

Abstract
Multidrug-resistant tuberculosis (MDR-TB) is a significant health problem in Panama. The extent to which such cases are the result of primary or acquired resistance and the strain families involved are unknown. We performed whole-genome sequencing of a collection of 66 clinical MDR isolates, along with 31 drug-susceptible isolates, that were isolated in Panama between 2001 and 2010; 78% of the MDR isolates belong to the Latin American-Mediterranean (LAM) family. Drug resistance mutations correlated well with drug susceptibility profiles. To determine the relationships among these strains and to better understand the acquisition of resistance mutations, a phylogenetic tree was constructed based on a genome-wide single-nucleotide polymorphism analysis. The phylogenetic tree shows that the isolates are highly clustered, with a single strain (LAM9-c1) accounting for nearly one-half of the MDR isolates (29/66 isolates). The LAM9-c1 strain was most prevalent among male patients of working age and was associated with high mortality rates. Members of this cluster all share identical mutations conferring resistance to isoniazid (KatG S315T mutation), rifampin (RpoB S531L mutation), and streptomycin (rrs C517T mutation). This evidence of primary resistance supports a model in which MDR-TB in Panama is driven by clonal expansion and ongoing transmission of several strains in the LAM family, including the highly successful MDR strain LAM9-c1. The phylogenetic analysis also shows that the LAM9-c1 strain is closely related to the KwaZulu-Natal (KZN) extensively drug-resistant TB strain identified in KwaZulu-Natal, South Africa. The LAM9-c1 and KZN strains likely arose from a recent common ancestor that was transmitted between Panama and South Africa and had the capacity to tolerate an accumulation of multiple resistance mutations.
INTRODUCTION
Outbreaks of drug-resistant tuberculosis (TB) in various regions around the world have been increasingly reported (1). Multidrug resistance (MDR), i.e., resistance to at least isoniazid (INH) and rifampin (RIF), is estimated to have a frequency of around 5% among TB cases globally. Extensively drug-resistant (XDR) strains, i.e., resistant to INH, RIF, fluoroquinolones, and one of three injectable second-line drugs (amikacin, kanamycin, or capreomycin), have been observed in South Africa, Russia, Eastern Europe, and East Asia (2–4). Multidrug resistance has been associated with strains belonging to nearly every Mycobacterium tuberculosis lineage (or strain family), including Beijing (5–8), Latin American-Mediterranean (LAM) (9), and Central Asian (CAS) (10, 11). In all cases investigated so far, multidrug resistance in M. tuberculosis appears to result from stepwise acquisition of mutations in individual resistance genes.
An open question is whether multidrug resistance spreads through primary resistance or is acquired in the patient. In some regions, there is evidence that drug resistance spreads by clonal expansion and transmission of a single strain with multiple resistance mutations (12, 13). In other regions, it appears that resistance mutations arose multiple times independently, suggesting repeated selection for resistance rather than transmission of a well-defined MDR clone (14, 15). Either model can have significant implications for local TB control policies. The success of an MDR-TB strain might require adaptive mutations that enable it to tolerate the presumed fitness cost associated with multiple resistance mutations. Although a subset of MDR strains appears to have putative compensatory mutations in RpoC (16), a systematic explanation for tolerance of resistance mutations in most other MDR-TB strains is currently lacking.
Epidemiological surveys of TB strain families and drug resistance in several Latin American countries, including Peru, Brazil, Trinidad and Tobago, French Guiana, Paraguay, and Venezuela (17–23), have shown that the LAM strain family predominates, accounting for around 50% of the cases throughout this region (24). Among Central American countries, the frequency of the LAM strain family was found to be 55% in Honduras (25), though the strain family appears to be less prevalent in Mexico (14.4%) (26). The overall prevalence of MDR-TB cases in Panama is unknown, since Panama has not conducted a national survey of antituberculosis drug resistance to date. Since 2001, 93 cases of MDR-TB have been reported to the Panama National Tuberculosis Program, and a majority of those strains have been isolated, characterized, and archived through the Mycobacteriology Department of the Laboratorio Central de Referencia de Salud Pública in Panama. The extent to which MDR-TB in Panama is driven by primary versus acquired resistance is currently unknown.
In this article, we report the analysis of 66 MDR-TB clinical isolates from Panama collected between 2001 and 2011 (approximately two-thirds of the reported cases). A majority of the MDR strains belong to the LAM TB strain family. Genome-wide single-nucleotide polymorphism (SNP) analysis was used to construct a phylogenetic tree in order to elucidate the relationships among the isolates. The phylogenetic analysis showed that nearly one-half of the MDR isolates (29 of 66 isolates) represent variants of a single MDR strain, LAM9-c1, with a distinct combination of mutations for resistance to isoniazid, rifampin, and streptomycin (STR), suggesting on-going transmission (primary resistance) in this region. Nonetheless, individual members of this phylogenetic cluster are continuing to acquire additional mutations for resistance to other drugs. We show that this LAM9-c1 strain is closely related to the KwaZulu-Natal (KZN) strain responsible for an outbreak of XDR TB in South Africa.
MATERIALS AND METHODS
M. tuberculosis isolates.
A set of 97 M. tuberculosis clinical isolates was obtained from the Mycobacteriology Department of the Laboratorio Central de Referencia de Salud Pública in Panama. These isolates consisted of 66 MDR-TB strains, which were coresistant to isoniazid (INH) and rifampin (RIF), complemented by a sample of 31 drug-susceptible strains from 2009 and 2010. All isolates were tested using the standard proportion method except for PanR1101, which was classified as MDR based on a lack of clinical response. Isolates were also tested for sensitivity to streptomycin (STR) and ethambutol (EMB).
Characterization and drug susceptibility testing.
Sputum samples were first decontaminated using a modified version of the methodology described by Petroff (27), according to the specifications of the Pan American Health Organization. The presumptive M. tuberculosis isolates were then characterized by biochemical assays, including the nitrate reduction test, niacin production test, and catalase inhibition assay performed at 68°C (28). Finally, drug susceptibility testing was done with the Canetti multiple-proportions method, using Lowenstein-Jensen (solid culture) medium and the following antibiotic concentrations: 0.2 μg/ml INH, 40 μg/ml RIF, 4 μg/ml streptomycin, and 2 μg/ml ethambutol. Identification of the isolates as M. tuberculosis was confirmed using the commercial AccuProbe hybridization assay (Gen-Probe), which has ≥99% sensitivity and specificity for the M. tuberculosis complex.
Whole-genome sequencing.
The genome of each isolate was sequenced using an Illumina Genome Analyzer IIx, as previously described (14). The samples were sequenced in paired-end mode, with a length of 36 to 54 bp for each read. Sequences were assembled by mapping reads to H37Rv (GenBank accession number NC_000962.2) as a reference genome. Genome sequences for all 66 MDR strains were deposited in GenBank. The sequencing data were used to determine the spoligotypes of the isolates by matching reads to the sequences of the spacers in the direct repeats region (29) and matching them to the SpolDB4 database (24).
Phylogenetic analysis.
A phylogenetic tree was constructed based on genome-wide SNP analysis using maximum parsimony (PHYLIP v. 3.66, provided by J. Felsenstein). A multisequence alignment was constructed from the genomes of the 97 isolates, along with the genome sequences for several published reference strains for comparison. We identified a total of 6,890 polymorphic sites at which we were confident that at least one of the strains was different from the others (where depth of coverage was at least 25% of the mean, and the majority nucleotide observed was represented in >70% of the reads; gaps and regions with clusters of SNPs were excluded).
Epidemiological analysis.
Epidemiological data for patients and national statistics were provided by the National TB Program in Panama, which is coordinated by Cecilia Arango and Clara Torres. Due to the retrospective nature of the study, the ability to conduct detailed tracing of contact relationships and transmission dynamics was limited.
Nucleotide sequence accession numbers.
The genome sequences of the 66 MDR isolates have been deposited in GenBank. Accession numbers are as follows: PanR0201, ANZG00000000; PanR0202, ATAS00000000; PanR0203, ATML00000000; PanR0205, ATMM00000000; PanR0206, ATMN00000000; PanR0207, ATMQ00000000; PanR0208, ATMO00000000; PanR0209, ATMP00000000; PanR0301, ATET00000000; PanR0304, ATES00000000; PanR0305, ATER00000000; PanR0306, ATEQ00000000; PanR0307, ATEP00000000; PanR0308, ATEO00000000; PanR0309, ATEN00000000; PanR0311, ATEM00000000; PanR0313, ATEL00000000; PanR0314, ATEK00000000; PanR0315, ATEJ00000000; PanR0316, ATEI00000000; PanR0317, ATEH00000000; PanR0401, ATEG00000000; PanR0402, ATEF00000000; PanR0403, ATEE00000000; PanR0404, ATED00000000; PanR0405, ATEC00000000; PanR0407, ATEB00000000; PanR0409, ATEA00000000; PanR0410, ATDZ00000000; PanR0411, ATDY00000000; PanR0412, ATDX00000000; PanR0501, ATRP00000000; PanR0503, ATRQ00000000; PanR0505, ATRR00000000; PanR0601, ATRS00000000; PanR0602, ATRT00000000; PanR0603, ATRU00000000; PanR0604, ATRV00000000; PanR0605, ATRW00000000; PanR0606, ATRX00000000; PanR0607, ATRY00000000; PanR0609, ATRZ00000000; PanR0610, ATSA00000000; PanR0611, ATSB00000000; PanR0702, ATSC00000000; PanR0703, ATSD00000000; PanR0704, ANNN00000000; PanR0707, ATSE00000000; PanR0708, ATSF00000000; PanR0801, ATSG00000000; PanR0802, ANZH00000000; PanR0803, ATSH00000000; PanR0804, ATSI00000000; PanR0805, ATSJ00000000; PanR0902, ATSK00000000; PanR0903, ATSL00000000; PanR0904, ATSM00000000; PanR0906, ATSN00000000; PanR0907, ATSO00000000; PanR0908, ATSP00000000; PanR0909, ATSQ00000000; PanR1005, ANZI00000000; PanR1006, ATST00000000; PanR1007, ATSR00000000; PanR1101, ATSS00000000.
RESULTS
Characteristics of patients and isolates.
A total of 97 isolates from TB patients in Panama were selected for phenotypic and genotypic analyses, including 66 MDR isolates and 31 drug-susceptible isolates selected for comparison (Table 1). All 66 MDR isolates were resistant to INH and RIF, and 47 (71%) were also resistant to streptomycin. The samples were obtained from patients in several health regions in Panama, including Colón, Panamá Metro, Chiriquí, Comarca Ngobe Buglé, San Miguelito, Bocas del Toro, Paname Este, Panamá Oeste, Veraguas, and Coclé. The sample included twice as many male patients as female patients, ranging in age from 14 to 81 years. Nearly all of the cases presented as pulmonary infections. Unlike in other regions where HIV coinfection is endemic (such as sub-Saharan Africa), most of the patients in this region were HIV negative (75% [39 of 52 patients tested]), and thus HIV coinfection is not implicated as a major factor associated with MDR-TB in this region.
Table 1.
Summary of isolates sequenced
| Characteristic | Drug-resistant | Drug-susceptiblea |
|---|---|---|
| Total no. of isolates | 66 | 31 |
| No. (%) of isolates with resistance to: | ||
| INF plus RIF | 66 (100) | 0 |
| STR | 48 (73) | 0 |
| EMB | 22 (33) | 0 |
| No. of cases by health region | ||
| Colón | 23 | 4 |
| Panamá Metro | 21 | 27 |
| Chiriquí | 8 | 0 |
| Comarca Ngobe Buglé | 3 | 0 |
| San Miguelito | 4 | 0 |
| Bocas del Toro | 2 | 0 |
| Panamá Oeste | 2 | 0 |
| Other (Paname Este, Veraguas, and Coclé) | 3 | 0 |
| No. (%) of patients by gender | ||
| Male | 44 (67) | 26 (84) |
| Female | 22 (33) | 5 (16) |
| No. of patients by age (yr) | ||
| 0–14 | 1 | 1 |
| 15–24 | 11 | 0 |
| 25–34 | 21 | 4 |
| 35–44 | 13 | 2 |
| 45–54 | 6 | 1 |
| 55–64 | 7 | 0 |
| ≥65 | 2 | 1 |
| No data | 5 | 22 |
| No. (%) of isolates by strain family | ||
| LAM | 52 (78.8) | 5 |
| PGG-1 | 2 (3.0) | 3 |
| T clade | 4 (6.0) | 12 |
| H clade | 5 (7.6) | 8 |
| X clade | 3 (4.5) | 2 |
| East African-Indian | 0 | 1 |
| No. (%) of patients by tuberculosis type | ||
| Pulmonary | 64 (97) | NA |
| Extrapulmonary | 2 (3) | NA |
| No. of patients by treatment outcome | ||
| Cured | 22 | NA |
| Died | 28 | NA |
| Default | 7 | NA |
| In treatment | 5 | NA |
| Untreated | 1 | NA |
| No data available | 4 | NA |
| No. of patients by HIV status | ||
| Seropositive | 12 | NA |
| Seronegative | 39 | NA |
| Not tested | 15 | NA |
NA, not available.
A majority of the isolates in the sample (57/97 isolates [58%]) belong to the LAM family. However, several other M. tuberculosis strain families were observed at lower frequencies (Table 1), including members of the X clade (5%), T clade (16%), and H clade (13%). Five isolates were labeled principle genetic group 1 (PGG-1), because their spoligotypes matched Beijing (2 isolates), CAS (1 isolate), and Manu (2 isolates), diverse members of this ancestral lineage. While LAM strains are prevalent in Latin American countries (20, 24), the LAM family is particularly highly represented among the drug-resistant Panamanian isolates; 78.8% of the drug-resistant strains (52/66 strains) are LAM.
The genome sequences of all 97 isolates were determined using next-generation sequencing. Depth of coverage and other sequencing statistics are presented in Table S1 in the supplemental material. Single-nucleotide polymorphisms (SNPs) and insertions/deletions between isolates were determined using multisequence alignment. The locations of the IS6110 transposon insertions were also identified. The number of copies of IS6110 in the genome of each isolate ranged between 1 and 16. There are two IS6110 insertions that appear to be common among all of the LAM strains but are not found in isolates from other strain families. One of these occurs in Rv3113, a phosphatase that is thought to be essential in vitro (30) and in vivo (31). Interestingly, the insertion in Rv3113 in 7 of the isolates is 2 nucleotides away from the position in the rest of the LAM isolates, implying that this insertion event occurred at least twice.
To gain insight into the population structure of these isolates, a phylogenetic tree was constructed for the MDR isolates based on a genome-wide set of 6,890 SNPs (Fig. 1). Several published reference strains (H37Rv, CDC1551, F11, HN878, and KZN_4207) were included in the phylogenetic analysis for comparison, and Mycobacterium bovis BCG was used as an out-group. In this phylogenetic tree, isolates were clustered in several families, which coincided with their grouping by spoligotype (see Table S1 in the supplemental material). The clusters can be identified broadly with strain families by spoligotype matching and more specifically by their association with known reference strains. The KZN_4207 and F11 strains clustered with the two largest LAM clusters, CDC1551 with isolates of the X clade, H37Rv with the T clade members, and the HN878 Beijing strain with the cluster representing PGG-1. For reference, a similar phylogenetic tree of the drug-susceptible isolates is provided in Fig. S1 in the supplemental material, showing that most of those isolates clustered with non-LAM clades (clades T, H, and X and PGG-1).
Fig 1.

Phylogenetic tree of the MDR isolates based on genome-wide SNP analysis (6,890 sites) with maximum parsimony. Branch lengths are proportional to the number of nucleotide differences. The clusters in boxes are identified with strain families based on spoligotype.
The MDR strains in the LAM family were found to be subdivided into four clusters. Members of the largest well-defined cluster (called c1 or LAM9-c1) have the LAM9 spoligotype (777777607760771) or a variant thereof, accounting for nearly one-half (29 isolates) of the drug-resistant strains in the sample. Cluster c2 (9 isolates) also has the LAM9 spoligotype, but members of this cluster are not resistant to streptomycin (see below). Cluster c3 (7 isolates) has the LAM3 spoligotype and is associated with the F11 reference strain. Cluster c4 is associated with the RDRio strain that was identified as a major cause of tuberculosis in Brazil, because the isolates in this cluster all have the characteristic 26-kb deletion of Rv3347 to Rv3354 (32). They also have the 3,649-bp deletion of Rv1992 to Rv1997 described for strain UT205 isolated from Colombia (33), suggesting a common origin for these strains. However, the RDRio strain is probably not frequent in Panama, because cluster c4 accounts for only 6% of the drug-resistant isolates in our sample.
Drug resistance patterns among MDR strains.
Drug resistance patterns among the MDR strains are consistent with mutations in individual genes associated with resistance to each drug (Table 2). The overall frequency of INH and RIF resistance mutations in this sample was described previously (34). The two most prevalent mutations for INH resistance observed among the 66 MDR isolates were the S315T mutation in KatG (70%, conferring high-level resistance) and the C−15T mutation in the inhA promoter (20%, conferring lower-level resistance but also ethionamide coresistance) (34). As noted previously (34), mutations at alternative (non-S315) residues in KatG appear to explain most of the remaining INH-resistant strains (8 of 10 strains) (see Table S2 in the supplemental material), as almost no mutations were observed in other genes associated with INH resistance (35, 36). The inhA promoter mutation was associated primarily with non-LAM MDR strains (8 of 14 isolates), whereas 50 of 52 LAM MDR strains had some nonsynonymous mutations in KatG (and only 3 had the inhA C−15T mutation). Thus, INH resistance in the LAM strain family is highly associated with mutations in KatG.
Table 2.
Summary of the most-frequent mutations associated with resistance to INH, RIF, and streptomycina
| Drug | Mutation | No. of isolates |
|---|---|---|
| Isoniazid | KatG S315T/G | 47 |
| InhA C−15T | 13 | |
| Other | 11 | |
| Total | 66b | |
| Rifampin | RpoB S531L | 52 |
| RpoB H526D/Y | 9 | |
| RpoB D516F/V | 4 | |
| Other | 1 | |
| Total | 66 | |
| Streptomycin | rrs C517T | 30 |
| rrs A514C | 1 | |
| RpsL K43R | 4 | |
| RpsL K88R | 1 | |
| Other | 11 | |
| Total | 47 |
The conventional Escherichia coli numbering system is used for mutation positions in RpoB and rrs.
There was some overlap in isoniazid resistance mutations.
As previously reported (34), all but one of the resistant strains had mutations in the RIF resistance-determining region of RpoB, which is commonly seen in other RIF-resistant strains (37). Most of the streptomycin-resistant strains had the nucleotide substitution C517T (30 isolates) or A514C (1 isolate) in rrs (the 16S rRNA), 5 isolates had K43R or K88R mutations in RpsL, and 11 isolates had nonsynonymous or frameshift mutations in GidB (38–40) (see Table S1 in the supplemental material). Although resistance to pyrazinamide (PZA) was not evaluated in this sample, multiple strains (36 isolates) exhibited mutations in pncA and none of the drug-susceptible strains did, suggesting frequent PZA coresistance (41). Strikingly, the mutations in PncA did not correlate with specific clusters but consisted of 15 distinct mutations scattered among individuals or small groups of isolates throughout the phylogenetic tree (see Table S1 in the supplemental material). This suggests that progenitors of the four LAM MDR clusters were not PZA resistant but many individuals within those clusters acquired PZA resistance independently. The gyrA locus (DNA gyrase) was examined for polymorphisms, and only 6 strains had mutations associated with fluoroquinolone resistance (four had the A90V mutation and two had the D94G mutation). Thus, most of these MDR strains should retain fluoroquinolone sensitivity, though this was not confirmed experimentally.
Certain resistance mutations were associated with clusters of isolates (Table 3), suggesting that they were inherited from a common ancestor (cluster progenitors). This clustering explains the prevalence of certain mutations in this sample. For example, the prevalence of S531L in this population (78%) can be explained by 6 independent mutational events, two of which were subsequently inherited by large clusters (c1, 29 isolates; c2, 9 isolates). Both clusters c1 and c2 have well-defined resistance genotypes (all members have KatG S315T and RpoB S531L mutations) and, in addition, all c1 isolates have the rrs C517T mutation. All members of cluster c3 have the H526D mutation in RpoB. Although all members of clusters c3 and c4 were INH resistant, several different INH resistance mutations were observed in these clusters.
Table 3.
Drug resistance polymorphisms associated with clusters in the phylogenetic tree
| Cluster | Total no. of strains | Polymorphism (no. of strains/total no.) |
||
|---|---|---|---|---|
| InhA or KatG | RpoB | rrs | ||
| LAM9-c1 | 29 | S315T (29/29) | S531L (29/29) | C517T (29/29) |
| LAM-c2 | 9 | S315T (9/9) | S531L (9/9) | Nonea |
| LAM-c3 | 7 | Variable (7/7) | H526D (7/7) | None |
| LAM-c4 | 4 | Variable (4/4) | Variable (4/4) | None |
One of 9 LAM-c2 strains had the rrs mutation A514C.
It was recently proposed that mutations in RpoA and RpoC could act as compensatory mutations for the fitness cost of RIF resistance mutations in RpoB (16). To test this hypothesis, we examined the sequences of RpoA and RpoC in the MDR isolates. None of the isolates had any nonsynonymous mutations in RpoA except for PanR0606 (with the RpoA mutation G31S). Furthermore, only one-quarter of the MDR strains (15 of 66 isolates) had a nonsynonymous mutation in RpoC in the region around amino acids 431 to 527 (Table 4). Strain PanR0304 had the RpoC mutation V483G and strain PanR0909 had the RpoC mutation L527V, both of which were previously reported for other RIF-resistant strains (16). The other mutations in RpoC were not previously reported and cover a total of 8 separate sites. Based on the phylogenetic analysis, the mutations were generally acquired subsequent to acquisition of RIF resistance mutations.
Table 4.
Nonsynonymous mutations in RpoC in the region around amino acid 500 (amino acids 431 to 527), previously implicated to possibly compensate for RpoB mutations
| Strain(s)a | Gene | Nonsynonymous mutation | Lineage (cluster) |
|---|---|---|---|
| 0316 | rpoC | Val431Met | X |
| 0301 | rpoC | Gly433Ser | X |
| 0902 | rpoC | Val483Ala | H |
| 0304 | rpoC | Val483Gly | LAM (c2) |
| 0303 | rpoC | Val486Glu | H |
| 0203 | rpoC | Ile491Thr | LAM (none) |
| 0402 | rpoC | Gly519Val | LAM (noneb) |
| 0909, 0207, 0314, 0607, 0609, 0803, 0904, 0601 | rpoC | Leu527Val | LAM (c1c) |
These are all MDR strains. No mutations were found in this region of RpoB in any of the drug-susceptible strains.
Associated with KZN strains.
A subcluster (single branch) consisting of 8 of 29 isolates in the LAM9-c1 cluster.
Identification of a major MDR cluster in the LAM family.
The LAM9-c1 cluster accounted for nearly one-half (29/66 isolates) of the MDR isolates in this sample. Strains in this cluster had the LAM9 spoligotype (777777607760771) or a variant thereof. Strains in this cluster were uniformly resistant to INH, RIF, and streptomycin. The LAM9-c1 cluster represents a distinct MDR strain that has persisted in the population and has predominated in the cases of MDR-TB in the Panama region over the past decade. The dates on which these 29 isolates were obtained spanned nearly the whole time period of the study (2002 to 2010). Isolates of the LAM9-c1 cluster were found most abundantly in Colón (66%) and Panamá Metro (21%) but also were represented as isolated cases in nearly all of the geographic regions from which samples were obtained (Fig. 2). This MDR strain was prevalent among individuals of productive (working) age (15 to 64 years) and was preferentially associated with male patients ≥35 years of age (odds ratio, 9.28; statistically significant by Fisher's exact test, P < 0.044) (Fig. 3). Over one-half of the patients infected with this strain died (15/29 patients [52%]), and this outcome was more frequent than for any other strain cluster, although the difference was not statistically significant, compared to the overall frequency of death among MDR cases, which was 44% (χ2 = 0.77, df = 1, P = 0.38).
Fig 2.
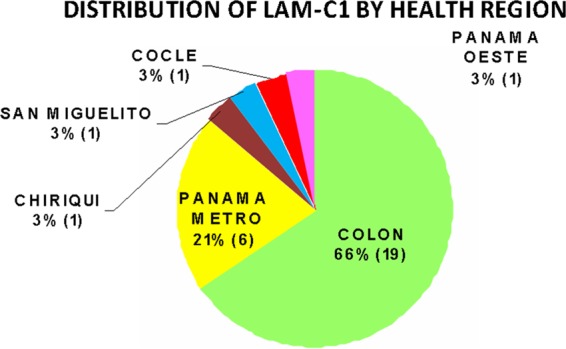
Geographic distribution of LAM9-c1 cases. Numbers in parentheses indicate the numbers of cases.
Fig 3.
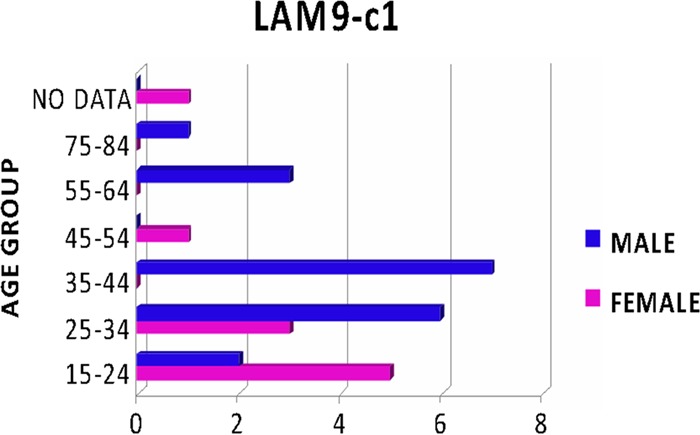
Association of the LAM9-c1 MDR strain with gender and age groups. Values shown on the x axis are numbers of cases.
The LAM9-c1 cluster can be distinguished from the other clusters in the LAM clade by a specific combination of resistance mutations, i.e., the KatG S315T, RpoB S531L, and rrs C517T mutations (Table 3). The S315T mutation in KatG provides high-level resistance to INH (42), and the S531L mutation in RpoB is the most frequently observed mutation for RIF resistance among clinical isolates (43). While each of these mutations is observed individually with high frequency in monoresistant strains, only one other MDR strain in the sample has exactly this combination of resistance mutations. The fact that all members of the LAM9-c1 cluster share this combination of three mutations is consistent with clonal expansion of a single drug-resistant strain. The long branch for this cluster in the dendrogram (Fig. 1) is defined by a set of 100 shared polymorphic sites found to be uniquely associated with LAM9-c1 isolates (see Table S3 in the supplemental material). These could be used as genetic markers in future epidemiological studies and/or could define unique phenotypic features of this strain.
The KZN_4207 reference strain is associated closely with the LAM9-c1 cluster in the dendrogram, implying that the Panamanian LAM9-c1 MDR strain is a variant of the KZN MDR/XDR strains identified in KwaZulu-Natal, South Africa (9, 12, 44). The KZN strains are in the LAM4 family and have a spoligotype very similar to that of LAM9. The genomic data support the relationship between these two strains. For example, both the KZN strains and the LAM9-c1 strains have a 2,606-bp deletion encompassing Rv0376 to Rv0378; the LAM-c2, LAM-c3, and LAM-c4 clusters do not have this deletion. However, it is important to note that the resistance mutations in the KZN MDR strain (strain V2475) and XDR strain (strain R506) are different from those observed in LAM9-c1. For example, V2475 and R506 have a deletion in gidB that explains resistance to streptomycin, instead of the C517T mutation in rrs as in LAM9-c1, and V2475 and R506 both have mutations in RpoB at amino acids other than S531. Both the LAM9-c1 and KZN strains have distinct sets of unique SNPs (shared among members of each cluster but not the other), so it cannot be said that LAM9-c1 evolved from KZN or vice versa. Nonetheless, the LAM9-c1 and KZN strains share a close common ancestor in the LAM family and have both developed multidrug resistance. However, none of the unique or shared SNPs in V2475 and R506 described previously (12) coincides with any of the 100 polymorphisms unique to the LAM9-c1 cluster (listed in Table S3 in the supplemental material), and a common polymorphism exclusively associated with drug-resistant strains has yet to be identified.
DISCUSSION
Phylogenetic analysis of this sample of 66 drug-resistant strains from Panama revealed that nearly one-half of the cases of MDR-TB over the past decade have been caused by a single circulating strain, LAM9-c1. The LAM9-c1 strain is a member of the LAM strain family, which is prevalent in South and Central America. The LAM9-c1 strain was identified in multiple health regions within Panama, primarily among men 25 to 64 years of age. The gender bias, which frequently is observed among TB cases, probably reflects sociocultural differences, in that men are more likely than women to work and interact with wider networks of people. While there is no direct evidence that LAM9-c1 is more virulent than other strains of M. tuberculosis, it is notable that over one-half (52%) of the patients infected with this strain ultimately died. The LAM9-c1 strain bears the resistance markers RpoB(S531L), KatG(S315T), and rrs(C517T), conferring resistance to INH, RIF, and streptomycin, respectively. Each of these mutations individually has among the lowest (but nonzero) fitness cost for each drug (36, 45, 46). Putative compensatory mutations in RpoC were found in only a fraction of the MDR isolates; therefore, a general molecular explanation for why these strains can tolerate the burden of resistance mutations remains unknown.
Among the LAM strains as a whole, we observed that INH resistance was almost exclusively associated with KatG mutations rather than inhA promoter mutations, including nonsynonymous and stop-codon mutations at sites other than S315. This association of KatG mutations with the LAM strain family is supported by previous findings (47) and possibly reflects lineage-specific resistance mutation pressures (e.g., epistatic interactions). Many individual isolates also had additional mutations in EmbB and PncA, associated with resistance to ethambutol and pyrazinamide, respectively. These latter mutations were more variable within clusters and likely result from the use of these drugs in combination therapy. Fortunately, few of the MDR isolates have resistance mutations in gyrA, suggesting that treatment with fluoroquinolones should still be clinically effective in this region.
Analysis of the genomic data shows that the LAM9-c1 MDR strain in Panama is highly related to the MDR/XDR strain in KwaZulu-Natal, South Africa (9). The phylogenetic analysis suggests that both the LAM9-c1 and KZN strains evolved from a common progenitor in the LAM family that might have had the capacity to develop drug resistance through some as-yet-unidentified compensatory mechanism, which was probably transmitted between South Africa and South America via international travel. Panama is recognized as a global trade center and experienced a large amount of immigration from Africa in the early part of the 20th century, during construction of the Panama Canal. Subsequent to the construction of the Panama Canal, the African population in Panama was decimated by three diseases, i.e., tuberculosis, diabetes, and cancer. The most recent migration of Africans to Panama was in the 1990s, including many Somalis escaping civil conflict. The genomic data do not establish the direction of transmission (despite the overall direction of migration, travel in the reverse direction also occurs). Because it is not the case that most of the SNPs in the KZN strain are a subset of the SNPs in the LAM9-c1 isolates or vice versa, it does not appear that either strain evolved directly from the other. Nonetheless, the two clusters of isolates share SNPs that are not found in the other LAM strains (e.g., c2, c3, and c4 clusters), suggesting they had a recent common ancestor. Unlike in South America, LAM strains are significantly less abundant in South Africa and represent a smaller proportion (10 to 30%) of TB cases (24, 48). In a recent study of the population structure of MDR-TB strains in KwaZulu-Natal (49), the LAM spoligotype was identified in 32% of the isolates, but >80% of those were of the LAM4 spoligotype (associated with KZN TB strains) and the rest had the LAM3 spoligotype. In contrast, the LAM9 spoligotype (associated with Panama MDR cluster c1) was not observed among 233 MDR isolates in KwaZulu-Natal and was observed for only 0.7% of the isolates from the Eastern Cape and the Western Cape combined (49). Thus, it appears that the occurrence of the two strains is mostly region specific, with LAM9-c1 in Panama and the KZN strain in KwaZulu-Natal (despite the fact that isolated cases of transmission to other countries have been reported [50]). However, it should be noted that some LAM9 MDR strains have been identified in other provinces near KZN, including Gauteng and Mpumalanga (51).
The clonal nature of the MDR-TB cases in Panama suggests that MDR-TB in this region is driven by ongoing transmission, rather than by repeated acquisition of mutations in independent hosts (which might result from medical nonadherence, for example). The significant clustering of isolates in the phylogenetic analysis provides strong evidence of primary resistance resulting from transmission of one of a few circulating MDR strains, dominated by LAM9-c1. Each cluster appears to have acquired a distinct signature of resistance mutations for the first-line drugs INH and RIF, as well as streptomycin (for LAM9-c1). The origin of these drug-resistant LAM strains could be due to the fact that directly observed therapy was not widely adopted as a standardized treatment regimen in Panama until around 2005. Some individual isolates have continued to acquire additional resistance mutations for other drugs such as pyrazinamide, reflecting the adaptability of these strains. The apparent ability of these strains to tolerate an accumulation of resistance mutations has likely contributed to the maintenance of these MDR strains within the population of Panama over a decade.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful for the direct or indirect collaboration of all of the members who make up the National TB Programme of the Ministry of Health of Panama (coordinated by Cecilia Arango and Clara Torres), all members in the 14 health regions, and Jaime Bravo, chief of the Mycobacteriology Department of the Laboratorio Central de Referencia de Salud Pública of the Instituto Conmemorativo Gorgas de Estudios de la Salud. Nilofar Mohaideen, Souvik Ghosh, and Saranya Sivandam provided assistance with sample preparation for sequencing, and Viktor Einarsson and Purvaja Narayanaswamy helped with genome assembly.
We report no conflicts of interest.
The study was approved by the institutional review board of the Johns Hopkins University School of Medicine and the Comité Nacional de Bioética de la Investigación of Panama.
This work was supported by a Johns Hopkins Center for Global Health faculty grant (to P.C.K.) and by the Robert A. Welch Foundation (grant A0015, to J.C.S.).
Footnotes
Published ahead of print 24 July 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JCM.01122-13.
REFERENCES
- 1.World Health Organization 2010. Multidrug and extensively drug-resistant TB (M/XDR-TB): 2010 global report on surveillance and response. Report WHO/HTM/TB/2010.3. World Health Organization, Geneva, Switzerland [Google Scholar]
- 2.Gandhi NR, Moll A, Sturm AW, Pawinski R, Govender T, Lalloo U, Zeller K, Andrews J, Friedland G. 2006. Extensively drug-resistant tuberculosis as a cause of death in patients co-infected with tuberculosis and HIV in a rural area of South Africa. Lancet 368:1575–1580 [DOI] [PubMed] [Google Scholar]
- 3.Casali N, Nikolayevskyy V, Balabanova Y, Ignatyeva O, Kontsevaya I, Harris SR, Bentley SD, Parkhill J, Nejentsev S, Hoffner SE, Horstmann RD, Brown T, Drobniewski F. 2012. Microevolution of extensively drug-resistant tuberculosis in Russia. Genome Res. 22:735–745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tang S, Zhang Q, Yu J, Liu Y, Sha W, Sun H, Fan L, Gu J, Hao X, Yao L, Xiao H. 2011. Extensively drug-resistant tuberculosis at a tuberculosis specialist hospital in Shanghai, China: clinical characteristics and treatment outcomes. Scand. J. Infect. Dis. 43:280–285 [DOI] [PubMed] [Google Scholar]
- 5.Frieden TR, Sherman LF, Maw KL, Fujiwara PI, Crawford JT, Nivin B, Sharp V, Hewlett D, Jr, Brudney K, Alland D, Kreisworth BN. 1996. A multi-institutional outbreak of highly drug-resistant tuberculosis: epidemiology and clinical outcomes. JAMA 276:1229–1235 [PubMed] [Google Scholar]
- 6.Glynn JR, Whiteley J, Bifani PJ, Kremer K, van Soolingen D. 2002. Worldwide occurrence of Beijing/W strains of Mycobacterium tuberculosis: a systematic review. Emerg. Infect. Dis. 8:843–849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mokrousov I, Jiao WW, Sun GZ, Liu JW, Valcheva V, Li M, Narvskaya O, Shen AD. 2006. Evolution of drug resistance in different sublineages of Mycobacterium tuberculosis Beijing genotype. Antimicrob. Agents Chemother. 50:2820–2823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Johnson R, Warren RM, van der Spuy GD, Gey van Pittius NC, Theron D, Streicher EM, Bosman M, Coetzee GJ, van Helden PD, Victor TC. 2010. Drug-resistant tuberculosis epidemic in the Western Cape driven by a virulent Beijing genotype strain. Int. J. Tuberc. Lung Dis. 14:119–121 [PubMed] [Google Scholar]
- 9.Pillay M, Sturm AW. 2007. Evolution of the extensively drug-resistant F15/LAM4/KZN strain of Mycobacterium tuberculosis in KwaZulu-Natal, South Africa. Clin. Infect. Dis. 45:1409–1414 [DOI] [PubMed] [Google Scholar]
- 10.Stavrum R, Myneedu VP, Arora VK, Ahmed N, Grewal HM. 2009. In-depth molecular characterization of Mycobacterium tuberculosis from New Delhi: predominance of drug resistant isolates of the ‘modern' (TbD1) type. PLoS One 4:e4540. 10.1371/journal.pone.0004540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hasan R, Jabeen K, Ali A, Rafiq Y, Laiq R, Malik B, Tanveer M, Groenheit R, Ghebremichael S, Hoffner S, Hasan Z. 2010. Extensively drug-resistant tuberculosis, Pakistan. Emerg. Infect. Dis. 16:1473–1475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ioerger TR, Koo S, No EG, Chen X, Larsen MH, Jacobs WR, Jr, Pillay M, Sturm AW, Sacchettini JC. 2009. Genome analysis of multi- and extensively-drug-resistant tuberculosis from KwaZulu-Natal, South Africa. PLoS One 4:e7778. 10.1371/journal.pone.0007778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gardy JL, Johnston JC, Ho Sui SJ, Cook VJ, Shah L, Brodkin E, Rempel S, Moore R, Zhao Y, Holt R, Varhol R, Birol I, Lem M, Sharma MK, Elwood K, Jones SJ, Brinkman FS, Brunham RC, Tang P. 2011. Whole-genome sequencing and social-network analysis of a tuberculosis outbreak. N. Engl. J. Med. 364:730–739 [DOI] [PubMed] [Google Scholar]
- 14.Ioerger TR, Feng Y, Chen X, Dobos KM, Victor TC, Streicher EM, Warren RM, Gey van Pittius NC, Van Helden PD, Sacchettini JC. 2010. The non-clonality of drug resistance in Beijing-genotype isolates of Mycobacterium tuberculosis from the Western Cape of South Africa. BMC Genomics 11:670. 10.1186/1471-2164-11-670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mlambo CK, Warren RM, Poswa X, Victor TC, Duse AG, Marais E. 2008. Genotypic diversity of extensively drug-resistant tuberculosis (XDR-TB) in South Africa. Int. J. Tuberc. Lung Dis. 12:99–104 [PubMed] [Google Scholar]
- 16.Comas I, Borrell S, Roetzer A, Rose G, Malla B, Kato-Maeda M, Galagan J, Niemann S, Gagneux S. 2012. Whole-genome sequencing of rifampicin-resistant Mycobacterium tuberculosis strains identifies compensatory mutations in RNA polymerase genes. Nat. Genet. 44:106–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zazueta-Beltran J, Leon-Sicairos N, Muro-Amador S, Flores-Gaxiola A, Velazquez-Roman J, Flores-Villasenor H, Canizalez-Roman A. 2011. Increasing drug resistance of Mycobacterium tuberculosis in Sinaloa, Mexico, 1997–2005. Int. J. Infect. Dis. 15:e272–e276. 10.1016/j.ijid.2011.01.001 [DOI] [PubMed] [Google Scholar]
- 18.Taype CA, Agapito JC, Accinelli RA, Espinoza JR, Godreuil S, Goodman SJ, Banuls AL, Shaw MA. 2012. Genetic diversity, population structure and drug resistance of Mycobacterium tuberculosis in Peru. Infect. Genet. Evol. 12:577–585 [DOI] [PubMed] [Google Scholar]
- 19.Brito RC, Mello FC, Andrade MK, Oliveira H, Costa W, Matos HJ, Lourenco MC, Rolla VC, Fonseca L, Ruffino Netto A, Kritski AL. 2010. Drug-resistant tuberculosis in six hospitals in Rio de Janeiro, Brazil. Int. J. Tuberc. Lung Dis. 14:24–33 [PubMed] [Google Scholar]
- 20.Aristimuno L, Armengol R, Cebollada A, Espana M, Guilarte A, Lafoz C, Lezcano MA, Revillo MJ, Martin C, Ramirez C, Rastogi N, Rojas J, de Salas AV, Sola C, Samper S. 2006. Molecular characterisation of Mycobacterium tuberculosis isolates in the First National Survey of Anti-tuberculosis Drug Resistance from Venezuela. BMC Microbiol. 6:90. 10.1186/1471-2180-6-90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baboolal S, Millet J, Akpaka PE, Ramoutar D, Rastogi N. 2009. First insight into Mycobacterium tuberculosis epidemiology and genetic diversity in Trinidad and Tobago. J. Clin. Microbiol. 47:1911–1914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guernier V, Sola C, Brudey K, Guegan JF, Rastogi N. 2008. Use of cluster-graphs from spoligotyping data to study genotype similarities and a comparison of three indices to quantify recent tuberculosis transmission among culture positive cases in French Guiana during a eight year period. BMC Infect. Dis. 8:46. 10.1186/1471-2334-8-46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Candia N, Lopez B, Zozio T, Carrivale M, Diaz C, Russomando G, de Romero NJ, Jara JC, Barrera L, Rastogi N, Ritacco V. 2007. First insight into Mycobacterium tuberculosis genetic diversity in Paraguay. BMC Microbiol. 7:75. 10.1186/1471-2180-7-75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brudey K, Driscoll JR, Rigouts L, Prodinger WM, Gori A, Al-Hajoj SA, Allix C, Aristimuno L, Arora J, Baumanis V, Binder L, Cafrune P, Cataldi A, Cheong S, Diel R, Ellermeier C, Evans JT, Fauville-Dufaux M, Ferdinand S, Garcia de Viedma D, Garzelli C, Gazzola L, Gomes HM, Guttierez MC, Hawkey PM, van Helden PD, Kadival GV, Kreiswirth BN, Kremer K, Kubin M, Kulkarni SP, Liens B, Lillebaek T, Ho ML, Martin C, Mokrousov I, Narvskaia O, Ngeow YF, Naumann L, Niemann S, Parwati I, Rahim Z, Rasolofo-Razanamparany V, Rasolonavalona T, Rossetti ML, Rusch-Gerdes S, Sajduda A, Samper S, Shemyakin IG, Singh UB, Somoskovi A, Skuce RA, van Soolingen D, Streicher EM, Suffys PN, Tortoli E, Tracevska T, Vincent V, Victor TC, Warren RM, Yap SF, Zaman K, Portaels F, Rastogi N, Sola C. 2006. Mycobacterium tuberculosis complex genetic diversity: mining the fourth international spoligotyping database (SpolDB4) for classification, population genetics and epidemiology. BMC Microbiol. 6:23. 10.1186/1471-2180-6-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rosales S, Pineda-Garcia L, Ghebremichael S, Rastogi N, Hoffner SE. 2010. Molecular diversity of Mycobacterium tuberculosis isolates from patients with tuberculosis in Honduras. BMC Microbiol. 10:208. 10.1186/1471-2180-10-208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Molina-Torres CA, Moreno-Torres E, Ocampo-Candiani J, Rendon A, Blackwood K, Kremer K, Rastogi N, Welsh O, Vera-Cabrera L. 2010. Mycobacterium tuberculosis spoligotypes in Monterrey, Mexico. J. Clin. Microbiol. 48:448–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Petroff SA. 1915. A new and rapid method for the isolation and cultivation of tubercle bacilli directly from the sputum and feces. J. Exp. Med. 21:38–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koneman EW. 1988. Color atlas and textbook of diagnostic microbiology, 3rd ed. J. B. Lippincott, Philadelphia, PA [Google Scholar]
- 29.Kamerbeek J, Schouls L, Kolk A, van Agterveld M, van Soolingen D, Kuijper S, Bunschoten A, Molhuizen H, Shaw R, Goyal M, van Embden J. 1997. Simultaneous detection and strain differentiation of Mycobacterium tuberculosis for diagnosis and epidemiology. J. Clin. Microbiol. 35:907–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sassetti CM, Rubin EJ. 2003. Genetic requirements for mycobacterial survival during infection. Proc. Natl. Acad. Sci. U. S. A. 100:12989–12994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lamichhane G, Tyagi S, Bishai WR. 2005. Designer arrays for defined mutant analysis to detect genes essential for survival of Mycobacterium tuberculosis in mouse lungs. Infect. Immun. 73:2533–2540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lazzarini LC, Huard RC, Boechat NL, Gomes HM, Oelemann MC, Kurepina N, Shashkina E, Mello FC, Gibson AL, Virginio MJ, Marsico AG, Butler WR, Kreiswirth BN, Suffys PN, Lapa ESJR, Ho JL. 2007. Discovery of a novel Mycobacterium tuberculosis lineage that is a major cause of tuberculosis in Rio de Janeiro, Brazil. J. Clin. Microbiol. 45:3891–3902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Isaza JP, Duque C, Gomez V, Robledo J, Barrera LF, Alzate JF. 2012. Whole genome shotgun sequencing of one Colombian clinical isolate of Mycobacterium tuberculosis reveals DosR regulon gene deletions. FEMS Microbiol. Lett. 330:113–120 [DOI] [PubMed] [Google Scholar]
- 34.Chia BS, Lanzas F, Rifat D, Herrera A, Kim EY, Sailer C, Torres-Chavolla E, Narayanaswamy P, Einarsson V, Bravo J, Pascale JM, Ioerger TR, Sacchettini JC, Karakousis PC. 2012. Use of multiplex allele-specific polymerase chain reaction (MAS-PCR) to detect multidrug-resistant tuberculosis in Panama. PLoS One 7:e40456. 10.1371/journal.pone.0040456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ramaswamy SV, Dou SJ, Rendon A, Yang Z, Cave MD, Graviss EA. 2004. Genotypic analysis of multidrug-resistant Mycobacterium tuberculosis isolates from Monterrey, Mexico. J. Med. Microbiol. 53:107–113 [DOI] [PubMed] [Google Scholar]
- 36.Pym AS, Saint-Joanis B, Cole ST. 2002. Effect of katG mutations on the virulence of Mycobacterium tuberculosis and the implication for transmission in humans. Infect. Immun. 70:4955–4960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ramaswamy S, Musser JM. 1998. Molecular genetic basis of antimicrobial agent resistance in Mycobacterium tuberculosis: 1998 update. Tuber. Lung Dis. 79:3–29 [DOI] [PubMed] [Google Scholar]
- 38.Okamoto S, Tamaru A, Nakajima C, Nishimura K, Tanaka Y, Tokuyama S, Suzuki Y, Ochi K. 2007. Loss of a conserved 7-methylguanosine modification in 16S rRNA confers low-level streptomycin resistance in bacteria. Mol. Microbiol. 63:1096–1106 [DOI] [PubMed] [Google Scholar]
- 39.Spies FS, da Silva PE, Ribeiro MO, Rossetti ML, Zaha A. 2008. Identification of mutations related to streptomycin resistance in clinical isolates of Mycobacterium tuberculosis and possible involvement of efflux mechanism. Antimicrob. Agents Chemother. 52:2947–2949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wong SY, Lee JS, Kwak HK, Via LE, Boshoff HI, Barry CE., III 2011. Mutations in gidB confer low-level streptomycin resistance in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 55:2515–2522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chiu YC, Huang SF, Yu KW, Lee YC, Feng JY, Su WJ. 2011. Characteristics of pncA mutations in multidrug-resistant tuberculosis in Taiwan. BMC Infect. Dis. 11:240. 10.1186/1471-2334-11-240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Morlock GP, Metchock B, Sikes D, Crawford JT, Cooksey RC. 2003. ethA, inhA, and katG loci of ethionamide-resistant clinical Mycobacterium tuberculosis isolates. Antimicrob. Agents Chemother. 47:3799–3805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hirano K, Abe C, Takahashi M. 1999. Mutations in the rpoB gene of rifampin-resistant Mycobacterium tuberculosis strains isolated mostly in Asian countries and their rapid detection by line probe assay. J. Clin. Microbiol. 37:2663–2666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Moodley P, Shah NS, Tayob N, Connolly C, Zetola N, Gandhi N, Friedland G, Sturm AW. 2011. Spread of extensively drug-resistant tuberculosis in KwaZulu-Natal province, South Africa. PLoS One 6:e17513. 10.1371/journal.pone.0017513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gagneux S, Long CD, Small PM, Van T, Schoolnik GK, Bohannan BJ. 2006. The competitive cost of antibiotic resistance in Mycobacterium tuberculosis. Science 312:1944–1946 [DOI] [PubMed] [Google Scholar]
- 46.Sander P, Springer B, Prammananan T, Sturmfels A, Kappler M, Pletschette M, Bottger EC. 2002. Fitness cost of chromosomal drug resistance-conferring mutations. Antimicrob. Agents Chemother. 46:1204–1211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dalla Costa ER, Ribeiro MO, Silva MS, Arnold LS, Rostirolla DC, Cafrune PI, Espinoza RC, Palaci M, Telles MA, Ritacco V, Suffys PN, Lopes ML, Campelo CL, Miranda SS, Kremer K, da Silva PE, Fonseca Lde S, Ho JL, Kritski AL, Rossetti ML. 2009. Correlations of mutations in katG, oxyR-ahpC and inhA genes and in vitro susceptibility in Mycobacterium tuberculosis clinical strains segregated by spoligotype families from tuberculosis prevalent countries in South America. BMC Microbiol. 9:39. 10.1186/1471-2180-9-39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Marais BJ, Victor TC, Hesseling AC, Barnard M, Jordaan A, Brittle W, Reuter H, Beyers N, van Helden PD, Warren RM, Schaaf HS. 2006. Beijing and Haarlem genotypes are overrepresented among children with drug-resistant tuberculosis in the Western Cape Province of South Africa. J. Clin. Microbiol. 44:3539–3543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chihota VN, Muller B, Mlambo CK, Pillay M, Tait M, Streicher EM, Marais E, van der Spuy GD, Hanekom M, Coetzee G, Trollip A, Hayes C, Bosman ME, Gey van Pittius NC, Victor TC, van Helden PD, Warren RM. 2012. Population structure of multi- and extensively drug-resistant Mycobacterium tuberculosis strains in South Africa. J. Clin. Microbiol. 50:995–1002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cooke GS, Beaton RK, Lessells RJ, John L, Ashworth S, Kon OM, Williams OM, Supply P, Moodley P, Pym AS. 2011. International spread of MDR TB from Tugela Ferry, South Africa. Emerg. Infect. Dis. 17:2035–2037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Said HM, Kock MM, Ismail NA, Mphahlele M, Baba K, Omar SV, Osman AG, Hoosen AA, Ehlers MM. 2012. Molecular characterization and second-line antituberculosis drug resistance patterns of multidrug-resistant Mycobacterium tuberculosis isolates from the northern region of South Africa. J. Clin. Microbiol. 50:2857–2862 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.