

Abstract
SUMMARY
Pathogenic bacteria commonly deploy enzymes to promote virulence. These enzymes can modulate the functions of host cell targets. While the actions of some enzymes can be very obvious (e.g., digesting plant cell walls), others have more subtle activities. Depending on the lifestyle of the bacteria, these subtle modifications can be crucially important for pathogenesis. In particular, if bacteria rely on a living host, subtle mechanisms to alter host cellular function are likely to dominate. Several bacterial virulence factors have evolved to use enzymatic deamidation as a subtle posttranslational mechanism to modify the functions of host protein targets. Deamidation is the irreversible conversion of the amino acids glutamine and asparagine to glutamic acid and aspartic acid, respectively. Interestingly, all currently characterized bacterial deamidases affect the function of the target protein by modifying a single glutamine residue in the sequence. Deamidation of target host proteins can disrupt host signaling and downstream processes by either activating or inactivating the target. Despite the subtlety of this modification, it has been shown to cause dramatic, context-dependent effects on host cells. Several crystal structures of bacterial deamidases have been solved. All are members of the papain-like superfamily and display a cysteine-based catalytic triad. However, these proteins form distinct structural subfamilies and feature combinations of modular domains of various functions. Based on the diverse pathogens that use deamidation as a mechanism to promote virulence and the recent identification of multiple deamidases, it is clear that this enzymatic activity is emerging as an important and widespread feature in bacterial pathogenesis.
INTRODUCTION
Many bacterial pathogens use diverse suites of virulence factors to contribute to pathogenicity. These virulence factors include toxins and type III effectors, which are proteins injected into host cells via specialized type III secretion systems. Effectors often modify eukaryotic host target proteins with posttranslational modifications that alter normal cellular function. Commonly described posttranslational modifications utilized by effectors include ubiquitination, acetylation, and AMPylation (1–3). Recently, enzymatic deamidation has emerged as a common posttranslational modification utilized by a broad range of bacterial pathogens of both plants and animals to alter the functions of host proteins. Deamidation is the replacement of an amide group with a carboxylate group (Fig. 1). Therefore, it converts glutamine and asparagine to glutamic acid and aspartic acid, respectively. This irreversible amino acid conversion results in an increase of approximately 1 Da in the mass of the target protein, an increase in the negative charge of the target protein, and the release of ammonia. Nonspecific deamidation can occur spontaneously as proteins age and are degraded (4). In contrast, specific enzymatic deamidation can regulate normal cellular functions, such as chemotaxis and protein turnover in prokaryotes, or disrupt eukaryotic host cell function during infection (5, 6). Here we focus on deamidases that contribute to bacterial virulence.
Fig 1.
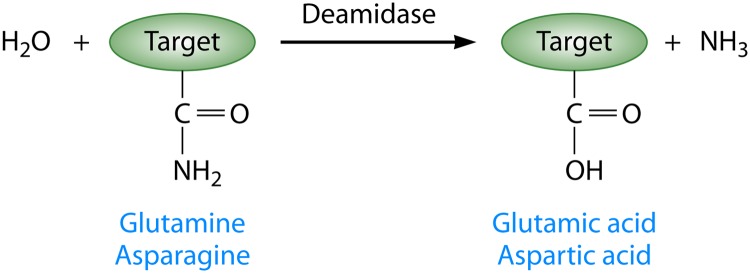
Schematic representation of enzymatic deamidation in proteins. Deamidases act on specific residues in the target protein. For all currently studied bacterial virulence factors, the targets of deamidation are glutamine side chains, which are converted to glutamic acids. However, it is also possible for the amide of the side chains of asparagines to be converted to aspartic acid. Deamidation results in an increase in the negative charge of the target protein, an increase of approximately 1 Da in the mass of the target protein, and the release of ammonia. Each of these outputs can be measured experimentally to characterize the activity of deamidases.
The topic of this review is the six currently known families of bacterial virulence factors that use deamidation to modulate host functions during infection (Table 1). Cytotoxic necrotizing factors (CNFs) are a family of deamidases from Escherichia coli (CNF1, -2, and -3) and Yersinia pseudotuberculosis (CNFY). The CNFs target a glutamine residue (either Gln63 or Gln61) in the switch II domain of GTPase proteins that is critical for function (7, 8). Deamidation of this glutamine leads to constitutive activation of the target GTPases, resulting in cytoskeletal rearrangements. Reorganization of the actin cytoskeleton is one mechanism used by invasive bacteria to promote entry into host cells (9, 10). BLF1 is a toxin from Burkholderia pseudomallei that is lethal to mice and tissue culture cells (11). BLF1 inhibits host protein synthesis via deamidation of eIF4A (11). VopC is a type III effector from Vibrio parahaemolyticus that deamidates and constitutively activates small GTPases (12). Pasteurella multocida toxin (PMT) is the major virulence factor of Pasteurella multocida, the causal agent of a variety of mammalian and avian diseases. PMT constitutively activates various G proteins, including Gαq/11, Gαi, and Gα12/13, via deamidation of a glutamine that is also in the switch II region (13). Cell cycle-inhibiting factors (Cifs) are type III effectors and cyclomodulins from multiple bacterial species. Cifs inhibit ubiquitination pathways by deamidating glutamine 40 of ubiquitin and the ubiquitin-like protein NEDD8 (14). OspI is a type III effector protein from Shigella flexneri that dampens host immune responses by deamidating UBC13 and disrupting the TRAF6-mediated signaling pathway (15). We review the details of each of these six families, specifically with respect to their three-dimensional structures and the impact that deamidation has on the functions of their host target proteins. We conclude that deamidation, as a nonreversible modification, is likely an “all or nothing” virulence switch to alter diverse cellular functions across diverse pathosystems.
Table 1.
Bacterial virulence factors that use deamidation to modify host proteins
| Protein(s) | Species | Target(s) | Effect | Key references |
|---|---|---|---|---|
| Cytotoxic necrotizing factors (CNFs) | E. coli, Yersinia pseudotuberculosis | Rho GTPases (RhoA, Rac, Cdc42) | Activation of Rho GTPases | 7, 8 |
| Burkholderia lethal factor 1 (BLF1) | Burkholderia pseudomallei | eIF4A | Inactivation of helicase activity of translation factor | 11 |
| VopC | Vibrio parahaemolyticus | Rho GTPases (Rac, Cdc42) | Activation of Rho GTPases | 12 |
| PMT | Pasteurella multocida | Heterotrimeric G proteins (Gαi, Gαq/11, and Gα12/13) | Activation of G protein signaling | 13 |
| Cycle-inhibiting factors (Cifs) | E. coli, Burkholderia pseudomallei, Yersinia pseudotuberculosis, Photorhabdus luminescens, Photorhabdus asymbiotica | NEDD8 and ubiquitin | Inactivation of ubiquitin-proteasome system | 14 |
| OspI | Shigella flexneri | UBC13 | Inactivation of E2 ubiquitin-conjugating activity of UBC13 | 15 |
CYTOTOXIC NECROTIZING FACTOR TOXINS
Cytotoxic Necrotizing Factor Toxins Deamidate Rho GTPases
CNF1 from E. coli was the first bacterial deamidase identified (7, 8). CNF1 is expressed in uropathogenic strains of E. coli (16). Following the discovery of CNF1, a number of homologs were identified in different strains of E. coli. For example, CNF2 is produced by enteropathogenic E. coli (EPEC) strains isolated from cows and sheep (17). The amino acid sequence of CNF2 is 85% identical to that of CNF1. CNF3 was isolated from a necrotoxigenic E. coli strain that infects sheep and goats (18). CNF3 shares 70% amino acid identity with CNF1. Interestingly, CNFs are not restricted to E. coli. Yersinia pseudotuberculosis produces CNFY, which is 61% identical to CNF1 (19). Furthermore, dermonecrotizing toxin (DNT) from Bordetella is a virulence factor that also shares sequence homology with CNF1. However, DNT behaves most efficiently as a transglutaminase in vitro, being active in the presence of polyamines (20, 21). Therefore, we do not include a detailed discussion of DNT here.
Schmidt et al. and Flatau et al. demonstrated that CNF1 converts Gln63 of the critical RhoA GTPase switch II region to glutamic acid via enzymatic deamidation (7, 8). Importantly, neither Gln29 nor Gln52, the other two glutamine residues in RhoA, were modified by CNF1 (7, 8). Deamidation of Gln63 triggers constitutive activation of RhoA, functionally mimicking the GTP-bound state. Furthermore, the RhoA Gln63Glu variant, which mimics the product of the deamidation reaction, phenocopies CNF1-treated RhoA upon microinjection into Vero cells (7, 8).
RhoA is not the only target of CNFs. CNF deamidases possess different GTPase substrate specificities. CNF1 can deamidate Rac and Cdc42 in vitro and Cdc42 in intact HeLa cells (8, 22). CNF2 can modify RhoA and Rac when coexpressed in E. coli (23). CNF3 can deamidate RhoA, Rac, and Cdc42 in intact HeLa cells treated with recombinant toxin (24). Although RhoA, Rac, and Cdc42 serve as substrates for CNFY deamidation in vitro, only RhoA could be deamidated by CNFY in intact cells (25).
Rho GTPases are molecular switches that cycle between an inactive, GDP-bound state and an active, GTP-bound state (26). The active state initiates a variety of downstream responses, including reorganization of the actin cytoskeleton. Rho GTPases that are unable to hydrolyze GTP are maintained in a constitutively active state. Activation of different members of the Rho GTPase family leads to specific effects on the cytoskeleton. RhoA activation leads to actin reorganization into stress fibers, Rac activation causes membrane ruffling, and Cdc42 activation causes the formation of filopodia (27–29). Treatment with CNF1 induces stress fiber formation, microspikes, membrane ruffles, and multinucleation in intact cells (7, 8, 22). CNF3 and CNFY also trigger strong formation of actin stress fibers in intact cells (24, 25). Surprisingly, there is no evidence of CNF3-triggered membrane ruffling or filopodium formation. It has been demonstrated that CNF1 is required for the invasion of uropathogenic E. coli into uroepithelial cell monolayers (9). This is consistent with observations of other bacterial pathogens, such as Salmonella, that attack the host cytoskeleton to facilitate bacterial entry (10).
The C Terminus of CNF1 Forms a Single Compact Catalytic Domain
CNFs are modular proteins. The N-terminal domains of these toxins are required for binding and entry into host cells via receptor-mediated endocytosis (30, 31). The structure of the catalytic C terminus of CNF1 (residues 720 to 1014) revealed a three-layered α/β/β sandwich, consisting of two mixed 5-stranded β-sheets flanked by two α-helices (Fig. 2A) (32). The structure of CNF1 identified a novel overall protein fold that contained an arrangement of residues in the active site reminiscent of the papain-like superfamily of enzymes, which includes the transglutaminase blood coagulation factor XIII and GMP synthetase (33). More recently, it was determined that CNF1 is also structurally similar to the Thermotoga maritima deamidase protein CheD and a bacterial protein of unknown function, YfiH (5). Papain-like superfamily enzymes typically contain a catalytic triad consisting of histidine, cysteine, and asparagine/aspartic acid. The cysteine and histidine form a thiolate-imidazolium pair that is critical for activating the cysteine nucleophile, with the asparagine/aspartic acid thought to be involved in orienting the histidine. CNF deamidases contain the invariant cysteine and histidine residues (34). Interestingly, in CNF1, valine is the third member of the catalytic triad, and this residue is involved in orienting the histidine through a main chain carbonyl hydrogen bond (32). Structure-based mutagenesis of conserved residues in the catalytic pocket identified Asn835 and Ser864, which are also required for CNF1 activity (Fig. 2A) (33). Furthermore, deletion of five individual loop regions surrounding the CNF1 catalytic pocket revealed that loops 8 and 9 are required for deamidation of RhoA (33). Most likely, Asn835 and Ser864 have indirect roles in catalysis and loops 8 and 9 are important for substrate recognition.
Fig 2.

Catalytic domains of diverse deamidases. (A) Catalytic domain of CNF1 from E. coli (Protein Data Bank [PDB] entry 1HQ0), with invariant catalytic residues Cys866 and His881 shown in purple. The main chain carbonyl of Val833 (also in purple) is involved in coordinating the imidazole ring of His881. Residues in pink are conserved residues important for CNF1 function, while loops 8 and 9 (likely important for substrate recognition) are shown in yellow. (B) BLF1 (PDB entry 3TU8) is a 23-kDa toxin and deamidase that forms a compact catalytic domain structurally related to those of CNF1 from E. coli and other papain-like superfamily enzymes. The residues that compose the catalytic triad (Cys94, His106, and Thr88) are shown in purple. α-Helices are shown in blue, β-sheets in green, and loops in gray. All structure figures were prepared with PyMol.
A peptide derived from RhoA residues 59 to 69 could be deamidated by CNF1, thus defining the minimal region of RhoA required for CNF1 deamidation (35). Mutations outside these key residues did not interfere with the ability of CNF1 to deamidate RhoA (35). This suggests that CNF1 interacts with a narrow surface of RhoA, and likely other GTPase substrates, and does not require a significant binding surface for substrate recognition.
BURKHOLDERIA LETHAL FACTOR 1 INHIBITS ACTIVITY OF TRANSLATION FACTOR eIF4A
Burkholderia lethal factor 1, formerly known as BPSL1549, is a potent toxin from Burkholderia pseudomallei. Melioidosis is a disease caused by infection with Burkholderia pseudomallei, a pathogen endemic in areas in Southeast Asia and northern Australia (36). It is associated with severe septicemia and can remain dormant in the host for decades. The molecular basis for the pathogenicity of B. pseudomallei is poorly understood.
BLF1 interacts with the human translation factor eIF4A in human cell lysates (11), suggesting that BLF1 might inhibit translation. This interaction was confirmed by coimmunoprecipitation. BLF1 reduces endogenous host cell protein synthesis and triggers increased stress granule formation, which is associated with translational blocks. In order to determine the outcome of the BLF1-eIF4A interaction, FLAG-tagged eIF4A was purified from human cells expressing BLF1. Subsequent mass spectrometric analysis of eIF4A revealed deamidation of Gln339 to Glu. To determine how deamidation of this residue altered eIF4A activity, the Gln339Glu variant was generated. Although this variant was capable of binding both RNA and ATP and maintained wild-type levels of ATPase activity, helicase activity was reduced by 47% (11).
The crystal structure of BLF1 revealed an α/β fold comprising a sandwich of two mixed β-sheets surrounded by loops and α-helices (Fig. 2B) (11). The β-sheet core of the catalytic pocket is structurally similar to that of the deamidase domain of CNF1, with a root mean square deviation (RMSD) of 3.9 Å over 170 residues (11). Interestingly, these deamidase domains share only 9% amino acid identity. BLF1 lacks the receptor binding and translocation domains of CNF1, an expected evolutionary innovation, since the intracellular lifestyle of B. pseudomallei renders these domains unnecessary. However, the invariant catalytic residues found in CNF1, i.e., cysteine and histidine, are conserved in the catalytic pocket. The third residue of the catalytic triad in BLF1 is a threonine. BLF1 deamidation of eIF4A and efficient inhibition of protein translation are dependent on the catalytic cysteine, as mutation of this residue to a serine renders BLF1 essentially nonfunctional (11).
VIBRIO PARAHAEMOLYTICUS TYPE III EFFECTOR VopC DEAMIDATES SMALL GTPases
A recent addition to the CNF family of deamidases is VopC from Vibrio parahaemolyticus. V. parahaemolyticus is an enteric pathogen that contains two type III secretion systems, namely, TTSS1 and TTSS2 (37); TTSS2 is encoded within a pathogenicity island. V. parahaemolyticus TTSS2 is necessary and sufficient for invasion of and replication in both HeLa cells and Caco-2 cells. Among the type III effectors encoded in the TTSS2 pathogenicity island is VopC, which shares 20% amino acid identity with the catalytic domain of CNF from E. coli. As VopC is a type III effector protein, it does not share the same mechanism for host cell delivery as CNF1. As a result, VopC and CNF1 have different N-terminal domains.
Both the catalytic cysteine and histidine residues of CNF1 are conserved in VopC (Cys220 and His235). Although a VopC structure has yet to be determined, homology modeling suggests that the carbonyl oxygen of Leu187 completes the papain-like catalytic triad. VopC deamidates and constitutively activates the small GTPases Rac and Cdc42 to facilitate host cell invasion (12). VopC is not able to activate RhoA. However, as noted above, differences in substrate specificity are common among CNF homologs. VopC activates Rac by deamidating Gln61 on the critical switch II domain. V. parahaemolyticus invasion of nonphagocytic HeLa cells is dependent on catalytically active VopC (12).
PASTEURELLA MULTOCIDA TOXIN
PMT Is a Multidomain Toxin, with Catalytic Activity Located at the C Terminus
Pasteurella multocida toxin (PMT) is a monomeric, 146-kDa enzyme. P. multocida causes severe diseases in animals and humans. It is associated with atrophic rhinitis in pigs and with dermatonecrosis and respiratory diseases, also known as snuffles, in cattle and rabbits (38, 39). In humans, P. multocida can cause dermonecrosis from bite wounds and bacteremia (40).
PMT is a multidomain toxin. The N-terminal region of PMT has sequence homology with the N termini of CNFs, suggesting that this region is also used for binding to the host cell surface. In fact, the first 506 N-terminal residues of PMT are sufficient to bind to cells and compete with full-length toxin in binding assays (41).
The C-terminal region of PMT contains the catalytic center of the enzyme and is sufficient for PMT-mediated toxicity when delivered into cells by electroporation (41). The crystal structure of the C-terminal region of PMT reveals a multidomain Trojan horse-like structure, with the feet, head, and body consisting of different domains (C1, C2, and C3) that possess different functions (Fig. 3A). The C1 domain, or the feet, consists of seven α-helices. The N-terminal α-helices of the C1 domain are structurally similar to the N-terminal domain of Clostridium difficile toxin B (42). In toxin B, these helices target the protein to the plasma membrane. Similarly, the first four α-helices of C1 are required to target the catalytic domain of PMT to the plasma membrane (42, 43). Deletion of the C1 domain causes a reduction but not complete loss of PMT function, suggesting that localization to the plasma membrane is necessary for full PMT-induced toxicity (42).
Fig 3.

Crystal structure of the C-terminal region of Pasteurella multocida toxin. (A) The crystal structure of the C-terminal region of PMT has been solved (residues 575 to 1285; PDB entry 2EBF). The catalytic region consists of several domains. The helical C1 domain (purple) forms the “feet” of the Trojan horse-like structure and contains membrane-targeting properties. The C2 “body” domain contains 2 α/β subdomains (shown in green and yellow) of unknown function. The domain responsible for the deamidase activity of PMT is the C3 “head” C-terminal domain (blue). (B) The crystal structure of the PMT catalytic domain reveals a disulfide bond between the catalytic cysteine, Cys1165, and Cys1159 in the binding pocket. Reduction of this disulfide bond is required for interaction of the catalytic triad and catalytic activity. His1205 and Asp1220 form the other residues of the triad. (C) The crystal structure of the catalytic domain of PMT(Cys1159Ser) (PDB entry 2EC5) demonstrates the position of the catalytic triad when the disulfide bond between Cys1159 and Cys1165 is reduced.
The function of the C2 domain, the body of the Trojan horse, is currently unknown. It is the largest section of the PMT C-terminal region and consists of two subdomains. Each subdomain contains typical α/β folds. Searches using the DALI server revealed possible structural homology between the second subdomain and phosphate-binding enzymes (43). Therefore, the C2 domain may be enzymatic or may be involved in binding target proteins in the host cell.
The C3 domain is the catalytic deamidase domain of PMT. Residues Cys1165 plus His1205 (which form the thiolate-imidazolium pair) and His1223 are required for PMT function (44, 45). Comparison with the structures of other members of the papain-like superfamily reveals that Asp1220 of PMT is the third residue of the catalytic triad. Mutation of any of these residues resulted in a complete loss of PMT function (43). Interestingly, the function of the catalytic cysteine requires the reduction of a disulfide bond with Cys1159 (Fig. 3B) (43). Reduction of the disulfide bond between Cys1159 and Cys1165 causes a rotation of the Cys1165 side chain toward the catalytic triad (Fig. 3C). Therefore, the disulfide bond has to be reduced for complete deamidase activity. Additionally, a Gln1225 residue in the catalytic cleft was shown to be required for deamidase activity. It has been postulated that this glutamine forms an oxyanion hole to stabilize the tetrahedral intermediate, similar to the role of Gln19 in papain (43).
PMT Deamidation of Heterotrimeric G Protein Families Affects Several Downstream Signaling Events
Unlike bacterial toxins that have high specificity for their host targets, such as the CNFs, PMT mediates its virulence effect through activation of multiple heterotrimeric G protein families. This leads to a plethora of changes in host signaling that lead to dysregulation of normal host cell homeostasis. A variety of cell systems and assays have been used to establish that the pleiotropic effects of PMT in target cells are due to the ability of PMT to activate multiple α subunits of diverse heterotrimeric G proteins (46–56). Heterotrimeric G proteins consist of four major families: Gαs, Gαi, Gαq/11, and Gα12/13. When PMT and Gαi proteins are coexpressed in E. coli, PMT modifies Gαi via deamidation (13). PMT converts the conserved Gln205 residue in the switch II region of Gαi to glutamic acid. PMT-mediated activation of Gαi proteins leads to the inhibition of adenylyl cyclase and reduction of cyclic AMP (cAMP) accumulation (Fig. 4) (49).
Fig 4.

PMT deamidates and activates heterotrimeric Gα proteins. The activation of diverse G protein-coupled receptor signal transduction pathways by PMT leads to several phenotypes that are disruptive to the host. PMT activates Gαi, Gαq/11, and Gα12/13 but not Gαs. PMT also triggers the release of Gβγ from the heterotrimeric complex, which leads to activation of the phosphoinositide-3-kinase (PI3K) pathway.
PMT activation of Gαq/11 proteins results in the stimulation of phospholipase Cβ (PLCβ) (Fig. 4). PLCβ catalyzes the hydrolysis of phosphatidylinositol-4,5-bisphosphate to inositol-1,4,5-triphosphate and diacylglycerol (DAG). It was originally postulated that PMT was capable of activating Gαq (56, 57). However, more recent in vitro expression assays coupled with a mass spectrometry output demonstrated that PMT is also able to deamidate Gα11 (58). PMT-induced Gα11 activation of PLCβ is weaker than the Gαq activation of PLCβ (59). Therefore, although PMT can deamidate and activate both Gαq and Gα11 in vitro, the relative contributions of these activities in vivo may differ. Regardless, activation of the Gαq/11 subfamily via PMT results in an increased release of the second messengers inositol-1,4,5-triphosphate and diacylglycerol, with a consequent increase in calcium mobilization. This leads to multiple calcium-mediated responses, such as activation of protein kinase C and secretion of chloride ions (46, 52, 55). PMT also stimulates the JAK/STAT pathway in a Gαq-dependent manner (48).
PMT also activates the small GTPase RhoA, resulting in reorganization of the actin cytoskeleton and formation of actin stress fibers (47, 54). PMT induces RhoA indirectly through activation of multiple heterotrimeric Gα protein families (Fig. 4). PMT-induced RhoA activation can occur through Gαq (55, 56) and Gα12/13 (50). A novel cyclic peptide that specifically inhibits Gαq partially blocked PMT-induced RhoA activation (50, 53). Additionally, PMT-induced RhoA activation was inhibited in Gα12/13-deficient HEK293m3 cells (50). However, it was not clear whether PMT functioned through Gα12, Gα13, or both. Recently, mass spectrometric analysis revealed that PMT can deamidate both Gα12 and Gα13 (58).
Finally, because Gα activation leads to the release of Gβγ from the heterotrimeric complex, PMT also stimulates increased signaling through Gβγ-specific effectors such as phosphoinositide-3-kinase (Fig. 4) (51).
CYCLE-INHIBITING FACTORS
Members of the Cif Family of Deamidases Are Found in Many Bacterial Pathogens
Enteropathogenic (EPEC) and enterohemorrhagic (EHEC) E. coli strains are causal agents of intestinal diarrhea (60, 61). EPEC is the cause of potentially fatal diarrhea in infants in developing countries. In contrast, highly infectious EHEC outbreaks occur most often in developed countries, most notably in North America, Japan, and Europe. Although the diseases caused by these two E. coli pathovars differ, they both cause the formation of attaching and effacing (A/E) lesions (60). The genes responsible for attaching and effacing lesions, the type III secretion system and type III effector genes, are carried on a 35-kb pathogenicity island called the locus of enterocyte effacement (LEE) (62). However, several effectors are non-LEE-encoded (Nle) proteins (63). Cif was the first Nle effector identified in EPEC and EHEC (64).
Cif homologs are encoded in multiple bacterial pathogen genomes and are all associated with horizontally transferred genetic elements (64, 65). Cif homologs from Burkholderia pseudomallei, Yersinia pseudotuberculosis, Photorhabdus luminescens, and Photorhabdus asymbiotica have been named CHBP, CHYP, CHPL, and CHPA, respectively. Interestingly, these proteins have low primary amino acid identities to Cif from E. coli: 56% for CHYP, 26% for CHBP, 23% for CHPL, and 26% for CHPA (65, 66).
Deamidation of NEDD8 by Cif Leads to Disruption of the Ubiquitin-Proteasome System
CifEc was shown to interact with NEDD8 in a yeast two-hybrid system, and this interaction was verified by in vitro pulldown and coexpression assays (14, 67, 68). Cif proteins modify NEDD8 and ubiquitin via deamidation (14). Ubiquitin and ubiquitin-like (UBL) proteins, such as NEDD8, control many cellular processes by targeting various proteins for degradation through the ubiquitin-proteasome system. Ligation of ubiquitin and UBLs such as NEDD8 to substrate proteins requires a series of three enzymes: an E1-activating enzyme, an E2-conjugating enzyme, and an E3 ligase. Cullin-RING ligases (CRLs) are a large superfamily of E3 ubiquitin ligases (69). The core CRL complex consists of cullin proteins, which are the scaffold proteins of CRLs and are modified by NEDD8, and a small RING protein.
In eukaryotic cells, cullins are continuously neddylated and deneddylated. The conjugation and removal of NEDD8 comprise an important cycling mechanism that regulates CRL activity. The COP9 signalosome (CSN), a conserved multiprotein complex with protease activity, is responsible for cullin deneddylation (69). When cullins are neddylated, they are maintained in an active, open conformation that allows substrate proteins to interact with the Rbx protein at the cullin C terminus (70). This flexible protein conformation and interaction lead to the ubiquitination and subsequent degradation of target proteins, some of which include important cell cycle regulators (Fig. 5A).
Fig 5.

Model of Cif inactivation of CRL activity and ubiquitination. (A) In uninfected cells, the COP9 signalosome (CSN) deneddylates CRLs. NEDD8 (yellow) conjugation and removal represent an important mechanism by which CRL activity is regulated. When CRLs are neddylated with wild-type (WT) NEDD8 (green), they are maintained in an open conformation. This allows substrates (blue) to bind the CRLs, become polyubiquitinated, and then be degraded. (B) In cells infected with bacteria that express Cif, deamidation of NEDD8 (yellow) may prevent CRL (green) from forming an open conformation. This prevents CRLs from associating with the substrate-binding module that allows downstream ubiquitination and degradation of substrate proteins (blue) to occur. This may lead to a reduction in the rate of deneddylation and a decrease in unmodified CRLs (red).
Cifs target residue Gln40 in NEDD8 and ubiquitin with exquisite selectivity (neither Gln39 nor Gln41 of NEDD8 is a target for deamidation) (14, 71). To date, recombinant CifEc, CifBp, and CifYp have all been shown to deamidate Gln40 of NEDD8 in vitro (14, 71). Ectopic expression of the Gln40Glu NEDD8 variant in HeLa cells phenocopies either the ectopic expression of Cifs or the effects of Cif during bacterial infection, demonstrating that this activity is responsible for the Cif-mediated cytopathic phenotype.
The deamidation of NEDD8 by Cif results in a reduction in the rate of cullin deneddylation (67, 68, 72). The Gln40Glu mutation in NEDD8 is sufficient to decrease the deneddylation rate by CSN in the absence of Cif (72). Gln40 is located close to the cullin-NEDD8 conjugation site. Deamidation of Gln40 in NEDD8 may prevent CSN-mediated deneddylation of CRLs through interference with the global NEDD8-induced conformational change of cullins (which may be necessary for CSN recognition) (Fig. 5B) (72). In contrast to previous studies, it was shown in yeast that CSN can remove deamidated NEDD8 from CRLs more efficiently than wild-type NEDD8 (73). This causes an increase in deneddylated CRLs. Additionally, it is appropriate to mention that deamidation of NEDD8 and subsequent effects on CRL activity have not been reconstituted in vitro. Therefore, the exact effect of Cif-mediated deamidation of NEDD8 remains unclear.
Cif causes cytopathic phenotypes such as rearrangement of the host cytoskeleton and the formation of actin stress fibers. However, the hallmark of Cif infection is the inhibition of cell cycle progression. These multiple cytopathic phenotypes can be attributed directly to disruption of the ubiquitin-protease system. Depending on the stage of the cells during infection, Cifs can arrest the cell cycle at either the G1/S transition or the G2/M transition (64, 74). G1/S and G2/M transitions require complexes that consist of a cyclin and a cyclin-dependent kinase (CDK). Inhibiting CRL activity prevents ubiquitination and degradation of cell cycle regulators such as p21 and p27, resulting in inactivation of cyclin-dependent kinases and cell cycle arrest (64, 74, 75). This triggers an increase in the inactive and phosphorylated CDKs, leading to cell cycle arrest (64).
Cifs also inhibit the degradation of a variety of other CRL substrates, such as pIκB, Cdt1, β-catenin, and RhoA (64). Therefore, inhibition of CRL activity can account for the multiple phenotypes observed during infection with bacterial species expressing Cifs.
Multiple Structures Reveal that Cif Proteins Are Members of the Papain-Like Superfamily
The crystal structures of CifEc, CHBP, CHYP, and CHPL have been solved (Fig. 6). Although the primary amino acid similarity among the homologs is low, the structures are highly conserved (76, 77). The disordered N termini provide type III secretion signals and are either not included in expressed proteins or not observed in the crystal structures. To date, the only crystal structure of CifEc includes a truncation of the first 100 amino acids (77). The overall fold of Cifs comprises a head-and-tail domain organization (Fig. 6A). The tail region, also known as the helical extension domain, is encoded in the N terminus of the protein. The head domain, also known as the globular core, is formed by the C terminus of the protein. The head domain consists of an antiparallel β-sheet surrounded by α-helices. This globular domain displays overall structural homology to the papain-like superfamily. Similar to the other deamidases covered in this review, the active sites of all Cifs contain an invariant Cys-His thiolate-imidazolium pair. The third residue of the catalytic triad in Cifs is a glutamine (Fig. 6B). Each of these catalytic residues is required for Cif-induced cytopathic effects, deamidation of NEDD8 and ubiquitin, and the resulting increase in stability of the cell cycle regulators p21 and p27 (14, 65, 66, 77).
Fig 6.

Crystal structures of cell cycle-inhibiting factors. (A) Crystal structures of Cif deamidases from E. coli (PDB entry 3EFY), B. pseudomallei (PDB entry 3GQM), P. luminescens (PDB entry 3GQJ), and Y. pseudotuberculosis (PDB entry 4F8C). Shown in blue are the α-helices of the core globular domain. In teal are the α-helices of the tail helical extension domain. The antiparallel β-sheet is shown in green, and the loops are colored gray, except for the occluding loop, which is colored black. The conserved catalytic residues histidine, cysteine, and glutamine are shown in purple. (B) Amino acids in the catalytic domain of CHBP are shown as purple sticks.
An interesting feature observed in the structures of Cifs is the so-called occluding loop. This loop was first identified in CifEc and is conserved across the Cif family (76, 77). The occluding loop partially blocks access to the catalytic site (substrate binding cleft of papain) and was hypothesized to be important in defining specificity for Cif substrates (Fig. 6B) (see below).
Cif Deamidases Were the First To Be Cocrystallized with Their Substrates
The crystal structures of CHYP, CHPL, and CHBP have been determined in complex with NEDD8 (71, 78). Additionally, the cocrystal structure of CHBP and ubiquitin has also been determined (71). The structures of these complexes reveal an extensive binding interface between Cif proteins and their ubiquitin/NEDD8 substrate (Fig. 7A), which involves both the head and tail domains of Cifs. Multiple alanine mutations in CHBP in the regions that interface with either the N-terminal β-hairpin or the residues adjacent to Gln40 in ubiquitin or NEDD8 were sufficient to abolish deamidation and Cif-induced cell cycle arrest (71). Furthermore, alanine mutations in the corresponding residues in ubiquitin and NEDD8 disrupted both the interaction and deamidation (71). Interestingly, all individual alanine mutations in CHYP (that were tested) were not sufficient to disrupt NEDD8 complex formation. Only mutations that introduced steric clashes (based on the complex structure) prevented binding (71).
Fig 7.

Crystal structure of the CifYp deamidase-NEDD8 complex. (A) The CHYP (α-helices are shown in blue, β-sheets in green, and loops in gray)-NEDD8 (red) complex consists of an extensive interaction surface (PDB entry 4FBJ). The catalytic Cys117 position is modeled based on the CHYP(Cys117Ala)-NEDD8 complex to demonstrate the proposed molecular mechanism of deamidation. (B) Close-up of the catalytic domain of CHYP showing that the deamidation target, Gln40 of NEDD8, is positioned near the Cif catalytic residues (shown in purple). (C) The proposed molecular mechanism of deamidation involves deprotonation of the cysteine by the imidazolium group of histidine (step 1) followed by a nucleophilic attack by the thiol group of the catalytic cysteine on the δ-carbon of glutamine (step 2).
There is very little change in the structures of Cifs bound to their substrates compared to the uncomplexed states (71, 76, 78). However, a significant reorientation of the flexible C-terminal tail of NEDD8 and ubiquitin is observed upon binding by Cifs. Interestingly, it is the occluding loop in Cifs that appears to be responsible for forcing this reorientation during substrate binding. Displacement of the flexible C-terminal tail is likely important for substrate recognition and for orienting Gln40 in the Cif catalytic pocket (71, 78). Consistent with this, deletion of the C-terminal domain of either ubiquitin or NEDD8 diminished deamidation by Cifs (76). Therefore, multiple areas within the extensive interface between Cif deamidases and their substrates are important for activity.
CHBP has a preference for NEDD8 as a substrate (14), and CHYP deamidation of NEDD8 is 10-fold more efficient than deamidation of ubiquitin (78). Molecular dynamic studies revealed that this efficiency difference may be due to decreased flexibility of NEDD8 in complex with Cif compared to ubiquitin (71). Residue Glu31 of NEDD8 interacts electrostatically with several residues in CHBP. The residue at the same position in ubiquitin is glutamine, which is not capable of electrostatic interactions. To determine whether this residue contributes to the difference in movement and deamidation efficiency, NEDD8(Glu31Gln) and ubiquitin(Gln31Glu) variants were generated. NEDD8(Glu31Gln) exhibited increased molecular dynamic motion in simulations, and deamidation efficiency in in vitro assays was comparable to that observed for wild-type ubiquitin. Conversely, ubiquitin(Gln31Glu) exhibited decreased molecular dynamic motion, bound more tightly to CHBP, and was deamidated at a level similar to that of wild-type NEDD8 (71). Thus, the Gln31 substrate residue may determine the difference in substrate deamidation efficiency by Cifs.
The data from the Cif-NEDD8 complex structures also suggest a molecular mechanism of enzymatic deamidation by Cifs (Fig. 7B and C). All solved structures of currently known enzymatic deamidases contain conserved papain-like catalytic residues and α/β folds similar to those in the papain-like superfamily. As a result, the molecular mechanism of deamidation is likely to be similar to the protease and transglutaminase reactions catalyzed by other superfamily members. During these reactions, the catalytic histidine residue deprotonates the cysteine sulfhydryl group to increase its ability to act as a nucleophile. This nucleophile then attacks the amide substrate, resulting in an acyl-enzyme intermediate that is resolved by hydrolysis. The catalytic histidine's imidazole ring is often coordinated by a hydrogen bond with a third residue, and this is ensures optimal orientation of the thiolate-imidazolium pair (Fig. 7B and C) (78).
OspI INHIBITS HOST IMMUNE RESPONSES BY DEAMIDATING AN E2-CONJUGATING ENZYME
Deamidation of Ubc13 by OspI Inhibits Host Inflammatory Responses
Shigella species are routinely found in patients suffering from severe diarrhea in developing countries (79). The severe symptoms associated with Shigella infections are due to the invasion and destruction of the colonic and rectal epithelium and a severe inflammatory reaction (79).
S. flexneri infections trigger changes to the host cell membrane that initiate the DAG–CBM complex–NF-κB signaling pathway. S. flexneri invades the host cell via macropinocytosis. It is then able to escape from the phagosome and into the cytoplasm through an unknown mechanism (80). The escape leads to the accumulation of host membrane components near the site of infection and to subsequent host signaling events that lead to inflammation. This likely occurs through the activation of phospholipases that hydrolyze plasma membrane phospholipids, such as DAG, which then activate protein kinases (81). Subsequently, protein kinases activate the complex of caspase recruitment domain (CARD), B-cell lymphoma 10 (BCL-10), and mucosa-associated lymphoid tissue lymphoma translocation gene 1 (MALT1). This complex is also referred to as the CBM complex. The CBM complex interacts with and activates tumor necrosis factor receptor-associated factor 6 (TRAF6). TRAF6 is an E3 ubiquitin ligase that interacts with Ubc13, an E2 ubiquitin-conjugating enzyme, to form lysine 63 (Lys63)-linked polyubiquitin chains (82). The presence of unconjugated Lys63 polyubiquitin chains activates the complex of transforming growth factor beta-activated kinase 1 (TAK1), TAK1-binding protein 1 (TAB1), and TAK1-binding protein 2 (TAB2). This leads to IκBα phosphorylation and degradation. This allows NF-κB to translocate into the nucleus, where it activates the expression of proinflammatory cytokine genes (82).
S. flexneri encodes a type III effector, OspI, which modulates this host response by deamidating Ubc13. Incubation of OspI with TRAF6, E2 ubiquitin-conjugating enzymes (UBC13 and UEV1A), and ubiquitin revealed that OspI causes a shift in the mobility of Ubc13 in SDS-PAGE gels. Mass spectrometry analysis revealed that Ubc13 is deamidated at residue Gln100 in the presence of OspI (15). The deamidation of Ubc13 Gln100 blocks its ubiquitin-conjugating activity, thereby inhibiting the TRAF6-DAG-CBM complex and subsequent NF-κB signaling. This is made evident by the increase in phosphorylation of the NF-κB inhibitor IκBα and consequent NF-κB translocation to the nucleus in HeLa cells infected with an S. flexneri ΔospI strain (15). Consistent with this result, NF-κB translocation to the nucleus is inhibited in the presence of OspI (15).
OspI Forms a Papain-Like Catalytic Pocket That Rearranges upon Binding Ubc13
The crystal structure of OspI reveals an α/β fold with four β-strands, seven α-helices, and one 310 helix (Fig. 8A) (15). OspI also shares structural homology with the papain-like superfamily, as noted above for PMT and the Cif family of deamidases. It is most closely related to the Pseudomonas syringae cysteine protease effector AvrPphB (83, 84).
Fig 8.

Crystal structures of unbound OspI and the OspI-Ubc13 complex. (A) OspI (α-helices are shown in blue, β-sheets in green, and loops in gray) (PDB entry 3B21) forms a compact catalytic domain structurally similar to those of papain-like superfamily enzymes. The residues in the catalytic triad (Cys62, His145, and Asp160) are shown in purple. The catalytic pocket is blocked by Asn61 in free OspI. (B) The OspI(Cys62Ala)-Ubc13 (red) complex includes an extensive interaction surface involving Ubc13 α-helix 1 and loops L1 and L2 (PDB entry 4IP3). (C) Close-up of the OspI(Cys62Ala)-Ubc13 complex showing Gln100 of Ubc13 positioned adjacent to the OspI catalytic center (purple; OspI residue 62 was modeled as a cysteine to demonstrate the nucleophilic attack [yellow dashes] by the thiol group of Cys62 on Gln100 in Ubc13).
Superimposition of the catalytic domain of AvrPphB with the OspI crystal structure revealed that the catalytic triad in OspI comprises Cys62, His145, and Asp160 (15). OspI inhibition of the DAG–CBM–NF-κB pathway is dependent on the catalytic residues, as mutation of any of these residues leads to the loss of OspI activity (15).
Recently, the crystal structure of the OspI(Cys62Ala)-Ubc13 complex was solved (Fig. 8B and C) (85). This structure revealed an extensive binding surface between OspI and Ubc13 that covers 11% of the exposed surface of OspI (85). A negatively charged region and a hydrophobic pocket of OspI bind α-helix 1 and the L1 and L2 loops of Ubc13, respectively. Analysis of point mutations in these regions revealed that both interfaces are required for deamidation of Ubc13 (85). In contrast to the case for Cifs, significant structural rearrangements occur in the catalytic pocket of OspI upon substrate binding (85). In uncomplexed OspI, the catalytic pocket is blocked by Asn61 (Fig. 8A). When OspI binds Ubc13, Asn61 rotates approximately 180 degrees. This leads to formation of a hydrogen bond with Asn54 and to repositioning of Ala62. The movement of these residues opens the pocket, allowing insertion of Gln100 from Ubc13 into the catalytic site. Superposition of the catalytic pockets of AvrPphB and Ubc13-bound OspI reveals an overlap of OspI Ala62 (Cys62 in the wild-type protein) and the catalytic cysteine of AvrPphB (85). This suggests that the remodeled OspI catalytic pocket (bound to Ubc13) contains the proper orientation for catalytic activity. Additionally, Phe95 and His96 of OspI reorient to form a hydrophobic pocket that interacts with Ubc13 loops L1 and L2 (85).
The OspI-Ubc13-complexed structure confirms that the molecular mechanism of OspI is similar to that of papain-like enzymes and that suggested for Cif/NEDD8 deamidation (Fig. 7C) (84–86). It is interesting that the catalytic cysteine is capable of forming disulfide bonds with Cys65 (Fig. 8C) (15). Although it has not been demonstrated that this disulfide bond affects the function and positioning of the catalytic cysteine, it is possible that it maintains the deamidase in a noncatalytic state under nonreducing conditions, as has been demonstrated for PMT (Fig. 3B and C) (43).
CONCLUSIONS
Recently, it was discovered that deamidation is a common mode of action used by several bacterial virulence factors to modify the functions of host proteins. Deamidation, as an irreversible modification of host proteins, offers many advantages to invading pathogens. Unlike the case of phosphorylation, host systems lack the ability to reverse the effects of deamidation. This makes deamidases particularly potent virulence factors. Characterized deamidases have been shown to modify specific residues on host proteins that are key to the host protein's function. Additionally, there is high specificity in the proteins that are targeted by deamidases, as can be seen in the ability of CNF1 to deamidate some, but not all, Rho GTPases. Although the deamidation modification is subtle and specific, the downstream impacts on host cell function are extensive. This is most evident in the case of cycle-inhibiting factors, which inhibit the ubiquitin-proteasome system, leading to both cell cycle arrest and cytoskeletal rearrangements. In fact, it is apparent that bacterial deamidase virulence factors have evolved to target key modulators of critical cell signaling systems in hosts.
However, our understanding of the role of deamidases in bacterial virulence is still in its infancy. Further elucidation of the molecular mechanisms and the interactions between deamidases and their substrates is important. To date, the use of enzymatic deamidation as a generic “housekeeping” mechanism for modifying protein function has been documented only for prokaryotes. Do eukaryotes also use this activity to modulate protein activity? Or is enzymatic deamidation in eukaryotic cells an innovation of bacterial pathogens mediated via translocated proteins? Future work would be aided greatly by new methods for easier identification of deamidation. Current methods require knowledge of the specific substrate of each deamidase in order for activity to be assayed. This differs from enzymes such as cysteine proteases, for which cleavage of generic substrates can easily be measured. Additionally, the presence of nonspecific deamidation in protein samples can make the identification of enzymatic deamidation difficult. Another important future direction is to define the molecular mechanisms by which deamidation specifically affects the functions of host cell targets. At present, our knowledge is restricted to observing the phenotypic outcomes of deamidation on particular targets during diverse steps in the lifestyles of diverse pathogens. It would be valuable to understand how deamidation alters function at the level of the modified protein. For example, G proteins are common targets of at least some of the deamidases discussed here. But is the phenotypic outcome of these events always merely an irreversible loss of catalytic function, or could the deamidated G protein now act as a dominant-negative variant on its normal cellular partners, amplifying the impact of the deamidation event to multiple output pathways, as noted above for PMT? Such understanding will be important, for example, in exploration of genome editing to target glutamine-to-glutamate mutations.
Regardless of the challenges of these studies, it is clear that tackling the molecular mechanisms of deamidation, as has been done for other posttranslational modifications used by bacterial pathogens, is essential for our understanding of bacterial pathogenesis and, in fact, for the normal host cell biology of their targets.
ACKNOWLEDGMENTS
This work was supported by a grant from the DOE to J.L.D. J.L.D. is an HHMI Investigator, and this work was funded by the Howard Hughes Medical Institute and the Gordon and Betty Moore Foundation (in part via grant GBMF3030 to J.L.D.). M.J.B. is supported by the BBSRC (United Kingdom; grants BB/J004553/1 and BB/F008732/1) and the John Innes Foundation.
Biographies

Erica J. Washington is currently a Ph.D. student in the lab of Jeff Dangl, in the Biology Department of the University of North Carolina at Chapel Hill. She received her B.S. in Biological Sciences from the University of Maryland, Baltimore County, in 2003. Her research interests include the molecular mechanisms of bacterial pathogenesis, with an interest in the structural biology of bacterial virulence factors. Her graduate research focuses on investigating the functions and structures of Pseudomonas syringae type III effectors. She also studies how these type III effector proteins manipulate plant immune responses to promote disease progression.

Mark J. Banfield, Ph.D., is a Project Leader in the Department of Biological Chemistry at the John Innes Centre, United Kingdom. Between 2002 and 2010, he was a Royal Society (United Kingdom) University Research Fellow, a position he first held at Bristol University (United Kingdom) and then Newcastle University (United Kingdom) before moving to the John Innes Centre. For the last 10 years, his research group has focused primarily on understanding the molecular mechanisms of host-translocated pathogen proteins, commonly known as effectors. His interests span both bacterial pathogens of mammals and bacterial, fungal, and oomycete pathogens of plants. He is especially interested in linking studies of protein structure to function and how this knowledge can be used to better understand pathogenesis. He has published many papers in this area, including studies of cycle-inhibiting factors (Cifs) and determining the first crystal structures of RXLR-type effectors from oomycetes.

Jeffery L. Dangl is the John N. Couch Distinguished Professor of Biology and an HHMI-GBMF Plant Sciences Investigator at the University of North Carolina at Chapel Hill. He is a geneticist recognized for his work on defining the molecular intricacies of the plant immune system and, recently, exploring the plant-associated microbiome. He graduated from Stanford University in 1981 and earned a doctorate in the Genetics Department of the Stanford University School of Medicine in 1986. He was an NSF Plant Molecular Biology Program postdoctoral fellow at the Max Planck Institute for Plant Breeding in Cologne, Germany, and thereafter a founding Group Leader of the associated Max Delbrueck Laboratory. He moved to UNC in 1995 and became the founding Associate Director of the Carolina Center for Genome Sciences in 2000. He was elected to membership in the U.S. National Academy of Sciences in 2007, the German National Academy of Sciences in 2003, and the American Academy of Microbiology in 2011.
REFERENCES
- 1. Cui J, Shao F. 2011. Biochemistry and cell signaling taught by bacterial effectors. Trends Biochem. Sci. 36:532–540 [DOI] [PubMed] [Google Scholar]
- 2. Ribet D, Cossart P. 2010. Pathogen-mediated posttranslational modifications: a re-emerging field. Cell 143:694–702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yarbrough ML, Orth K. 2009. AMPylation is a new post-translational modiFICation. Nat. Chem. Biol. 5:378–379 [DOI] [PubMed] [Google Scholar]
- 4. Robinson AB, Rudd CJ. 1974. Deamidation of glutaminyl and asparaginyl residues in peptides and proteins. Curr. Top. Cell. Regul. 8:247–295 [DOI] [PubMed] [Google Scholar]
- 5. Chao X, Muff TJ, Park SY, Zhang S, Pollard AM, Ordal GW, Bilwes AM, Crane BR. 2006. A receptor-modifying deamidase in complex with a signaling phosphatase reveals reciprocal regulation. Cell 124:561–571 [DOI] [PubMed] [Google Scholar]
- 6. Striebel F, Imkamp F, Sutter M, Steiner M, Mamedov A, Weber-Ban E. 2009. Bacterial ubiquitin-like modifier Pup is deamidated and conjugated to substrates by distinct but homologous enzymes. Nat. Struct. Mol. Biol. 16:647–651 [DOI] [PubMed] [Google Scholar]
- 7. Flatau G, Lemichez E, Gauthier M, Chardin P, Paris S, Fiorentini C, Boquet P. 1997. Toxin-induced activation of the G protein p21 Rho by deamidation of glutamine. Nature 387:729–733 [DOI] [PubMed] [Google Scholar]
- 8. Schmidt G, Sehr P, Wilm M, Selzer J, Mann M, Aktories K. 1997. Gln 63 of Rho is deamidated by Escherichia coli cytotoxic necrotizing factor-1. Nature 387:725–729 [DOI] [PubMed] [Google Scholar]
- 9. Doye A, Mettouchi A, Bossis G, Clement R, Buisson-Touati C, Flatau G, Gagnoux L, Piechaczyk M, Boquet P, Lemichez E. 2002. CNF1 exploits the ubiquitin-proteasome machinery to restrict Rho GTPase activation for bacterial host cell invasion. Cell 111:553–564 [DOI] [PubMed] [Google Scholar]
- 10. Galan JE, Zhou D. 2000. Striking a balance: modulation of the actin cytoskeleton by Salmonella. Proc. Natl. Acad. Sci. U. S. A. 97:8754–8761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cruz-Migoni A, Hautbergue GM, Artymiuk PJ, Baker PJ, Bokori-Brown M, Chang CT, Dickman MJ, Essex-Lopresti A, Harding SV, Mahadi NM, Marshall LE, Mobbs GW, Mohamed R, Nathan S, Ngugi SA, Ong C, Ooi WF, Partridge LJ, Phillips HL, Raih MF, Ruzheinikov S, Sarkar-Tyson M, Sedelnikova SE, Smither SJ, Tan P, Titball RW, Wilson SA, Rice DW. 2011. A Burkholderia pseudomallei toxin inhibits helicase activity of translation factor eIF4A. Science 334:821–824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhang L, Krachler AM, Broberg CA, Li Y, Mirzaei H, Gilpin CJ, Orth K. 2012. Type III effector VopC mediates invasion for Vibrio species. Cell Rep. 1:453–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Orth JH, Preuss I, Fester I, Schlosser A, Wilson BA, Aktories K. 2009. Pasteurella multocida toxin activation of heterotrimeric G proteins by deamidation. Proc. Natl. Acad. Sci. U. S. A. 106:7179–7184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cui J, Yao Q, Li S, Ding X, Lu Q, Mao H, Liu L, Zheng N, Chen S, Shao F. 2010. Glutamine deamidation and dysfunction of ubiquitin/NEDD8 induced by a bacterial effector family. Science 329:1215–1218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sanada T, Kim M, Mimuro H, Suzuki M, Ogawa M, Oyama A, Ashida H, Kobayashi T, Koyama T, Nagai S, Shibata Y, Gohda J, Inoue J, Mizushima T, Sasakawa C. 2012. The Shigella flexneri effector OspI deamidates UBC13 to dampen the inflammatory response. Nature 483:623–626 [DOI] [PubMed] [Google Scholar]
- 16. Caprioli AFV, Roda LG, Ruggeri FM, Zona C. 1983. Partial purification and characterization of an Escherichia coli toxic factor that induces morphological cell alterations. Infect. Immun. 39:1300–1306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Oswald E, De Rycke J, Guillot JF, Boivin R. 1989. Cytotoxic effect of multinucleation in HeLa cell cultures associated with the presence of Vir plasmid in Escherichia coli strains. FEMS Microbiol. Lett. 49:95–99 [DOI] [PubMed] [Google Scholar]
- 18. Orden JA, Dominguez-Bernal G, Martinez-Pulgarin S, Blanco M, Blanco JE, Mora A, Blanco J, Blanco J, de la Fuente R. 2007. Necrotoxigenic Escherichia coli from sheep and goats produce a new type of cytotoxic necrotizing factor (CNF3) associated with the eae and ehxA genes. Int. Microbiol. 10:47–55 [PubMed] [Google Scholar]
- 19. Lockman HA, Gillespie RA, Baker BD, Shakhnovich E. 2002. Yersinia pseudotuberculosis produces a cytotoxic necrotizing factor. Infect. Immun. 70:2708–2714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Schmidt G, Goehring UM, Schirmer J, Uttenweiler-Joseph S, Wilm M, Lohmann M, Giese A, Schmalzing G, Aktories K. 2001. Lysine and polyamines are substrates for transglutamination of Rho by the Bordetella dermonecrotic toxin. Infect. Immun. 69:7663–7670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Masuda M, Betancourt L, Matsuzawa T, Kashimoto T, Takao T, Shimonishi Y, Horiguchi Y. 2000. Activation of rho through a cross-link with polyamines catalyzed by Bordetella dermonecrotizing toxin. EMBO J. 19:521–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lerm M, Selzer J, Hoffmeyer A, Rapp UR, Aktories K, Schmidt G. 1999. Deamidation of Cdc42 and Rac by Escherichia coli cytotoxic necrotizing factor 1: activation of c-Jun N-terminal kinase in HeLa cells. Infect. Immun. 67:496–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sugai M, Hatazaki K, Mogami A, Ohta H, Peres SY, Herault F, Horiguchi Y, Masuda M, Ueno Y, Komatsuzawa H, Suginaka H, Oswald E. 1999. Cytotoxic necrotizing factor type 2 produced by pathogenic Escherichia coli deamidates a Gln residue in the conserved G-3 domain of the Rho family and preferentially inhibits the GTPase activity of RhoA and Rac1. Infect. Immun. 67:6550–6557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Stoll T, Markwirth G, Reipschlager S, Schmidt G. 2009. A new member of a growing toxin family— Escherichia coli cytotoxic necrotizing factor 3 (CNF3). Toxicon 54:745–753 [DOI] [PubMed] [Google Scholar]
- 25. Hoffmann C, Pop M, Leemhuis J, Schirmer J, Aktories K, Schmidt G. 2004. The Yersinia pseudotuberculosis cytotoxic necrotizing factor (CNFY) selectively activates RhoA. J. Biol. Chem. 279:16026–16032 [DOI] [PubMed] [Google Scholar]
- 26. Heasman SJ, Ridley AJ. 2008. Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat. Rev. Mol. Cell Biol. 9:690–701 [DOI] [PubMed] [Google Scholar]
- 27. Nobes CD, Hall A. 1995. Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell 81:53–62 [DOI] [PubMed] [Google Scholar]
- 28. Ridley AJ, Hall A. 1992. The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell 70:389–399 [DOI] [PubMed] [Google Scholar]
- 29. Ridley AJ, Paterson HF, Johnston CL, Diekmann D, Hall A. 1992. The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell 70:401–410 [DOI] [PubMed] [Google Scholar]
- 30. Lemichez E, Flatau G, Bruzzone M, Boquet P, Gauthier M. 1997. Molecular localization of the Escherichia coli cytotoxic necrotizing factor CNF1 cell-binding and catalytic domains. Mol. Microbiol. 24:1061–1070 [DOI] [PubMed] [Google Scholar]
- 31. Contamin S, Galmiche A, Doye A, Flatau G, Benmerah A, Boquet P. 2000. The p21 Rho-activating toxin cytotoxic necrotizing factor 1 is endocytosed by a clathrin-independent mechanism and enters the cytosol by an acidic-dependent membrane translocation step. Mol. Biol. Cell 11:1775–1787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Buetow L, Flatau G, Chiu K, Boquet P, Ghosh P. 2001. Structure of the Rho-activating domain of Escherichia coli cytotoxic necrotizing factor 1. Nat. Struct. Biol. 8:584–588 [DOI] [PubMed] [Google Scholar]
- 33. Buetow L, Ghosh P. 2003. Structural elements required for deamidation of RhoA by cytotoxic necrotizing factor 1. Biochemistry 42:12784–12791 [DOI] [PubMed] [Google Scholar]
- 34. Schmidt G, Selzer J, Lerm M, Aktories K. 1998. The Rho-deamidating cytotoxic necrotizing factor 1 from Escherichia coli possesses transglutaminase activity. Cysteine 866 and histidine 881 are essential for enzyme activity. J. Biol. Chem. 273:13669–13674 [DOI] [PubMed] [Google Scholar]
- 35. Flatau G, Landraud L, Boquet P, Bruzzone M, Munro P. 2000. Deamidation of RhoA glutamine 63 by the Escherichia coli CNF1 toxin requires a short sequence of the GTPase switch 2 domain. Biochem. Biophys. Res. Commun. 267:588–592 [DOI] [PubMed] [Google Scholar]
- 36. Wiersinga WJ, van der Poll T, White NJ, Day NP, Peacock SJ. 2006. Melioidosis: insights into the pathogenicity of Burkholderia pseudomallei. Nat. Rev. Microbiol. 4:272–282 [DOI] [PubMed] [Google Scholar]
- 37. Broberg CA, Calder TJ, Orth K. 2011. Vibrio parahaemolyticus cell biology and pathogenicity determinants. Microbes Infect. 13:992–1001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Felix R, Fleisch H, Frandsen PL. 1992. Effect of Pasteurella multocida toxin on bone resorption in vitro. Infect. Immun. 60:4984–4988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mushin R, Schoenbaum M. 1980. A strain of Pasteurella multocida associated with infections in rabbit colonies. Lab. Anim. 14:353–356 [DOI] [PubMed] [Google Scholar]
- 40. Garcia VF. 1997. Animal bites and Pasteurella infections. Pediatr. Rev. 18:127–130 [DOI] [PubMed] [Google Scholar]
- 41. Pullinger GD, Sowdhamini R, Lax AJ. 2001. Localization of functional domains of the mitogenic toxin of Pasteurella multocida. Infect. Immun. 69:7839–7850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kamitani S, Kitadokoro K, Miyazawa M, Toshima H, Fukui A, Abe H, Miyake M, Horiguchi Y. 2010. Characterization of the membrane-targeting C1 domain in Pasteurella multocida toxin. J. Biol. Chem. 285:25467–25475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kitadokoro K, Kamitani S, Miyazawa M, Hanajima-Ozawa M, Fukui A, Miyake M, Horiguchi Y. 2007. Crystal structures reveal a thiol protease-like catalytic triad in the C-terminal region of Pasteurella multocida toxin. Proc. Natl. Acad. Sci. U. S. A. 104:5139–5144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ward PN, Miles AJ, Sumner IG, Thomas LH, Lax AJ. 1998. Activity of the mitogenic Pasteurella multocida toxin requires an essential C-terminal residue. Infect. Immun. 66:5636–5642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Orth JH, Blocker D, Aktories K. 2003. His1205 and His1223 are essential for the activity of the mitogenic Pasteurella multocida toxin. Biochemistry 42:4971–4977 [DOI] [PubMed] [Google Scholar]
- 46. Hennig B, Orth J, Aktories K, Diener M. 2008. Anion secretion evoked by Pasteurella multocida toxin across rat colon. Eur. J. Pharmacol. 583:156–163 [DOI] [PubMed] [Google Scholar]
- 47. Lacerda HM, Lax AJ, Rozengurt E. 1996. Pasteurella multocida toxin, a potent intracellularly acting mitogen, induces p125FAK and paxillin tyrosine phosphorylation, actin stress fiber formation, and focal contact assembly in Swiss 3T3 cells. J. Biol. Chem. 271:439–445 [DOI] [PubMed] [Google Scholar]
- 48. Orth JH, Aktories K, Kubatzky KF. 2007. Modulation of host cell gene expression through activation of STAT transcription factors by Pasteurella multocida toxin. J. Biol. Chem. 282:3050–3057 [DOI] [PubMed] [Google Scholar]
- 49. Orth JH, Fester I, Preuss I, Agnoletto L, Wilson BA, Aktories K. 2008. Activation of Gαi and subsequent uncoupling of receptor-Gαi signaling by Pasteurella multocida toxin. J. Biol. Chem. 283:23288–23294 [DOI] [PubMed] [Google Scholar]
- 50. Orth JH, Lang S, Taniguchi M, Aktories K. 2005. Pasteurella multocida toxin-induced activation of RhoA is mediated via two families of Gα proteins, Gαq and Gα12/13. J. Biol. Chem. 280:36701–36707 [DOI] [PubMed] [Google Scholar]
- 51. Preuss I, Kurig B, Nurnberg B, Orth JH, Aktories K. 2009. Pasteurella multocida toxin activates Gβγ dimers of heterotrimeric G proteins. Cell. Signal. 21:551–558 [DOI] [PubMed] [Google Scholar]
- 52. Staddon JM, Chanter N, Lax AJ, Higgins TE, Rozengurt E. 1990. Pasteurella multocida toxin, a potent mitogen, stimulates protein kinase C-dependent and -independent protein phosphorylation in Swiss 3T3 cells. J. Biol. Chem. 265:11841–11848 [PubMed] [Google Scholar]
- 53. Takasaki J, Saito T, Taniguchi M, Kawasaki T, Moritani Y, Hayashi K, Kobori M. 2004. A novel Gαq/11-selective inhibitor. J. Biol. Chem. 279:47438–47445 [DOI] [PubMed] [Google Scholar]
- 54. Thomas W, Pullinger GD, Lax AJ, Rozengurt E. 2001. Escherichia coli cytotoxic necrotizing factor and Pasteurella multocida toxin induce focal adhesion kinase autophosphorylation and Src association. Infect. Immun. 69:5931–5935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wilson BA, Zhu X, Ho M, Lu L. 1997. Pasteurella multocida toxin activates the inositol triphosphate signaling pathway in Xenopus oocytes via Gαq-coupled phospholipase C-β1. J. Biol. Chem. 272:1268–1275 [DOI] [PubMed] [Google Scholar]
- 56. Zywietz A, Gohla A, Schmelz M, Schultz G, Offermanns S. 2001. Pleiotropic effects of Pasteurella multocida toxin are mediated by Gq-dependent and -independent mechanisms. Involvement of Gq but not G11. J. Biol. Chem. 276:3840–3845 [DOI] [PubMed] [Google Scholar]
- 57. Orth JH, Lang S, Aktories K. 2004. Action of Pasteurella multocida toxin depends on the helical domain of Gαq. J. Biol. Chem. 279:34150–34155 [DOI] [PubMed] [Google Scholar]
- 58. Orth JH, Fester I, Siegert P, Weise M, Lanner U, Kamitani S, Tachibana T, Wilson BA, Schlosser A, Horiguchi Y, Aktories K. 2013. Substrate specificity of Pasteurella multocida toxin for α subunits of heterotrimeric G proteins. FASEB J. 27:832–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kamitani S, Ao S, Toshima H, Tachibana T, Hashimoto M, Kitadokoro K, Fukui-Miyazaki A, Abe H, Horiguchi Y. 2011. Enzymatic actions of Pasteurella multocida toxin detected by monoclonal antibodies recognizing the deamidated α subunit of the heterotrimeric GTPase Gq. FEBS J. 278:2702–2712 [DOI] [PubMed] [Google Scholar]
- 60. Croxen MA, Finlay BB. 2010. Molecular mechanisms of Escherichia coli pathogenicity. Nat. Rev. Microbiol. 8:26–38 [DOI] [PubMed] [Google Scholar]
- 61. Kaper JB, Nataro JP, Mobley HL. 2004. Pathogenic Escherichia coli. Nat. Rev. Microbiol. 2:123–140 [DOI] [PubMed] [Google Scholar]
- 62. McDaniel TK, Jarvis KG, Donnenberg MS, Kaper JB. 1995. A genetic locus of enterocyte effacement conserved among diverse enterobacterial pathogens. Proc. Natl. Acad. Sci. U. S. A. 92:1664–1668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Dean P, Kenny B. 2009. The effector repertoire of enteropathogenic E. coli: ganging up on the host cell. Curr. Opin. Microbiol. 12:101–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Marches O, Ledger TN, Boury M, Ohara M, Tu X, Goffaux F, Mainil J, Rosenshine I, Sugai M, De Rycke J, Oswald E. 2003. Enteropathogenic and enterohaemorrhagic Escherichia coli deliver a novel effector called Cif, which blocks cell cycle G2/M transition. Mol. Microbiol. 50:1553–1567 [DOI] [PubMed] [Google Scholar]
- 65. Jubelin G, Chavez CV, Taieb F, Banfield MJ, Samba-Louaka A, Nobe R, Nougayrede JP, Zumbihl R, Givaudan A, Escoubas JM, Oswald E. 2009. Cycle inhibiting factors (CIFs) are a growing family of functional cyclomodulins present in invertebrate and mammal bacterial pathogens. PLoS One 4:e4855. 10.1371/journal.pone.0004855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Yao Q, Cui J, Zhu Y, Wang G, Hu L, Long C, Cao R, Liu X, Huang N, Chen S, Liu L, Shao F. 2009. A bacterial type III effector family uses the papain-like hydrolytic activity to arrest the host cell cycle. Proc. Natl. Acad. Sci. U. S. A. 106:3716–3721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Jubelin G, Taieb F, Duda DM, Hsu Y, Samba-Louaka A, Nobe R, Penary M, Watrin C, Nougayrede JP, Schulman BA, Stebbins CE, Oswald E. 2010. Pathogenic bacteria target NEDD8-conjugated cullins to hijack host-cell signaling pathways. PLoS Pathog. 6:e1001128. 10.1371/journal.ppat.1001128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Morikawa H, Kim M, Mimuro H, Punginelli C, Koyama T, Nagai S, Miyawaki A, Iwai K, Sasakawa C. 2010. The bacterial effector Cif interferes with SCF ubiquitin ligase function by inhibiting deneddylation of Cullin1. Biochem. Biophys. Res. Commun. 401:268–274 [DOI] [PubMed] [Google Scholar]
- 69. Wei N, Serino G, Deng XW. 2008. The COP9 signalosome: more than a protease. Trends Biochem. Sci. 33:592–600 [DOI] [PubMed] [Google Scholar]
- 70. Duda DM, Borg LA, Scott DC, Hunt HW, Hammel M, Schulman BA. 2008. Structural insights into NEDD8 activation of cullin-RING ligases: conformational control of conjugation. Cell 134:995–1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Yao Q, Cui J, Wang J, Li T, Wan X, Luo T, Gong YN, Xu Y, Huang N, Shao F. 2012. Structural mechanism of ubiquitin and NEDD8 deamidation catalyzed by bacterial effectors that induce macrophage-specific apoptosis. Proc. Natl. Acad. Sci. U. S. A. 109:20395–20400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Boh BK, Ng MY, Leck YC, Shaw B, Long J, Sun GW, Gan YH, Searle MS, Layfield R, Hagen T. 2011. Inhibition of cullin RING ligases by cycle inhibiting factor: evidence for interference with Nedd8-induced conformational control. J. Mol. Biol. 413:430–437 [DOI] [PubMed] [Google Scholar]
- 73. Toro TB, Toth JI, Petroski MD. 2013. The cyclomodulin cycle inhibiting factor (CIF) alters cullin neddylation dynamics. J. Biol. Chem. 288:14716–14726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Taieb F, Nougayrede JP, Watrin C, Samba-Louaka A, Oswald E. 2006. Escherichia coli cyclomodulin Cif induces G2 arrest of the host cell cycle without activation of the DNA-damage checkpoint-signalling pathway. Cell. Microbiol. 8:1910–1921 [DOI] [PubMed] [Google Scholar]
- 75. Samba-Louaka A, Nougayrede JP, Watrin C, Jubelin G, Oswald E, Taieb F. 2008. Bacterial cyclomodulin Cif blocks the host cell cycle by stabilizing the cyclin-dependent kinase inhibitors p21 and p27. Cell. Microbiol. 10:2496–2508 [DOI] [PubMed] [Google Scholar]
- 76. Crow A, Race PR, Jubelin G, Varela Chavez C, Escoubas JM, Oswald E, Banfield MJ. 2009. Crystal structures of Cif from bacterial pathogens Photorhabdus luminescens and Burkholderia pseudomallei. PLoS One 4:e5582. 10.1371/journal.pone.0005582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Hsu Y, Jubelin G, Taieb F, Nougayrede JP, Oswald E, Stebbins CE. 2008. Structure of the cyclomodulin Cif from pathogenic Escherichia coli. J. Mol. Biol. 384:465–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Crow A, Hughes RK, Taieb F, Oswald E, Banfield MJ. 2012. The molecular basis of ubiquitin-like protein NEDD8 deamidation by the bacterial effector protein Cif. Proc. Natl. Acad. Sci. U. S. A. 109:E1830–E1838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Schroeder GN, Hilbi H. 2008. Molecular pathogenesis of Shigella spp.: controlling host cell signaling, invasion, and death by type III secretion. Clin. Microbiol. Rev. 21:134–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Dupont N, Lacas-Gervais S, Bertout J, Paz I, Freche B, Van Nhieu GT, van der Goot FG, Sansonetti PJ, Lafont F. 2009. Shigella phagocytic vacuolar membrane remnants participate in the cellular response to pathogen invasion and are regulated by autophagy. Cell Host Microbe 6:137–149 [DOI] [PubMed] [Google Scholar]
- 81. Rawlings DJ, Sommer K, Moreno-Garcia ME. 2006. The CARMA1 signalosome links the signalling machinery of adaptive and innate immunity in lymphocytes. Nat. Rev. Immunol. 6:799–812 [DOI] [PubMed] [Google Scholar]
- 82. Takeuchi O, Akira S. 2010. Pattern recognition receptors and inflammation. Cell 140:805–820 [DOI] [PubMed] [Google Scholar]
- 83. Shao F, Merritt PM, Bao Z, Innes RW, Dixon JE. 2002. A Yersinia effector and a Pseudomonas avirulence protein define a family of cysteine proteases functioning in bacterial pathogenesis. Cell 109:575–588 [DOI] [PubMed] [Google Scholar]
- 84. Zhu M, Shao F, Innes RW, Dixon JE, Xu Z. 2004. The crystal structure of Pseudomonas avirulence protein AvrPphB: a papain-like fold with a distinct substrate-binding site. Proc. Natl. Acad. Sci. U. S. A. 101:302–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Fu P, Zhang X, Jin M, Xu L, Wang C, Xia Z, Zhu Y. 2013. Complex structure of OspI and Ubc13: the molecular basis of Ubc13 deamidation and convergence of bacterial and host E2 recognition. PLoS Pathog. 9:e1003322. 10.1371/journal.ppat.1003322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Storer AC, Menard R. 1994. Catalytic mechanism in papain family of cysteine peptidases. Methods Enzymol. 244:486–500 [DOI] [PubMed] [Google Scholar]