13.6 ± 0.498 | 6 |
| Ser | 9.58 ± 0.467 | 6 | Ser | 4.91 ± 0.074 | 2 |
| Ala | 3.95 ± 0.550 | 3 | Ala | 4.43 ± 0.127 | 2 |
The values were obtained through repeated measurements (n = 3).
Kinetic parameters.
Lineweaver-Burk plots of the initial rates of epimerase activity for l-Ile or d-allo-Ile showed typical straight lines for both reaction directions (see the data at https://www.dropbox.com/s/h4084ry9b3406ji/Supplemental%20materials%20for%20JB00709-13.pdf). From the lines, the Km for l-Ile and Vmax were determined to be 5.00 mM and 153 μmol·mg−1·min−1, respectively, and in the reverse direction, the Km for d-allo-Ile and Vmax were 13.2 mM and 286 μmol·mg−1·min−1, respectively. In addition, the kcat was calculated from the Vmax and molecular mass of the enzyme, and then the kcat/Km ratio was also calculated (Table 4).
Table 4.
Kinetic parameters of the isoleucine 2-epimerase reactiona
| Reaction | Km (mM) | kcat (s−1) | kcat/Km (s−1·mM−1) |
|---|---|---|---|
| l-Ile→d-allo-Ile | 5.00 ± 0.080 | 502 ± 16.2 | 101 ± 4.86 |
| d-allo-Ile→l-Ile | 13.2 ± 0.644 | 939 ± 26.8 | 71.4 ± 5.52 |
The values were obtained through repeated measurements (n = 3).
Effect of PLP and divalent metal ions on enzyme activity.
Amino acid racemases are generally categorized into two groups, PLP dependent or PLP independent, with the latter requiring neither cofactors nor metals (24, 25). To determine which group the recombinant enzyme belonged to, the effect of specific PLP inhibitors on the isoleucine 2-epimerase activity was examined. The epimerase activity was totally blocked by the inhibitors at a concentration of 1 mM (see the URL given above). In addition, the enzyme was almost fully inactivated by the first dialysis against buffer containing 50 mM hydroxylamine, after which about 75% of the activity was recovered by a subsequent dialysis against buffer containing 1 mM PLP. Furthermore, when the enzyme was dialyzed against PLP-free buffer, about 97% of the activity was lost, but about 90% of the enzyme activity was recovered by a second dialysis against buffer containing 1.0 mM PLP (see the URL given above). We also investigated the effects of various divalent metal ions (Ca2+, Mg2+, Sr2+, Mn2+, Cu2+, Ni2+, Co2+, Fe2+, and Zn2+) on the isoleucine 2-epimerase activity. When added as chloride salts to the reaction mixture, these divalent metal ions had almost no effect on the enzyme activity (data not shown).
Effect of pH and temperature on the enzyme activity and stability.
The highest activity for the l-Ile→d-allo-Ile reaction was obtained at around pH 5.0, and the highest activity for the reverse reaction was obtained at around pH 6.0 (Fig. 3). In addition, the highest activity for the l-Ile→d-allo-Ile reaction was obtained at around 50°C, and that for the reverse reaction was obtained at around 45°C (Fig. 4). Furthermore, the enzyme retained about 80% of its activity after incubation for 30 min at pHs ranging from 5.0 to 11.0 (Fig. 5A). The enzyme also retained more than 80% of its activity after incubation for 30 min at 60°C (Fig. 5B).
Fig 3.
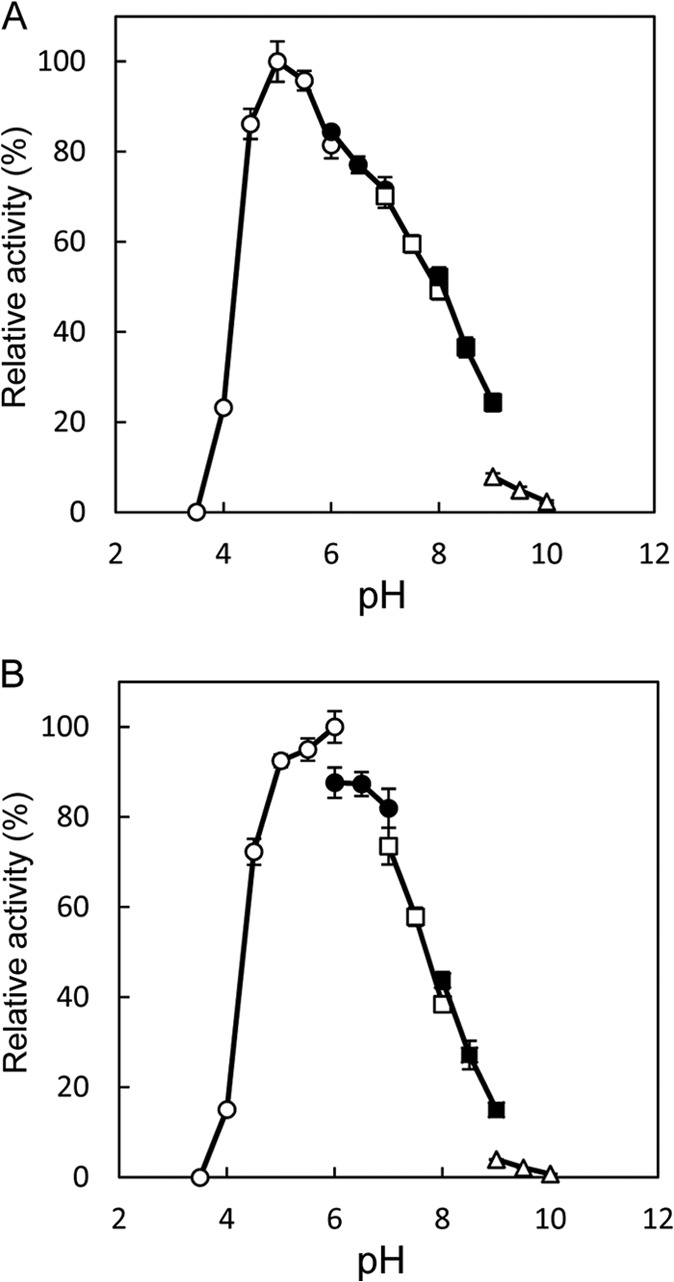
Effect of pH on the isoleucine 2-epimerase activity with l-Ile (A) and d-allo-Ile (B) as the substrates. The buffers used were sodium citrate (pH 3.5 to 6.0) (open circles), MES (morpholineethanesulfonic acid)-NaOH (pH 6.0 to 7.0) (solid circles), HEPES-NaOH (pH 7.0 to 8.0) (open squares), TAPS {3-([2-hydroxy-1,1-bis(hydroxymethyl)ethyl]-amino)-1-propanesulfonic acid}-NaOH (pH 8.0 to 9.0) (solid squares), and CHES (N-cyclohexyl-2-aminoethanesulfonic acid)-NaOH (pH 9.0 to 10.0) (open triangles). Enzyme activities are expressed as percentages relative to 156 μmol·mg−1·min−1 (A) and 233 μmol·mg−1·min−1 (B), which are shown as 100%. All values were obtained through repeated measurements (n = 3).
Fig 4.
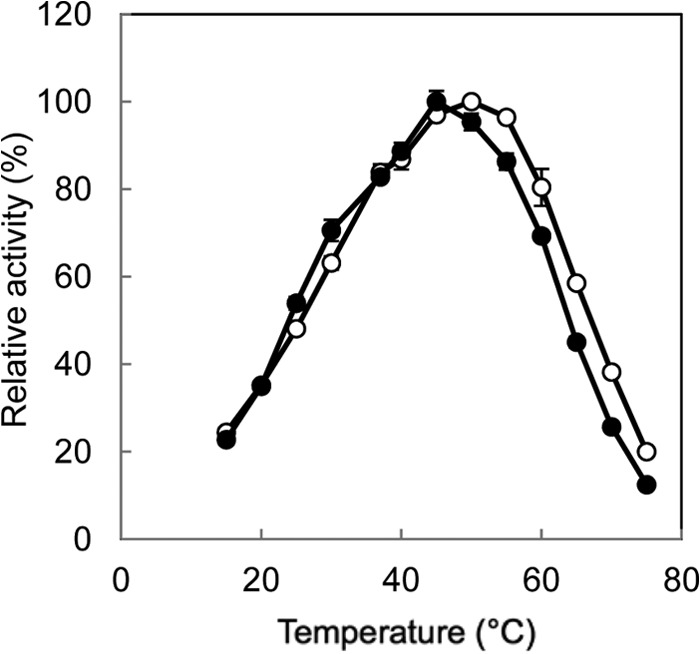
Effect of temperature on the isoleucine 2-epimerase activity with l-Ile (open circles) and d-allo-Ile (solid circles) as the substrates. Enzyme activities are expressed as percentages relative to 178 μmol·mg−1·min−1 (l-Ile as the substrate) and 267 μmol·mg−1·min−1 (d-allo-Ile as the substrate), which are shown as 100%. All values were obtained through repeated measurements (n = 3).
Fig 5.
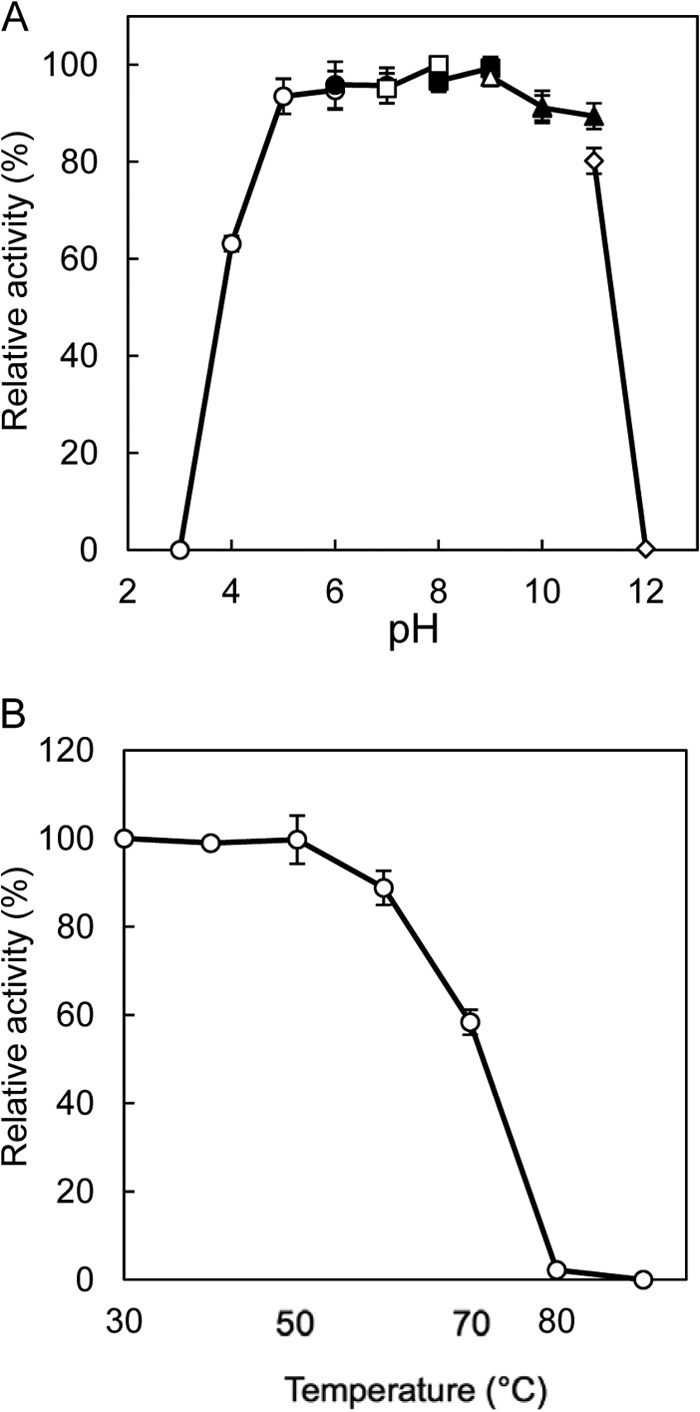
Effects of pH (A) and temperature (B) on the stability of the recombinant enzyme. The buffers used were sodium citrate (pH 3.0 to 6.0) (open circles), MES-NaOH (pH 6.0 to 7.0) (solid circles), HEPES-NaOH (pH 7.0 to 8.0) (open squares), TAPS-NaOH (pH 8.0 to 9.0) (solid squares), CHES-NaOH (pH 9.0 to 10.0) (open triangles), CAPS (N-cyclohexyl-3-aminopropanesulfonic acid)-NaOH (pH 10.0 to 11.0) (solid triangles), and Na2HPO4-NaOH (pH 11.0 to 12.0) (open diamonds). Enzyme activities are expressed as percentages relative to 149 μmol·mg−1·min−1, which is shown as 100%. The values were obtained through repeated measurements (n = 3).
Homology search of the isoleucine 2-epimerase in lactic acid bacteria.
We searched for L. buchneri isoleucine 2-epimerase homologous genes in lactic acid bacteria and found that 26 species of Lactobacillus, 11 species of Leuconostoc, 8 species of Streptococcus, 7 species of Enterococcus, 2 species of Weissella, and one species of Oenococcus have homologs showing more than 30% identity to the L. buchneri enzyme (Table 5). In particular, Lactobacillus hilgardii, Lactobacillus parafarraginis, and Oenococcus oeni have homologs showing more than 80% identity to L. buchneri enzyme.
Table 5.
Lactic acid bacteria having L. buchneri isoleucine 2-epimerase homologs
| Lactic acid bacterium | GI no. of homologous amino acid sequencea | Identity (%)b |
|---|---|---|
| Lactobacillus | ||
| L. hilgardii | 489646465 | 89 |
| L. parafarraginis | 495486032 | 88 |
| L. farciminis | 497706818 | 75 |
| L. versmoldensis | 498310982 | 73 |
| L. pentosus | 489734613 | 70 |
| L. plantarum | 254556614 | 70 |
| L. plantarum subsp. plantarum | 308180559 | 70 |
| L. gastricus | 494193600 | 61 |
| L. acidpiscis | 498181006 | 60 |
| L. vaginalis | 489813945 | 60 |
| L. murinus | 490152569 | 60 |
| L. malefermentans | 498305994 | 59 |
| L. suebicus | 498307063 | 59 |
| L. animalis | 498373997 | 59 |
| L. pobuzihii | 516478760 | 59 |
| L. mucosae | 493546858 | 58 |
| L. vini | 498266185 | 57 |
| L. sanfranciscensis | 347533884 | 57 |
| L. reuteri | 518082091 | 56 |
| L. fructivorans | 497708861 | 56 |
| L. fermentum | 501675571 | 56 |
| L. florum | 496457402 | 53 |
| L. mali | 489784559 | 34 |
| L. ruminis | 489788176 | 33 |
| L. parafarraginis | 495486664 | 31 |
| L. brevis subsp. gravesensis | 490601567 | 30 |
| Leuconostoc | ||
| L. carnosum | 407718645 | 60 |
| L. mesenteroides subsp. cremoris | 490267966 | 60 |
| L. citreum | 170016824 | 59 |
| L. mesenteroides subsp. mesenteroides | 116617671 | 59 |
| L. fallax | 497693964 | 58 |
| L. kimchii | 296110669 | 58 |
| L. pseudomesenteroides | 491049189 | 58 |
| L. lactis | 497687862 | 58 |
| L. inhae | 498068665 | 58 |
| L. gelidum | 406600315 | 57 |
| L. gasicomitatum | 300173640 | 56 |
| Enterococcus | ||
| E. columbae | 510807871 | 32 |
| E. casseliflavus | 491369323 | 31 |
| E. avium | 510805436 | 31 |
| E. faecium | 514879442 | 31 |
| E. pallens | 498453561 | 30 |
| E. gilvus | 498476463 | 30 |
| E. malodoratus | 498433498 | 30 |
| Streptococcus | ||
| S. ratti | 489179634 | 31 |
| S. sanguinis | 488987151 | 31 |
| S. gordonii Challis | 157151527 | 31 |
| S. infantarius subsp. infantarius | 379704932 | 30 |
| S. lutetiensis | 527329769 | 30 |
| S. mutans | 488198523 | 30 |
| S. ferus | 516665549 | 30 |
| S. equinus | 490351159 | 30 |
| Oenococcus oeni | 488906786 | 86 |
| Weissella | ||
| W. thailandensis | 341821030 | 54 |
| W. paramesenteroides | 488917328 | 53 |
GI, GenInfo identifier in National Center for Biotechnology Information.
Amino acid sequence identity with the isoleucine 2-epimerase from L. buchneri JCM 1115.
DISCUSSION
In this study, prominent accumulation of d-Leu, d-allo-Ile, and d-Val was observed in the culture medium of L. otakiensis JCM 15040, and a BCAA racemase activity was detected in the crude extract from this bacterium. To our knowledge, this is the first reported observation of such production of d-BCAAs in lactic acid bacteria and the first example of a BCAA racemase with isoleucine 2-epimerase activity in any organism. Until now, alanine racemase (AlaR) from Pseudomonas putida (26) was the only racemase known to act on l-Ile, but that enzyme shows much less activity toward l-Ile than l-Ala (relative activity, 38.7%). This suggests L. otakiensis may express a novel amino acid racemase. Because the sequencing of the L. otakiensis genome is incomplete, we purified the protein exhibiting isoleucine 2-epimerase activity from the cell extract and determined the N-terminal amino acid sequence of the purified enzyme. Then from a homology search based on that N-terminal amino acid sequence, we identified two putative GABA-AT genes from L. buchneri NRRL B-30929 and CD034. Unfortunately, we were not able to obtain either of those strains from any culture collections of microorganisms. As an alternative, we selected L. buchneri JCM 1115 for study and found that it exhibited a pattern of time-dependent accumulation of d-Leu, d-allo-Ile, and d-Val in the culture medium that was similar to the pattern seen with L. otakiensis. We then cloned the putative GABA-AT gene from L. buchneri JCM 1115 and expressed it in recombinant E. coli. Notably, the purified recombinant enzyme exhibited strong isoleucine 2-epimerase activity but not GABA-AT activity.
When we characterized the recombinant isoleucine 2-epimerase from L. buchneri JCM 1115 in detail, we found that the enzyme preferentially catalyzes epimerization between l-Ile and d-allo-Ile and also catalyzes racemizations of nonpolar amino acids, including Leu and Val. This suggests this is the first enzyme catalyzing racemization of BCAAs as their main substrate in all the organisms. In addition, both optimum pHs for epimerization are fairly acidic (pH 5.0 to 6.0). In contrast, with the exception for proline racemase (27), other amino acid racemases have pH optima around 8.0. Another difference between this enzyme and other racemases is its tetrameric structure; most reported amino acid racemases exist as monomers or dimers (28–34). Furthermore, the isoleucine 2-epimerase is unique among PLP-dependent enzymes. Racemases are generally divided into two groups: PLP dependent and PLP independent (24, 25). Our examination of the effects of PLP inhibitors and PLP supplementation on the isoleucine 2-epimerase activity shows this enzyme to be in the PLP-dependent group. All PLP-dependent enzymes whose structures have been solved are presently classified into five fold types, I to V, based on similarities in their primary and secondary structures (35–37). There is no correlation between fold type and reaction specificity, however. The PLP-dependent racemases identified so far have been classified into fold types I, II, and III, and the bacterial PLP-dependent racemases are included in fold types I and III (38). We examined the amino acid sequence homology of L. buchneri isoleucine 2-epimerase with a BLAST search of the Protein Data Bank and found that the enzyme was classified into the fold type I of PLP-dependent enzymes. Until now, bacterial racemases classified into the fold type I of PLP-dependent enzymes has been only α-amino-ε-caprolactam racemase from Achromobacter obae (38). Thus, the isoleucine 2-epimerase is the second example of the bacterial racemase belonging to fold type I, and this amino acid sequence is informative and sheds light to structure-function relationship of bacterial PLP-dependent racemases in fold type I. Collectively, the characteristics summarized above indicate that the isoleucine 2-epimerase from L. buchneri is a novel type of amino acid racemase.
In the case of many racemase reactions, the kcat/Km ratio for l-amino acid equals that for d-amino acid, because the equilibrium constant, Keq, of racemization is 1 in the Haldane equation. However, in this isoleucine 2-epimerase reaction, the value of Keq with l-Ile as a substrate was 1.31 (Fig. 2), and the value of (kcat/Km for l-Ile)/(kcat/Km for d-allo-Ile) was 1.41 (Table 4). Such values (>1) are probably due to the fact that the reaction is epimerization but not racemization. The Km values of the isoleucine 2-epimerase were 5.00 mM for l-Ile and 13.2 mM for d-allo-Ile. These values are relatively high compared with their estimated concentrations in the bacterial cells, but the similar Km values have been reported in bacterial AlaRs (39–42) and glutamate racemase (29). Until now, the details about the production mechanisms of d-BCAAs in Lactobacillus species have been unclear. However, in this study, we identified the novel amino acid racemase that preferentially acts on BCAAs and found the wide distribution of the homologous gene in lactic acid bacteria. That may be informative to further investigations for a novel d-BCAA synthetic pathway in L. buchneri, L. otakiensis, and other lactic acid bacteria.
ACKNOWLEDGMENTS
This work was supported by a grant for Promotion of Basic Research Activities for Innovate Bioscience from the Bio-Oriented Technology Research Advancement Institution (BRAIN) and JSPS KAKENHI grant no. 2402734.
Footnotes
Published ahead of print 13 September 2013
REFERENCES
- 1.Ghuysen JM. 1961. Data on the structure of disaccharide-peptide complexes liberated from the wall of Micrococcus lysodeikticus by the action of beta(1–4)N-acetylhexosaminidases. Biochim. Biophys. Acta 47:561–568 [DOI] [PubMed] [Google Scholar]
- 2.Hancock R. 1960. The amino acid composition of the protein and cell wall of Staphylococcus aureus. Biochim. Biophys. Acta 37:42–46 [DOI] [PubMed] [Google Scholar]
- 3.Veiga P, Piquet S, Maisons A, Furlan S, Courtin P, Chapot-Chartier MP, Kulakauskas S. 2006. Identification of an essential gene responsible for D-Asp incorporation in the Lactococcus lactis peptidoglycan crossbridge. Mol. Microbiol. 62:1713–1724 [DOI] [PubMed] [Google Scholar]
- 4.Bellais S, Arthur M, Dubost L, Hugonnet JE, Gutmann L, van Heijenoort J, Legrand R, Brouard JP, Rice L, Mainardi JL. 2006. Aslfm, the d-aspartate ligase responsible for the addition of d-aspartic acid onto the peptidoglycan precursor of Enterococcus faecium. J. Biol. Chem. 281:11586–11594 [DOI] [PubMed] [Google Scholar]
- 5.Grohs P, Gutmann L, Legrand R, Schoot B, Mainardi JL. 2000. Vancomycin resistance is associated with serine-containing peptidoglycan in Enterococcus gallinarum. J. Bacteriol. 182:6228–6232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.De Jonge BL, Gage D, Xu N. 2002. The carboxyl terminus of peptidoglycan stem peptides is a determinant for methicillin resistance in Staphylococcus aureus. Antimicrob. Agents Chemother. 46:3151–3155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reynolds PE, Courvalin P. 2005. Vancomycin resistance in enterococci due to synthesis of precursors terminating in d-alanyl-d-serine. Antimicrob. Agents Chemother. 49:21–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sieradzki K, Tomasz A. 1996. A highly vancomycin-resistant laboratory mutant of Staphylococcus aureus. FEMS Microbiol. Lett. 142:161–166 [DOI] [PubMed] [Google Scholar]
- 9.Lam H, Oh DC, Cava F, Takacs CN, Clardy J, de Pedro MA, Waldor MK. 2009. d-amino acids govern stationary phase cell wall remodeling in bacteria. Science 325:1552–1555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kolodkin-Gal I, Romero D, Cao S, Clardy J, Kolter R, Losick R. 2010. d-amino acids trigger biofilm disassembly. Science 328:627–629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yorifuji T, Ogata K, Soda K. 1971. Arginine racemase of Pseudomonas graveolens. I. Purification, crystallization, and properties. J. Biol. Chem. 246:5085–5092 [PubMed] [Google Scholar]
- 12.Lim YH, Yokoigawa K, Esaki N, Soda K. 1993. A new amino acid racemase with threonine α-epimerase activity from Pseudomonas putida: purification and characterization. J. Bacteriol. 175:4213–4217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ohshima T, Sakuraba H. 1986. Purification and characterization of malate dehydrogenase from the phototrophic bacterium, Rhodopseudomonas capsulata. Biochim. Biophys. Acta 869:171–177 [Google Scholar]
- 14.Aswad DW. 1984. Determination of d- and l-aspartate in amino acid mixtures by high-performance liquid chromatography after derivatization with a chiral adduct of o-phthaldialdehyde. Anal. Biochem. 137:405–409 [DOI] [PubMed] [Google Scholar]
- 15.Hashimoto A, Nishikawa T, Oka T, Takahashi K, Hayashi T. 1992. Determination of free amino acid enantiomers in rat brain and serum by high-performance liquid chromatography after derivatization with N-tert-butyloxycarbonyl-l-cysteine and o-phthaldialdehyde. J. Chromatogr. 582:41–48 [DOI] [PubMed] [Google Scholar]
- 16.Laemmli UK. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685 [DOI] [PubMed] [Google Scholar]
- 17.Bradford MM. 1976. A rapid and sensitive method for quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248–254 [DOI] [PubMed] [Google Scholar]
- 18.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J. Mol. Biol. 215:403–410 [DOI] [PubMed] [Google Scholar]
- 19.Yonaha K, Suzuki K, Toyama S. 1985. 4-Aminobutyrate: 2-oxoglutarate aminotransferase of Streptomyces griseus: purification and properties. Eur. J. Biochem. 146:101–106 [DOI] [PubMed] [Google Scholar]
- 20.Andersen G, Andersen B, Dobritzsch D, Schnackerz KD, Piškur J. 2007. A gene duplication led to specialized γ-aminobutyrate and β-alanine aminotransferase in yeast. FEBS J. 274:1804–1817 [DOI] [PubMed] [Google Scholar]
- 21.Scott EM, Jakoby WB. 1959. Soluble γ-aminobutyric-glutamic transaminase from Pseudomonas fluorescens. J. Biol. Chem. 234:932–936 [PubMed] [Google Scholar]
- 22.Der Garabedian PA, Lotti AM, Vermeersch JJ. 1986. 4-Aminobutyrate: 2-oxoglutarate aminotransferase from Candida. Purification and properties. Eur. J. Biochem. 156:589–596 [DOI] [PubMed] [Google Scholar]
- 23.Mayer J, Cook AM. 2009. Homotaurine metabolized to 3-sulfopropanoate in Cupriavidus necator H16: enzymes and genes in a patchwork pathway. J. Bacteriol. 191:6052–6058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Soda K, Esaki N. 1994. Pyridoxal enzymes acting on d-amino acids. Pure Appl. Chem. 66:709–714 [Google Scholar]
- 25.Yoshimura T, Esaki N. 2003. Amino acid racemases: functions and mechanisms. J. Biosci. Bioeng. 96:103–109 [DOI] [PubMed] [Google Scholar]
- 26.Liu JL, Liu XQ, Shi YW. 2012. Expression, purification, and characterization of alanine racemase from Pseudomonas putida YZ-26. World J. Microbiol. Biotechnol. 28:267–274 [DOI] [PubMed] [Google Scholar]
- 27.Chamond N, Grégoire C, Coatnoan N, Rougeot C, Freitas-Junior LH, da Silveira JF, Degrave WM, Minoprio P. 2003. Biochemical characterization of proline racemase from the human protozoan parasite Trypanosoma cruzi and definition of putative protein signatures. J. Biol. Chem. 278:15484–15494 [DOI] [PubMed] [Google Scholar]
- 28.Ju J, Xu S, Furukawa Y, Zhang Y, Misono H, Minamino T, Namba K, Zhao B, Ohnishi K. 2011. Correlation between catalytic activity and monomer-dimer equilibrium of bacterial alanine racemases. J. Biochem. 149:83–89 [DOI] [PubMed] [Google Scholar]
- 29.Ashiuchi M, Tani K, Soda K, Misono H. 1998. Properties of glutamate racemase from Bacillus subtilis IFO 3336 producing poly-γ-glutamate. J. Biochem. 123:1156–1163 [DOI] [PubMed] [Google Scholar]
- 30.Inagaki K, Tanizawa K, Tanaka H, Soda K. 1987. Purification and characterization of amino acid racemase with very broad substrate specificity from Aeromonas caviae. Agric. Biol. Chem. 51:173–180 [Google Scholar]
- 31.Chen HP, Lin CF, Lee YJ, Tsay SS, Wu SH. 2000. Purification and properties of ornithine racemase from Clostridium sticklandii. J. Bacteriol. 182:2052–2054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Okada H, Yohda M, Giga-Hama Y, Ueno Y, Ohdo S, Kumagai H. 1991. Distribution and purification of aspartate racemase in lactic acid bacteria. Biochim. Biophys. Acta 1078:377–382 [DOI] [PubMed] [Google Scholar]
- 33.Strísovský K, Jirásková J, Barinka C, Majer P, Rojas C, Slusher BS, Konvalinka J. 2003. Mouse brain serine racemase catalyzes specific elimination of l-serine to pyruvate. FEBS Lett. 535:44–48 [DOI] [PubMed] [Google Scholar]
- 34.Grishin NV, Phillips MA, Goldsmith EJ. 1995. Modeling of the spatial structure of eukaryotic ornithine decarboxylases. Protein Sci. 4:1291–1304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Soda K, Yoshimura T, Esaki N. 2001. Stereospecificity for the hydrogen transfer of pyridoxal enzyme reactions. Chem. Rec. 1:373–384 [DOI] [PubMed] [Google Scholar]
- 36.Eliot AC, Kirsch JF. 2004. Pyridoxal phosphate enzymes: mechanistic, structural, and evolutionary considerations. Annu. Rev. Biochem. 73:383–415 [DOI] [PubMed] [Google Scholar]
- 37.Yoshimura T, Goto M. 2008. d-Amino acids in the brain: structure and function of pyridoxal phosphate-dependent amino acid racemases. FEBS J. 275:3527–3537 [DOI] [PubMed] [Google Scholar]
- 38.Okazaki S, Suzuki A, Mizushima T, Kawano T, Komeda H, Asano Y, Yamane T. 2009. The novel structure of a pyridoxal 5′-phosphate-dependent fold-type I racemase, α-amino-ε-caprolactam racemase from Achromobacter obae. Biochemistry 48:941–950 [DOI] [PubMed] [Google Scholar]
- 39.Badet B, Walsh C. 1985. Purification of an alanine racemase from Streptococcus faecalis and analysis of its inactivation by (1-aminoethyl)phosphonic acid enantiomers. Biochemistry 24:1333–1341 [DOI] [PubMed] [Google Scholar]
- 40.Yokoigawa K, Hirasawa R, Ueno H, Okubo Y, Umesako S, Soda K. 2001. Gene cloning and characterization of alanine racemases from Shigella dysenteriae, Shigella boydii, Shigella flexneri, and Shigella sonnei. Biochem. Biophys. Res. Commun. 288:676–684 [DOI] [PubMed] [Google Scholar]
- 41.Ju J, Xu S, Wen J, Li G, Ohnishi K, Xue Y, Ma Y. 2009. Characterization of endogenous pyridoxal 5′-phosphate-dependent alanine racemase from Bacillus pseudofirmus OF4. J. Biosci. Bioeng. 107:225–229 [DOI] [PubMed] [Google Scholar]
- 42.Yokoigawa K, Kawai H, Endo K, Lim YH, Esaki N, Soda K. 1993. Thermolabile alanine racemase from a psychotroph, Pseudomonas fluorescens: purification and properties. Biosci. Biotechnol. Biochem. 57:93–97 [DOI] [PubMed] [Google Scholar]