

Abstract
Studying the relationships between gut microbiota, human health, and diseases is a major challenge that generates contradictory results. Most studies draw conclusions about the gut repertoire using a single biased metagenomics approach. We analyzed 16 different stool samples collected from healthy subjects who were from different areas, had metabolic disorders, were immunocompromised, or were treated with antibiotics at the time of the stool collection. The analyses performed included Gram staining, flow cytometry, transmission electron microscopy (TEM), quantitative real-time PCR (qPCR) of the Bacteroidetes and Firmicutes phyla, and pyrosequencing of the 16S rRNA gene amplicons targeting the V6 region. We quantified 1010 prokaryotes per gram of feces, which is less than was previously described. The Mann-Whitney test revealed that Gram-negative proportions of the prokaryotes obtained by Gram staining, TEM, and pyrosequencing differed according to the analysis used, with Gram-negative prokaryotes yielding median percentages of 70.6%, 31.0%, and 16.4%, respectively. A comparison of TEM and pyrosequencing analyses highlighted a difference of 14.6% in the identification of Gram-negative prokaryotes, and a Spearman test showed a tendency toward correlation, albeit not significant, in the Gram-negative/Gram-positive prokaryote ratio (ρ = 0.3282, P = 0.2146). In contrast, when comparing the qPCR and pyrosequencing results, a significant correlation was found for the Bacteroidetes/Firmicutes ratio (ρ = 0.6057, P = 0.0130). Our study showed that the entire diversity of the human gut microbiota remains unknown because different techniques generate extremely different results. We found that to assess the overall composition of bacterial communities, multiple techniques must be combined. The biases that exist for each technique may be useful in exploring the major discrepancies in molecular studies.
INTRODUCTION
Unraveling the relationships between gut microbiota, human health, and disease is a major challenge for scientists (1–3). The gut composition varies with physiological conditions, such as age (4, 5) or geographical origin (6, 7), and external factors, such as dietary habits (8) and the use of antibiotics (9–11) or probiotics (12). Moreover, a causative relationship between gut composition and diseases, such as obesity (13–15), has recently been suggested. Several methods have been used to study the diversity of the gut microbiota. Microscopy, culture, and deep-sequencing methodologies have yielded contradictory results. Because many species of bacteria cannot be easily cultured, most notably anaerobic bacteria (16, 17), a discrepancy known as “the great plate count anomaly” exists (18). Gram staining may lead to bacterial misidentification due to the Gram stain variability of some bacteria (19). The improved resolution of electron microscopy has allowed for an expansion of knowledge about viruses (20) and bacteria (21). This technique plays a role in the clinical diagnosis of viral infections but has limited applications in bacterial diagnosis. It is also used in cellular research to study the structure and function of cells (22) and in environmental research to study the soil microflora (23, 24). Based on environmental microbiology models (25), a renewed interest in culture methods has recently occurred (26, 27). A study performed in our laboratory used 212 different culture conditions (microbial culturomics) on 3 different samples and compared the results with those from pyrosequencing (27). We found that 85% of the culture sequence was not recovered by deep sequencing. Finally, the results of metagenomic studies are often contradictory (28).
Indeed, there are many biases inherent to each technique. Molecular techniques targeting the 16S rRNA gene have revolutionized our knowledge of microbial diversity (29). However, several biases occurring during DNA extraction and purification (30) and during PCR amplification, due to various primer efficiencies (31–33), have been reported, leading to the incorrect representation of the actual abundance of microbial communities. New high-throughput sequencing methods, such as pyrosequencing, are currently the most common methods used to describe microbial ecosystems. However, many factors can influence the ability of this technique to efficiently detect different taxa (34). For example, universal primers are known to be biased against Actinobacteria (35), and the proportion of phyla detected depends on the hypervariable regions targeted in the 16S rRNA gene (36). Furthermore, there is a depth bias, and bacterial populations at concentrations of <106 CFU/ml are not detected by pyrosequencing (27).
Here, as a large part of a gut microbiota study (3, 10, 27), we report an analysis of 16 different stool samples obtained from healthy or ill individuals from different locales to avoid sample biases and to study different profiles. The samples were collected from patients suffering from metabolic disorders or infectious diseases or from patients treated with broad-spectrum antibiotics. We compared the bacterial compositions using a large variety of techniques, including Gram staining, flow cytometry, transmission electron microscopy (TEM), quantitative real-time PCR (qPCR), and the 454 FLX Titanium pyrosequencing of 16S rRNA gene amplicons; the use of some of these methodologies in other studies is unknown.
MATERIALS AND METHODS
Stool sample collection.
Sixteen stool specimens were collected from 16 different subjects (see Table S1 in the supplemental material). Twelve samples were collected from patients from Marseille, France, and four were collected from patients in French Polynesia. Six patients were affected by metabolic disorders: one woman had anorexia nervosa and a body mass index (BMI) of 10.4 kg/m2 (sample SRA062687), two patients were overweight (men with BMIs of 24.0 kg/m2 [sample SRS387498] and 27.4 kg/m2 [sample SRS385004]), two patients were obese (37) (men with BMIs of 31.0 kg/m2 [sample SRS387497] and 33.0 kg/m2 [sample SRS387496]), and one young woman was morbidly obese and had a BMI of 48.2 kg/m2 (sample SRA049748). Two patients were hospitalized in the intensive care unit (ICU) and were treated with antibiotics (samples SRA058885 and SRA058885). The stool sample collected from the first patient (sample 4) was collected at the end of an ICU stay during which he received cocktails of antibiotics, and the other stool sample was collected from a second patient after receiving a 10-day course of imipenem (sample 5). Eight patients were HIV positive: six patients had HIV-1 (samples SRA062846, SRS387488, SRS387490, SRS387491, SRS385014, and SRS387493), one patient had HIV-2 (sample SRS387489), and one woman was an elite controller (38) (sample SRS387495). We collected 10 control stool samples to compare 2 different counting methods. Each fecal sample was aliquoted into 1-g samples at the time of the collection and frozen at −80°C upon receipt. No clinical manifestations of diarrhea were observed in any patient. Patient consents were obtained. The study was approved by the local ethics committee of the Institut Fédératif de Recherche 48 (IFR48) under agreement no. 09-022, Marseille, France.
Gram staining.
Approximately 1 g of each stool was suspended in 9 ml of phosphate-buffered saline (PBS), and 10-fold serial dilutions were made in PBS with vigorous shaking between dilutions. Samples at 3 different dilutions (dilutions ranged from 10−4 to 10−6) were analyzed using the following protocol: smears of 10 μl of the dilution were spread on a slide, and after rapid heat fixation of the smears, the slides were Gram stained with a slide stainer/cytocentrifuge (Aerospray; Wescor, Inc., Logan, UT). The repartition and morphology of the cells were examined by counting 10 microscopic fields with an ×100 objective in oil immersion on a microscope (model DM1000; Leica, Wetzlar, Germany) immediately after Gram staining. The results expressed in our study are those obtained with the 10−4 dilution, as was recommended by the initial studies analyzing the fecal flora (16, 17).
Quantification using a Kova slide.
Each sample was analyzed in triplicate. We used a calibrated chamber of 1 μl (Kova slide) to quantify the total bacteria in the samples. A dilute crystal violet solution (dilution, 10−3) was added to the 10−4 dilution of the stool samples (vol/vol). We counted the bacteria in one square at ×40 and multiplied the results by 100 to obtain the bacterial concentration in 1 μl. The results are expressed as the number of bacteria per gram of stool weight. A dilution range from 10−4 to 10−6 was performed for 10 control samples, and we quantified the bacteria observed in each dilution using the Kova slide.
Quantification by flow cytometry.
The absolute number of bacterial cells (measured in cells/μl) in each sample was calculated using the following equation: (number of cells counted/number of CytoCount beads counted) × CytoCount concentration (1,100 beads/μl) × dilution factor (39). The parameters were adjusted using diluted samples with or without beads to the following settings: forward scatter (FSC) 250 nm, SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate) 174 nm, FSC photomultiplier tube (PMT) 500 nm, Pacific Blue 313 nm (to visualize the 4′,6-diamidino-2-phenylindole [DAPI] stain), fluorescein isothiocyanate (FITC) 426 nm, and allophycocyanin (APC) 500 nm. Flow cytometry (40) was performed in triplicate with 3 different dilutions (from 10−4 to 10−6) for the 10 control samples and in triplicate with the 10−4 dilution for the 16 patient samples. To 500 μl of each dilution, 500 μl of Triton 0.1% in PBS was added to permeabilize the bacterial cell walls. The cells were pelleted at 13,000 × g for 5 min in a microcentrifuge tube and resuspended in a 1-ml volume of PBS. Each sample was stained with 1 μl of DAPI dye (1 μg/μl concentration) (Invitrogen, Life Technologies, Saint Aubin, France). The samples were incubated for a minimum of 30 min at room temperature in the dark, and 25 μl of CytoCount beads was added before processing the samples on the cytometer in triplicate. The total number of recorded events was 10,000 for cell counting using a BD LSRFortessa cell analyzer.
The data were analyzed using the BD FACSDiva 6.2 software. We created a one-dimensional gate in the histogram for cells stained with DAPI and another one for liquid-containing fluorescent beads. The number of events using each gate was calculated, and we calculated the number of bacteria in 1 ml of sample by following the equation detailed above.
Transmission electron microscopy.
Before examining the samples using transmission electron microscopy, each stool sample was fixed twice (first fixation with glutaraldehyde for bacterial proteins, second fixation with OsO2 for bacterial lipids) to prevent the loss of cellular material during dehydration or while cutting the resin block.
For each sample, we selected a minimum of 500 bacterial pictures (one picture corresponded to one bacterium), and we examined the bacterial surface layer organization. Next, we quantified how many bacteria had a Gram-positive-type prokaryote cell wall, which was characterized by a thick cell wall and no outer membrane (see Fig. S1B in the supplemental material), or a Gram-negative-type prokaryote cell wall, which was characterized by the presence of an outer membrane structure at the external surface and a relatively thin peptidoglycan layer (see Fig. S1A in the supplemental material). Some prokaryotes, such as Mycobacteria or Archaea (41), have cell envelopes that are different from those of both Gram-positive and Gram-negative structures (see Fig. S1C in the supplemental material), and a few bacteria could not be categorized into either group because their cell wall structures were unique and difficult to characterize (23) (see Fig. S1D in the supplemental material).
Real-time quantitative PCR assays.
DNA was extracted from the stools using a NucleoSpin tissue minikit (Macherey-Nagel, Hoerdt, France), as described in Dridi et al. (42). The design of the probes and primers for the Bacteroidetes and Firmicutes divisions were previously described by Armougom and Raoult (43) (see Table S2 in the supplemental material), and the Methanobrevibacter smithii probe and primers were designed using the Primer3 v0.4.0 software (44). Chimeric fragments were used for the quantification of Bacteroidetes, Firmicutes, and M. smithii (45). Real-time PCR was performed with a Stratagene Mx3000 system (Agilent, Santa Clara, CA) using QuantiTect PCR mix (Qiagen, Courtaboeuf, France) (45). The extracted DNA was analyzed as undiluted and also diluted at 1:10 and 1:100 to minimize the presence of inhibitors. The samples were analyzed in triplicate. For each cycle threshold, a range calibration was performed to determine the quantity of bacteria in the samples.
16S rRNA pyrosequencing.
Fecal DNA was extracted from stool samples using a NucleoSpin tissue minikit (Macherey-Nagel, Hoerdt, France) following a previously described protocol (42). An approximately 400-nucleotide (nt) region of the 16S rRNA gene was amplified by PCR using the primers 917F (5′-GAATTGACGGGGRCCC) and 1391R (5′-GACGGGCGGTGWGTRCA). These primers were selected because they can amplify the hypervariable region V6 (950 to 1,080 bp), which displays high variability (36, 46), and they can produce an amplicon length equivalent to the average length of the reads produced by the GS FLX Titanium. This high-throughput sequencing was performed on the 16 stool samples either by bidirectional sequencing (samples 2, 3, and 4) or unidirectional sequencing (samples 1, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, and 16).
The taxonomic read classification to the phylum level was performed using a BLASTn algorithm analysis, with a sequence identity and coverage threshold of 90%.
Pyrosequencing data analysis.
The 16S pyrosequencing data were processed using the mothur package 1.5 (47, 48). No ambiguous bases (“N”) and only one mismatch were allowed in the read and primer sequences, respectively. The quality-read trimming used a moving window of 50 bp and required that the average quality score over the region did not drop to <35. The trimmed reads were dereplicated and aligned using the Silva bacteria reference alignment provided through the mothur website (http://www.mothur.org/). The multiple sequence alignment was filtered using a minimum read length of 200 bp. In addition, a preclustering step (49) was performed before chimera detection with the chimera.uchime tool in mothur. A distance matrix was built based on the multiple alignment strategy, and operational taxonomic unit (OTU) clustering was performed at 97% sequence identity. We used the RDP classifier tool and the RDP training set 9 database (http://www.mothur.org/) to assemble the taxonomic classification from the phylum to the genus levels for each representative OTU sequence. We did not analyze the stool sample composition at the species level. The relative abundance of reads per phylum was deduced from this classification, and we calculated the proportion of bacteria belonging to each of the following phyla: Bacteroidetes, Proteobacteria, Deinococcus-Thermus, Fusobacteria, Cyanobacteria, Tenericutes, candidate division TM7, Synergistetes, Lentisphaerae, and Verrucomicrobia, all which contain Gram-negative bacteria, and Firmicutes and Actinobacteria, which contain Gram-positive bacteria. M. smithii could not be targeted using this method because the V6 hypervariable region is not detected in Archaea. We corrected the number of reads from the major phyla (Bacteroidetes, Firmicutes, Actinobacteria, Proteobacteria, and Verrucomicrobia) obtained by pyrosequencing using the mean number of 16S rRNA gene copies for these phyla.
Statistical analysis.
To validate our counting method, we correlated 2 different counting methods using a Kova slide and flow cytometry (using Spearman's ρ and two-tailed P values; a significant correlation was defined as one with a P value of <0.05) and analyzed the differences in prokaryote concentrations with both methods using a Mann-Whitney test (the concentrations obtained with both methods were considered significantly different when the P value was <0.05). Next, we compared and correlated the techniques in pairs (Gram staining versus TEM and TEM versus pyrosequencing) using a correlation test between the concentrations obtained for the Gram-negative and Gram-positive prokaryotes. Then, we analyzed differences in the percentage of Gram-positive and Gram-negative bacteria observed by Gram staining, TEM, and pyrosequencing using a Mann-Whitney test (the percentages obtained were considered significantly different when the P value was <0.05). The quantitative PCR and pyrosequencing techniques were correlated using a Spearman test to analyze the concentrations of the Firmicutes and Bacteroidetes phyla. The statistical tests and graphics were generated using GraphPad Prism 4.
RESULTS
Prokaryote counting.
The number of prokaryotes per gram of feces ranged from 6.30 × 109 (sample 15) to 9.60 × 1010 (sample 8) using the Kova slide and from 2.61 × 109 (sample 4) to 1.49 × 1011 (sample 1) using flow cytometry (see Table S3A in the supplemental material). The concentration of prokaryotes per gram of feces obtained by examining the 10−4 dilution of the 10 control stool samples ranged from 3.15 × 1010 (sample E) to 8.55 × 1010 (sample F) using the Kova slide and from 5.62 × 109 (sample C) to 9.11 × 1010 (sample F) using flow cytometry (see Table S3B in the supplemental material). We concluded that the average number of prokaryotes per gram of feces is approximately 1010.
Gram-positive and Gram-negative prokaryotes counted using Gram staining and TEM.
Gram-positive prokaryotes predominated in only three samples as assessed by Gram staining (Fig. 1A), accounting for 48.82% (1.64 × 109) in sample 4, 75.27% (3.46 × 1010) in sample 6, and 77.0% (5.66 × 109) in sample 15 of the prokaryotes present. In 13 samples, Gram-negative prokaryotes represented the majority of the organisms observed (Fig. 1B; see also Table S6B in the supplemental material), ranging from 68.35% (4.18 × 109) in sample 5 to 58.82% (8.78 × 1010) in sample 1 of the prokaryotes present. The ratio of Gram-negative to Gram-positive prokaryotes ranged from 0.30 in sample 15 to 5.08 in sample 13 (Fig. 2), with a median Gram-negative-to-Gram-positive prokaryote ratio of 2.31 ± 1.59. Using TEM, we observed that sample 2 was the only sample with a predominance of Gram-negative prokaryotes (Fig. 1B), accounting for 56.0% (1.80 × 1010) of the prokaryotes present. For the other 15 samples, Gram-positive prokaryotes predominated, ranging from 52.0% (1.72 × 109) in sample 4 to 51.0% (7.61 × 1010) in sample 1. The ratio of Gram-negative to Gram-positive prokaryotes ranged from 0.21 in sample 14 to 1.27 in sample 2 (Fig. 2), with a median Gram-negative-to-Gram-positive prokaryotes ratio of 0.51 ± 2.28. In conclusion, the number of Gram-negative prokaryotes was overestimated by Gram staining compared to TEM in several samples.
Fig 1.

Concentrations of Gram-positive (A) and Gram-negative (B) prokaryotes obtained by Gram staining, transmission electron microscopy, and pyrosequencing.
Fig 2.
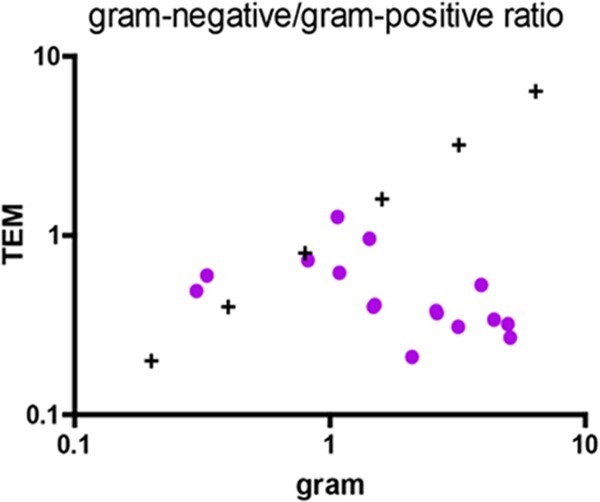
Ratio of the Gram-negative to Gram-positive prokaryotes by Gram staining and transmission electron microscopy (TEM) (Spearman's ρ = −0.7265, P value [two-tailed] = 0.0014).
Correction by 16S rRNA gene copy number of major species composing the gut microbiota.
On the basis of previous studies (50, 51) we assessed 16S rRNA gene copy numbers of major species composing the gut microbiota. The 16S rRNA gene copy number obtained for the 10 Firmicutes species varied from 4 (for Enterococcus faecalis, Veillonella parvula, and Ruminococcus albus) to 10 (for Clostridium difficile), and the mean 16S rRNA gene copy number for the Firmicutes species was 5.7. The 16S rRNA gene copy number for the 8 Bacteroidetes species varied from 2 (for Alistipes finegoldii) to 7 (for Bacteroides vulgatus and Parabacteroides distasonis), and the mean 16S rRNA gene copy number for the Bacteroidetes species was 4.9. The 16S rRNA gene copy number for the 10 Actinobacteria species varied from 1.0 (for Atopobium parvulum) to 5.0 (for Bifidobacterium adolescentis), and the mean 16S rRNA gene copy number for the Actinobacteria species was 2.8. The 16S rRNA gene copy number for the 10 Proteobacteria species varied from 2 (for Helicobacter pylori) to 10 (for Aeromonas veronii), and the mean 16S rRNA gene copy number for the Proteobacteria species was 6.7 (see Table S4A in the supplemental material). The 16S rRNA gene copy number for M. smithii was 2.
Bacteroidetes, Firmicutes, and M. smithii quantification using real-time quantitative PCR.
For all samples, the Bacteroidetes 16S rRNA gene copy numbers ranged from 7.02 × 107/g of feces in sample 9 to 1.80 × 1010/g of feces in sample 6. The Firmicutes 16S rRNA gene copy numbers ranged from 1.11 × 108/g of feces in sample 5 to 2.26 × 1012/g of feces in sample 2 (see Tables S6A and S6B in the supplemental material). The ratio of Bacteroidetes to Firmicutes ranged from 0.001 (in samples 2 and 13) to 1.559 in sample 1 (Fig. 3). The M. smithii 16S rRNA gene copy numbers ranged from 1.05 × 106/g of feces in sample 1 to 2.28 × 109/g of feces in sample 14 (see Table S4B in the supplemental material). Only 2 samples (1 and 5) had a predominance of Bacteroidetes compared to Firmicutes. Conversely, the 14 other specimens had a higher concentration of Firmicutes than Bacteroidetes.
Fig 3.
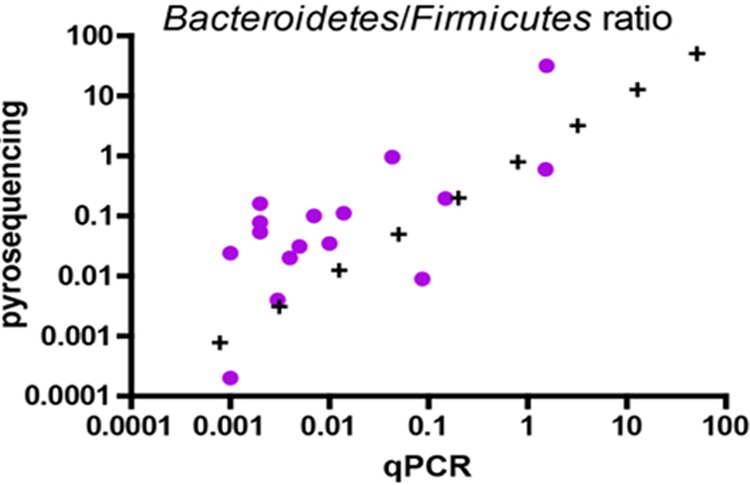
Ratio of Bacteroidetes to Firmicutes (Spearman's ρ = 0.6057, P value [two-tailed] = 0.0130).
Pyrosequencing.
Pyrosequencing of the 16 samples generated a number of reads, ranging from 11,531 (sample 16) to 225,965 (sample 7) (see Table S5 in the supplemental material). After correction with the 16S rRNA gene copy number, the Gram-positive bacterial concentration ranged from 7.95 × 107/g (sample 5) to 5.24 × 109/g (sample 2), and the Gram-negative bacterial concentration ranged from 4.01 × 107/g (sample 2) to 2.61 × 1010/g (sample 1). The ratio of Gram-negative to Gram-positive bacteria ranged from 0.01 (sample 2) to 34.32 (sample 1). However, without correction by the 16S rRNA gene copy number, Firmicutes quantification ranged from 4.52 × 108 in sample 5 to 2.92 × 1010 in sample 2, and the Bacteroidetes quantification ranged from 6.41 × 106 in sample 2 to 7.85 ×1010 in sample 1. The ratio of Bacteroidetes to Firmicutes ranged from 0.0001 (sample 2) to 31.711 (sample 1). In addition, in all samples except for sample 1, a portion of the total reads ranging from 0.01% (sample 9) to 16.58% (sample 14) of the total reads could not be assigned at the phylum level and are therefore noted as “others” (see Table S5 in the supplemental material).
Comparison of techniques.
A comparison of the prokaryote concentrations obtained using flow cytometry and Kova slide counting on the 10 control stools at 3 different dilutions indicated a significant correlation between the methods (Spearman's ρ = 0.5295, P = 0.0026) (Fig. 4), and the Mann-Whitney test indicated that the bacterial concentrations were not significantly different between the techniques (P = 0.8505). When comparing the prokaryote concentrations obtained using morphological tools, a significant correlation existed between Gram staining and TEM (Spearman's ρ = 0.8035, P = 0.0002 for Gram-positive prokaryotes, and Spearman's ρ = 0.8176, P = 0.0001 for Gram-negative prokaryotes) (see Fig. S2B in the supplemental material), and this was confirmed by an analysis of the Gram-negative-to-Gram-positive prokaryote ratio (Spearman's ρ = −0.7265; P = 0.0014) (Fig. 2). A comparison of the TEM and pyrosequencing techniques for analyzing prokaryote concentrations showed a tendency toward a correlation; the results were not significant for Gram-negative prokaryote concentrations (Spearman's ρ = 0.3143, P = 0.2539), but there was a significant correlation for the Gram-positive prokaryote concentration (Spearman's ρ = 0.8551, P < 0.0001). Similarly, the comparison of the Gram-negative/Gram-positive prokaryote ratio yielded a Spearman's ρ of 0.3282 (P = 0.2146), which indicates a tendency toward correlation that was not significant. The comparison of the molecular techniques (qPCR and pyrosequencing) for determining the Firmicutes concentrations showed a tendency toward correlation, but this result was also not significant (Spearman's ρ = 0.4912, P = 0.0534), and no significant correlation was observed for the Bacteroidetes concentration (Spearman's ρ = 0.05887, P = 0.8286) (see Fig. S2A in the supplemental material). Moreover, a significant correlation existed between the Bacteroidetes/Firmicutes ratios generated by the two tests, yielding a Spearman's ρ of 0.6052 (P = 0.0130) (Fig. 3).
Fig 4.
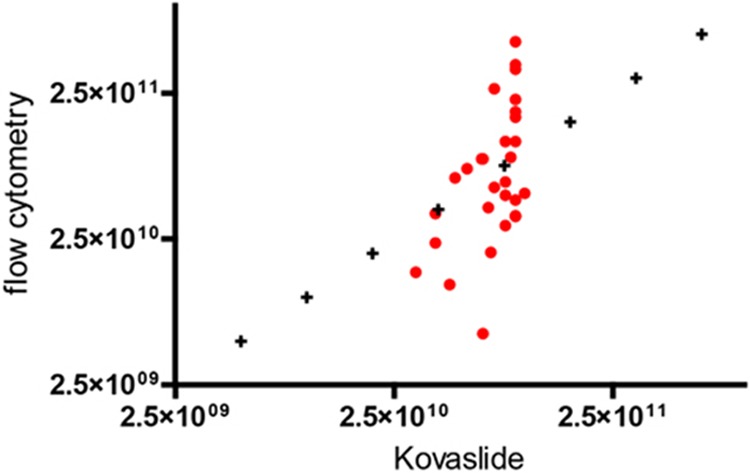
Correlation analysis performed on prokaryote concentrations obtained using the Kova slide and flow cytometry methods in 10 control stool samples. Each sample was quantified using 3 different dilutions (Spearman's ρ = 0.5295, P value [two-tailed] = 0.0026).
Deficit in apparently Gram-negative-type prokaryotes.
Comparing the percentages of the Gram-negative-type prokaryotes obtained by Gram staining, TEM, and pyrosequencing, Fig. 5 shows that the techniques identified 3 distinct populations that were confirmed by the Mann-Whitney test. The percentages of Gram-negative-type prokaryotes identified using Gram staining ranged from 23.0% (sample 15) to 86.5% (sample 16), with an average percentage of 64.6%. Using TEM, the percentages ranged from 21.0% (samples 12 and 13) to 56.0% (sample 2), with an average percentage of 33.3%. Using pyrosequencing, the percentages of Gram-negative-type prokaryotes ranged from 0.69% in sample 2 to 97.2% in sample 1, with an average percentage of 27.9%. Moreover, the medians calculated for the Gram-negative-type prokaryote percentages were 70.6% using Gram staining (Fig. 6A), 31.0% using TEM, and 16.4% using pyrosequencing (Fig. 6B). The Gram stain is known to overestimate Gram-negative bacteria due to the decolorization of some Gram-positive species (52) that causes them to appear as Gram variable or Gram negative, and electron microscopy is the methodology commonly used to assess cell wall types (22). We compared the percentages obtained for Gram-negative-type prokaryotes by TEM and pyrosequencing and observed a difference of 14.6% in the median percentages of the Gram-negative-type prokaryotes that were identified using pyrosequencing (Fig. 6B).
Fig 5.
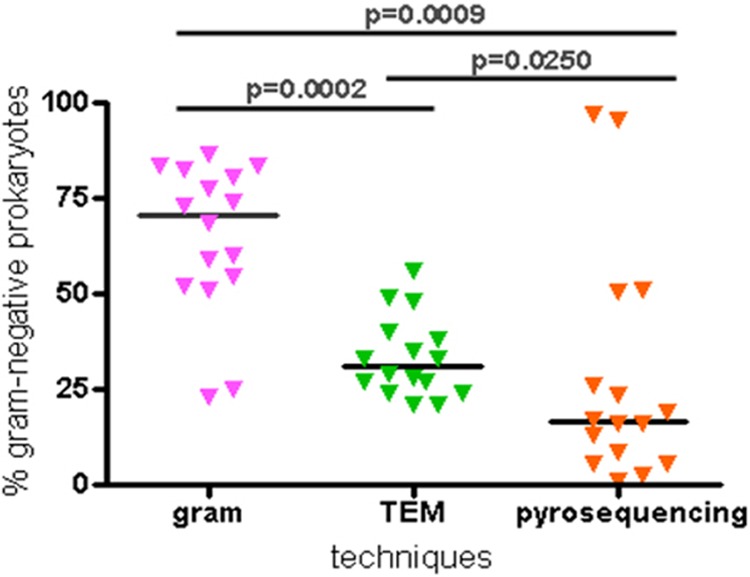
Mann-Whitney test performed on percentages of Gram-negative-type prokaryotes. The median values are represented by dark horizontal lines.
Fig 6.

Representation of median percentages using Gram staining versus TEM (A) and pyrosequencing versus TEM (B). The shaded areas in panel B highlight the median difference between the pyrosequencing and TEM techniques.
DISCUSSION
We are confident in our results because flow cytometry is a rapid and reliable method to quantify bacteria in fecal samples (39, 40, 53), and Gram staining is a technique that is commonly used in clinical bacteriology to differentiate Gram-positive and Gram-negative bacteria (52); it was also the first method used in initial studies of the gut microbiota (16, 17). Transmission electron microscopy has been used for bacterial applications since the late 1960s (22). The protocol used for real-time quantitative PCR was previously described in studies from our laboratory (43, 45). Finally, we used the pyrosequencing of 16S rRNA gene amplicons targeting the hypervariable region V6 that has been widely used to study the gut microbiota (36). All of these techniques were performed under stringent conditions based on the manufacturer's recommendations, and triplicates of each sample were analyzed by flow cytometry and quantitative PCR. To the best of our knowledge, this is the first study to compare the results of 5 different methods performed on 16 different stool samples. To consolidate our analysis and to be free from sample biases, we collected samples from different geographic origins and patients with different diseases.
Metagenomic studies are increasingly being used to describe the human gut microbiota (1, 46, 50, 54), and they produce a very large number of sequences. These studies tend to overestimate bacterial concentrations because they are based on the number of 16S rRNA gene copies, and reports based on these studies calculate bacterial concentrations of 1013 to 1014. As of yet, very few studies have compared the quantitative estimates obtained by high-throughput sequencing data and qPCR results (55–58). Moreover, many studies performed over the past 70 years have reported fecal bacterial concentrations on the order of 1011 bacteria per gram of feces (16, 17, 59). In this study, we found that the concentrations of prokaryotes determined using flow cytometry and the Kova slide methods were lower, on the order of 1010 prokaryotes per gram of feces. We did not observe a decrease in fecal bacterial concentrations in patients who were treated with antibiotics. Actually, it appears that the influence of antibiotics on the gut microbiota is unpredictable (60). Other studies did not report a significant decrease in bacterial biomass after antibiotic intake (61), while others revealed that the gut microbiota composition was affected in only a subset of cases (62). The methods used to quantify fecal samples are varied and might lead to an overestimation of bacterial numbers; for example, the Petroff-Hausser counting chamber (16) does not distinguish between bacteria and debris. We imagine that the lower prokaryote concentrations observed during our study are the result of incorrect observations using the microscope because the Kova slide did not allow for overlay bacterial examination, and the DAPI staining used for flow cytometry is known to underestimate bacterial abundance in natural samples (63).
We compared two morphological tools and found a significant correlation between the prokaryote concentrations by use of both methods. However, the bacterial percentages were significantly different between the methods, and the median Gram-negative/Gram-positive ratio was 2.31 using Gram staining and 0.51 using TEM. Gram staining is a fundamental technique for the identification of unknown pathogens, but it lacks robustness due to the aberrant Gram staining of some bacteria (52) that leads to an overestimation of the Gram-negative population. However, TEM can distinguish between bi- or trilamellar cell walls (22) and is more accurate than Gram staining for discriminating between Gram-negative and Gram-positive bacteria. Although TEM can be difficult to interpret in some cases (23), this technique is a useful reference tool for characterizing bacteria.
A number of biases occur during DNA extraction (30) and throughout the steps of the molecular biological approaches. For example, differential PCR amplification depends on the primers selected (31, 32), and variation occurs depending on the platform sequencing technologies and software analysis pipelines used (64). These methods appear to neglect a portion of the Gram-negative bacteria that were described in the first culture studies as the major constituents of the fecal microbiota (16). We show here that pyrosequencing resulted in a gap in the detection of the Gram-negative-type prokaryotes, despite the fact that the reads obtained by this technique were corrected by the 16S rRNA gene copy number. While we know that the final number of reads depends on the G+C content of the sample library (65), this discrepancy might be explained by the biases that are associated with multitemplate PCRs that lead to errors in the mean relative abundance of each detected OTU (65).
By comparing 5 different methods to describe the gut microbiota composition regarding Gram-positive and Gram-negative bacteria, we found that some techniques gave extremely different results, and we believe that molecular studies neglect a large part of the unknown Gram-negative bacteria. The reductionist approach (66), which uses only one experimental model in some studies (54, 67), might interfere with the actual determination of the gut microbiota diversity and thus introduce a bias (68). Indeed, results based on pyrosequencing techniques suggesting that 3 robust clusters (“enterotypes”) characterize the human gut microbiota (7), that there are 2 enterotypes, obese and nonobese (54), or that there are associations with diet (69) may be less clear than previously thought. Moreover, some scientists now contend that there is a continuum or gradient of species that are characteristic of the human gut microbiota (70). In conclusion, multidisciplinary approaches for characterizing the gut microbiota show a major discrepancy between morphological and molecular studies, and it is therefore important to perform multiple techniques to assess the overall bacterial diversity in the gut.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge P. Weber and G. Audoly for help with the transmission electron microscopy and flow cytometry measurements.
Footnotes
Published ahead of print 24 July 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JCM.00473-13.
REFERENCES
- 1.Turnbaugh PJ, Ley RE, Hamady M, Fraser-Liggett CM, Knight R, Gordon JI. 2007. The human microbiome project. Nature 449:804–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Raoult D. 2012. Human microbiota. [Corrected.] Clin. Microbiol. Infect. 18(Suppl 4):1. 10.1111/j.1469-0691.2012.03915.x [DOI] [PubMed] [Google Scholar]
- 3.Lagier JC, Million M, Hugon P, Armougom F, Raoult D. 2012. Human gut microbiota: repertoire and variations. Front. Cell Infect. Microbiol. 2:136. 10.3389/fcimb.2012.00136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Palmer C, Bik EM, DiGiulio DB, Relman DA, Brown PO. 2007. Development of the human infant intestinal microbiota. PLoS Biol. 5:e177. 10.1371/journal.pbio.0050177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Claesson MJ, Cusack S, O'Sullivan O, Greene-Diniz R, de Weerd H, Flannery E, Marchesi JR, Falush D, Dinan T, Fitzgerald G, Stanton C, van Sinderen D, O'Connor M, Harnedy N, O'Connor K, Henry C, O'Mahony D, Fitzgerald AP, Shanahan F, Twomey C, Hill C, Ross RP, O'Toole PW. 2011. Composition, variability, and temporal stability of the intestinal microbiota of the elderly. Proc. Natl. Acad. Sci. U. S. A. 108(Suppl 1):4586–4591. 10.1073/pnas.1000097107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee S, Sung J, Lee J, Ko G. 2011. Comparison of the gut microbiotas of healthy adult twins living in South Korea and the United States. Appl. Environ. Microbiol. 77:7433–7437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arumugam M, Raes J, Pelletier E, Le Paslier D, Yamada T, Mende DR, Fernandes GR, Tap J, Bruls T, Batto JM, Bertalan M, Borruel N, Casellas F, Fernandez L, Gautier L, Hansen T, Hattori M, Hayashi T, Kleerebezem M, Kurokawa K, Leclerc M, Levenez F, Manichanh C, Nielsen HB, Nielsen T, Pons N, Poulain J, Qin J, Sicheritz-Ponten T, Tims S, Torrents D, Ugarte E, Zoetendal EG, Wang J, Guarner F, Pedersen O, de Vos WM, Brunak S, Doré J, Weissenbach J, Ehrlich SD, Bork P. 2011. Enterotypes of the human gut microbiome. Nature 473:174–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bäckhed F, Ding H, Wang T, Hooper LV, Koh GY, Nagy A, Semenkovich CF, Gordon JI. 2004. The gut microbiota as an environmental factor that regulates fat storage. Proc. Natl. Acad. Sci. U. S. A. 101:15718–15723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sullivan A, Edlund C, Nord CE. 2001. Effect of antimicrobial agents on the ecological balance of human microflora. Lancet Infect. Dis. 1:101–114 [DOI] [PubMed] [Google Scholar]
- 10.Dubourg G, Lagier JC, Armougom F, Robert C, Hamad I, Brouqui P, Raoult D. 2013. The gut microbiota of a patient with resistant tuberculosis is more comprehensively studied by culturomics than by metagenomics. Eur. J. Clin. Microbiol. Infect. Dis. 32:637–645 [DOI] [PubMed] [Google Scholar]
- 11.Dubourg G, Lagier JC, Armougom F, Robert C, Audoly G, Papazian L, Raoult D. 2013. High-level colonisation of the human gut by Verrucomicrobia following broad-spectrum antibiotic treatment Int. J. Antimicrob. Agents 41:149–155 [DOI] [PubMed] [Google Scholar]
- 12.Million M, Maraninchi M, Henry M, Armougom F, Richet H, Carrieri P, Valero R, Raccah D, Vialettes B, Raoult D. 2011. Obesity-associated gut microbiota is enriched in Lactobacillus reuteri and depleted in Bifidobacterium animalis and Methanobrevibacter smithii. Int. J. Obes. (Lond.) 36:817–825 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 13.Raoult D. 2008. Obesity pandemics and the modification of digestive bacterial flora. Eur. J. Clin. Microbiol. Infect. Dis. 27:631–634 [DOI] [PubMed] [Google Scholar]
- 14.DiBaise JK, Zhang H, Crowell MD, Krajmalnik-Brown R, Decker GA, Rittmann BE. 2008. Gut microbiota and its possible relationship with obesity. Mayo Clin. Proc. 83:460–469 [DOI] [PubMed] [Google Scholar]
- 15.Ley RE, Bäckhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI. 2005. Obesity alters gut microbial ecology. Proc. Natl. Acad. Sci. U. S. A. 102:11070–11075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gossling J, Slack JM. 1974. Predominant Gram-positive bacteria in human feces: numbers, variety, and persistence. Infect. Immun. 9:719–729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van Houte J, Gibbons RJ. 1966. Studies of the cultivable flora of normal human feces. Antonie van Leeuwenhoek 32:212–222 [DOI] [PubMed] [Google Scholar]
- 18.Staley JT, Konopka A. 1985. Measurement of in situ activities of nonphotosynthetic microorganisms in aquatic and terrestrial habitats. Annu. Rev. Microbiol. 39:321–346 [DOI] [PubMed] [Google Scholar]
- 19.Beveridge TJ. 1990. Mechanism of Gram variability in select bacteria. J. Bacteriol. 172:1609–1620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Horne RW, Wildy P. 1963. Virus structure revealed by negative staining. Adv. Virus Res. 10:101–170 [DOI] [PubMed] [Google Scholar]
- 21.Isalska BJ, Curry A, Stanbridge TN, Tweddle D, Caul EO. 1996. Electron microscopy and serological features of a patient with Q fever prosthetic valve endocarditis. J. Clin. Pathol. 49:679–681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Curry A, Appleton H, Dowsett B. 2006. Application of transmission electron microscopy to the clinical study of viral and bacterial infections: present and future. Micron 37:91–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Amako K, Takade A, Taniai H, Yoshida S. 2008. Electron microscopic examination of uncultured soil-dwelling bacteria. Microbiol. Immunol. 52:265–269 [DOI] [PubMed] [Google Scholar]
- 24.Bae HC, Cota-Robles EH, Casida LE., Jr 1972. Microflora of soil as viewed by transmission electron microscopy. Appl. Microbiol. 23:637–648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kaeberlein T, Lewis K, Epstein SS. 2002. Isolating “uncultivable” microorganisms in pure culture in a simulated natural environment. Science 296:1127–1129 [DOI] [PubMed] [Google Scholar]
- 26.Goodman AL, Kallstrom G, Faith JJ, Reyes A, Moore A, Dantas G, Gordon JI. 2011. Extensive personal human gut microbiota culture collections characterized and manipulated in gnotobiotic mice. Proc. Natl. Acad. Sci. U. S. A. 108:6252–6257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lagier JC, Armougom F, Million M, Hugon P, Pagnier I, Robert C, Bittar F, Fournous G, Gimenez G, Maraninchi M, Trape JF, Koonin EV, La SB, Raoult D. 2012. Microbial culturomics: paradigm shift in the human gut microbiome study. Clin. Microbiol. Infect. 18:1185–1193 [DOI] [PubMed] [Google Scholar]
- 28.Angelakis E, Armougom F, Million M, Raoult D. 2012. The relationship between gut microbiota and weight gain in humans. Future Microbiol. 7:91–109 [DOI] [PubMed] [Google Scholar]
- 29.Hugenholtz P, Goebel BM, Pace NR. 1998. Impact of culture-independent studies on the emerging phylogenetic view of bacterial diversity. J. Bacteriol. 180:4765–4774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yuan S, Cohen DB, Ravel J, Abdo Z, Forney LJ. 2012. Evaluation of methods for the extraction and purification of DNA from the human microbiome. PLoS One 7:e33865. 10.1371/journal.pone.0033865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hong S, Bunge J, Leslin C, Jeon S, Epstein SS. 2009. Polymerase chain reaction primers miss half of rRNA microbial diversity. ISME J. 3:1365–1373 [DOI] [PubMed] [Google Scholar]
- 32.Gonzalez JM, Portillo MC, Belda-Ferre P, Mira A. 2012. Amplification by PCR artificially reduces the proportion of the rare biosphere in microbial communities. PLoS One 7:e29973. 10.1371/journal.pone.0029973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Frank JA, Reich CI, Sharma S, Weisbaum JS, Wilson BA, Olsen GJ. 2008. Critical evaluation of two primers commonly used for amplification of bacterial 16S rDNA genes. Appl. Environ. Microbiol. 74:2461–2470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Amend AS, Seifert KA, Bruns TD. 2010. Quantifying microbial communities with 454 pyrosequencing: does read abundance count? Mol. Ecol. 19:5555–5565 [DOI] [PubMed] [Google Scholar]
- 35.Farris MH, Olson JB. 2007. Detection of Actinobacteria cultivated from environmental samples reveals bias in universal primers. Lett. Appl. Microbiol. 45:376–381 [DOI] [PubMed] [Google Scholar]
- 36.Claesson MJ, Wang Q, O'Sullivan O, Greene-Diniz R, Cole JR, Ross RP, O'Toole PW. 2010. Comparison of two next-generation sequencing technologies for resolving highly complex microbiota composition using tandem variable 16S rRNA gene regions. Nucleic Acids Res. 38:e200. 10.1093/nar/gkq873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.World Health Organization 2013. Obesity and overweight: fact sheet no. 311. World Health Organization, Geneva, Switzerland: http://www.who.int/mediacentre/factsheets/fs311/en/ [Google Scholar]
- 38.Okulicz JF, Lambotte O. 2011. Epidemiology and clinical characteristics of elite controllers. Curr. Opin. HIV AIDS 6:163–168 [DOI] [PubMed] [Google Scholar]
- 39.Khan MMT, Pyle BH, Camper AK. 2010. Specific and rapid enumeration of viable but nonculturable and viable-culturable Gram-negative bacteria by using flow cytometry. Appl. Environ. Microbiol. 76:5088–5096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.van der Waaij LA, Mesander G, Limburg PC, van der Waaij D. 1994. Direct flow cytometry of anaerobic bacteria in human feces. Cytometry 16:270–279 [DOI] [PubMed] [Google Scholar]
- 41.Ellen AF, Zolghadr B, Driessen AMJ, Albers SV. 2010. Shaping the archaeal cell envelope. Archaea 2010:608243. 10.1155/2010/608243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dridi B, Henry M, El Khéchine A, Raoult D, Drancourt M. 2009. High prevalence of Methanobrevibacter smithii and Methanosphaera stadtmanae detected in the human gut using an improved DNA detection protocol. PLoS One 4:e7063. 10.1371/journal.pone.0007063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Armougom F, Raoult D. 2008. Use of pyrosequencing and DNA barcodes to monitor variations in Firmicutes and Bacteroidetes communities in the gut microbiota of obese humans. BMC Genomics 9:576. 10.1186/1471-2164-9-576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rozen S, Skaletsky H. 2000. Primer3 on the WWW for general users and for biologist programmers. Methods Mol. Biol. 132:365–386 [DOI] [PubMed] [Google Scholar]
- 45.Armougom F, Henry M, Vialettes B, Raccah D, Raoult D. 2009. Monitoring bacterial community of human gut microbiota reveals an increase in Lactobacillus in obese patients and methanogens in anorexic patients. PLoS One 4:e7125. 10.1371/journal.pone.0007125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Andersson AF, Lindberg M, Jakobsson H, Bäckhed F, Nyrén P, Engstrand L. 2008. Comparative analysis of human gut microbiota by barcoded pyrosequencing. PLoS One 3:e2836. 10.1371/journal.pone.0002836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF. 2009. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75:7537–7541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schloss PD, Gevers D, Westcott SL. 2011. Reducing the effects of PCR amplification and sequencing artifacts on 16S rRNA-based studies. PLoS One 6:e27310. 10.1371/journal.pone.0027310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huse SM, Welch DM, Morrison HG, Sogin ML. 2010. Ironing out the wrinkles in the rare biosphere through improved OTU clustering. Environ. Microbiol. 12:1889–1898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, Mende DR, Li J, Xu J, Li S, Li D, Cao J, Wang B, Liang H, Zheng H, Xie Y, Tap J, Lepage P, Bertalan M, Batto JM, Hansen T, Le Paslier D, Linneberg A, Nielsen HB, Pelletier E, Renault P, Sicheritz-Ponten T, Turner K, Zhu H, Yu C, Li S, Jian M, Zhou Y, Li Y, Zhang X, Li S, Qin N, Yang H, Wang J, Brunak S, Doré J, Guarner F, Kristiansen K, Pedersen O, Parkhill J, Weissenbach J, et al. 2010. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464:59–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, Gill SR, Nelson KE, Relman DA. 2005. Diversity of the human intestinal microbial flora. 308:1635–1638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fenollar F, Raoult D. 2000. Comparison of a commercial disk test with vancomycin and colimycin susceptibility testing for identification of bacteria with abnormal gram staining reactions. Eur. J. Clin. Microbiol. Infect. Dis. 19:33–38 [DOI] [PubMed] [Google Scholar]
- 53.Vaahtovuo J, Korkeamäki M, Munukka E, Viljanen MK, Toivanen P. 2005. Quantification of bacteria in human feces using 16S rRNA-hybridization, DNA-staining and flow cytometry. J. Microbiol. Methods 63:276–286 [DOI] [PubMed] [Google Scholar]
- 54.Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, Sogin ML, Jones WJ, Roe BA, Affourtit JP, Egholm M, Henrissat B, Heath AC, Knight R, Gordon JI. 2009. A core gut microbiome in obese and lean twins. Nature 457:480–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Arboleya S, Ang L, Margolles A, Yiyuan L, Dongya Z, Liang X, Solís G, Fernández N, de Los Reyes-Gavilán CG, Gueimonde M. 2012. Deep 16S rRNA metagenomics and quantitative PCR analyses of the premature infant fecal microbiota. Anaerobe 18:378–380 [DOI] [PubMed] [Google Scholar]
- 56.Zhang H, Parameswaran P, Badalamenti J, Rittmann BE, Krajmalnik-Brown R. 2011. Integrating high-throughput pyrosequencing and quantitative real-time PCR to analyze complex microbial communities. Methods Mol. Biol. 733:107–128 [DOI] [PubMed] [Google Scholar]
- 57.Murray DC, Bunce M, Cannell BL, Oliver R, Houston J, White NE, Barrero RA, Bellgard MI, Haile J. 2011. DNA-based faecal dietary analysis: a comparison of qPCR and high throughput sequencing approaches. PLoS One 6:e25776. 10.1371/journal.pone.0025776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.van den Bogert B, de Vos WM, Zoetendal EG, Kleerebezem M. 2011. Microarray analysis and barcoded pyrosequencing provide consistent microbial profiles depending on the source of human intestinal samples. Appl. Environ. Microbiol. 77:2071–2080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Moore WE, Holdeman LV. 1974. Human fecal flora: the normal flora of 20 Japanese-Hawaiians. Appl. Microbiol. 27:961–979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Million M, Lagier JC, Yahav D, Paul M. 2013. Gut bacterial microbiota and obesity. Clin. Microbiol. Infect. 19:305–313 [DOI] [PubMed] [Google Scholar]
- 61.Robinson CJ, Young VB. 2010. Antibiotic administration alters the community structure of the gastrointestinal microbiota. Gut Microbes 1:279–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sekirov I, Tam NM, Jogova M, Robertson ML, Li Y, Lupp C, Finlay BB. 2008. Antibiotic-induced perturbations of the intestinal microbiota alter host susceptibility to enteric infection. Infect. Immun. 76:4726–4736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Seo EY, Ahn TS, Zo YG. 2010. Agreement, precision, and accuracy of epifluorescence microscopy methods for enumeration of total bacterial numbers. Appl. Environ. Microbiol. 76:1981–1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Smith BC, McAndrew T, Chen Z, Harari A, Barris DM, Viswanathan S, Rodriguez AC, Castle P, Herrero R, Schiffman M, Burk RD. 2012. The cervical microbiome over 7 years and a comparison of methodologies for its characterization. PLoS One 7:e40425. 10.1371/journal.pone.0040425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pinto AJ, Raskin L. 2012. PCR biases distort bacterial and archaeal community structure in pyrosequencing datasets. PLoS One 7:e43093. 10.1371/journal.pone.0043093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fang FC, Casadevall A. 2011. Reductionistic and holistic science. Infect. Immun. 79:1401–1404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ley RE, Turnbaugh PJ, Klein S, Gordon JI. 2006. Microbial ecology: human gut microbes associated with obesity. Nature 444:1022–1023 [DOI] [PubMed] [Google Scholar]
- 68.Raoult D. 2010. Technology-driven research will dominate hypothesis-driven research: the future of microbiology. Future Microbiol. 5:135–137. 10.2217/fmb.09.119 [DOI] [PubMed] [Google Scholar]
- 69.Wu GD, Chen J, Hoffmann C, Bittinger K, Chen YY, Keilbaugh SA, Bewtra M, Knights D, Walters WA, Knight R, Sinha R, Gilroy E, Gupta K, Baldassano R, Nessel L, Li H, Bushman FD, Lewis JD. 2011. Linking long-term dietary patterns with gut microbial enterotypes. Science 334:105–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jeffery IB, Claesson MJ, O'Toole PW, Shanahan F. 2012. Categorization of the gut microbiota: enterotypes or gradients? Nat. Rev. Microbiol. 10:591–592 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.