

Abstract
Two distinct but interrelated pheromone-signaling systems, LuxI/LuxR and AinS/AinR, positively control bioluminescence in Vibrio fischeri. Although each system generates an acyl-homoserine lactone (AHL) signal, the protein sequences of LuxI/LuxR and AinS/AinR are unrelated. AinS and LuxI generate the pheromones N-octanoyl-AHL (C8-AHL) and N-3-oxo-hexanoyl-AHL (3OC6-AHL), respectively. LuxR is a transcriptional activator that responds to 3OC6-AHL, and to a lesser extent to C8-AHL. AinR is hypothesized to respond to C8-AHL and, based on homology to Vibrio harveyi LuxN, to mediate the repression of a Qrr regulatory RNA. However, a ΔainR mutation decreased luminescence, which was not predicted based on V. harveyi LuxN, raising the possibility of a distinct regulatory mechanism for AinR. Here we show that ainR can complement a luxN mutant, suggesting functional similarity. Moreover, in V. fischeri, we observed ainR-dependent repression of a Pqrr-lacZ transcriptional reporter in the presence of C8-AHL, consistent with its hypothesized regulatory role. The system appears quite sensitive, with a half-maximal effect on a Pqrr reporter at 140 pM C8-AHL. Several other AHLs with substituted and unsubstituted acyl chains between 6 and 10 carbons also displayed an AinR-dependent effect on Pqrr-lacZ; however, AHLs with acyl chains of four carbons or 12 or more carbons lacked activity. Interestingly, 3OC6-AHL also affected expression from the qrr promoter, but this effect was largely luxR dependent, indicating a previously unknown connection between these systems. Finally, we propose a preliminary explanation for the unexpected luminescence phenotype of the ΔainR mutant.
INTRODUCTION
Vibrio fischeri is a valuable model for studying pheromone signaling (PS), and its PS-mediated control of luminescence was a fundamental discovery in the field of bacterial cell-cell communication (1). V. fischeri uses three entwined PS systems to control the luxICDABEG luminescence operon (2), which is induced during infection of symbiotic hosts and serves as a colonization factor (3–5). One of these PS systems is the widespread AI-2 signaling system (6, 7), underpinned by the LuxS pheromone synthase along with receptors LuxQ and LuxP; however, AI-2 exerts relatively little effect on bioluminescence under the conditions tested (8). The two major PS controls of luminescence use acylhomoserine lactone (AHL) signals. The first AHL system discovered was LuxI/LuxR (9, 10). LuxI is a signal synthase that generates N-3-oxo-hexanoyl-homoserine lactone (3OC6-AHL) (11). At a threshold concentration, this membrane-permeative autoinducer AHL binds to LuxR (9, 12, 13). 3OC6-AHL-LuxR then binds the lux box located within the intergenic region between luxR and luxI and activates luxICDABEG transcription.
It was later discovered that V. fischeri has a second AHL system comprised of AinS and AinR, whose protein sequences bear no resemblance to those of LuxI/LuxR (8, 14–17). AinS generates N-octanoyl-homoserine lactone (C8-AHL), which can bind LuxR directly, although it is a weaker activator than 3OC6-AHL and can inhibit 3OC6-AHL-mediated activation (17, 18). It is thought that a major role for C8-AHL involves sensing by AinR, which then converges with AI-2 signaling, acting via LuxU, LuxO, Hfq, and the regulatory RNA Qrr to increase levels of the transcriptional regulator LitR (17, 19–21).
Homologs of LitR are widespread in the Vibrionaceae and are similarly controlled via pheromone receptors and Qrr regulatory RNAs. Such LitR homologs include HapR in Vibrio cholerae, OpaR in Vibrio parahaemolyticus, and SmcR in Vibrio vulnificus. The LitR homolog in Vibrio harveyi is LuxR, and unlike LitR, it directly regulates bioluminescence. Through an unfortunately confusing twist of nomenclature, V. harveyi LuxR bears no structural similarity to V. fischeri LuxR. Indeed, the organisms above generally lack AHL systems similar to V. fischeri LuxI/LuxR. LitR homologs are often called PS “master regulators,” but in V. fischeri, LuxI/LuxR is the PS system that directly controls bioluminescence, with LitR playing a role by activating transcription of luxR (19).
The proposed function of AinR in the V. fischeri signaling cascade (Fig. 1) has been inferred largely by homology to V. harveyi LuxN (22–25). LuxN phosphorylates and dephosphorylates LuxU, and LuxU-P initiates a cascade involving LuxO, resulting in activation of Qrr regulatory RNAs, which posttranscriptionally repress expression of the PS master regulator. LuxN′s cognate pheromone N-d-3-hydroxybutanoyl homoserine lactone (AI-1) decreases its kinase activity, initiating a shift toward the unphosphorylated form of LuxU, less Qrr, and more master regulator (24). This system has the noteworthy trait that a signal receptor mutant (e.g., ΔluxN) does not mimic the lack of signal and in fact has the opposite effect. This perhaps counterintuitive effect is illustrated with respect to the predicted model of AinR function in Fig. 1. Interestingly, a ΔainR mutation resulted in dimmer luminescence (26), which is the opposite of that predicted by the current model. However, this observation alone does not rule out AinR's having the predicted effect on Qrr.
Fig 1.

Model of AinR-mediated pheromone signaling in V. fischeri. This model is drawn largely based on the function of the homolog LuxN in V. harveyi, although in that bacterium, the homolog of LitR is the direct regulator of luminescence. Differences in font size, arrow thickness, and numbers of gene products shown are meant to reflect differences in activation or abundance. An asterisk highlights the fact that a ΔainR mutant actually displays dimmer bioluminescence than the wild type (26). This simplified model omits any input from AI-2 and LuxQ into the shared regulatory cascade.
Signal cross talk might further entwine the Ain and Lux systems. Given that AinS's product, C8-AHL, is recognized by LuxR, and that C8-AHL's net effect depends on the availability of 3OC6-AHL, we were interested in whether these two AHLs might also have interrelated effects on AinR-mediated signaling. Moreover, because AinR apparently shares a signaling pathway with the widespread pheromone AI-2, we wondered whether AinR would similarly sense AHL signals from a broad array of bacteria or have more a specific AHL range, like LuxR (18).
Given the central position of AinR in our current model of the V. fischeri PS circuitry and the lack of experimental evidence for its function, the goals of this study were (i) to determine whether C8-AHL signals through AinR to direct the predicted net decrease of transcription from the qrr promoter, (ii) to test the range of AHL pheromones to which AinR directs this response, and (iii) to measure the relative sensitivity of AinR for these cognate AHLs.
MATERIALS AND METHODS
Bacteria, growth media, and reagents.
Bacterial strains are listed and briefly described in Table 1. V. fischeri ES114 was the wild-type strain used throughout (27). Plasmids were transformed into Escherichia coli strain DH5α (28) or, in the case of plasmids with the R6K origin of replication, into strain DH5αλpir (29). E. coli was grown in LB medium (30), and V. fischeri was grown in LBS medium (31) or SWTO medium (32). Solid media were prepared with 15 mg ml−1 agar. For selection of E. coli, chloramphenicol (Cam) and kanamycin (Kan) were added to LB at final concentrations of 20 and 40 μg ml−1, respectively. For selection of V. fischeri on LBS, Cam, erythromycin (Erm), and Kan were used at concentrations of 2, 5, and 100 μg ml−1, respectively. 3OC6-AHL and C8-AHL were obtained from Sigma-Aldrich (St. Louis, MO), and N-butanoyl (C4)-AHL, N-3-hydroxy-butanoyl (3OHC4)-AHL, N-hexanoyl (C6)-AHL, N-heptanoyl (C7)-AHL, N-3-hydroxy-octanoyl (3OH-C8)-AHL, N-3-oxo-octanoyl (3OC8)-AHL, N-decanoyl (C10)-AHL, N-dodecanoyl (C12)-AHL, N-tetradecanoyl (C14)-AHL, N-oxo-tetradecanoyl (3OC14)-AHL, N-cis-tetradec-9Z-enoyl (cis-C14-9Z-enoyl)-AHL, and N-octadecanoyl (C18)-AHL were obtained from Cayman Chemical (Ann Arbor, MI). Ethyl acetate-dissolved AHLs were added in defined amounts to flasks, and the solvent was evaporated overnight. AHLs were then redissolved to specific concentrations in SWTO medium and diluted as necessary.
Table 1.
Bacterial strains, plasmids, and oligonucleotides used in this study
| Strain, plasmid, or oligonucleotide | Relevant characteristics or sequencea | Source or reference |
|---|---|---|
| E. coli strains | ||
| CC118λpir | Δ(ara-leu) araD Δlac74 galE galK phoA20 thi-1 rpsE rpsB argE(Am) recA λpir | 34 |
| DH5α | ϕ80dlacZΔM15 Δ(lacZYA-argF)U169 deoR supE44 hsdR17 recA1 endA1 gyrA96 thi-1 relA1 | 28 |
| DH5αλpir | DH5α lysogenized with λpir | 29 |
| V. harveyi TL183 | ΔluxN ΔluxQ ΔcqsS | B. Bassler |
| V. fischeri strains | ||
| DC22 | C8-AHL bioreporter: ES114 ΔainS ΔluxR-luxI, mutant luxR (MJ1 T33A R67M S116A M135I), PluxI-luxCDABEG | D. Colton |
| ES114 | Wild-type isolate from E. scolopes | 27 |
| JB18 | ES114 litR::ermR | This study |
| JHK001 | ES114 ΔainSR luxI | This study |
| JHK003 | ES114 ΔainR | This study |
| JHK007 | ES114 ΔainS ΔluxIR PluxI-luxCDABEG | This study |
| JHK008 | ES114 ΔainR litR::ermR | This study |
| JHK009 | ES114 ΔainS luxI ΔluxQ | This study |
| JHK010 | ES114 ΔainSR ΔluxIR PluxI-luxCDABEG | This study |
| NL55 | ES114 ΔainSR | 38 |
| NL60 | ES114 ΔainS | 38 |
| NL63 | ES114 ΔainS luxI | This study |
| VCW2G7 | ES114 luxI (frameshift mutation) | 17 |
| Plasmidsb | ||
| pAKD701 | Promoterless lacZ, pES213, R6Kγ oriTRP4, kanR | 33 |
| pAS61 | ainR in pVSV105; pES213, R6Kγ oriTRP4, camR | This study |
| pDC44 | ΔluxIR PluxI-luxCDABEG allele; ColE1 R6Kγ oriTRP4, camR | D. Colton |
| pEVS104 | Conjugative helper plasmid; R6Kγ oriTRP4 kanR | 34 |
| pHK20 | Pqrr-lacZ reporter in pAKD701; pES213, R6Kγ oriTRP4 kanR | This study |
| pVCW2A6 | luxI frameshift allele; ColE1 oriTRP4 camR | 17 |
| pJLB123 | Porf1-luxR in pVSV104; pES213, oriTRP4 R6Kγ kanR | 32 |
| pNL62 | ΔainS allele; ColE1 R6Kγ oriTRP4 kanR, ermR | N. Lyell |
| pTM268 | Pqrr1-gfp, mCherry, pES213, R6Kγ oriTRP4 camR | 21 |
| pVAR29 | ΔluxQ allele; R6Kγ oriTRP4 camR ccdB araC | 26 |
| pVAR62 | ΔainR allele; R6Kγ oriTRP4 camR ccdB araC | 26 |
| pVSV104 | pES213, R6Kγ oriTRP4 kanR lacZα | 47 |
| pVSV105 | pES213, R6Kγ oriTRP4 camR lacZα | 47 |
| Oligonucleotidesc | ||
| ASAinRF | TACCTAGGATGTTAACTACTTTACCTAAAG | This study |
| ASAinRR | ATCCTAGGAATAAATTATCGAAGTAGCC | This study |
| pr_HK25 | GGGGGCATGCGGCTTCTACTGCAGCATCAATTGA | This study |
| pr_HK26 | GGGGGCTAGCAAAGGGTCAATATACCTATTGCAGGG | This study |
Drug resistance genes: camR, chloramphenicol resistance (cat); ermR, erythromycin resistance; kanR, kanamycin resistance (aph).
All alleles cloned in this study are from V. fischeri strain ES114. The replication origin(s) of each vector is listed as R6Kγ, ColE1, and/or pES213. Plasmids based on pES213 are stable and do not require antibiotic selection for maintenance (47).
All oligonucleotides are shown 5′ to 3′. Underlined regions are restriction enzyme recognition sites.
Molecular genetics and sequence analyses.
Plasmids were constructed using standard techniques and are described briefly in Table 1. DNA ligase and restriction enzymes were obtained from New England BioLabs (Beverly, MA). Oligonucleotides used for PCR and cloning are listed in Table 1 and were synthesized by Integrated DNA Technologies (Coralville, IA). PCR was conducted with Phusion high-fidelity DNA polymerase (Finnzymes, Finland) in an iCycler (Bio-Rad Laboratories, Hercules, CA). Plasmids used for cloning were isolated with the GenElute plasmid miniprep kit (Sigma-Aldrich, Inc., St. Louis, MO). DNA was repurified between cloning steps with the DNA Clean and Concentrator-5 kit (Zymo Research, Orange, CA). Cloned PCR products were sequenced at the University of Michigan DNA Sequencing Core Facility, and sequences were analyzed using the Lasergene Core suite (DNASTAR, Madison, WI).
To generate the Pqrr-lacZ transcriptional reporter plasmid pHK20, 450 bp upstream of V. fischeri qrr was PCR amplified using primers pr_HK25 and pr_HK26. The resulting amplicon was digested with SphI and NheI and cloned into the SphI and NheI sites in the promoterless-lacZ vector parent pAKD701 (33). To generate the ainR-containing shuttle vector pAS61, ainR was PCR amplified from ES114 using primers ASAinRF and ASAinRR, and the amplicon was digested with AvrII and ligated into XbaI-digested pVSV105.
Mutant construction.
Mutant alleles were transferred from E. coli into V. fischeri on plasmids by triparental matings using the conjugative helper plasmid pEVS104 (34) in strain CC118λpir (35). Recombinational insertion and marker exchange were identified by screening for antibiotic resistance, and putative mutants were tested by PCR. Marker exchange using plasmids pVAR29 and pVAR62 was facilitated by an arabinose-inducible toxin system on the vector sequence as previously described (26, 36). Resulting mutants are listed in Table 1. The litR::ermR mutant JB18 is phenotypically indistinguishable from the litR::ermR mutant JB19 (32) and was constructed in the same way except that the ermR cassette was in the opposite orientation. To construct the ΔainSR luxI mutant JHK001, plasmid pVCW2A6 (17) bearing luxI with the frame-shifting 4-bp insertion allele originally on pHV200I− (37) was exchanged into the ΔainSR strain NL55 (38). The ΔainS allele in pNL62 was exchanged into VCW2G7 (luxI) to generate strain NL63. To add luxR mutations to NL63 (ΔainS luxI) and JHK001 (ΔainSR luxI), the ΔluxRI::PluxI-luxC allele on pDC44 was exchanged into these parent strains to generate JHK007 and JHK010, respectively. To generate the ΔainS luxI ΔluxQ mutant JHK009, the ΔluxQ allele on pVAR29 (26) was exchanged into NL63 (ΔainS luxI). The ΔainR allele in pVAR62 was exchanged into wild-type strain ES114 and JB18 (litR::ermR) to generate JHK003 and JHK008, respectively.
Luminescence and fluorescence measurements.
Overnight V. fischeri cultures were diluted 1:1,000 in 25 ml of SWTO in 125-ml flasks and then incubated at 24°C with shaking (200 rpm). Samples (500 μl) were removed at various times, and optical density at 595 nm (OD595) was measured with a BioPhotometer (Brinkman Instruments, Westbury, NY). Samples were aerated by rapid shaking, and relative luminescence was measured immediately with a Glomax TD-20/20 luminometer (Promega, Madison, WI) (3). Specific luminescence reported is the relative luminescence per OD595 unit.
Fluorescence expressed from the gfp-derived qrr reporter on plasmid pTM268 was measured with a Synergy 2 plate reader (Biotek, Winooski, VT). Overnight cultures were diluted 1:100 in clear-bottomed, black-walled microtiter plates containing SWTO, with or without AHL, and measured at regular intervals for green fluorescent protein (GFP) using an excitation/emission wavelength pair of 485 nm/540 nm. Green fluorescence from the Pqrr-gfp reporter on pTM268 was normalized against red fluorescence from the constitutive Ptet-mCherry gene, also on the plasmid, to arrive at the specific fluorescence reported (GFP/mCherry). The EC50, defined as the AHL concentration at which the effect on the Pqrr-gfp reporter was half maximal, was determined based on curve fitting using SigmaPlot's “ligand binding” function (Systat Software, San Jose, CA).
Transcriptional lacZ reporter assays.
V. fischeri strains harboring the Pqrr-lacZ transcriptional reporter plasmid pHK20 or the promoterless-lacZ vector parent pAKD701 were grown overnight in LBS and subcultured 1:300 into 24-well microtiter plates containing 1.5 ml fresh SWTO per well, with or without AHL, and incubated at 24°C with shaking at 200 rpm. Cells were collected at an OD595 of ∼2.5 by centrifugation, the supernatant was discarded, and cell pellets were stored overnight at −80°C. β-Galactosidase assays were performed as previously described (3). EC50s were determined with the Pqrr-lacZ reporter based on curve fitting using SigmaPlot's “ligand binding” function (Systat Software).
C8-AHL bioassays.
C8-AHL accumulation was assessed for strains grown at 24°C with shaking in 125-ml flasks containing 15 ml of SWTO. The ΔainS mutant NL60 was included as a negative control. At an OD595 of ∼2.5, cells were pelleted, supernatants were filter sterilized, and an equal volume of acidified ethyl acetate (1:1,000 glacial acetic acid/ethyl acetate) was added. The mixture was incubated on a rotary shaker at 55 rpm for 30 min, 3 ml of the organic phase were removed and added to sterile glass beakers, the ethyl acetate was evaporated, and extracts were dissolved in 3 ml SWTO. A 200-μl portion of extract dissolved in SWTO was added to a 96-well microplate. Wells were then inoculated with the C8-AHL bioreporter strain DC22, which lacks the luxI and ainS pheromone synthase genes and in which expression of luxCDABEG is activated by a mutant LuxR (LuxRMJ1 T33A R67M S116A M135I) that is responsive to C8-AHL but not to 3OC6-AHL (39). C8-AHL concentration was determined by measuring the luminescence of DC22 in a Synergy 2 plate reader (BioTek) and comparing it to that of C8-AHL standards.
RESULTS
To begin testing the prediction that V. fischeri AinR functions similarly to LuxN from V. harveyi, we first complemented a V. harveyi luxN mutant with ainR. We used V. harveyi strain TL183 (ΔluxN ΔluxQ ΔcqsS), which has mutations in all three known pheromone receptors that act on LuxU in this bacterium, to minimize any chance of AinR-independent signaling in the transconjugants. The introduction of V. fischeri ainR led to decreased luminescence in this ΔluxN (and ΔluxQ ΔcqsS) V. harveyi mutant, but brighter luminescence was restored by adding C8-AHL (Fig. 2). The parental vector lacking ainR did not have these effects. Thus, ainR appeared to functionally replace luxN in V. harveyi but responded to the C8-AHL signal generated by AinS in V. fischeri.
Fig 2.
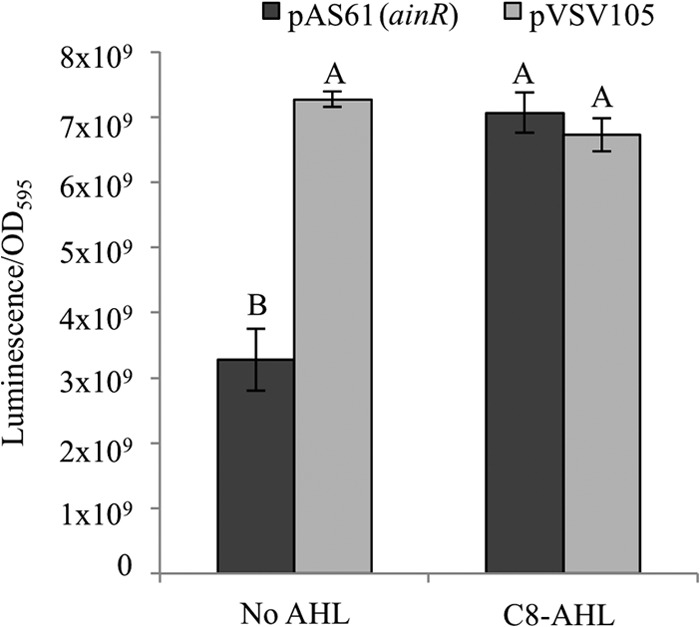
Complementation of a V. harveyi luxN mutant with ainR. Peak specific luminescence of the V. harveyi pheromone sensor mutant TL183 (ΔcqsS ΔluxN ΔluxQ), grown in SWTO, with or without 1 μM C8-AHL, and either bearing ainR from V. fischeri on plasmid pAS61 (dark gray) or carrying the parental shuttle vector pVSV105 (light gray) as a control is shown. Peak specific luminescence was observed at an OD595 of ∼1. Uppercase letters shared between bars indicate no statistically significant difference (P > 0.5), whereas different letters indicate significant difference (P < 0.0005), based on a one-way analysis of variance (ANOVA) and post hoc testing using Tukey's honestly significant difference test. Data are from one representative experiment of three, each with three biological replicates. Error bars represent standard errors (n = 3).
To more directly test whether AinR has the predicted effect on transcription of qrr in V. fischeri (Fig. 1), we generated a Pqrr-lacZ transcriptional reporter using the sequence upstream of qrr (also called qrr1) from V. fischeri as the source of the promoter and assayed its activity under different conditions. Experiments were performed in a genetic background lacking both luxI and ainS, to eliminate endogenous AHL production and allow us to control which potential signals were present. We found that in the absence of AHL, the reporter showed lower activity in the ainR mutant, and that in the presence of ainR, adding 1 μM C8-AHL, 3OC6-AHL, or both lowered Pqrr-lacZ reporter activity (Fig. 3A). Providing ainR in trans to the ΔainR mutant restored higher levels of Pqrr-lacZ reporter expression in the absence of AHL, and this effect could be reversed by the addition of C8-AHL (Fig. 3B). However, as discussed below, when ainR was added in trans to the ΔainR mutant, the effect of 3OC6-AHL on the qrr reporter was not fully complemented (Fig. 3B).
Fig 3.
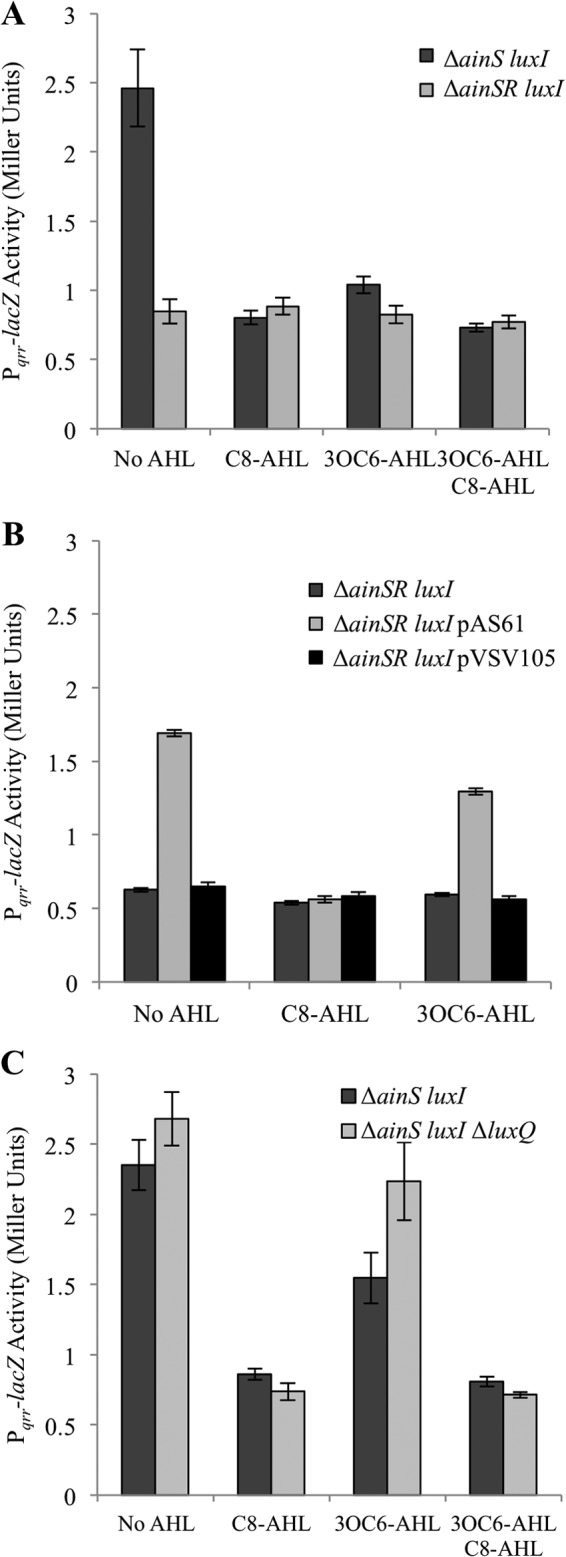
ainR-dependent and AHL-dependent effects on a qrr transcriptional reporter. Each panel shows β-galactosidase activity expressed from the Pqrr-lacZ transcriptional reporter in cultures grown on SWTO with a 1 μM concentration of each AHL indicated. (A) Reporter activity in mutants NL63 (ΔainS luxI) or JHK001 (ΔainSR luxI). (B) Reporter activity in JHK001 (ΔainSR luxI) with or without pAS61 (ainR) or its empty parent vector pVSV105. (C) Reporter activity in NL63 (ΔainS luxI) or JHK009 (ΔainS luxI ΔluxQ) in response to different AHL treatments. Data in each panel are from a single representative from among three independent experiments, and bars represent standard errors (A, n = 4; B and C, n = 3).
The ainR- and AHL-dependent effects on the qrr reporter were reproducible and statistically significant (P < 0.02), but they represented relatively modest 2- to 3-fold changes in LacZ activity. We considered the possibility that AI-2-mediated signaling through LuxQ might dampen or obscure AinR-mediated effects on qrr, because LuxQ- and AinR-mediated signaling are thought to converge at LuxU. However, we found no difference in the magnitude of such AHL-driven control of the Pqrr-lacZ reporter in a ΔluxQ mutant background (Fig. 3C). Thus, for our purpose of detecting ainR-dependent changes in qrr, there seemed to be no advantage in introducing the ΔluxQ allele.
The data above are consistent with the model of AinR function presented in Fig. 1, but the 1 μM C8-AHL used was at the upper end of what might be considered physiologically relevant. We therefore tested the response of the Pqrr-gfp reporter on pTM268 to different doses of C8-AHL. C8-AHL concentrations from 1 μM to 50 pM were sufficient to decrease Pqrr-gfp activity, while concentrations below 50 pM were not significantly different than the negative control lacking added AHL (Fig. 4). The EC50 for C8-AHL, defined as the concentration at which its effect on Pqrr-gfp was half maximal, was estimated at 140 pM for the data in Fig. 4. Similar EC50s were determined in repetitions of this experiment.
Fig 4.
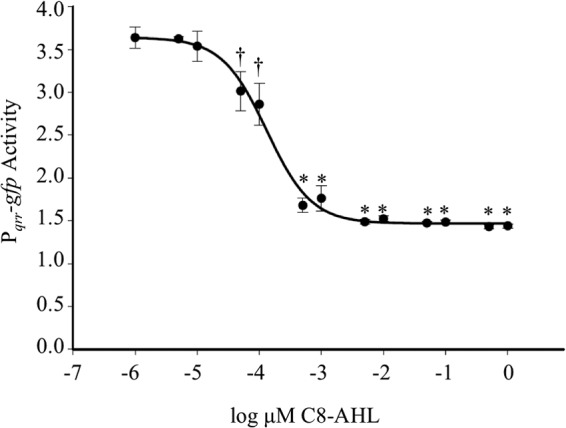
Effect of C8-AHL dose on qrr transcriptional reporter. The AHL pheromone synthase mutant NL63 (ΔainS luxI) harboring the Pqrr-gfp reporter plasmid pTM268 was exposed to C8-AHL concentrations ranging from 0 μM to 1 μM. GFP expression was normalized to the red fluorescence expressed from a constitutive mCherry gene on the same plasmid. Curve fitting and associated EC50 calculation were performed using SigmaPlot (EC50 = 140 pM; R2 = 0.9582). Statistically significant decreases in reporter activity are indicated by daggers (P < 0.05) and asterisks (P < 0.01).
Having established ainR-dependent effects on Pqrr-lacZ by both the C8-AHL and 3OC6-AHL pheromones produced by V. fischeri, we next tested a variety of other AHL pheromones in this assay. AHLs with acyl chain lengths deviating from C8-AHL by ±2 carbons, regardless of substitution at the third carbon of the acyl chain (unsubstituted or oxo- or hydroxy-substituted), significantly depressed reporter activity in an ainR-dependent manner (Fig. 5) (P < 0.01). AHLs with 4-carbon acyl chains or with acyl chains 12 carbons or longer did not affect reporter activity, regardless of substitution.
Fig 5.

Effects of a variety of AHL molecules on ainR-dependent signaling. β-Galactosidase activity expressed from the Pqrr-lacZ transcriptional reporter on pHK20, harbored by NL63 (ΔainS luxI) or JHK001 (ΔainSR luxI), is shown. Cultures were grown in SWTO with 1 μM dissolved AHL. Data from one representative experiment of three independent experiments are shown. Asterisks indicate statistically significantly lower activity compared to the “No AHL” treatment as determined by Student's t test (P < 0.01). Error bars represent standard errors (n = 3).
Although C6-AHL, 3OC6-AHL, C7-AHL, 3OHC8-AHL, and C10-AHL each displayed ainR-dependent depression of Pqrr-lacZ activity, for the results shown in Fig. 5, these AHLs had been tested at the relatively high concentration of 1 μM. By testing the reporter response as a function of AHL dose, over a series of serial 10-fold dilutions, we found that C6-AHL and C10-AHL were the least active, eliciting a response only at 1 μM (Table 2). 3OC6-AHL was the next weakest in activity and significantly affected the reporter only down to 100 nM (Table 2). C7-AHL was active at 10 nM, while 3OC8-AHL and 3OHC8-AHL were active down to 1 nM (Table 2). The EC50s for these AHLs followed similar trends, although by this measure 3OHC8-AHL was more active than 3OC8-AHL (Table 2). None of these AHLs was as active as C8-AHL in this reporter system; however, those with the chain lengths most similar to that of C8-AHL were closest in activity.
Table 2.
Sensitivity of ainR-dependent effects on the Pqrr-lacZ reporter to different AHL molecules
| AHL | AHL concn (nM) |
|
|---|---|---|
| EC50a | Minimum stimulatory doseb | |
| C6-AHL | 114 | 1,000 |
| 3OC6-AHL | 29 | 100 |
| C7-AHL | 11 | 10 |
| 3OC8-AHL | 8 | 1 |
| 3OHC8-AHL | 0.8 | 1 |
| C10-AHL | 476 | 1,000 |
EC50s were generated from dose-response assays using NL63 (ΔainS luxI) harboring the Pqrr-lacZ reporter pHK20 grown in SWTO. AHL was supplemented in 10-fold steps over a range of 1 μM to 1 pM. Data are from a single representative experiment of three independent experiments.
Lowest tested AHL concentration that elicited a significantly lower (P < 0.05) Pqrr-lacZ activity than the treatment lacking AHL.
We next tested whether any of the noninducing or relatively weakly inducing AHLs could interfere with AinR-dependent C8-AHL-mediated signaling, as is the case in some other AHL-dependent PS systems. In these experiments we tested the effect of adding other AHLs along with 1 nM C8-AHL, a relatively low C8-AHL concentration that nonetheless influenced the qrr reporter (Fig. 4). Under these conditions, adding a 1 μM concentration of the noninducing AHLs, which included C4-AHL, 3OHC4-AHL, C12-AHL, 3OC14-AHL, cis-C14-9Z-enoyl-AHL, and C18-AHL, had no discernible effect on C8-AHL-mediated signaling (data not shown). Thus, even in a 1,000-fold molar excess, these noninducers did not interfere with signaling by the cognate pheromone C8-AHL. For relatively weakly inducing AHLs, including C6-AHL, 3OC6-AHL, C7-AHL, 3OHC8-AHL, and C10-AHL, we added different concentrations both at and just below the threshold where they individually displayed an effect on the Pqrr-lacZ reporter (Table 2). We reasoned that these AHLs probably bind AinR at or below the concentration at which they detectably affect the qrr reporter, and if they bind AinR well but are poor at influencing AinR's kinase activity, they might effectively interfere with C8-AHL signaling at concentrations where they themselves have a modest or undetectable effect on the qrr reporter. However, we again saw no inhibition of C8-AHL-mediated signaling (data not shown).
The results above show an ainR-dependent effect of a variety of AHLs on Pqrr-lacZ expression, and we speculated that our data reflected AinR acting as a receptor for these AHLs. We were especially intrigued that AinR might recognize the LuxI product, 3OC6-AHL; however, the assays described above were done in genetic backgrounds that retained the AHL-dependent activator LuxR, which is the cognate 3OC6-AHL receptor. We therefore tested all the AHLs that were active in our assays (Fig. 5 and Table 2) in a luxR mutant background. A 1 μM concentration of each of these AHLs was again sufficient to decrease qrr reporter activity in the presence of luxR (Fig. 6A, dark bars). However, deletion of luxR eliminated the effect of 3OC6-AHL on qrr reporter activity, while the effects of all other AHLs were the same regardless of the presence or absence of luxR (Fig. 6A, light bars; also data not shown). Complementing the luxR mutant with luxR in trans on pJLB123 restored 3OC6-AHL-dependent reduction of Pqrr reporter activity, whereas the vector alone did not (data not shown). Due to the shared antibiotic resistance markers of pJLB123 (luxR), pVSV104 (parent of pJLB123), and the Pqrr-lacZ transcriptional reporter plasmid pHK20, which was used for Fig. 6A, for complementation we used the Pqrr-gfp reporter pTM268, which was used in generating the data for Fig. 4 with results similar to those obtained with the Pqrr-lacZ reporter.
Fig 6.
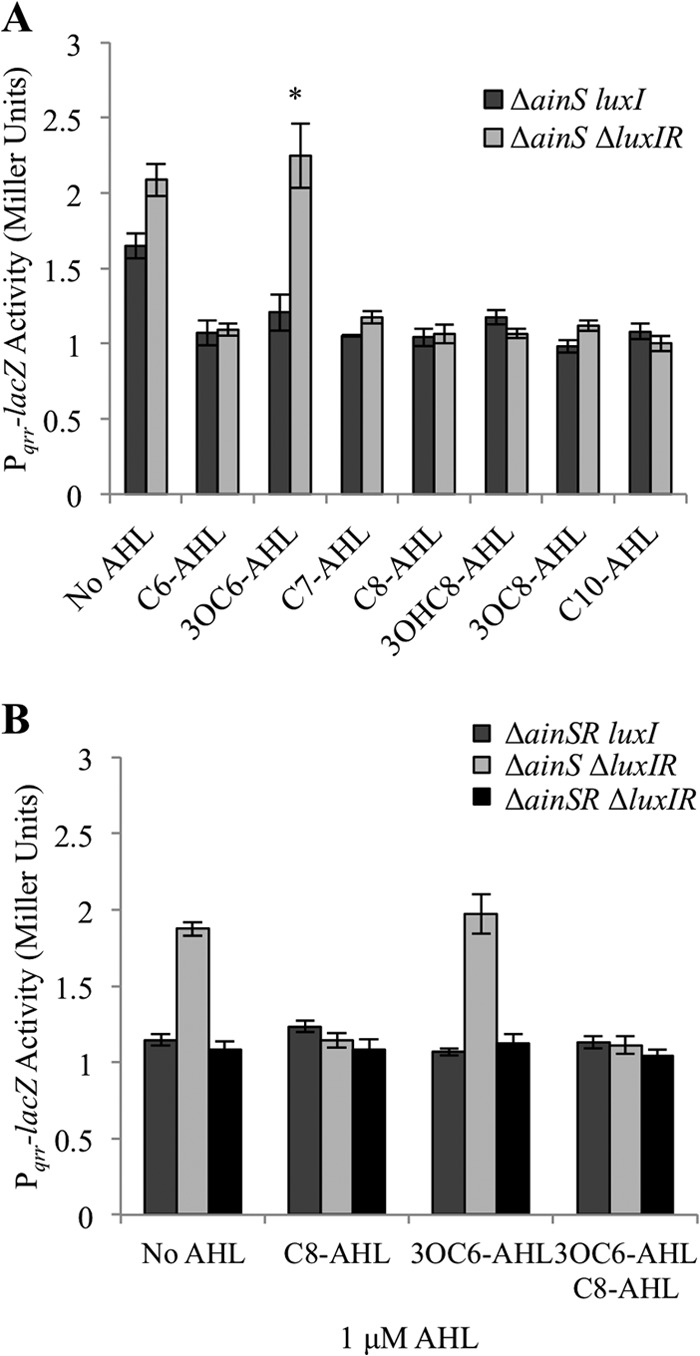
The effect of 3OC6-AHL on Pqrr-lacZ activity is dependent on luxR. β-Galactosidase activity expressed from the Pqrr-lacZ reporter on pHK20, from cultures grown in SWTO with a 1 μM concentration of each AHL, is indicated. Data in each panel are from a single representative experiment of three independent experiments, and error bars represent standard errors (n = 3). (A) Reporter activity in NL63 (ΔainS luxI) and JHK007 (ΔainS ΔluxIR). The asterisk indicates a statistically significant difference between the two strains (P < 0.02). (B) Reporter activity in mutants JHK001 (ΔainSR luxI), JHK007 (ΔainS ΔluxIR), and JHK010 (ΔainSR ΔluxIR).
We were curious whether 3OC6-AHL-LuxR may act on qrr independently of a 3OC6-AHL-AinR-dependent effect (Fig. 3A and B). If there were two different mechanisms by which 3OC6-AHL affected qrr, one that is AinR dependent and another that is LuxR dependent, then we might find an additive reduction in qrr reporter activity if the luxR and ainR mutations were combined in a single strain. However, this was not the case, as deletion of both ainR and luxR resulted in reporter activity that was not significantly different (P > 0.05) from that of the ainR mutant (Fig. 6B). Thus, these data are consistent with AinR and LuxR acting in the same pathway to affect qrr in response to 3OC6-AHL.
Finally, we sought to investigate why the ΔainR mutation leads to dimmer luminescence (26), when (i) the opposite might reasonably be predicted by the model in Fig. 1, (ii) our data above support the predicted relationship between ainR and qrr, including lower qrr transcription in the ainR mutant, and (iii) Miyashiro et al. already confirmed that loss of qrr results in brighter (not dimmer) luminescence (21). Ray and Visick (26) observed that the ΔainR mutation did not affect luminescence when excess C8-AHL was added, and we therefore tested C8-AHL accumulation by ΔainR mutants. Evidence suggests a LitR-mediated positive-feedback mechanism for the AinS/AinR system (8), so we tested C8-AHL accumulation in both wild-type and litR mutant backgrounds. Consistent with the previous report that a transcriptional reporter showed LitR activation of ainSR (8), C8-AHL levels were reduced in a litR mutant (Fig. 7). More importantly, we found that an in-frame deletion of ainR modestly but significantly (P < 0.05) reduced C8-AHL accumulation in the wild-type and litR mutant backgrounds (Fig. 7). As discussed further below, these results, together with the C8-AHL amendment experiment of Ray and Visick (26), suggest that the ΔainR mutation results in dimness because this allele causes decreased C8-AHL levels.
Fig 7.
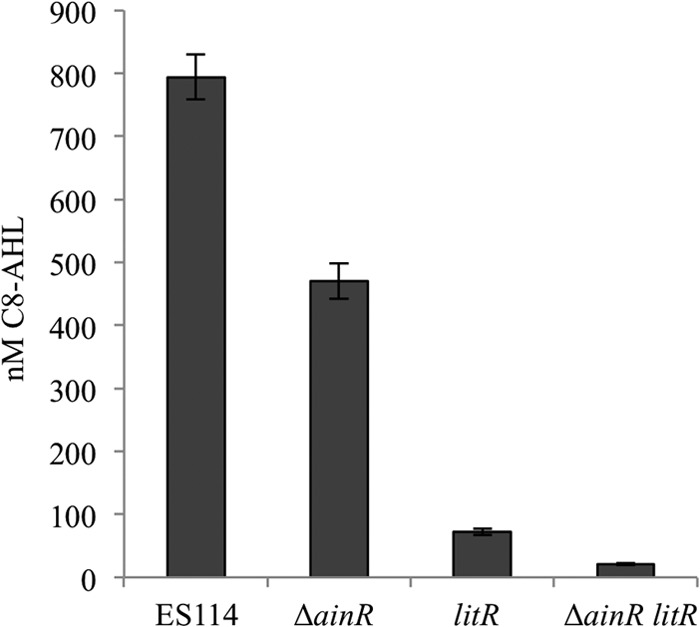
C8-AHL accumulation is reduced in ΔainR mutants. C8-AHL was extracted and its concentration estimated from cultures of ES114, the ΔainR mutant JHK003, the litR mutant JB18, or the litR ΔainR mutant JHK008. The C8-AHL synthase mutant NL60 (ΔainS) served as a negative control and had no detectable activity (data not shown). Cultures were grown to an OD595 of ∼2.5 in SWTO, and C8-AHL levels were determined by bioassays of ethyl acetate-extracted cultures. Error bars indicate standard errors (n = 3).
DISCUSSION
Many bacteria possess multiple PS systems, which may allow distinct population density-dependent regulons or hierarchical activation of pheromone-dependent phenotypes. V. fischeri possesses three PS systems, and it primarily uses two distinct and structurally unrelated AHL-dependent systems, LuxI/LuxR and AinS/AinR, to control the induction of luminescence in its mutualistic light-organ symbiosis with the squid Euprymna scolopes. While LuxI, LuxR, and AinS have been studied in some detail, the role of AinR has been largely speculative, based on its homology to the V. harveyi AHL receptor LuxN. Unlike V. fischeri, V. harveyi is not known to enter monospecies light organ symbioses, it lacks LuxI/LuxR homologs, and it contains only one AHL system. Given that the activities of AinR and LuxN may be shaped differently by the distinct lifestyles and PS circuitry of these bacteria, we investigated the function of AinR. Our results indicate that AinR does mediate control over the Qrr regulatory RNA in V. fischeri, as previously predicted (Fig. 1). Thus, the unexpected luminescence phenotype of a ΔainR mutant does not reflect a fundamental difference in AinR's placement in the PS regulatory circuitry. Interestingly, we found that AinR-dependent control responds to a range of AHLs and is exquisitely sensitive to C8-AHL, and we discovered a new regulatory connection between the Lux and Ain systems.
High sensitivity of AinR toward C8-AHL.
The sensitivity of AinR to C8-AHL is unusual among characterized AHL sensor proteins. The ainR-dependent effect on a Pqrr reporter was half-maximal (EC50) at ∼140 pM, while concentrations approaching 50 pM were still sufficient to decrease reporter activity (Fig. 4). TraR from Agrobacterium tumefaciens can elicit responses to similar subnanomolar levels of AHL, at least when overexpressed (40, 41), but sensitivity in the 10 nM range or higher appears to be more common for AHL receptors, including LuxR's response to 3OC6-AHL (42). In V. harveyi, the EC50 for a LuxN-dependent response to its cognate pheromone AI-1 was 23 nM (23), although the response measured was downstream of Qrr, rather than a qrr reporter itself as we used. Thus, the difference in EC50 may reflect the phenotype and/or its position in the PS circuitry. However, the two studies suggest that AinR is a hundredfold more sensitive to its cognate pheromone than is LuxN. Interestingly, Swem et al. showed that a S184N mutation made LuxN more sensitive to AI-1, reducing the EC50 for AI-1 to 11 nM (23), and an alignment of LuxN and AinR indicated that the native AinR has an N at that residue.
Relatively low specificity for C8-AHL.
The high sensitivity of AinR toward C8-AHL may come at a cost of low specificity. We found that AHLs with substituted or unsubstituted acyl chains of 6 to 10 carbons affected Pqrr reporter activity (Fig. 5), and inactivity was seen only with acyl chains of four carbons or more than 12 carbons. In general, the further the deviation from the C8 chain length, the less effective the AHL in our assay (Table 2). Although C8-AHL was the most active pheromone in our assays, other AHLs with seven or eight carbons showed activity at low concentrations, in the 1 to 10 nM range. In comparison, LuxR seems more selective and less sensitive than AinR (18). LuxN is also regarded as selective for AI-1, and its sensitivity and selectivity for other AHLs should be of interest, affording an opportunity for comparison with AinR.
Alternative models to explain the evolution of AinR and its properties.
It is interesting to consider what selective pressure(s) might have driven AinR evolution toward high sensitivity and low specificity. Although LuxI/LuxR appears important for persistence within the host, initial activation of luminescence and other colonization factors is under the control of AinS and C8-AHL (17). These observations led Lupp et al. to propose a model of sequential PS systems, with Ain functioning at moderate cell densities and jump starting the Lux system to function at higher densities (17). In this model, AinR could serve as a sentinel receptor, and accordingly it may have evolved a high sensitivity to detect small amounts of C8-AHL as cells approach moderate density. Other traits of AinR, including the relatively broad range of signals detected and the surprising lack of apparent inhibition by noncognate AHLs, may be a coincidental consequence of evolution toward high sensitivity.
Alternatively, AinR might have evolved to detect a broad array of AHLs, including those from other bacteria. Such signaling could be important in environments for V. fischeri outside the light organ, where AinR may serve as a multisignal receptor, funneling information about the bacterial community into the PS network. This hypothesis is consistent with the current thinking that the Ain system converges with the widespread bacterial signal AI-2. The AI-2 and Ain systems control the LitR regulon, which is distinct from the LuxR regulon, providing an opportunity for distinct responses by V. fischeri either to a community or to itself. On the other hand, LuxN also converges with AI-2 signaling in V. harveyi, yet LuxN is generally thought of as a self-specific receptor. Moreover, in V. fischeri there is significant cross talk between the Lux and Ain systems, which does not suggest such distinct functions. AinR has evolved rapidly between V. fischeri strains (43), and we may gain insight into the selective pressures that have shaped it by evaluating a reconstructed ancestral AinR or AinR in isolates from environments with no known light organ hosts.
Cross talk between Lux and Ain.
One motivation behind this study was the potential to discover new interconnections between Lux and Ain. C8-AHL/LuxR cross talk had been established, but reciprocal signaling between LuxI-synthesized 3OC6-AHL and AinR would further entwine these systems. We initially suspected that AinR detected 3OC6-AHL, because 3OC6-AHL led to decreased Pqrr reporter activity in an ainR-dependent manner (Fig. 3). However, we subsequently found that this effect was also dependent on luxR, which encodes the 3OC6-AHL receptor (Fig. 6). Although we cannot rule out perception of 3OC6-AHL by AinR, based on our data we speculate that 3OC6-AHL-LuxR mediates repression of Pqrr transcription in a manner that requires the presence of ainR.
The mechanism underlying this additional layer of PS cross talk is unknown, but it could involve direct or indirect LuxR-mediated regulation of ainSR. Such a mechanism would be consistent with our observation that deleting both ainR and luxR did not additively reduce Pqrr activity (Fig. 6). One simple model would be that 3OC6-AHL-LuxR acts as a repressor of the ainSR promoter. Although LuxR is best known as a transcriptional activator, it can act as a repressor (44). Moreover, Gilson et al. identified a putative lux box binding site for LuxR upstream of ainS and suggested that it overlaps a −35 promoter element (45), consistent with LuxR acting as a repressor. Subsequently, Antunes et al. did not identify ainSR as a member of the LuxR regulon during a screen for up- and downregulation in response to 3OC6-AHL addition (46); however, unlike the experiments of Antunes et al., our study was conducted in a luxI ainS mutant background lacking endogenous AHL production, which might reveal effects on ainSR regulation that would be obscured by C8-AHL and positive feedback in the Ain system. It may be relevant to note that when a plasmid-borne copy of ainR was provided in trans to the ΔainR mutant, the effect of C8-AHL on the qrr reporter was complemented fully, while the effect of 3OC6-AHL was not (Fig. 3B). Perhaps when ainR is expressed from a nonnative promoter in multicopy, repression of ain by 3OC6-AHL-LuxR is lost. Alternative mechanisms are possible, too, including 3OC6-AHL-LuxR-mediated repression of luxU or luxO, the effects of which would be dependent on the presence of AinR to initiate the cascade that phosphorylates LuxU and LuxO.
Insight into the dim luminescence of the ΔainR mutant.
Although it was not the main focus of our study, our results help explain the apparently paradoxical observation that luxN and ainR mutants in V. harveyi and V. fischeri, respectively, have similar effects on Qrr but opposite effects on luminescence. The luminescence defect in an ainR strain was rescued by adding C8-AHL (26), and we have now shown that the ΔainR allele results in decreased C8-AHL output, in either wild-type or litR mutant backgrounds. Consistent with the idea that the ΔainR allele causes decreased luminescence because of lowered C8-AHL levels, in an ainS mutant background lacking C8-AHL, an additional ainR mutation has the effect predicted in Fig. 1 of enhancing luminescence (data not shown). The reason C8-AHL affects luminescence even without its AinR receptor probably relates to LuxR and its activation by C8-AHL. Ultimately, bioluminescence in V. fischeri is activated by AHL-LuxR, and previous studies overwhelmingly suggest that in strain ES114, C8-AHL drives LuxR-dependent activation, especially in broth cultures (5, 16, 17, 19). Although C8-AHL is a weaker activator of LuxR, in ES114 it accumulates to much higher levels than does 3OC6-AHL in broth culture (2). In V. harveyi, the homolog of V. fischeri LitR is the direct regulator of luminescence, and there is no analogous additional layer of AHL-dependent regulation. It seems likely that the dimness of the ΔainR mutant is due to a lack of C8-AHL-LuxR activation of the lux genes, a mechanism for which there is no parallel in a V. harveyi luxN mutant.
Although a mechanism awaits investigation, we speculate that the in-frame deletion of ainR may destabilize the ainS message. The ainS and ainR genes are adjacent on the same strand and separated by 11 bp, with no obvious terminator between them, suggesting that they may be transcriptionally linked, resulting in one polycistronic mRNA. Moreover, the lack of ainR does not result in dimmer luminescence when either ainS or ainR is expressed ectopically from a plasmid, so this effect is evident only when the two are in cis (data not shown). Interestingly, there is a large inverted repeat within ainR, which we further speculate may be responsible for stabilizing the ainS portion of the transcript, perhaps by blocking 3′-5′ exonuclease digestion. If this is correct, such a mechanism could allow cotranscription of ainSR but subsequently change the stoichiometry of the partners favoring the AinS synthase over the AinR receptor.
Areas for future research.
Although AinR fits in a regulatory circuit similar to that of LuxN, it seems that as an interface between the AHL signal and LuxU there may be significant differences between LuxN and AinR. Other researchers have made great strides in dissecting the structure and function of LuxN, and we believe that comparative analyses including AinR will be enlightening. Similarly, we are intrigued by the divergence of AinR between V. fischeri isolates from different environments (43), and further investigation could elucidate selective pressures on AinS/AinR for V. fischeri in different hosts. Finally, we are interested in the regulatory control of AinS/AinR expression, and our results have opened new research avenues, particularly with respect to our discovery of LuxR-mediated regulation and the potential for posttranscriptional control.
ACKNOWLEDGMENTS
We thank Deanna Colton, Jeffrey Bose, and Noreen Lyell for technical assistance, Alecia Septer for technical assistance and review of the manuscript, Valerie Ray and Karen Visick for helpful discussions and for providing pVAR29 and pVAR62, and Jianping Cong and Bonnie Bassler for providing the Vibrio harveyi luxN cqsS luxQ mutant TL183.
The National Science Foundation supported this research under grant IOS-1121106. J.H.K. was supported with funds awarded by the University of Georgia Presidential Graduate Fellows Program.
Footnotes
Published ahead of print 20 September 2013
REFERENCES
- 1.Hastings JW, Greenberg EP. 1999. Quorum sensing: the explanation of a curious phenomenon reveals a common characteristic of bacteria. J. Bacteriol. 181:2667–2668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stabb EV, Schaefer A, Bose JL, Ruby EG. 2008. Quorum signaling and symbiosis in the marine luminous bacterium Vibrio fischeri, p 233–250 In Winans SC, Bassler BL. (ed), Chemical communication among bacteria. ASM Press, Washington, DC [Google Scholar]
- 3.Bose JL, Rosenberg CS, Stabb EV. 2008. Effects of luxCDABEG induction in Vibrio fischeri: enhancement of symbiotic colonization and conditional attenuation of growth in culture. Arch. Microbiol. 190:169–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Koch EJ, Miyashiro TI, McFall-Ngai MJ, Ruby EG. Features governing symbiont persistence in the squid-vibrio association. Mol. Ecol., in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Visick KL, Foster J, Doino J, McFall-Ngai M, Ruby EG. 2000. Vibrio fischeri lux genes play an important role in colonization and development of the host light organ. J. Bacteriol. 182:4578–4586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Federle MJ, Bassler BL. 2003. Interspecies communication in bacteria. J. Clin. Invest. 112:1291–1299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pereira CS, Thompson JA, Xavier KB. 2013. AI-2-mediated signalling in bacteria. FEMS Microbiol. Rev. 37:156–181 [DOI] [PubMed] [Google Scholar]
- 8.Lupp C, Ruby EG. 2004. Vibrio fischeri LuxS and AinS: comparative study of two signal synthases. J. Bacteriol. 186:3873–3881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Engebrecht J, Nealson K, Silverman M. 1983. Bacterial bioluminescence: isolation and genetic analysis of functions from Vibrio fischeri. Cell 32:773–781 [DOI] [PubMed] [Google Scholar]
- 10.Engebrecht J, Silverman M. 1984. Identification of genes and gene products necessary for bacterial bioluminescence. Proc. Natl. Acad. Sci. U. S. A. 81:4154–4158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eberhard A, Burlingame AL, Eberhard C, Kenyon GL, Nealson KH, Oppenheimer NJ. 1981. Structural identification of autoinducer of Photobacterium fischeri luciferase. Biochemistry 20:2444–2449 [DOI] [PubMed] [Google Scholar]
- 12.Kaplan HB, Greenberg EP. 1985. Diffusion of autoinducer is involved in regulation of the Vibrio fischeri luminescence system. J. Bacteriol. 163:1210–1214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Urbanowski ML, Lostroh CP, Greenberg EP. 2004. Reversible acyl-homoserine lactone binding to purified Vibrio fischeri LuxR protein. J. Bacteriol. 186:631–637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kuo A, Blough NV, Dunlap PV. 1994. Multiple N-acyl-l-homoserine lactone autoinducers of luminescence in the marine symbiotic bacterium Vibrio fischeri. J. Bacteriol. 176:7558–7565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kuo A, Callahan SM, Dunlap PV. 1996. Modulation of luminescence operon expression by N-octanoyl-l-homoserine lactone in ainS mutants of Vibrio fischeri. J. Bacteriol. 178:971–976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lupp C, Ruby EG. 2005. Vibrio fischeri uses two quorum-sensing systems for the regulation of early and late colonization factors. J. Bacteriol. 187:3620–3629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lupp C, Urbanowski M, Greenberg EP, Ruby EG. 2003. The Vibrio fischeri quorum-sensing systems ain and lux sequentially induce luminescence gene expression and are important for persistence in the squid host. Mol. Microbiol. 50:319–331 [DOI] [PubMed] [Google Scholar]
- 18.Schaefer AL, Hanzelka BL, Eberhard A, Greenberg EP. 1996. Quorum sensing in Vibrio fischeri: probing autoinducer-LuxR interactions with autoinducer analogs. J. Bacteriol. 178:2897–2901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fidopiastis PM, Miyamoto CM, Jobling MG, Meighen EA, Ruby EG. 2002. LitR, a new transcriptional activator in Vibrio fischeri, regulates luminescence and symbiotic light organ colonization. Mol. Microbiol. 45:131–143 [DOI] [PubMed] [Google Scholar]
- 20.Miyamoto CM, Lin YH, Meighen EA. 2000. Control of bioluminescence in Vibrio fischeri by the LuxO signal response regulator. Mol. Microbiol. 36:594–607 [DOI] [PubMed] [Google Scholar]
- 21.Miyashiro T, Wollenberg MS, Cao X, Oehlert D, Ruby EG. 2010. A single qrr gene is necessary and sufficient for LuxO-mediated regulation in Vibrio fischeri. Mol. Microbiol. 77:1556–1567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Henke JM, Bassler BL. 2004. Three parallel quorum-sensing systems regulate gene expression in Vibrio harveyi. J. Bacteriol. 186:6902–6914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Swem LR, Swem DL, Wingreen NS, Bassler BL. 2008. Deducing receptor signaling parameters from in vivo analysis: LuxN/AI-1 quorum sensing in Vibrio harveyi. Cell 134:461–473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Timmen M, Bassler BL, Jung K. 2006. AI-1 influences the kinase activity but not the phosphatase activity of LuxN of Vibrio harveyi. J. Biol. Chem. 281:24398–24404 [DOI] [PubMed] [Google Scholar]
- 25.Bassler BL, Wright M, Showalter RE, Silverman MR. 1993. Intercellular signalling in Vibrio harveyi: sequence and function of genes regulating expression of luminescence. Mol. Microbiol. 9:773–786 [DOI] [PubMed] [Google Scholar]
- 26.Ray VA, Visick KL. 2012. LuxU connects quorum sensing to biofilm formation in Vibrio fischeri. Mol. Microbiol. 86:954–970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boettcher KJ, Ruby EG. 1990. Depressed light emission by symbiotic Vibrio fischeri of the sepiolid squid Euprymna scolopes. J. Bacteriol. 172:3701–3706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hanahan D. 1983. Studies on transformation of Escherichia coli with plasmids. J. Mol. Biol. 166:557–580 [DOI] [PubMed] [Google Scholar]
- 29.Dunn AK, Martin MO, Stabb EV. 2005. Characterization of pES213, a small mobilizable plasmid from Vibrio fischeri. Plasmid 54:114–134 [DOI] [PubMed] [Google Scholar]
- 30.Miller JH. 1992. A short course in bacterial genetics. Cold Spring Harbor Laboratory Press, New York, NY [Google Scholar]
- 31.Stabb EV, Reich KA, Ruby EG. 2001. Vibrio fischeri genes hvnA and hvnB encode secreted NAD+-glycohydrolases. J. Bacteriol. 183:309–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bose JL, Kim U, Bartkowski W, Gunsalus RP, Overley AM, Lyell NL, Visick KL, Stabb EV. 2007. Bioluminescence in Vibrio fischeri is controlled by the redox-responsive regulator ArcA. Mol. Microbiol. 65:538–553 [DOI] [PubMed] [Google Scholar]
- 33.Dunn AK, Stabb EV. 2008. Genetic analysis of trimethylamine N-oxide reductases in the light organ symbiont Vibrio fischeri ES114. J. Bacteriol. 190:5814–5823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stabb EV, Ruby EG. 2002. RP4-based plasmids for conjugation between Escherichia coli and members of the Vibrionaceae. Methods Enzymol. 358:413–426 [DOI] [PubMed] [Google Scholar]
- 35.Herrero M, De Lorenzo V, Timmis KN. 1990. Transposon vectors containing non-antibiotic resistance selection markers for cloning and stable chromosomal insertion of foreign genes in Gram-negative bacteria. J. Bacteriol. 172:6557–6567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Le Roux F, Binesse J, Saulnier D, Mazel D. 2007. Construction of a Vibrio splendidus mutant lacking the metalloprotease gene vsm by use of a novel counterselectable suicide vector. Appl. Environ. Microbiol. 73:777–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pearson JP, Gray KM, Passador L, Tucker KD, Eberhard A, Iglewski BH, Greenberg EP. 1994. Structure of the autoinducer required for expression of Pseudomonas aeruginosa virulence genes. Proc. Natl. Acad. Sci. U. S. A. 91:197–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lyell NL, Dunn AK, Bose JL, Stabb EV. 2010. Bright mutants of Vibrio fischeri ES114 reveal conditions and regulators that control bioluminescence and expression of the lux operon. J. Bacteriol. 192:5103–5114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Collins CH, Leadbetter JR, Arnold FH. 2006. Dual selection enhances the signaling specificity of a variant of the quorum-sensing transcriptional activator LuxR. Nat. Biotechnol. 24:708–712 [DOI] [PubMed] [Google Scholar]
- 40.Su S, Khan SR, Farrand SK. 2008. Induction and loss of Ti plasmid conjugative competence in response to the acyl-homoserine lactone quorum-sensing signal. J. Bacteriol. 190:4398–4407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhu J, Chai Y, Zhong Z, Li S, Winans SC. 2003. Agrobacterium bioassay strain for ultrasensitive detection of N-acylhomoserine lactone-type quorum-sensing molecules: detection of autoinducers in Mesorhizobium huakuii. Appl. Environ. Microbiol. 69:6949–6953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Collins CH, Arnold FH, Leadbetter JR. 2005. Directed evolution of Vibrio fischeri LuxR for increased sensitivity to a broad spectrum of acyl-homoserine lactones. Mol. Microbiol. 55:712–723 [DOI] [PubMed] [Google Scholar]
- 43.Bose JL, Wollenberg MS, Colton DM, Mandel MJ, Septer AN, Dunn AK, Stabb EV. 2011. Contribution of rapid evolution of the luxR-luxI intergenic region to the diverse bioluminescence outputs of Vibrio fischeri strains isolated from different environments. Appl. Environ. Microbiol. 77:2445–2457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Egland KA, Greenberg EP. 2000. Conversion of the Vibrio fischeri transcriptional activator, LuxR, to a repressor. J. Bacteriol. 182:805–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gilson L, Kuo A, Dunlap PV. 1995. AinS and a new family of autoinducer synthesis proteins. J. Bacteriol. 177:6946–6951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Antunes LC, Schaefer AL, Ferreira RB, Qin N, Stevens AM, Ruby EG, Greenberg EP. 2007. Transcriptome analysis of the Vibrio fischeri LuxR-LuxI regulon. J. Bacteriol. 189:8387–8391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dunn AK, Millikan DS, Adin DM, Bose JL, Stabb EV. 2006. New rfp- and pES213-derived tools for analyzing symbiotic Vibrio fischeri reveal patterns of infection and lux expression in situ. Appl. Environ. Microbiol. 72:802–810 [DOI] [PMC free article] [PubMed] [Google Scholar]