

Abstract
High-risk (HR) human papillomavirus (HPV)-associated carcinogenesis is driven mainly by the overexpression of E7 and E6 oncoproteins following viral DNA integration and the concomitant loss of the E2 open reading frame (ORF). However, the integration of HR-HPV DNA is not systematically observed in cervical cancers. The E2 protein acts as a transcription factor that governs viral oncogene expression. The methylation of CpGs in the E2-binding sites (E2BSs) in the viral long control region abrogates E2 binding, thus impairing the E2-mediated regulation of E7/E6 transcription. Here, high-resolution melting (HRM)–PCR was developed to quantitatively analyze the methylation statuses of E2BS1, E2BS2, and the specificity protein 1 (Sp1)-binding site in 119 HPV16-positive cervical smears. This is a rapid assay that is suitable for the analysis of cervical samples. The proportion of cancer samples with methylated E2BS1, E2BS2, and Sp1-binding site CpGs was 47%, whereas the vast majority of samples diagnosed as being within normal limits, low-grade squamous intraepithelial lesions (LSIL), or high-grade squamous intraepithelial lesions (HSIL) harbored unmethylated CpGs. Methylation levels varied widely, since some cancer samples harbored up to 60% of methylated HPV16 genomes. A pyrosequencing approach was used as a confirmation test and highlighted that quantitative measurement of methylation can be achieved by HRM-PCR. Its prognostic value deserves to be investigated alone or in association with other biomarkers. The reliability of this single-tube assay offers great opportunities for the investigation of HPV16 methylation in other HPV-related cancers, such as head and neck cancers, which are a major public health burden.
INTRODUCTION
Certain Alphapapillomavirus species classified as high-risk human papillomaviruses (HR-HPVs) have been recognized as the etiologic factors of invasive cervical carcinoma worldwide (1, 2). HR-HPV infections are frequent in young women but they are generally transient, with an estimated mean duration of incident infection of 16 months (3). Only persistent HR-HPV infections are associated with an increased risk of developing high-grade cervical lesions or cancer of the cervix (4–6). Persistent HR-HPV infections may be associated with microscopic abnormalities called low-grade squamous intraepithelial lesions (LSIL). These LSIL present a high rate of regression following HPV clearance but may progress toward high-grade squamous intraepithelial lesions (HSIL) if HR-HPV infection persists. Upon diagnosis, HSIL are treated mostly by excisional procedures to avoid the risk of progression toward invasive cancer (for a review, see reference 7).
Among the HR-HPV types, HPV16 exhibits the highest persistence rate (8). Moreover, HPV16 is associated with an increased risk of developing precancerous and cancerous lesions (9) and is considered the most carcinogenic PV in humans (10). This is probably why HPV16 prevalence increases with the severity of cervical lesions (11), reaching 61% in invasive cervical carcinoma worldwide (12).
HPV-associated carcinogenesis requires the continuous overexpression of the two main viral oncoproteins E7 and E6, which interact with many cellular proteins leading to the induction and the maintenance of a transformed phenotype in infected cells (for a review, see reference 13). E7 and E6 expression is regulated by the viral E2 protein through the early promoter (named p97 for HPV16) located in the long control region (LCR) of the PV genome. This promoter harbors four specific E2-binding sites (E2BSs) sharing the consensus sequence 5′-ACCG(N)4CGGT-3′ (14, 15). The three sites proximal to the TATA box have been shown to be involved in E2-mediated repression of the promoter activity, and E2BS1 and E2BS2 are probably the main sites to achieve this effect (for a review, see reference 16). From a mechanistic point of view, the binding of E2 to E2BS1 and E2BS2 induces a displacement of transcription activators, such as specificity protein 1 (Sp1) and TATA binding protein (TBP), from their binding sites, leading to a repression of E7 and E6 expression from the early promoter (17, 18). HR-HPV DNA integration has been described as a key step in the carcinogenesis process, since it generally results in the disruption of the E2 open reading frame (ORF) (19, 20).
Epigenetic alterations, and particularly aberrant DNA methylation, are frequently associated with tumor progression. Methylation of DNA mainly occurs on cytosines located in CpG dinucleotides and leads to gene silencing after chromatin structure remodeling. Both the HPV16 E7 and E6 proteins seem to increase DNA methyltransferase (DNMT) activity in vitro (21). Moreover, increased methylation of cellular tumor suppressor genes, such as retinoic acid receptor beta (rarb), death-associated protein kinase 1 (dapk1), and cell adhesion molecule 1 (camd1), is frequently observed in cervical cancer samples (22). Viral DNA sequences, including HPV genomes, can also undergo methylation by cellular DNMT (23, 24).
The HPV16 genome contains 112 CpG sites and its methylome has been described (25–27). The CpG dinucleotides located in the L1/L2 region of the HPV16 genome are preferentially methylated during disease progression (25, 26, 28, 29), and a recent study suggested the clinical value of methylation for the identification of prevalent and incident high-grade lesions (27).
Several studies aimed to describe methylation on the HPV16 LCR and the therein-contained E2BS in clinical samples, but the results are very conflicting. Some studies reported decreased E2BS methylation during lesion progression (30–32) or a trend of a lower risk of incident precancerous and cancerous lesions for patients with high E2BS methylation (33, 34). On the contrary, methylation of the five CpGs located in the E2BS1 and E2BS2 and Sp1-binding site (Sp1BS) has frequently been associated with cervical cancer (28, 29, 35–38), and a gradual increase in methylation during lesion progression toward cancer has been reported in some studies (29, 36, 37, 39). Nevertheless, most of these studies did not include the full spectrum of cervical disease progression from normal to cancer. Furthermore, various quantitative and nonquantitative techniques have been used to study HPV16 DNA methylation. Some studies have been conducted on fresh-frozen tissues or microdissected formalin-fixed paraffin-embedded (FFPE) biopsy samples, whereas others have been performed on exfoliated cervical cells; this may explain discrepant results.
We report here a simple and robust high-resolution melting (HRM)–PCR that permits the quantitation of the methylation levels of the five CpGs located in the two most important HPV16 E2BSs involved in E7/E6 regulation, namely, E2BS1 and E2BS2, and in the Sp1BS. After the optimization and validation steps, this single-tube assay was used to explore E2BS1, E2BS2, and Sp1BS methylation in samples previously identified to harbor HPV16 and that are representative of the natural history of cervical cancer. The HRM analysis reveals that methylation of the target CpG is specific for cancer samples.
MATERIALS AND METHODS
Cell lines.
The human HPV16-positive CaSki and SiHa cell lines were obtained from the ATCC (Manassas, VA) and maintained at 37°C (in 5% CO2) in complete RPMI medium or Dulbecco's modified Eagle medium (DMEM) (Lonza, Levallois-Perret, France), respectively, supplemented with 10% fetal bovine serum (FBS) (Lonza), 5 × 104 U/liter penicillin-streptomycin (Lonza), and 2 mM l-glutamine (Lonza).
Clinical samples.
Samples were collected at the endo-ectocervical junction with a cytobrush and conserved in specimen transport medium (Digene; Qiagen, Courtaboeuf, France) or in PreservCyt (Cytyc, Marlborough, MA) until routine HR-HPV testing with the Digene Hybrid Capture 2 (HC2) high-risk HPV DNA test (Qiagen). Samples were stored into a biobank for which a declaration of preparation and storage of human samples for research use was sent to the “Ministère de l'Enseignement Supérieur et de la Recherche” (no. DC-2008-850).
Samples positive for HR-HPV DNA were used for DNA extraction with the QIAamp DNA minikit (Qiagen). The presence of HPV16 DNA was assessed by quantitative PCR as previously described (40).
One hundred nineteen HPV16-positive samples with cytological abnormalities representative of the natural history of cervical cancer were selected. When available, the histological diagnosis obtained from a subsequent biopsy sample or cervical resection was retrieved from the medical record. For 13 samples, the histological and cytological diagnoses were discrepant and the histological diagnosis was retained. Thirty-seven samples were finally diagnosed as within normal limits (WNL), 30 as low-grade intraepithelial lesions (LSIL), 35 as high-grade intraepithelial lesions (HSIL), and 17 as invasive carcinomas. Among the carcinomas, five were diagnosed as adenocarcinomas and 11 as squamous cell carcinomas (SCC); all of them were early-stage tumors (stage I to II).
DNA bisulfite conversion.
Bisulfite conversion of DNA (500 ng) was performed using the Cells-to-CpG bisulfite conversion kit (Life Technologies, Villebon-sur-Yvette, France). Briefly, DNA was denatured at 50°C for 10 min with the denaturation reagent. Then, 100 μl of reconstituted conversion reagent was added to the denatured DNA and the solution was incubated for 2 cycles of 30 min at 65°C and 1.5 min at 95°C followed by 30 min at 65°C. Bisulfite-converted DNA was purified on columns provided with the kit. Several washes were realized to remove salts and sulfonic groups. Converted DNA was eluted in 40 μl of elution buffer.
HPV16 E2BS HRM-PCR.
We set up a methylation-independent PCR that targets a sequence of HPV16 3′-LCR localized from nucleotide (nt) 6 to nt 115 (NCBI accession no. NC_001526.2 [41]). This stretch contains two CpGs located in the proximal E2BS1 (nt 52 and 58), two CpGs located in the proximal E2BS2 (nt 37 and 43), and one CpG in the Sp1BS site (nt 31). The primers 16E2BS_For and 16E2BS_Rev are listed in Table 1 and were purchased from Eurogentec (Angers, France). For convenience, this assay is referred to as HPV16 E2BS HRM-PCR.
Table 1.
Primer and target sequences
| Target or primer | Nucleotide sequence (5′ to 3′) | Length (bp) |
|---|---|---|
| 16 E2BS DNA sequence | AATAATTCATGTATAAAACTAAGGGCGTAACCGAAATCGGTTGAACCGAAACCGGTTAGTATAAAAGCAGACATTTTATGCACCAAAAGAGAACTGCAATGTTTCAGGAC | 110 |
| 16E2BS bisulfite converted sequence | AATAATTTATGTATAAAATTAAGGGTGTAATTGAAATTGGTTGAATTGAAATTGGTTAGTATAAAAGTAGATATTTTATGTATTAAAAGAGAATTGTAATGTTTTAGGAT | 110 |
| 16E2BS_For primer | AATAATTTATGTATAAAATTAAGGG | 25 |
| 16E2BS_Rev primer | ATCCTAAAACATTACAATTCTCTTTT | 26 |
| 16E2BS pyrosequencing analysis sequence | YGTAATYGAAATYGGTTGAATYGAAATYGGTT | 32 |
| 16E2BS pyrosequencing dispensation order | ATCGTGACTCGTATCGTGTATCGTATCG | 28 |
HRM-PCR experiments were run on the 7500 Fast real-time PCR system (Life Technologies). The reaction was performed in a final volume of 20 μl containing Melt Doctor HRM master mix 1× (Life Technologies) and 300 nM each 16E2BS primer. After an initial denaturation step at 95°C for 10 min, 40 amplification cycles were performed (95°C for 15 s, 52° for 20 s, and 60°C for 1 min). The high-resolution melting curve stage was performed with a 1% ramping from 50°C to 95°C. HRM curves were analyzed using the HRM 2.0 software (Life Technologies). Raw data were normalized for fluorescence intensity by the software algorithms in order to establish normalized HRM curves.
Serial dilutions (1:10) of CaSki cell DNA were also used to evaluate the repeatability and efficiency of the PCR.
Construction of pE2BS.0/5 and pE2BS.5/5 plasmids.
Bisulfite-converted DNA from SiHa cells (harboring unmethylated HPV16 E2BSs) and from CaSki cells (harboring mostly methylated HPV16 E2BSs) was amplified by PCR in a final volume of 50 μl containing 1× Green GoTaq Flexi buffer, 1.25 mM MgCl2, 200 nM deoxynucleoside triphosphate (dNTP), 500 nM each primer, and 1.25 U GoTaq Hot Start polymerase (Promega, Charbonnières, France). The thermal cycling used was an initial denaturation step of 5 min at 94°C followed by 40 amplification cycles (94°C for 30 s, 50°C for 30 s, and 72°C for 30 s) and a final elongation step of 7 min at 72°C. After purification with the QIAEXII gel extraction kit (Qiagen), PCR products were cloned into the pGEM-T Easy vector (Promega). Subcloning Efficiency DH5α competent cells (Life Technologies) were transformed with 15 ng of recombinant vector by heat shock and grown in a selection medium. Plasmids from multiple colonies were sequenced to confirm the presence of the insert. The plasmid with five T residues at positions 31, 37, 43, 52, and 58 (corresponding to unmethylated DNA) was named pE2BS.0/5, and the plasmid with five C residues at positions 31, 37, 43, 52, and 58 (corresponding to methylated DNA) was named pE2BS.5/5.
pE2BS.0/5 and pE2BS.5/5 were mixed in different proportions to mimic samples harboring 0 to 100% methylated CpGs with 10% increments. Six replicates of each mix were analyzed to assess the sensitivity and repeatability of the technique.
Standards for methylation quantification.
Standards consisted of artificial samples containing known amounts of methylated E2BS DNA in a background of unmethylated target. To obtain a fully methylated DNA matrix, 1 μg of CaSki cell DNA was treated with 4 units of methylase SssI (New England BioLabs, Évry, France) in a final volume of 50 μl containing 1× NE buffer and 160 μM S-adenosylmethionine over 4 h at 37°C followed by 20 min at 65°C. The QIAEXII gel extraction kit (Qiagen) was used to purify and concentrate the modified DNA. For the unmethylated matrix, pBR322-HPV16 plasmids that contained the HPV16 whole genome were used. Standards were prepared to mimic samples with 0, 10, 25, 50, 75, and 100% methylated target. They were used to validate the overall HRM-PCR method, including the bisulfite conversion step.
Standards were run in triplicate in parallel with clinical samples to quantify the level of methylation of the five CpG dinucleotides of interest. Considering the unmethylated standard (0%) as the reference, difference plots were calculated in order to obtain a linear regression of the standard curves that can be used to quantify methylation (42).
HPV16 E2BS pyrosequencing for methylation analysis.
Bisulfite-converted DNA from CaSki cells, standards, and clinical samples (5 μl) were amplified by PCR in a final volume of 25 μl containing PyroMark PCR master mix (1×) (Qiagen), CoralLoad (1×) (Qiagen), 200 nM 16E2BS_For primer, and 200 nM 16E2BS_Rev primer tagged with biotin at their 5′ end (Biotin TEG 569.61; Eurogentec). After an initial denaturation step at 95°C for 15 min, 40 amplification cycles (95°C for 20 s, 52°C for 30 s, and 72°C for 20 s) were performed, followed by a final elongation step at 72°C for 5 min. Pyrosequencing was applied to each sample in duplicate. Ten microliters of PCR product was immobilized on Streptavidin Sepharose HP beads (Qiagen) and purified using the PyroMark Q24 vacuum workstation (Qiagen), according to the manufacturer's instructions. After denaturation (80°C for 2 min), PCR products were annealed to 300 nM 16E2BS_For primer, used as a sequencing primer, and subjected to pyrosequencing using the PyroMark Q24 instrument (Qiagen) and the PyroMark Gold Q24 reagents (Qiagen). The nucleotide dispensation order and the analysis sequence (Table 1) were determined with the PyroMark Q24 software 2.0.6. Negative-control nucleotides were automatically incorporated, and a cytosine was added at position 8 to check the efficiency of bisulfite conversion. Pyrograms were analyzed in “CpG assay” mode in order to quantify the methylation percentage of the 5 CpGs. The cutoff value used to distinguish methylated and unmethylated samples was calculated as the mean methylation percentage plus three standard deviations from samples that clearly harbor unmethylated targets. The cutoff value for the global methylation of the five CpGs was set at 4.1%.
Data analyses.
The intra-assay repeatability of HPV16 E2BS PCR was assessed by calculating the mean, standard deviation (SD), and variance from cycles of quantification (Cq) from six or eight replicates. The reproducibility of HPV16 E2BS PCR for the accurate quantification of methylation was assessed by calculating the mean, SD, and coefficients of variation (CV) of standard difference plot peak height from eight independent experiments.
RESULTS
Analytical characteristic of the HPV16 E2BS PCR.
CaSki and SiHa cellular DNA treated with sodium bisulfite was successfully amplified by the HPV16 E2BS PCR, rendering a unique amplicon after gel electrophoresis (data not shown). Furthermore, no amplification product was observed when the HPV16 E2BS PCR was carried out with DNA from HPV-negative cells (C-33A) or from cells harboring HPV18, HPV31, HPV33, or HPV45, indicating that the PCR was specific (data not shown).
The analysis of eight replicates of bisulfite-converted CaSki cell DNA diluted from 105 to 1 copies/μl showed that the HPV16 E2BS PCR was highly repeatable. Indeed, Cq variances were <0.2, independent of HPV16 DNA concentration. Moreover, the linear dynamic range covered four orders of magnitude, from 105 to 10 copies/μl. The efficiency calculated from these experiments was 1.94 ± 0.06 (mean ± SD). Finally, six out of eight replicates with the lowest concentration (1 copy/μl) were amplified, and the analytical sensitivity was set at 10 copies/μl, corresponding to a Cq of 33 (Table 2). Since HRM analysis required enough fluorescence intensity and high repeatability between replicates, the Cq limit for HRM analysis was set at 33 to avoid misinterpretation of the HRM curves.
Table 2.
Intra-assay repeatability of HPV16 E2BS PCR
| Cq valuea | HPV16 E2BS PCR results (copies/μl) at theoretical concn of: |
|||||
|---|---|---|---|---|---|---|
| 105 | 104 | 103 | 102 | 10 | 1 | |
| Mean | 18.53 | 22.45 | 26 | 29.26 | 32.83 | 36.09 |
| SD | 0.42 | 0.31 | 0.19 | 0.39 | 0.30 | 0.30 |
| Variance | 0.18 | 0.1 | 0.03 | 0.15 | 0.09 | 0.09 |
Mean, SD, and variance of Cq were calculated from 8 replicates of serial dilutions of bisulfite-converted CaSki cell DNA (or 6 replicates for the dilution theoretically containing 1 copy/μl).
Performance of the HPV16 E2BS HRM-PCR for methylation assessment.
Mixes with different proportions of pE2BS.0/5 (corresponding to 0% methylated target) and pE2BS.5/5 (corresponding to 100% methylated target) were run to assess the performance of the HPV16 E2BS HRM-PCR to discriminate between differentially methylated targets.
As expected, each derivative HRM curve obtained from pE2BS.0/5 and pE2BS.5/5 exhibited a single peak, with melting temperature (Tm) values of 68°C and 70°C, respectively (Fig. 1A), contrary to derivative curves obtained from other mixes that exhibited two peaks at 68°C and 70°C with variable fluorescence intensity (data not shown). Blinded HRM analysis of mixes defined six profiles according to the shape of normalized melting curves (Fig. 1B). Samples with 0% and 10% methylated targets were clearly discriminated, as they presented different melting curves (Fig. 1B, profiles 1 and 2). The third profile grouped samples with 20% and 30% methylated targets, and the fourth profile grouped those with 40%, 50%, and 60% methylated targets. The fifth and the sixth profiles grouped mixes with 70% to 80% and 90% to 100% methylated targets, respectively. Furthermore, the six replicates of each mix were classified in the same group, suggesting good repeatability of the HRM analysis (Fig. 1B).
Fig 1.
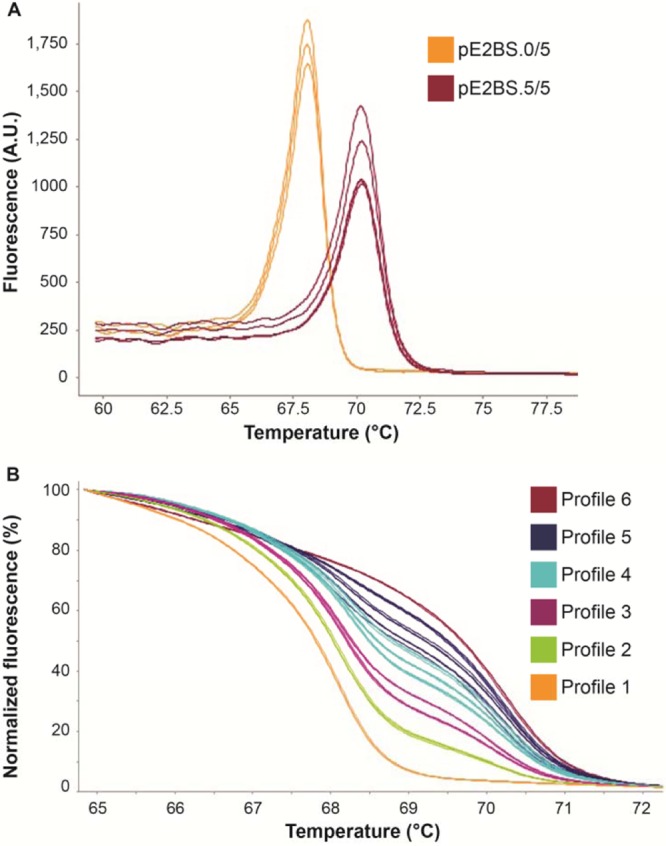
Intra-assay validation of HPV16 E2BS HRM analysis. HRM curves were obtained after analysis of pE2BS.0/5 and pE2BS.5/5 alone or mixed in different proportions. (A) Derivative melt curves derived from pE2BS.0/5 representing unmethylated target and from pE2BS.5/5 representing fully methylated target (measured in AU [arbitrary units]). (B) Normalized HRM curves derived from mixes of pE2BS.0/5 and pE2BS.5/5 and their assignation to six HRM profiles. For clarity, only representative triplicates of the 10 mixes are shown. Profile 1, 0% methylation; profile 2, 10% methylation; profile 3, 20% to 30% methylation; profile 4, 40% to 60% methylation; profile 5, 70% to 90% methylation; and profile 6, 100% methylation.
HRM analysis of standardized DNA mixes permits the methylation level to be quantified.
In order to quantify methylation levels, standards with known proportions of methylated target DNA were run in parallel with clinical samples. These standards consisted of unmethylated pBR322-HPV16 and in vitro methylated CaSki cell DNA whose methylation patterns had been previously confirmed by HRM and cloning-sequencing (data not shown).
After bisulfite conversion, eight replicates of standards containing 0%, 10%, 25%, 50%, 75%, and 100% methylated target were assayed with the HPV16 E2BS HRM-PCR. Again, six HRM profiles were defined corresponding to the six standards, and the eight replicates were correctly assigned to their corresponding profiles, showing good repeatability of the technique (Fig. 2A).
Fig 2.
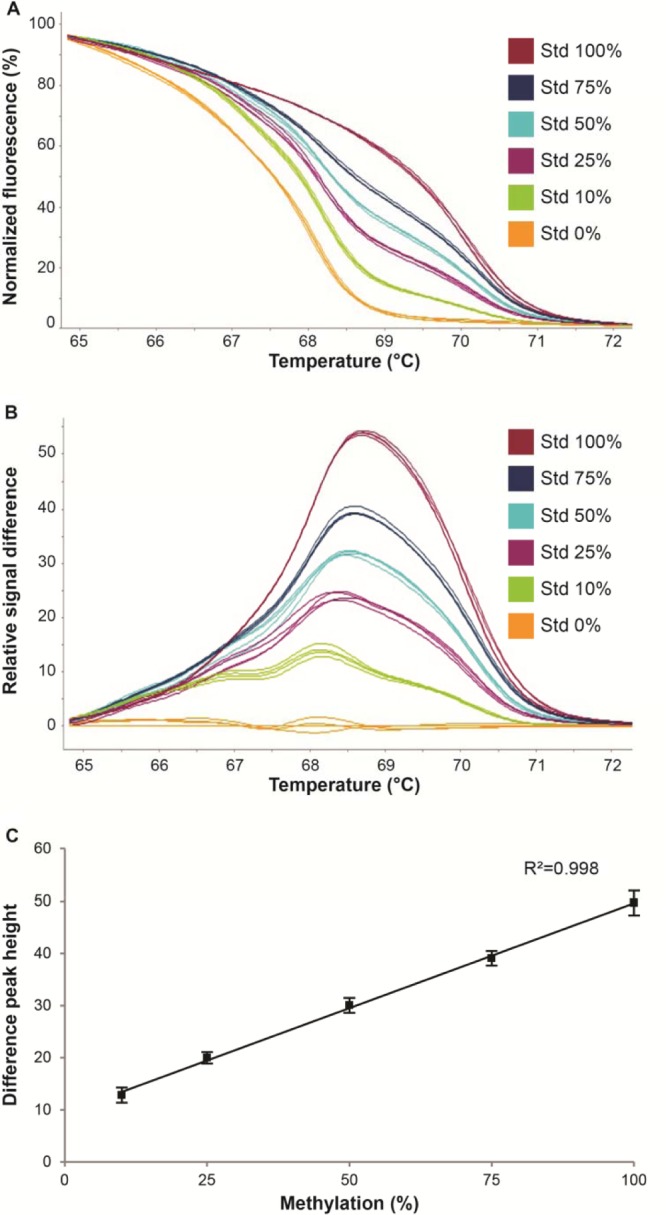
Standards to quantify the methylation levels of HPV16 E2BS1, E2BS2, and Sp1BS. (A) Normalized HRM curves obtained from eight replicates of standard mixes (Std) containing 0, 10, 25, 50, 75, and 100% methylated target. For clarity, only four out of eight representative replicates are shown. (B) Representative difference plots derived from four replicates of standards, defining Std 0% as the reference. (C) Linear regression plot of the standard curves against percent methylation. Data were plotted as the means of the difference peak height obtained in eight independent experiments ± the standard deviation (SD).
The peak-height differences (Fig. 2B) derived from standards run in eight independent experiments were plotted against methylation percentage in order to generate a linear regression (Fig. 2C) (42). The correlation coefficient, R2 = 0.998, indicates a perfect linearity across 10 and 100% methylation. Moreover, minimal variations were observed between the eight experiments. Furthermore, the reproducibility of the HPV16 E2BS HRM-PCR was very good, with a CV of ≤11% for all standards tested (Table 3). Linear regression analysis can thus be applied for accurate determination of DNA methylation levels in clinical samples exhibiting difference plot heights between 12.8 and 49.6 arbitrary fluorescence units. The average methylation level of CaSki cell DNA, as calculated according to this method, was 87%.
Table 3.
Interassay reproducibility of HPV16 E2BS HRM-PCR for methylation-level determinationa
| DNA sample characteristicsb | Results at Std % of: |
Results for CaSki cells | ||||
|---|---|---|---|---|---|---|
| 10 | 25 | 50 | 75 | 100 | ||
| Mean difference peak heights | 12.8 | 20.0 | 30.1 | 39.1 | 49.6 | 43.9 |
| SD | 1.5 | 1.1 | 1.4 | 1.4 | 2.3 | 2.0 |
| CV (%) | 11 | 6 | 5 | 4 | 5 | 5 |
Triplicates of standards were analyzed in 8 independent experiments. Difference plots were generated for each standard using the unmethylated standard (Std) 0% as the reference.
Mean, SD, and CV were calculated from difference in peak heights to evaluate the reproducibility of the method.
E2BS1, E2BS2, and Sp1BS are specifically methylated in cervical cancers.
We quantitatively analyzed methylation status differences in HPV16 E2BS1, E2BS2, and Sp1BS from 119 HPV16-positive cervical smears with different cytological diagnoses, which are globally representative of the natural history of cervical cancer. HRM analysis failed for 33 samples due to a low quantity of target DNA. Indeed, Cq values following HPV16 E2BS PCR were >33 in these samples. For the remaining 86 cervical samples, bisulfite-converted DNA was successfully analyzed by HRM-PCR. Based on the comparison of HRM profiles, 77 samples were assigned to the 0% methylation profile, five to the 10% methylation profile, two to the 25% methylation profile, and two to the 75% methylation profile (Table 4). Interestingly, most if not all samples with a within normal limits, LSIL, or HSIL cytology did not exhibit methylation of the targeted CpG sites, since only one LSIL sample harbored methylated target CpGs. Conversely, 47% of the cancer samples exhibited methylated CpG sites. For samples exhibiting different peak heights in the linear regression range (12.8 to 49.6), methylation levels were accurately calculated and varied from 10% to 60% (Table 5). Moreover, for five out of eight cancer samples with methylated CpG, the subsequent biopsy samples revealed histological features of adenocarcinoma.
Table 4.
Clinical samples with unmethylated or methylated E2BS1, E2BS2, and Sp1BS according to cytological diagnosisa
| Diagnosis (n) | Results at E2BS methylation level (%) of: |
||||
|---|---|---|---|---|---|
| 0 | >0 to 10 | >10 to 25 | >25 to 50 | >50 to 75 | |
| WNL (21) | 21 | 0 | 0 | 0 | 0 |
| LSIL (23) | 22 | 0 | 1 | 0 | 0 |
| HSIL (25) | 25 | 0 | 0 | 0 | 0 |
| Carcinomas (17) | 9 | 5 | 1 | 0 | 2 |
HRM profiles derived from clinical sample DNA were compared to those derived from standards and assigned to the most similar profile. The number of samples assigned to each profile among WNL, LSIL, HSIL, and carcinoma samples is shown.
Table 5.
Methylation levels of HPV16 E2BS1, E2BS2, and Sp1BS determined by HRM-PCR and pyrosequencing
| Sample type, diagnosis | n | HPV16 E2BS HRM-PCR methylation level (%) | Pyrosequencing methylation level (%) of CpG sites at nt position: |
||||
|---|---|---|---|---|---|---|---|
| 31 | 37 | 43 | 52 | 58 | |||
| Standard % | |||||||
| 0 | 0 | 2 | 2 | 2 | 3 | 3 | |
| 10 | 10 | 10 | 10 | 10 | 12 | 12 | |
| 25 | 25 | 24 | 24 | 25 | 26 | 26 | |
| 50 | 50 | 47 | 47 | 49 | 50 | 50 | |
| 75 | 75 | 67 | 67 | 70 | 70 | 71 | |
| 100 | 100 | 94 | 95 | 97 | 97 | 98 | |
| CaSki cell DNA | 87 | 93 | 89 | 98 | 96 | 98 | |
| Cervical smears | |||||||
| WNL | 13 | 0 | 2.2 | 1.2 | 1.3 | 2 | 1.5 |
| LSIL | 21 | 0 | 2.1 | 1.4 | 1.5 | 2.1 | 1.8 |
| 1 | 24 | 25 | 25 | 28 | 28 | 28 | |
| HSIL | 20 | 0 | 1.8 | 1.8 | 1.5 | 2.6 | 2.2 |
| Cervical smears, carcinoma | 9 | 0 | 2.2 | 1.7 | 1.9 | 2.4 | 2.4 |
| 1 | 0–10 | 6 | 4 | 5 | 6 | 7 | |
| 1 | 0–10 | 8 | 4 | 5 | 10 | 11 | |
| 1 | 0–10 | 18 | 17 | 20 | 21 | 22 | |
| 1 | 0–10 | 7 | 7 | 8 | 9 | 8 | |
| 1 | 0–10 | 3 | 3 | 4 | 7 | 7 | |
| 1 | 25 | 40 | 38 | 37 | 39 | 35 | |
| 1 | 59 | 80 | 80 | 89 | 84 | 89 | |
| 1 | 61 | 72 | 72 | 78 | 78 | 79 | |
To confirm our results obtained by HRM-PCR, we used a pyrosequencing approach to reanalyze the standards and the 86 cervical samples. Among clinical samples, 16 could not be analyzed by pyrosequencing. Thirteen samples were not amplified by PCR and three samples were not properly converted with sodium bisulfite, which may lead to an erroneous assessment of methylation levels. Apart from these samples that have not been reassessed because no more DNA was available, and with a cutoff value set at 4.1% for pyrosequencing positivity, one hundred percent agreement was obtained between the two techniques. Furthermore, quantitative data obtained with the two techniques were very similar (Table 5). Nevertheless, for three samples with the highest methylation level, the HPV16 E2BS HRM-PCR resulted in a lower estimation of methylation than pyrosequencing. Interestingly, we noted that methylation level was very consistent between the five CpG sites with no position effect. From a clinical point of view, with a cutoff set at 10%, targeted CpG site methylation determined with HPV16 E2BS HRM-PCR allowed for the identification of women with a cervical cancer with a specificity of 97% and a sensitivity of 44%.
DISCUSSION
HPV16 E2BS HRM-PCR is a reliable method for methylation assessment in clinical samples.
Several studies have described the methylation status of the HPV16 E2BSs in cervical samples. Different assays have been used, more or less quantitative, more or less labor-intensive, and at times requiring a large amount of DNA. Here, we report a rapid, quantitative, and reliable single-tube assay based on DNA bisulfite conversion and E2BS methylation-independent real-time PCR followed by HRM analysis, allowing for the determination of the methylation levels of five CpGs localized in HPV16 E2BS1, E2BS2, and Sp1BS. We show that the HPV16 E2BS PCR is specific and highly reproducible, with Cq variances of <0.2, independently of the target DNA concentration (from 105 to 10 copies of HPV16 genome per reaction mixture).
As for the methylation assessment of the five CpGs in the long control region, HPV16 E2BS HRM-PCR allows for accurate discrimination between methylated and unmethylated targets. Indeed, following HRM analysis, the eight replicates of pE2BS.0/5 and pE2BS.5/5 mixed in different proportions were systematically attributed to the same group. Furthermore, the use of standards comprising known proportions of methylated target (CaSki cell DNA treated with methylase SssI) mixed with unmethylated pBR322-HPV16 plasmid allowed us to achieve a linear regression plot of standard curves from 10% to 100% methylation. Reproducibility of the HPV16 E2BS HRM-PCR is very good, since CV never exceeded 11% for each standard. Thus, the peak height plot derived from standards can be readily applied for accurate quantification of the global methylation of the five CpGs. To further demonstrate the quantitative aspect of the HPV16 E2BS HRM-PCR, all standards were tested using pyrosequencing as a confirmation test. An excellent agreement between HRM analysis and pyrosequencing results confirms the robustness of our assay. Finally, we set a limit Cq value of 33 for HRM analysis. Indeed, for amplification with Cq of >33, methylated and unmethylated targets cannot be discriminated by HRM. This needs to be taken into account when the amount of starting DNA is small (i.e., from a small number of cells or from microdissected tissues), especially because bisulfite modification frequently leads to the degradation of DNA (43).
Methylation of HPV16 E2BS1, E2BS2, and Sp1BS is specifically associated with cervical carcinomas.
Analysis of HPV16 DNA methylation in our samples with the HPV16 E2BS HRM-PCR reveals a specific methylation pattern in cancers. Indeed, 47% of smears with a diagnosis of cancer exhibited methylation of the five target CpGs, whereas virtually all targeted CpGs in WNL, LSIL, and HSIL samples were unmethylated. As for the only LSIL samples that harbored methylated CpGs, no histological diagnosis or follow-up data could be retrieved from the medical record. Thus, we cannot exclude that the lesion severity could have been underestimated by cytological analysis.
Taken together, our results confirm data from previous studies reporting a higher frequency of HPV16 promoter methylation in cancer samples than in precancerous lesions or normal samples (28, 35–37). However, the proportion of cancer samples with methylated E2BSs varied from 20% to 90%, likely highlighting the variability in the methodology used in the different studies (44). For example, Snellenberg and collaborators (37) used a Luminex xMAP system coupled to a methylation-independent PCR approach and communicated a methylation frequency of 69% for the E2BS1 and of 90% for the E2BS2 in SCC samples. These high frequencies are probably related to the extremely high sensitivity of their technique (0.5 to 1%). In the same line, Bhattacharjee and collaborators (35) found up to 85% of cancer samples with methylated E2BS1 and E2BS2 by performing a direct sequencing of bisulfite-treated DNA. This contrasts with the study by Kalantari and colleagues (28) that used a cloning sequencing approach in which they reported that only one out of five cancers presented a methylated promoter. As for precancerous lesions, we found a low methylation frequency (2%) of the five target CpGs, and normal samples harbored only unmethylated CpGs. Several studies also reported a very low methylation frequency in cervical dysplasia (28, 30, 32, 33, 36). This contrasts with the work of Mazumder and collaborators (31), who describe methylated CpG in 50% of LSIL-HSIL. These authors used microdissected histological samples to select tissue with at least 60% abnormal epithelial cells, an approach that is not well suited for routine use.
The HPV16 E2BS HRM-PCR described here also allows for a quantitative assessment of HPV16 multiple-copy methylation. In our sample series, the average methylation level was low (2.3%), with only two out of nine samples showing >50% of HPV16 E2BS1, E2BS2, and Sp1BS carrying methylated CpGs. The overall results are consistent with previously published data reporting average methylation levels from 4.3% to 8% (32) and 0.2% to 5% (45), regardless of the cytological diagnosis. Even with a highly sensitive method, Snellenberg and collaborators (37) also found methylation levels of <10% in the majority of samples, except for cervical carcinoma biopsy samples.
Interestingly, the comparison between the quantitative assessment of all five CpG methylation levels by HRM and of each individual CpG by pyrosequencing revealed no differences. This confirms the robustness of HRM for quantitative methylation analysis. We propose that HPV16 E2BS HRM-PCR alone is sufficient to evaluate the methylation status of HPV16 E2BS1, E2BS2, and Sp1BS from cervical smears. Pyrosequencing analysis also showed that the five CpGs always displayed similar methylation levels, indicating no position effect, as was previously described (37, 45). This suggests a concerted regulation of DNA methylation at the E2BS1, E2BS2, and Sp1BS CpG sites. Nevertheless, a differential methylation pattern of the HPV16 genome has been described, with the HPV16 L1 ORF being 5 to 10 times more methylated than the LCR in high-grade lesions and cancer samples (27, 28, 45). Turan and collaborators (46) hypothesized that the L1 ORF may be preferentially methylated, while the accessibility of CpGs located in the LCR to DNMTs may be hampered by transcription factors. Furthermore, it has been shown that transcription factors might also favor the recruitment of DNMT to specific gene loci. For example, the phosphorylation of RelA/p65 promotes DNMT-1 recruitment to chromatin and represses the transcription of the tumor metastasis suppressor gene, BRMS1 (47).
HPV16 E2BS1, E2BS2, and Sp1BS methylation in the carcinogenesis process.
The analytical importance of the E2BS methylation status arises from its involvement in E7 and E6 expression regulation. The overexpression of E7/E6 during HPV-associated transforming infection may be associated with the integration of the viral DNA into the cellular genome. Integration usually occurs in the early-late boundary of the PV genome and often implies the partial or complete ablation of the E2 ORF (19, 20). Consequently, transcription from early promoter is no longer repressed, resulting in an overexpression of E7/E6. Integration seems to be an early phenomenon, as it has been documented in cervical smears with no cellular abnormalities (48, 49). Nevertheless, the proportion of samples harboring integrated viral genomes (pure or mixed with episomal forms) increases with the severity of lesions (40, 50, 51), and integration is associated with an increased risk of lesion progression (50, 52). Surprisingly, some studies revealed that cervical cancer samples harbor no integrated forms, only HPV16 episomes (40, 50, 53). Moreover, multiple copies of the viral genome can integrate as head-to-tail concatemers. This integration pattern allows for functional E2 mRNA expression in cells, as observed in the CaSki cell line (54). Taken together, these data suggest that mechanisms other than integration can also lead to E7/E6 overexpression and cellular transformation. Since methylation of the CpG dinucleotides contained in HPV16 E2BSs inhibits E2 binding to viral DNA and impairs its regulatory functions (26, 55, 56), their methylation may be involved in E7/E6 expression deregulation. The technique described here will also address these questions at the basic level of PV biology, for instance, the hitherto poorly understood mechanistic interplay between integration and methylation that ultimately controls oncogene expression. In our samples, the integration level has been estimated by measuring the E2/E6 ratio, as described by Peitsaro and collaborators (49). Unfortunately, we have not observed an association between methylation and integration, even if none of the samples harboring fully integrated HPV16 genomes, including cancer samples, exhibited methylated target CpGs (data not shown).
In the present study, the methylation statuses of the E2BS3 and E2BS4 were not examined because these sites do not appear to play a major role in the transcriptional repression induced by E2 (for a review, see reference 16). However, a recent study by Vinokurova and von Knebel Doeberitz (38) showed a specific and high methylation of E2BS4 (named E2BS1 in their paper) linked with a transforming infection characterized by p16 overexpression, whereas no methylated CpG was observed in permissive infection that allowed the HPV life cycle to continue.
HPV16 E2BS1, E2BS2, and Sp1BS methylation clinical value.
HPV16 E2BS1, E2BS2, and Sp1BS methylation appear to be late events in cervical carcinogenesis, as they were observed only in invasive cancer samples. We demonstrate here that the methylation of HPV16 E2BS1, E2BS2, and Sp1BS analyzed from cervical smears by HRM allows for the identification of women with cervical cancers among those who are infected by HPV16 with a good specificity and a limited sensitivity. Several previous studies showed that methylation was higher in high-grade lesions than in normal samples and/or low-grade lesions (29, 36, 37). The results reported here support the limited interest of HPV16 E2BS1, E2BS2, and Sp1BS methylation in cervical cancer screening, since neither prevalent nor incident high-grade lesions can be identified with this assay.
Nevertheless, the prognostic value of HPV16 E2BS methylation likely deserves to be explored, either alone or in association with other biomarkers in cancer samples. For example, Mazumder and collaborators (31) showed that in addition to integration, the absence of HPV16 LCR methylation was associated with a poor prognosis in cancer patients. In our series, all the cancer samples were early-stage tumors (Federation of Gynecology and Obstetrics [FIGO] stage I or II), which are known to have a better outcome than late-stage tumors. Remarkably, five out seven of the cancers with methylated CpGs were adenocarcinomas. Whether this apparent association between histological type and methylation statuses of E2BS1, E2BS2, and Sp1BS may be due to the small series of samples analyzed, it also may be hypothesized that specific methylation patterns drive the specific mechanism of HPV16-associated carcinogenesis according to HPV-infected epithelium types.
In conclusion, HRM-PCR allows for the accurate assessment of the methylation statuses of HPV16 E2BS1, E2BS2, and Sp1BS CpG sites. This versatile approach may be easily adapted to target other DNA loci and other PV genotypes. It would be interesting to transfer this method to the HPV16 L1 gene, which is a good biomarker candidate for differential methylation status, as reviewed by Clarke and collaborators (44). In both males and females, HR-HPV infection is associated with several anogenital cancers and with a subset of head and neck cancers for which little is known about viral DNA methylation. We propose here that HRM-PCR might allow for the efficient screening of samples to rapidly determine the clinical relevance of HPV DNA methylation in terms of diagnostic, prognostic, and predictive values.
ACKNOWLEDGMENTS
We thank K. Le Bail Carval for providing us with some clinical samples and F. Poncet (Genomic platform, FED4234, Besançon, France), who performed the Sanger sequencing analysis. We also thank E. M. de Villiers (DKFZ, Heidelberg, Germany) for kindly providing us with the HPV16 plasmid.
E. Jacquin was the recipient of a predoctoral scholarship from the Conseil Régional de Franche-Comté. Part of this work was supported by grants from La Ligue Contre le Cancer (Comité du Doubs and CCIR-GE), the Conseil Régional de Franche-Comté and the University of Franche-Comte (BQR), and L'Association Française des Femmes Diplômées des Universités (AFFDU, Besançon group).
Footnotes
Published ahead of print 17 July 2013
REFERENCES
- 1.Bosch FX, Manos MM, Muñoz N, Sherman M, Jansen AM, Peto J, Schiffman MH, Moreno V, Kurman R, Shah KV. 1995. Prevalence of human papillomavirus in cervical cancer: a worldwide perspective. International Biological Study on Cervical Cancer (IBSCC) Study Group. J. Natl. Cancer Inst. 87:796–802 [DOI] [PubMed] [Google Scholar]
- 2.Walboomers JM, Jacobs MV, Manos MM, Bosch FX, Kummer JA, Shah KV, Snijders PJ, Peto J, Meijer CJ, Muñoz N. 1999. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J. Pathol. 189:12–19 [DOI] [PubMed] [Google Scholar]
- 3.Richardson H, Kelsall G, Tellier P, Voyer H, Abrahamowicz M, Ferenczy A, Coutlée F, Franco EL. 2003. The natural history of type-specific human papillomavirus infections in female university students. Cancer Epidemiol. Biomarkers Prev. 12:485–490 [PubMed] [Google Scholar]
- 4.Ho GY, Burk RD, Klein S, Kadish AS, Chang CJ, Palan P, Basu J, Tachezy R, Lewis R, Romney S. 1995. Persistent genital human papillomavirus infection as a risk factor for persistent cervical dysplasia. J. Natl. Cancer Inst. 87:1365–1371 [DOI] [PubMed] [Google Scholar]
- 5.Dalstein V, Riethmuller D, Prétet JL, Le Bail Carval K, Sautière JL, Carbillet JP, Kantelip B, Schaal JP, Mougin C. 2003. Persistence and load of high-risk HPV are predictors for development of high-grade cervical lesions: a longitudinal French cohort study. Int. J. Cancer 106:396–403 [DOI] [PubMed] [Google Scholar]
- 6.Woodman CB, Collins S, Winter H, Bailey A, Ellis J, Prior P, Yates M, Rollason TP, Young LS. 2001. Natural history of cervical human papillomavirus infection in young women: a longitudinal cohort study. Lancet 357:1831–1836 [DOI] [PubMed] [Google Scholar]
- 7.Moscicki AB, Schiffman M, Kjaer S, Villa LL. 2006. Chapter 5: updating the natural history of HPV and anogenital cancer. Vaccine 24(Suppl 3):S3/42–S3/51. 10.1016/j.vaccine.2012.05.089 [DOI] [PubMed] [Google Scholar]
- 8.Bulkmans NW, Berkhof J, Bulk S, Bleeker MC, van Kemenade FJ, Rozendaal L, Snijders PJ, Meijer CJ, POBASCAM Study Group 2007. High-risk HPV type-specific clearance rates in cervical screening. Br. J. Cancer 96:1419–1424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Khan MJ, Castle PE, Lorincz AT, Wacholder S, Sherman M, Scott DR, Rush BB, Glass AG, Schiffman M. 2005. The elevated 10-year risk of cervical precancer and cancer in women with human papillomavirus (HPV) type 16 or 18 and the possible utility of type-specific HPV testing in clinical practice. J. Natl. Cancer Inst. 97:1072–1079 [DOI] [PubMed] [Google Scholar]
- 10.Bouvard V, Baan R, Straif K, Grosse Y, Secretan B, El Ghissassi F, Benbrahim-Tallaa L, Guha N, Freeman C, Galichet L, Cogliano V, WHO International Agency for Research on Cancer Monograph Working Group 2009. A review of human carcinogens–part B: biological agents. Lancet Oncol. 10:321–322 [DOI] [PubMed] [Google Scholar]
- 11.Guan P, Howell-Jones R, Li N, Bruni L, de Sanjosé S, Franceschi S, Clifford GM. 2012. Human papillomavirus types in 115,789 HPV-positive women: a meta-analysis from cervical infection to cancer. Int. J. Cancer 131:2349–2359 [DOI] [PubMed] [Google Scholar]
- 12.de Sanjose S, Quint WG, Alemany L, Geraets DT, Klaustermeier JE, Lloveras B, Tous S, Felix A, Bravo LE, Shin HR, Vallejos CS, de Ruiz PA, Lima MA, Guimera N, Clavero O, Alejo M, Llombart-Bosch A, Cheng-Yang C, Tatti SA, Kasamatsu E, Iljazovic E, Odida M, Prado R, Seoud M, Grce M, Usubutun A, Jain A, Suarez GA, Lombardi LE, Banjo A, Menéndez C, Domingo EJ, Velasco J, Nessa A, Chichareon SC, Qiao YL, Lerma E, Garland SM, Sasagawa T, Ferrera A, Hammouda D, Mariani L, Pelayo A, Steiner I, Oliva E, Meijer CJ, Al-Jassar WF, Cruz E, Wright TC, Puras A, et al. 2010. Human papillomavirus genotype attribution in invasive cervical cancer: a retrospective cross-sectional worldwide study. Lancet Oncol. 11:1048–1056 [DOI] [PubMed] [Google Scholar]
- 13.McLaughlin-Drubin ME, Munger K. 2009. Oncogenic activities of human papillomaviruses. Virus Res. 143:195–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Phelps WC, Howley PM. 1987. Transcriptional trans-activation by the human papillomavirus type 16 E2 gene product. J. Virol. 61:1630–1638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Romanczuk H, Thierry F, Howley PM. 1990. Mutational analysis of cis elements involved in E2 modulation of human papillomavirus type 16 P97 and type 18 P105 promoters. J. Virol. 64:2849–2859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thierry F. 2009. Transcriptional regulation of the papillomavirus oncogenes by cellular and viral transcription factors in cervical carcinoma. Virology 384:375–379 [DOI] [PubMed] [Google Scholar]
- 17.Tan SH, Leong LE, Walker PA, Bernard HU. 1994. The human papillomavirus type 16 E2 transcription factor binds with low cooperativity to two flanking sites and represses the E6 promoter through displacement of Sp1 and TFIID. J. Virol. 68:6411–6420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bellanger S, Tan CL, Xue YZ, Teissier S, Thierry F. 2011. Tumor suppressor or oncogene? A critical role of the human papillomavirus (HPV) E2 protein in cervical cancer progression. Am. J. Cancer Res. 1:373–389 [PMC free article] [PubMed] [Google Scholar]
- 19.Kalantari M, Karlsen F, Kristensen G, Holm R, Hagmar B, Johansson B. 1998. Disruption of the E1 and E2 reading frames of HPV 16 in cervical carcinoma is associated with poor prognosis. Int. J. Gynecol. Pathol. 17:146–153 [DOI] [PubMed] [Google Scholar]
- 20.Arias-Pulido H, Peyton CL, Joste NE, Vargas H, Wheeler CM. 2006. Human papillomavirus type 16 integration in cervical carcinoma in situ and in invasive cervical cancer. J. Clin. Microbiol. 44:1755–1762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leonard SM, Wei W, Collins SI, Pereira M, Diyaf A, Constandinou-Williams C, Young LS, Roberts S, Woodman CB. 2012. Oncogenic human papillomavirus imposes an instructive pattern of DNA methylation changes which parallel the natural history of cervical HPV infection in young women. Carcinogenesis 33:1286–1293 [DOI] [PubMed] [Google Scholar]
- 22.Wentzensen N, Sherman ME, Schiffman M, Wang SS. 2009. Utility of methylation markers in cervical cancer early detection: appraisal of the state-of-the-science. Gynecol. Oncol. 112:293–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Danos O, Katinka M, Yaniv M. 1980. Molecular cloning, refined physical map and heterogeneity of methylation sites of papilloma virus type 1a DNA. Eur. J. Biochem. 109:457–461 [DOI] [PubMed] [Google Scholar]
- 24.Fernandez AF, Esteller M. 2010. Viral epigenomes in human tumorigenesis. Oncogene 29:1405–1420 [DOI] [PubMed] [Google Scholar]
- 25.Brandsma JL, Sun Y, Lizardi PM, Tuck DP, Zelterman D, Haines GK, III, Martel M, Harigopal M, Schofield K, Neapolitano M. 2009. Distinct human papillomavirus type 16 methylomes in cervical cells at different stages of premalignancy. Virology 389:100–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fernandez AF, Rosales C, Lopez-Nieva P, Graña O, Ballestar E, Ropero S, Espada J, Melo SA, Lujambio A, Fraga MF, Pino I, Javierre B, Carmona FJ, Acquadro F, Steenbergen RD, Snijders PJ, Meijer CJ, Pineau P, Dejean A, Lloveras B, Capella G, Quer J, Buti M, Esteban JI, Allende H, Rodriguez-Frias F, Castellsague X, Minarovits J, Ponce J, Capello D, Gaidano G, Cigudosa JC, Gomez-Lopez G, Pisano DG, Valencia A, Piris MA, Bosch FX, Cahir-McFarland E, Kieff E, Esteller M. 2009. The dynamic DNA methylomes of double-stranded DNA viruses associated with human cancer. Genome Res. 19:438–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mirabello L, Schiffman M, Ghosh A, Rodriguez AC, Vasiljevic N, Wentzensen N, Herrero R, Hildesheim A, Wacholder S, Scibior-Bentkowska D, Burk RD, Lorincz AT. 2013. Elevated methylation of HPV16 DNA is associated with the development of high grade cervical intraepithelial neoplasia. Int. J. Cancer 132:1412–1422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kalantari M, Calleja-Macias IE, Tewari D, Hagmar B, Lie K, Barrera-Saldana HA, Wiley DJ, Bernard HU. 2004. Conserved methylation patterns of human papillomavirus type 16 DNA in asymptomatic infection and cervical neoplasia. J. Virol. 78:12762–12772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sun C, Reimers LL, Burk RD. 2011. Methylation of HPV16 genome CpG sites is associated with cervix precancer and cancer. Gynecol. Oncol. 121:59–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Badal V, Chuang LS, Tan EH, Badal S, Villa LL, Wheeler CM, Li BF, Bernard HU. 2003. CpG methylation of human papillomavirus type 16 DNA in cervical cancer cell lines and in clinical specimens: genomic hypomethylation correlates with carcinogenic progression. J. Virol. 77:6227–6234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mazumder Indra D, Singh RK, Mitra S, Dutta S, Chakraborty C, Basu PS, Mondal RK, Roychoudhury S, Panda CK. 2011. Genetic and epigenetic changes of HPV16 in cervical cancer differentially regulate E6/E7 expression and associate with disease progression. Gynecol. Oncol. 123:597–604 [DOI] [PubMed] [Google Scholar]
- 32.Patel DA, Rozek LS, Colacino JA, Van Zomeren-Dohm A, Ruffin MT, Unger ER, Dolinoy DC, Swan DC, Onyekwuluje J, DeGraffinreid CR, Paskett ED. 2012. Patterns of cellular and HPV 16 methylation as biomarkers for cervical neoplasia. J. Virol. Methods 184:84–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Piyathilake CJ, Macaluso M, Alvarez RD, Chen M, Badiga S, Edberg JC, Partridge EE, Johanning GL. 2011. A higher degree of methylation of the HPV 16 E6 gene is associated with a lower likelihood of being diagnosed with cervical intraepithelial neoplasia. Cancer 117:957–963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xi LF, Jiang M, Shen Z, Hulbert A, Zhou XH, Lin YY, Kiviat NB, Koutsky LA. 2011. Inverse association between methylation of human papillomavirus type 16 DNA and risk of cervical intraepithelial neoplasia grades 2 or 3. PLoS One 6:e23897. 10.1371/journal.pone.0023897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bhattacharjee B, Sengupta S. 2006. CpG methylation of HPV 16 LCR at E2 binding site proximal to P97 is associated with cervical cancer in presence of intact E2. Virology 354:280–285 [DOI] [PubMed] [Google Scholar]
- 36.Ding DC, Chiang MH, Lai HC, Hsiung CA, Hsieh CY, Chu TY. 2009. Methylation of the long control region of HPV16 is related to the severity of cervical neoplasia. Eur. J. Obstet. Gynecol. Reprod. Biol. 147:215–220 [DOI] [PubMed] [Google Scholar]
- 37.Snellenberg S, Schütze DM, Claassen-Kramer D, Meijer CJ, Snijders PJ, Steenbergen RD. 2012. Methylation status of the E2 binding sites of HPV16 in cervical lesions determined with the Luminex xMAP system. Virology 422:357–365 [DOI] [PubMed] [Google Scholar]
- 38.Vinokurova S, von Knebel Doeberitz M. 2011. Differential methylation of the HPV 16 upstream regulatory region during epithelial differentiation and neoplastic transformation. PLoS One 6:e24451. 10.1371/journal.pone.0024451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hong D, Ye F, Lu W, Hu Y, Wan X, Chen Y, Xie X. 2008. Methylation status of the long control region of HPV 16 in clinical cervical specimens. Mol. Med. Rep. 1:555–560 [PubMed] [Google Scholar]
- 40.Saunier M, Monnier-Benoit S, Mauny F, Dalstein V, Briolat J, Riethmuller D, Kantelip B, Schwarz E, Mougin C, Prétet JL. 2008. Analysis of human papillomavirus type 16 (HPV16) DNA load and physical state for identification of HPV16-infected women with high-grade lesions or cervical carcinoma. J. Clin. Microbiol. 46:3678–3685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Seedorf K, Krämmer G, Dürst M, Suhai S, Röwekamp WG. 1985. Human papillomavirus type 16 DNA sequence. Virology 145:181–185 [DOI] [PubMed] [Google Scholar]
- 42.Tse MY, Ashbury JE, Zwingerman N, King WD, Taylor SAM, Pang SC. 2011. A refined, rapid and reproducible high resolution melt (HRM)-based method suitable for quantification of global LINE-1 repetitive element methylation. BMC Res. Notes 4:565. 10.1186/1756-0500-4-565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Warnecke PM, Stirzaker C, Song J, Grunau C, Melki JR, Clark SJ. 2002. Identification and resolution of artifacts in bisulfite sequencing. Methods 27:101–107 [DOI] [PubMed] [Google Scholar]
- 44.Clarke MA, Wentzensen N, Mirabello L, Ghosh A, Wacholder S, Harari A, Lorincz A, Schiffman M, Burk RD. 2012. Human papillomavirus DNA methylation as a potential biomarker for cervical cancer. Cancer Epidemiol. Biomarkers Prev. 21:2125–2137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sun LL, Cao DY, Yang JX, Li H, Zhou XR, Song ZQ, Cheng XM, Chen J, Shen K. 2012. Population-based case-control study on DAPK1, RAR-β2 and MGMT methylation in liquid-based cytology. Arch. Gynecol. Obstet. 285:1433–1439 [DOI] [PubMed] [Google Scholar]
- 46.Turan T, Kalantari M, Calleja-Macias IE, Cubie HA, Cuschieri K, Villa LL, Skomedal H, Barrera-Saldaña HA, Bernard HU. 2006. Methylation of the human papillomavirus-18 L1 gene: a biomarker of neoplastic progression? Virology 349:175–183 [DOI] [PubMed] [Google Scholar]
- 47.Liu Y, Mayo MW, Nagji AS, Smith PW, Ramsey CS, Li D, Jones DR. 2012. Phosphorylation of RelA/p65 promotes DNMT-1 recruitment to chromatin and represses transcription of the tumor metastasis suppressor gene BRMS1. Oncogene 31:1143–1154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kulmala SM, Syrjanen SM, Gyllensten UB, Shabalova IP, Petrovichev N, Tosi P, Syrjänen KJ, Johansson BC. 2006. Early integration of high copy HPV16 detectable in women with normal and low grade cervical cytology and histology. J. Clin. Pathol. 59:513–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Peitsaro P, Johansson B, Syrjanen S. 2002. Integrated human papillomavirus type 16 is frequently found in cervical cancer precursors as demonstrated by a novel quantitative real-time PCR technique. J. Clin. Microbiol. 40:886–891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tonon SA, Picconi MA, Bos PD, Zinovich JB, Galuppo J, Alonio LV, Teyssie AR. 2001. Physical status of the E2 human papilloma virus 16 viral gene in cervical preneoplastic and neoplastic lesions. J. Clin. Virol. 21:129–134 [DOI] [PubMed] [Google Scholar]
- 51.Cricca M, Morselli-Labate AM, Venturoli S, Ambretti S, Gentilomi GA, Gallinella G, Costa S, Musiani M, Zerbini M. 2007. Viral DNA load, physical status and E2/E6 ratio as markers to grade HPV16 positive women for high-grade cervical lesions. Gynecol. Oncol. 106:549–557 [DOI] [PubMed] [Google Scholar]
- 52.Hudelist G, Manavi M, Pischinger KI, Watkins-Riedel T, Singer CF, Kubista E, Czerwenka KF. 2004. Physical state and expression of HPV DNA in benign and dysplastic cervical tissue: different levels of viral integration are correlated with lesion grade. Gynecol. Oncol. 92:873–880 [DOI] [PubMed] [Google Scholar]
- 53.Fujii T, Masumoto N, Saito M, Hirao N, Niimi S, Mukai M, Ono A, Hayashi S, Kubushiro K, Sakai E, Tsukazaki K, Nozawa S. 2005. Comparison between in situ hybridization and real-time PCR technique as a means of detecting the integrated form of human papillomavirus 16 in cervical neoplasia. Diagn. Mol. Pathol. 14:103–108 [DOI] [PubMed] [Google Scholar]
- 54.Schmitt M, Pawlita M. 2011. The HPV transcriptome in HPV16 positive cell lines. Mol. Cell. Probes 25:108–113 [DOI] [PubMed] [Google Scholar]
- 55.Thain A, Jenkins O, Clarke AR, Gaston K. 1996. CpG methylation directly inhibits binding of the human papillomavirus type 16 E2 protein to specific DNA sequences. J. Virol. 70:7233–7235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kim K, Garner-Hamrick PA, Fisher C, Lee D, Lambert PF. 2003. Methylation patterns of papillomavirus DNA, its influence on E2 function, and implications in viral infection. J. Virol. 77:12450–12459 [DOI] [PMC free article] [PubMed] [Google Scholar]