

Abstract
Electrospray ionization mass spectrometry (ESI-MS) analysis of reverse transcription (RT)-PCR amplicons from human respiratory samples allows for broad pathogen identification approximately 8 h after collection. We investigated the performance characteristics of a high-throughput RT-PCR-coupled ESI-MS assay for distinguishing biothreat (BT) agents from common bacterial, fungal, and viral respiratory pathogens in bronchoalveolar lavage (BAL) fluid specimens from subjects with suspected respiratory infections. In a retrospective case series, 202 BAL fluid specimens were collected at the Johns Hopkins Hospital between August 2010 and February 2011 from patients with suspected acute respiratory infections. Samples were processed using standard bacterial, viral, and fungal testing in the clinical microbiology laboratory as part of routine care and then were blindly spiked with either water or nucleic acids from BT organisms (Bacillus anthracis, Yersinia pestis, Francisella tularensis, Brucella spp., Burkholderia spp., and Rickettsia prowazekii) and tested by RT-PCR–ESI-MS. The sensitivities and specificities of RT-PCR–ESI-MS versus standard clinical methods were as follows: for mock BT DNA, 98.5% sensitivity (95% confidence interval [CI], 94.2 to 99.7%) and 100% specificity (95% CI, 93.1 to 100.0%); for bacterial pathogens, 81.8% sensitivity (95% CI, 74.3 to 87.6%) and 73.6% specificity (95% CI, 64.2 to 81.4%); for viral pathogens, 93.3% sensitivity (95% CI, 66.0 to 99.7%) and 97.3% specificity (95% CI, 89.7 to 99.5%); for fungal pathogens, 42.6% sensitivity (95% CI, 29.5 to 56.7%) and 97.8% specificity (95% CI, 91.8 to 99.6%). Our data suggest that RT-PCR–ESI-MS is a useful adjunct to standard culture protocols for rapid detection of both BT and common respiratory pathogens; further study is required for assay validation, especially for fungal detection, and potential implementation.
INTRODUCTION
Biothreat (BT) agents are among the most feared of mass terrorism weapons (1–3). In 1970, the World Health Organization estimated that the destruction caused by a theoretical attack with 50 kg of aerosolized Bacillus anthracis, Yersinia pestis, or Francisella tularensis could incur between 150,000 and 250,000 incapacitating casualties and between 19,000 and 100,000 fatalities (4). A more recent analysis by the US Congressional Office of Technology Assessment predicted that 100 kg of Bacillus anthracis has the potential to cause up to 3 million deaths, a mortality rate that would match the predicted lethality of a hydrogen bomb (5).
Early recognition of a BT attack is critical to prevent subsequent waves of infected patients but, because initial symptoms are nonspecific and resemble common respiratory infections, rapid identification of the specific pathogen is challenging (6–8). Current algorithms for the detection and identification of BT agents can be time-consuming, as they rely on culture-based methods followed by referral to specialized state and/or national reference laboratories for confirmatory testing (9). However, advances in molecular diagnostics in the past decade have led to the introduction of significantly more-rapid assays, principally using sensitive and specific nucleic acid amplification tests (NAATs), which provide pathogen identification and genotyping capabilities previously not possible with traditional culture-based methods (10). Although a variety of NAATs have been designed and validated for BT detection, the majority of currently available assays are optimized to detect a narrow range of targets (8, 11). This technical limitation necessitates a high index of clinical suspicion for a particular agent, limiting practical utility in real-world practice and hindering widespread integration into routine patient care settings.
New broad-range PCR assays present a potential solution to this challenge, as they exploit highly conserved bacterial, fungal, and viral genes such as heat shock proteins and RNA polymerases for rapid identification of a large number of target pathogens (both common and rare). Using primers that target conserved genomic motifs flanking variable regions, broad-range assays can generate a diverse array of species-specific amplicons while employing relatively few PCRs. These species-specific amplicons can subsequently be differentiated with sequencing or other genotyping technologies to identify unambiguously a wide array of organisms (12). Several broad-range PCR assays have already been developed (13) but, as of yet, none has been evaluated for BT detection in samples from patients with suspected acute respiratory infections.
As an alternative to previously validated individual NAATs, in this study we describe the application of broad-range reverse transcription (RT)-PCR coupled to electrospray ionization mass spectrometry (ESI-MS) for detection of both BT and naturally occurring organisms in clinical respiratory samples. The assay includes broad-range primers that target viral, bacterial, and fungal pathogens, in addition to a select array of genus-specific primers dedicated to BT detection. The amplicons that result from the broad-range amplification are analyzed with ESI-MS, which precisely measures the molecular masses of the PCR products to derive unambiguous base compositions (xAxGxCxT). Comparison against a database of previously characterized organisms allows “triangulation” of these base compositions to identify unknown pathogens down to the genus, species, and in some cases even strain level (14–16). The RT-PCR–ESI-MS platform has been investigated previously for bloodstream infections and general viral respiratory testing (17–20), but it has never been studied in the context of BT detection in clinical respiratory samples.
This RT-PCR–ESI-MS assay was designed to optimize performance characteristics lacking in previously described NAATs and therefore may have application for improving the management of undifferentiated infections, particularly in the context of unsuspected biothreats. Spectrometric analysis does not require prior knowledge of an organism's nucleic acid sequence, which permits detection of novel organisms (either naturally occurring or genetically engineered) without the use of sequencing techniques (21–23). Importantly, detection is sufficiently sensitive to allow recognition of multiple products from a single PCR, enabling analysis of polymicrobial sample matrices with differentiation of BT and common organisms (19, 20). Finally, the primers utilized represent multiple broad categories of clinically relevant organisms, including viruses, bacteria, fungi, and BT agents, so that BT testing can be incorporated into routine testing for common pathogens in clinical samples with RT-PCR–ESI-MS. The power to identify viral, bacterial, and fungal causes of respiratory infections rapidly has implications for early directed antimicrobial therapy, especially targeting fastidious or BT organisms.
Given its design advantages, RT-PCR–ESI-MS may be able to provide accurate detection of both BT and common pathogens in respiratory samples. The aim of this study was to characterize this research assay's performance for detection of common respiratory pathogens and BT agents by challenging the assay with bronchoalveolar lavage (BAL) fluid specimens collected from patients with suspected respiratory infections and blindly spiked with BT DNA. RT-PCR–ESI-MS sensitivity and specificity were evaluated for BT, bacterial, viral, and fungal agents using BT spiking patterns, established reference culture methods, and suspension microarray microbiology protocols as gold standard comparators.
MATERIALS AND METHODS
Study setting and sample selection.
This study was conducted at a large tertiary care inner-city hospital. We used waste residual bronchoalveolar lavage (BAL) fluid derived from specimens that had been collected from patients with acute respiratory symptoms, upon presentation or during their hospital stay, as part of routine clinical care. Samples were processed by the clinical microbiology laboratory with standard protocols between 3 August 2010 and 17 February 2011 and subsequently were stored at −80°C until processing for research purposes. Positive and negative samples, as determined by the reference culture and suspension microarray methods, were collected from the clinical microbiology laboratory by a study coordinator between 4 August 2010 and 19 February 2011 for evaluation with the research assay. Subsets of samples were selected by a study coordinator to create a panel of BAL fluid specimens enriched for a variety of organisms, including specimens containing polymicrobial infections. A second study coordinator randomly spiked 6 to 8 fg of either Bacillus anthracis DNA (n = 40), Yersinia pestis DNA (n = 40), Francisella tularensis DNA (n = 40), or 0.1 mM EDTA (negative spiking; n = 66) into the respiratory samples prior to RT-PCR–ESI-MS testing. Additionally, 6 to 8 fg of Rickettsia prowazekii DNA (n = 5), Brucella DNA (n = 5), and Burkholderia DNA (n = 5) was evaluated with RT-PCR–ESI-MS. The rationale for utilizing 6 to 8 fg of genomic DNA from either Bacillus anthracis, Francisella tularensis, Rickettsia prowazekii, Brucella spp., or Burkholderia spp. was to challenge RT-PCR–ESI-MS by allowing BT recovery to be obscured by normal microorganisms or coinfecting organisms in the sample. Additionally, a 6- to 8-fg spike of genomic DNA represents a range of concentrations approximating 1 to 70 genomic copies of each organism, which would challenge the lower limit of detection for RT-PCR–ESI-MS. Both the specimen reference results and the identity of the BT spike were masked prior to the spiking. Samples were collected, deidentified, and processed in accordance with a study protocol approved by the Johns Hopkins University Institutional Review Board.
Standard diagnostic testing in the microbiology laboratory.
If BAL fluid specimens were sent for standard-of-care virological testing, then they were tested with the RT-PCR Luminex xTAG respiratory viral panel (RVP) assay (the standard assay used at this hospital) for adenovirus, influenza viruses, parainfluenza viruses (PIVs), human metapneumoviruses (hMPVs), coronavirus, and respiratory syncytial virus (RSV). Although the xTAG RVP assay is capable of detecting rhinoviruses, rhinoviruses were not included in the comparison because the respiratory virus surveillance (RVS) 2.5 kit cannot detect rhinoviruses. For bacteriological testing, aliquots of BAL fluid were subjected to Gram staining and 10 μl was used for plating on each of the following media: trypticase soy agar supplemented with 5% sheep blood (Remel, Lenexa, KS), chocolate agar (Remel), MacConkey agar (Remel), and standard and selective buffered charcoal yeast extract agar (BBL, Sparks, MD) for recovery of Legionella spp. Sabouraud agar plates (Remel) with and without gentamicin (BBL), Mycosel agar (BBL), brain heart infusion agar with gentamicin (BBL), and CHROMagar Candida were also inoculated routinely for recovery of fungal pathogens. After subculture, colony morphology was used as the basis for additional phenotypic testing, based on established protocols detailed in the Manual of Clinical Microbiology (24). Organisms that are part of the normal microbiota but also are known to cause infection (Staphylococcus spp., Haemophilus spp., and Haemophilus, Actinobacillus actinomycetemcomitans, Cardiobacterium hominis, Eikenella corrodens, and Kingella [HACEK] organisms) were not considered a cause of infection if they were detected by reference culture techniques at concentrations of ≤1,000 CFU/ml in the setting of heavy normal microbiota (≥10,000 CFU/ml) (25).
Research assay sample processing.
When all routine microbiological testing was complete, residual BAL fluid samples were kept at 4°C for 24 h in case additional testing was requested by the treating physician. After this 24-h hold, the samples were considered waste because they were no longer relevant to clinical care and they were taken from the clinical laboratory to the research laboratory for storage at −80°C until processing for nucleic acid extraction. After thawing, viral nucleic acids were extracted from 300 μl of remnant BAL fluid using a Thermo Scientific (Waltham, MA) KingFisher robot with an Ambion (ABI, Foster City, CA) MagMAX viral extraction protocol. Bacterial and fungal DNA was extracted from 100-μl aliquots of BAL fluid using a Roche MagNA Pure LC robot (Roche Molecular Diagnostics) with the DNA isolation kit III protocol. Extraction samples were eluted into 200 μl of elution buffer (0.1 mM EDTA). All samples were processed by a dedicated investigator who was blinded to the clinical virology, bacteriology, and mycology laboratory results, as well as the identity of the BT spike, at the time of processing.
RT-PCR.
Three previously developed assay kits were used in parallel for this study, i.e., the PLEX-ID respiratory virus surveillance (RVS) 2.5 kit, the PLEX-ID bacterial antibiotic resistance and Candida (BAC) assay, and the PLEX-ID biodefense (BD) bacterial and viral surveillance kit (Ibis Biosciences, Carlsbad, CA). The RVS 2.5 kit utilizes 16 primer pairs to detect and to subtype 6 groups of viruses (RSV, influenza virus A and B, PIV 1 to 3, adenovirus A to F, coronaviruses, and hMPVs). The BAC kit includes 18 broad-range primer pairs that can identify over 400 species of bacteria and yeast in addition to the presence of the mecA (methicillin resistance), vanA/B (vancomycin resistance), and kpc (carbapenem resistance) genes. The BD kit includes 36 primers (3 of which are broad-range primers shared with the BAC kit) and is designed to identify 17 category A and B BT agents, including Bacillus anthracis, Yersinia pestis, and Francisella tularensis, and to distinguish them from near neighbors. However, because no viral BT nucleic acids were available for this study, the 8 primers for variola virus, Ebola virus, H5N1 influenza A virus, and Venezuelan equine encephalitis virus detection were not used.
RT-PCRs were performed with 50-μl reaction mixtures with 3 U of AmpliTaq Gold (Applied Biosystems, Foster City, CA), 2 U Superscript III (Invitrogen Corp., Carlsbad, CA), and 400 ng T4 gene 32 protein (Roche Diagnostics Corp., Indianapolis, IN). PCRs for BAC and RT-PCRs for RVS 2.5 assays were performed as described previously (14, 17–20). The following RT-PCR cycling conditions were used for BD assays: 60°C for 5 min, 4°C for 10 min, 55°C for 45 min, 95°C for 10 min, and 8 cycles of 95°C for 30 s, 48°C for 30 s, and 72°C for 30 s, with the 48°C annealing temperature increasing 0.9°C each cycle. The PCR was then continued with 37 additional cycles of 95°C for 15 s, 56°C for 20 s, and 72°C for 20 s. The RT-PCR cycle ended with a final extension at 72°C for 2 min, followed by a 4°C hold.
Electrospray ionization mass spectrometry.
RT-PCR products were analyzed with the Ibis T5000 universal biosensor platform (Ibis Biosciences, Inc., Carlsbad, CA). Each PCR mixture underwent an automated weak anion exchange protocol for desalting and purification (26). Accurate (61 ppm), high-resolution (m/Δm of 100,000, full width at half-maximum) mass spectra were acquired for the purified DNA products using established high-throughput ESI-MS protocols (27). Comparisons of mass measurements of complementary single-stranded oligonucleotides resulted in unambiguous base composition data. An internal PCR calibrant present in every PCR well at 100 molecules/well served as an internal positive control and as a comparator for estimating the number of genomes per well in the starting pathogen concentration. Using the internal calibrant, the quantities of the BT spike and common pathogens were estimated and recorded for polymicrobial analysis.
Data for previously characterized viral, bacterial, and fungal specimens have established a reference library of base composition signatures unique to each pathogen. The confidence for a match between an unknown pathogen and a reference pathogen in the database was calculated by the Ibis Track software package (Ibis Biosciences, Carlsbad, CA) as a correlation, which took into account information such as the relative number of genomes per well, the number of expected PCRs agreeing with a given identification, and the base composition similarity between the reference and unknown samples. All detections with values of >0.85 were reported; scores of <0.85 were considered indeterminate, in accordance with the manufacturer's recommendations (Ibis Biosciences, Carlsbad, CA). After a list of organism detections with >85% confidence was established, the findings were unblinded and RT-PCR–ESI-MS results were compared with spiking patterns and standard microbiological testing results to determine sensitivities and specificities. Figure 1 details the sample-processing workflow.
Fig 1.
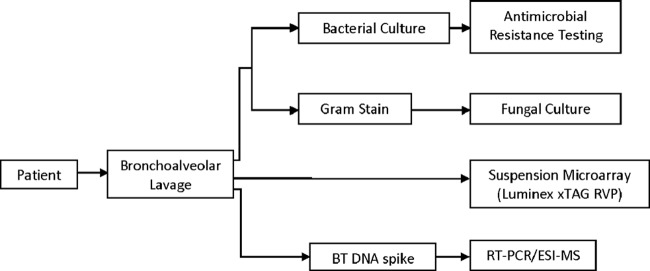
Reference microbiological testing and RT-PCR–ESI-MS workflow.
Statistical analysis.
For the primary analysis of RT-PCR–ESI-MS performance in the detection of BT agents in clinical samples, identifications made with >85% confidence using the BD assay were compared with BT spiking patterns. Sensitivities and specificities were calculated, with 95% confidence intervals (CIs) based on exact binomial probabilities. Secondary analysis of RT-PCR–ESI-MS performance in the detection of common pathogens used standard clinical microbiological tests as reference tests and also used a minimum confidence cutoff value of 85%. Results that were in agreement for the two methods were considered concordant, and exact binomial probabilities were used to calculate 95% confidence intervals.
RESULTS
Specimens and pathogens.
Between 4 August 2010 and 19 February 2011, 400 BAL fluid specimens were collected as residual samples from the clinical microbiology laboratory. Of these samples, 202 BAL fluid specimens were selected for inclusion in this study, of which 52.0% (105/202 specimens) were positive for at least one common pathogen and 37% (75/202 specimens) were polymicrobial samples containing two or more organisms. For purposes of this study, polymicrobial infections were defined as infections that contained ≥2 of the following: heavy respiratory flora (≥10,000 CFU/ml), a bacterial pathogen, a viral pathogen, or a fungal pathogen. Overall, 38 distinct common pathogens were represented. A total of 135 samples were blindly spiked with BT DNA, while 67 were blindly spiked with blanks (0.1 mM EDTA); all samples were processed with the BD assay. All BAL fluid specimens underwent reference bacteriological testing and therefore were processed with the BAC assay. Forty-seven percent of BAL fluid specimens (92/202 specimens) underwent reference virological testing (RVP testing was performed for all of these specimens) ordered by the clinician at the time of initial sample collection. In addition to the BD and BAC assays, 94 BAL fluid specimens were processed with the RVS 2.5 kit. The diagnostic algorithm used by the microbiology laboratory and the experimental workflow are outlined in Fig. 1.
The identity and distribution within this sample set of the organisms spiked and detected with routine reference protocols are detailed in Table 1. The sensitivity of the research assay for each pathogen and the average level (genomes per well) at which each pathogen was detected are shown. Table 2 outlines the overall sensitivity of the research assay for detecting BT, bacterial, viral, and fungal organisms, and the comparison of sensitivities for detection in monomicrobial versus polymicrobial backgrounds is shown in Table 3.
Table 1.
Distribution of pathogens detected by reference testing and BT agents spiked into specimensa
| Organism | No. detected by RT-PCR–ESI-MS/no. detected by reference test (%)b | No. of false-positives | No. missed in polymicrobial samples/no. in polymicrobial samplesc | Quantity detected (no. of genomes/well) (mean ± SD) |
|---|---|---|---|---|
| Bacillus anthracis (spiked) | 40/40 (100) | 0/13 | 80.5 ± 36.5 | |
| Francisella tularensis (spiked) | 38/40 (96.3) | 1/17 | 117.5 ± 108.3 | |
| Yersinia pestis (spiked) | 40/40 (100) | 0/11 | 115.2 ± 71.6 | |
| Brucella spp. (spiked) | 5/5 (100) | 82.6 ± 10.2 | ||
| Rickettsia prowazekii (spiked) | 5/5 (100) | 64.8 ± 12.6 | ||
| Burkholderia spp. (spiked) | 5/5 (100) | 458.2 ± 261.0 | ||
| All spiked organisms | 115.8 ± 114.4 | |||
| Achromobacter spp. | 1/1 (100) | 0/1 | 59 | |
| Acinetobacter baumannii | 1/1 (100) | 0/1 | 110 | |
| Bordetella bronchiseptica | 1/1 (100) | 0/1 | 571 | |
| Burkholderia cepacia | 2/2 (100) | 109.0 ± 118.8 | ||
| Citrobacter freundii | 2/2 (100) | 0/2 | 329.5 ± 132.2 | |
| Citrobacter koseri | 0/1 (0) | 1/1 | ||
| Corynebacterium propinquum | 1/1 (100) | 1 | 194 | |
| Corynebacterium striatum | 10/11 (90.9) | 1 | 0/5 | 5,620 ± 5,581 |
| Enterobacter aerogenes | 7/7 (100) | 0/6 | 520.7 ± 382.1 | |
| Enterobacter cloacae | 2/3 (66.7) | 1/3 | 29 ± 41.1 | |
| Enterococcus faecalis | 5/7 (71.4) | 7 | 1/5 | 168.9 ± 378.2 |
| Non-faecalis Enterococcus | 1/1 (100) | 1/1 | 259 | |
| Escherichia coli | 4/5 (80.0) | 1/4 | 208.0 ± 294.2 | |
| Haemophilus influenzae | 12/14 (85.7) | 2/13 | 247.2 ± 325.2 | |
| Klebsiella oxytoca | 1/1 (100) | 0/1 | 1,729 | |
| Klebsiella pneumoniae | 5/5 (100) | 0/3 | 57.2 ± 61.5 | |
| Nocardia asteroides | 1/1 (100) | 2,133 | ||
| Proteus mirabilis | 0/1 (0) | 1/1 | ||
| Pseudomonas aeruginosa | 19/25 (76.0) | 6/15 | 440.1 ± 940.5 | |
| Non-aeruginosa Pseudomonas | 1/1 (100) | 0/1 | 76 | |
| Serratia marcescens | 3/4 (75.0) | 1/2 | 129.5 ± 135.2 | |
| Non-marcescens Serratia | 1/1 (100) | 0/1 | 130 | |
| Staphylococcus aureus | 25/33 (75.8) | 6/18 | 191.64 ± 314.7 | |
| Stenotrophomonas maltophilia | 6/7 (85.7) | 1/5 | 625.0 ± 764.3 | |
| Streptococcus group B | 0/2 (0) | 2/2 | ||
| Streptococcus group C | 0/1 (0) | 1/1 | ||
| Streptococcus pneumoniae | 5/5 (100) | 1 | 0/4 | 626.2 ± 823.8 |
| Coronavirus | 2 | |||
| Human metapneumovirus | 4/4 (100) | 0/2 | NAd | |
| Influenza A | 6/6 (100) | 1 | NA | |
| Parainfluenza virus 1 | 1/2 (50) | 1/2 | NA | |
| Respiratory syncytial virus | 3/3 (100) | 0/1 | NA | |
| Candida spp. | 18/31 (58.1) | 2 | 7/18 | 165.8 ± 304.8 |
| Aspergillus spp. | 6/12 (50.0) | 1 | 6/10 | 55.7 ± 123.9 |
| Penicillium spp. | 0/6 (0) | 6/6 | 0 | |
| Microascus spp. | 0/1 (0) | 1/1 | 0 | |
| Coccidioides immitis | 1/1 (100) | 0/1 | 10 |
RT-PCR–ESI-MS performance is detailed by agent.
A positive reference detection was defined as a positive result by standard culture or RVP testing.
Polymicrobial infections were defined as infections that contained ≥2 of the following: heavy respiratory flora (≥10,000 CFU/ml), a bacterial pathogen, a viral pathogen, or a fungal pathogen.
NA, not applicable.
Table 2.
Summary of sensitivity, specificity, and concordance for detection of BT and common pathogens
| Detection | No. of samples processed | Concordance with standard method (%) | No. identified as positive by RT-PCR–ESI-MS/no. identified as positive by reference test (% [95% CI]) | No. identified as negative by RT-PCR–ESI-MS/no. identified as negative by reference test (% [95% CI]) |
|---|---|---|---|---|
| Biothreat agents | 202 | 99 | 133/135 (98.5 [94.2–99.7]) | 66/66 (100.0 [93.1–100.0]) |
| Common bacterial organisms | 202 | 77.1 | 117/143 (81.8 [74.3–87.6]) | 81/110 (73.6 [64.2–81.4]) |
| Common viral organisms | 89 | 96.6 | 14/15 (93.3 [66.0–99.7]) | 72/74 (97.3 [89.7–99.5]) |
| Common fungal organisms | 145 | 80.0 | 23/54 (42.6 [29.5–56.7]) | 92/94 (97.8 [91.8–99.6]) |
Table 3.
RT-PCR–ESI-MS detection performance with monomicrobial versus polymicrobial samplesa
| Background | No. of samples identified as positive by RT-PCR–ESI-MS/no. of samples identified as positive by reference test (% [95% CI]) |
|||
|---|---|---|---|---|
| BT agents | Bacterial pathogens | Viral pathogens | Fungal pathogens | |
| Monomicrobial (no other organisms) | 82/83 (98.8 [92.3–99.9]) | 35/38 (92.1 [77.5–97.9]) | 10/10 (100 [65.5–100]) | 10/15 (66.7 [38.7–87.0]) |
| Polymicrobial (≥1 other organism) | 51/52 (98.1 [88.4–99.9]) | 83/105 (79.1 [69.8–86.1]) | 4/5 (80.0 [29.9–98.9]) | 14/38 (36.8 [22.3–54.0]) |
Normal flora detected at <10,000 CFU/ml was not considered a background organism.
BT detection.
The sensitivity and specificity of RT-PCR–ESI-MS for the BT spikes were 98.5% (95% CI, 94.2 to 99.7%) and 100.0% (95% CI, 93.1 to 100.0%), respectively (Table 2). Further analysis was performed to determine whether the sensitivity and specificity varied based on interference from an endogenous polymicrobial matrix. After comparison of reference testing and spiking patterns, it was found that BT DNA had been included in 52 samples that were culture or PCR positive for two or more organisms. Rates of BT detection in the presence or absence of endogenous organisms were not significantly different (P = 0.30) (Table 3), and overall concordance with the spiking pattern was 99% (Table 2). The average quantity of DNA detected by RT-PCR–ESI-MS quantification from a 6- to 8-fg spike was 116 genomes per well (Table 1).
Samples BVBT0337 and BVBT0346 were spiked with Francisella tularensis DNA, but detection was not made by the BD assay. These samples represented the two BT spikes not recovered by RT-PCR–ESI-MS. Both samples had endogenous organisms present before BT spiking. Sample BVBT0337 was positive by both culture and our research assay for Klebsiella oxytoca (>10,000 CFU/ml by culture and 1,729 genomes/well by RT-PCR–ESI-MS) and Enterococcus faecalis (5,200 CFU/ml by culture and 13 genomes/well by RT-PCR–ESI-MS), while sample BVBT0346 was positive for influenza A with both the Luminex RVP test and our research assay.
Common bacterial, viral, and fungal pathogen detection.
A total of 212 naturally occurring pathogens, representing 38 species, were detected by reference culture methods and were included in this study (Table 1). The sensitivity and specificity for each category of pathogen (bacterial, viral, and fungal) are detailed below and presented in Table 2.
For common bacterial pathogen detection, the RT-PCR–ESI-MS sensitivity was 81.8% (95% CI, 74.3 to 87.6%) and the specificity was 73.6% (95% CI, 64.2 to 81.4%). Overall concordance between bacterial culture and RT-PCR–ESI-MS results was 77.1% (Table 2). Nine samples contained bacteria detected by culture that were missed by RT-PCR–ESI-MS, and 8 samples contained bacteria detected by RT-PCR–ESI-MS that were not present in cultures. The most common bacterial pathogen missed by the BAC assay was Pseudomonas aeruginosa; 29.4% of the samples (5/17 samples) positive for P. aeruginosa were called false-negative samples (Table 1). Two of the missed specimens concurrently grew heavy mixed respiratory microbiota (≥10,000 CFU/ml) in cultures, while the other 3 cases were reported in the setting of polymicrobial infections. The most common bacterial pathogen detected by the BAC assay when culture results were negative was Enterococcus faecalis. Of 10 positive RT-PCR–ESI-MS detections, only 3 had concordant culture results positive for E. faecalis (Table 1).
In all, 15 viral pathogens were detected by RVP testing in 89 BAL fluid specimens. Ninety-three percent of samples (14/15 samples) were positive for a concordant virus by RT-PCR–ESI-MS, resulting in a sensitivity of 93.3% (95% CI, 66.0 to 99.7%) (Table 2). The only false-negative sample was a sample that tested positive for parainfluenza 1 with the RVP assay but was negative by RT-PCR–ESI-MS. Two samples were positive for coronavirus by RT-PCR–ESI-MS but were found to be negative for viral pathogens by RVP testing, leading to a specificity of 97.3% (95% CI, 89.7 to 99.5%). The overall concordance rate was 96.6%.
A total of 145 BAL fluid specimens were cultured for fungal pathogens after initial Gram staining findings. Of these, 31 were reported as positive for yeast other than Cryptococcus neoformans. No further species identification was performed by the clinical mycology laboratory. RT-PCR–ESI-MS detected a Candida species in 58.1% of these samples (18/31 samples) for a sensitivity of 58.1% (assuming that Candida was the yeast for which the species was not identified). Aspergillus was cultured in 12 samples; of these, 6 cases were detected by RT-PCR–ESI-MS. One sample also was culture positive for Coccidioides immitis, which was concordant with our assay results. The overall rate of agreement for fungal culture results was 80.0%, with a sensitivity of 42.6% (95% CI, 29.5 to 56.7%) and a specificity of 97.8% (95% CI, 91.8 to 99.6%) (Table 2).
Polymicrobial samples.
Seventy-five of the BAL fluid specimens included in this study contained 2 or more naturally occurring organisms, as determined by reference testing. For the purpose of this analysis, normal microbiota quantified at <10,000 CFU/ml was excluded as a contributing naturally occurring organism. Of these samples, 53 contained mixtures of only bacteria, 18 contained mixtures of fungal pathogens with Gram-positive or Gram-negative bacteria, 2 contained mixtures of fungal pathogens, 1 contained a mixture of a virus with Gram-negative bacteria, and 1 contained a mixture of a virus with a fungal pathogen.
The sensitivities of BT, bacterial, viral, and fungal detection in polymicrobial samples versus monomicrobial samples are detailed in Table 3. The sensitivity for bacterial detection was significantly lower in polymicrobial samples (92.1% in monomicrobial backgrounds versus 79.1% in polymicrobial backgrounds [P < 0.05]) but not that for fungal detection (66.7% versus 36.8% [P = 0.130]).
DISCUSSION
This hospital-based retrospective pilot study was designed to define the performance characteristics of a broad-range RT-PCR–ESI-MS platform for the detection of both common and BT pathogens in clinical respiratory samples. Blind spiking with BT DNA was used as a surrogate for infection in residual BAL fluid specimens. Our study showed that this all-in-one platform was able to identify mock BT pathogens correctly, with 99.0% concordance with BT spiking patterns. Furthermore, common bacterial pathogens were detected with 77.1% concordance with conventional microbiology protocols, viral pathogens with 96.6% concordance, and yeast with 80.0% concordance. These results corroborate conclusions from previous studies applying RT-PCR–ESI-MS to organism detection in clinical samples (14, 20), suggesting that this platform could be a useful clinical adjunct to standard microbiology laboratory protocols for BT detection. The platform can deliver robust results and may be able to influence the early recognition of and response to a BT attack.
The central finding of this study is that RT-PCR–ESI-MS can provide accurate detection of BT organisms in clinical samples despite potential interference from complicated endogenous polymicrobial matrices. In addition, the broad-range capabilities of this platform provide diagnostic utility for common pathogens. A platform that incorporates high-fidelity BT surveillance with routine diagnostic assessment of pathogens has both clinical and public health implications, as application of NAATs for BT detection may no longer require a high level of suspicion for individual agents for testing. This information can guide immediate treatment decisions and prevention strategies (i.e., mass prophylactic treatment or vaccination) to avoid further cases of infection (15), significantly reducing morbidity and mortality rates.
The quantities of BT DNA spiked into specimens were intentionally kept low in this study and were calculated to be ≤100 genomic copies of the organism/well. In the majority of cases, ESI-MS analysis detected those concentrations, although the instrument indicated an average concentration detected of 116 ± 114 genomes/well. These levels of BT DNA were significantly lower than the quantities of organisms found in naturally occurring infections. For comparison, Pseudomonas aeruginosa infections were detected by RT-PCR–ESI-MS at an average of 440.1 ± 940.5 genomes/well, and Corynebacterium striatum infections were detected at an average of 5,620 ± 5,581 genomes/well. The BT spike quantities were intentionally kept at low genome copy numbers, to ensure that there was potential for normal microbiota or coinfecting agents to overwhelm the BT spike and to obscure BT recovery by RT-PCR–ESI-MS, as well as to challenge the lower limit of detection for RT-PCR–ESI-MS. Despite these challenges, BT identification was highly accurate, even in specimens with multiple naturally occurring respiratory organisms (Table 3). Because no live BT organisms were used in this study, these data represent our best approximation of the polymicrobial specimen matrices in a real BT attack; a real-world application of this technology would assume a validated nucleic acid extraction protocol for BT agents. True clinical validation would require further testing with live organisms in a biocontainment facility.
In addition to BT infections, common respiratory infections have nonspecific symptoms that do not readily suggest a causative pathogen (28). Usually, empirical antibiotic therapy is initiated upon suspicion of a respiratory infection, but fungal and viral pathogens can cause severe symptoms that would not be expected to respond to antibiotic therapy (29). In undifferentiated infections with multiple possible causative agents, broad-range platforms such as RT-PCR–ESI-MS might be advantageous for rapid identification of pathogens that may require different selected therapy. Sample BVBT0225 was a particularly illustrative case. In this case, the microbiology standard RVP assay was positive for PIV 3, fungal cultures were positive for yeast, and bacterial cultures revealed only normal respiratory microbiota. The RT-PCR–ESI-MS research assay detected both Candida krusei and PIV 3, but results were available within 7 h, compared with the standard processing times of 24 h for RVP results and 2 days for fungal culture results from the clinical microbiology laboratory.
A similar principle applies to fastidious organisms that may require long incubation periods before detection in culture, as this platform can provide faster results. As an example, sample BVBT0252 was positive for Nocardia of unidentified species in 1 of 4 culture tubes after 4 weeks of culture (data not shown), while PCR–ESI-MS identified the sample as positive for Nocardia within 6 h. Similarly, sample BVBT0200 was positive for Coccidioides immitis by culture after a 6-day incubation period (data not shown), but PCR–ESI-MS detected Coccidioides immitis in 6 h.
Although the RT-PCR–ESI-MS platform demonstrated the capacity to distinguish polymicrobial infections, analysis of samples that contained multiple organisms revealed a significantly higher rate of false-negative bacterial detections than in single-agent samples (Table 3). This observation was likely the effect of interference from the background polymicrobial matrices, which can compete for PCR reagents and obscure pathogen identification (an inherent limitation of PCR-based methods). Decreased polymicrobial sensitivity was statistically significant for bacterial detection but not for BT detection. These findings were likely related to inherent characteristics of the assay design, namely, that the BD plate used for BT detection consisted of 36 primers, 25 of which were dedicated to specific genus and species differentiation of 17 high-priority BT organisms. These primers were not designed to be as broad range as the primers used in the BAC assay, which relied more heavily on universally conserved genomic targets such as 16S rRNA and 23S rRNA genes to characterize up to 400 species of bacteria. When universally conserved primer targets are used, competition for primers and enzymes can result in preferential amplification of more-populous organisms over other organisms, and simultaneous pathogen detection may be reliable only when the original concentrations of the organisms are of similar orders of magnitude. This problem became particularly pronounced in fungal detection; the BAC primers were designed for Candida detection, which led to the low overall sensitivity and high specificity observed in this study for fungal detection.
Despite the potential clinical gains this broad-range NAAT offers, we noted various logistical barriers that must be addressed before RT-PCR–ESI-MS can be implemented in clinical settings. The Ibis T5000 ESI-MS system used in this study was the first automated model available for research-use-only testing and was found to be prone to mechanical malfunctions. Additionally, as with many diagnostic platforms of this nature, the initial costs associated with deployment of a RT-PCR–ESI-MS platform are substantial and can pose an additional barrier to routine implementation in clinical laboratories. Also, while results of this study suggest that the use of RT-PCR–ESI-MS with standard culture protocols would represent a significant improvement over current methods, the sensitivity and specificity for common bacterial and fungal detection are still suboptimal and do not warrant the use of RT-PCR–ESI-MS as a stand-alone diagnostic testing platform. Future studies that build on these data may include different or modified primer sequences to better target commonly encountered respiratory pathogens.
In conclusion, the RT-PCR–ESI-MS platform demonstrated potential diagnostic utility for biothreat and common pathogen detection, with results comparable to those of a variety of reference microbiological assays. As a platform for BT surveillance, RT-PCR–ESI-MS rapidly detected mock BT DNA in clinical samples with nearly 100% accuracy despite being challenged with interference from naturally occurring polymicrobial matrices, as well as the lower limit of detection for RT-PCR–ESI-MS. Further validation in specialized biosafety laboratories utilizing intact organisms spiked into clinical samples or inoculated into model organisms may ultimately justify placing RT-PCR–ESI-MS systems in key strategic locations for BT surveillance. These results warrant further prospective evaluation of the RT-PCR–ESI-MS platform for broad-range diagnostic evaluations, with future studies focusing on elucidating the true clinical benefits of rapid PCR-based results for patient management decisions and addressing the associated challenges of integrating such technologies into routine care.
ACKNOWLEDGMENTS
Justin Hardick, Richard Rothman, Samuel Yang, Stephen Peterson, Billie Jo Masek, Karen C. Carroll, and Charlotte A. Gaydos received funds from Ibis Biosciences/Abbott for prior research projects. Funding was provided by the National Center for the Study of Preparedness and Catastrophic Event Response (project domain 2010-ST_061 PA0001, grant 108822), the Middle Atlantic Region Center of Excellence for Biodefense and Emerging Infectious Diseases Research Program (NIAID grant U54 AI057168), and the Doris Duke Charitable Foundation.
Footnotes
Published ahead of print 31 July 2013
REFERENCES
- 1.Dennis DT, Inglesby TV, Henderson DA, Bartlett JG, Ascher MS, Eitzen E, Fine AD, Friedlander AM, Hauer J, Layton M, Lillibridge SR, McDade JE, Osterholm MT, O'Toole T, Parker G, Perl TM, Russell PK, Tonat K, Working Group on Civilian Biodefense 2001. Tularemia as a biological weapon: medical and public health management. JAMA 285:2763–2773 [DOI] [PubMed] [Google Scholar]
- 2.Inglesby TV, Dennis DT, Henderson DA, Bartlett JG, Ascher MS, Eitzen E, Fine AD, Friedlander AM, Hauer J, Koerner JF, Layton M, McDade J, Osterholm MT, O'Toole T, Parker G, Perl TM, Russell PK, Schoch-Spana M, Tonat K, Working Group on Civilian Biodefense 2000. Plague as a biological weapon: medical and public health management. JAMA 283:2281–2290 [DOI] [PubMed] [Google Scholar]
- 3.Inglesby TV, O'Toole T, Henderson DA, Bartlett JG, Ascher MS, Eitzen E, Friedlander AM, Gerberding J, Hauer J, Hughes J, McDade J, Osterholm MT, Parker G, Perl TM, Russell PK, Tonat K, Working Group on Civilian Biodefense 2002. Anthrax as a biological weapon, 2002: updated recommendations for management. JAMA 287:2236–2252 [DOI] [PubMed] [Google Scholar]
- 4.World Health Organization 1970. Health aspects of chemical and biological weapons. World Health Organization, Geneva, Switzerland [Google Scholar]
- 5.Office of Technology Assessment 1993. Proliferation of weapons of mass destruction: assessing the risks. OTA-ISC-559 Office of Technology Assessment, Washington, DC [Google Scholar]
- 6.Kaufman Z, Aharonowitz G, Dichtiar R, Green MS. 2006. Estimating the usual prevalence and incidence of acute illness in the community: implications for pandemic influenza and bioterrorism preparedness. Isr. Med. Assoc. J. 8:563–567 [PubMed] [Google Scholar]
- 7.Khan AS, Levitt AM, Sage M. 2000. Biological and chemical terrorism: strategic plan for preparedness and response: recommendations of the CDC Strategic Planning Workgroup. MMWR Recomm. Rep. 49(RR-4):1–14 [PubMed] [Google Scholar]
- 8.Taitt CR, Malanoski AP, Lin B, Stenger DA, Ligler FS, Kusterbeck AW, Anderson GP, Harmon SE, Shriver-Lake LC, Pollack SK, Lennon DM, Lobo-Menendez F, Wang Z, Schnur JM. 2008. Discrimination between biothreat agents and ‘near neighbor' species using a resequencing array. FEMS Immunol. Med. Microbiol. 54:356–364 [DOI] [PubMed] [Google Scholar]
- 9.Sewell DL. 2003. Laboratory safety practices associated with potential agents of biocrime or bioterrorism. J. Clin. Microbiol. 41:2801–2809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sintchenko V, Gallego B. 2009. Laboratory-guided detection of disease outbreaks: three generations of surveillance systems. Arch. Pathol. Lab. Med. 133:916–925 [DOI] [PubMed] [Google Scholar]
- 11.Cirino NM, Musser KA, Egan C. 2004. Multiplex diagnostic platforms for detection of biothreat agents. Expert Rev. Mol. Diagn. 4:841–857 [DOI] [PubMed] [Google Scholar]
- 12.Maiwald M. 2004. Broad-range PCR for detection and identification of bacteria, p 379–390 In Persing DH, Tenover FC, Versalovic J, Tang Y-W, Unger ER, Relman DA, White TJ. (ed), Molecular microbiology: diagnostic principles and practice. ASM Press, Washington, DC [Google Scholar]
- 13.Baldwin CD, Howe GB, Sampath R, Blyn LB, Matthews H, Harpin V, Hall TA, Drader JJ, Hofstadler SA, Eshoo MW, Rudnick K, Studarus K, Moore D, Abbott S, Janda JM, Whitehouse CA. 2009. Usefulness of multilocus polymerase chain reaction followed by electrospray ionization mass spectrometry to identify a diverse panel of bacterial isolates. Diagn. Microbiol. Infect. Dis. 63:403–408 [DOI] [PubMed] [Google Scholar]
- 14.Chen KF, Rothman RE, Ramachandran P, Blyn L, Sampath R, Ecker DJ, Valsamakis A, Gaydos CA. 2011. Rapid identification viruses from nasal pharyngeal aspirates in acute viral respiratory infections by RT-PCR and electrospray ionization mass spectrometry. J. Virol. Methods 173:60–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ecker DJ, Sampath R, Massire C, Blyn LB, Hall TA, Eshoo MW, Hofstadler SA. 2008. Ibis T5000: a universal biosensor approach for microbiology. Nat. Rev. Microbiol. 6:553–558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ecker JA, Massire C, Hall TA, Ranken R, Pennella TT, Agasino Ivy C, Blyn LB, Hofstadler SA, Endy TP, Scott PT, Lindler L, Hamilton T, Gaddy C, Snow K, Pe M, Fishbain J, Craft D, Deye G, Riddell S, Milstrey E, Petruccelli B, Brisse S, Harpin V, Schink A, Ecker DJ, Sampath R, Eshoo MW. 2006. Identification of Acinetobacter species and genotyping of Acinetobacter baumannii by multilocus PCR and mass spectrometry. J. Clin. Microbiol. 44:2921–2932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen KF, Blyn L, Rothman RE, Ramachandran P, Valsamakis A, Ecker D, Sampath R, Gaydos CA. 2011. Reverse transcription polymerase chain reaction and electrospray ionization mass spectrometry for identifying acute viral upper respiratory tract infections. Diagn. Microbiol. Infect. Dis. 69:179–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jeng K, Gaydos CA, Blyn LB, Yang S, Won H, Matthews H, Toleno D, Hsieh YH, Carroll KC, Hardick J, Masek B, Kecojevic A, Sampath R, Peterson S, Rothman RE. 2012. Comparative analysis of two broad-range PCR assays for pathogen detection in positive-blood-culture bottles: PCR-high-resolution melting analysis versus PCR-mass spectrometry. J. Clin. Microbiol. 50:3287–3292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kaleta EJ, Clark AE, Cherkaoui A, Wysocki VH, Ingram EL, Schrenzel J, Wolk DM. 2011. Comparative analysis of PCR-electrospray ionization/mass spectrometry (MS) and MALDI-TOF/MS for the identification of bacteria and yeast from positive blood culture bottles. Clin. Chem. 57:1057–1067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kaleta EJ, Clark AE, Johnson DR, Gamage DC, Wysocki VH, Cherkaoui A, Schrenzel J, Wolk DM. 2011. Use of PCR coupled with electrospray ionization mass spectrometry for rapid identification of bacterial and yeast bloodstream pathogens from blood culture bottles. J. Clin. Microbiol. 49:345–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Deyde VM, Sampath R, Garten RJ, Blair PJ, Myers CA, Massire C, Matthews H, Svoboda P, Reed MS, Pohl J, Klimov AI, Gubareva LV. 2010. Genomic signature-based identification of influenza A viruses using RT-PCR/electro-spray ionization mass spectrometry (ESI-MS) technology. PLoS One 5:e13293. 10.1371/journal.pone.0013293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jeng K, Massire C, Zembower TR, Deyde VM, Gubareva LV, Hsieh YH, Rothman RE, Sampath R, Penugonda S, Metzgar D, Blyn LB, Hardick J, Gaydos CA. 2012. Monitoring seasonal influenza A evolution: rapid 2009 pandemic H1N1 surveillance with a reverse transcription-polymerase chain reaction/electro-spray ionization mass spectrometry assay. J. Clin. Virol. 54:332–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Metzgar D, Baynes D, Myers CA, Kammerer P, Unabia M, Faix DJ, Blair PJ. 2010. Initial identification and characterization of an emerging zoonotic influenza virus prior to pandemic spread. J. Clin. Microbiol. 48:4228–4234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Versalovic J, Carroll KC, Funke G, Jorgensen JH, Landry ML, Warnock DW. (ed). 2011. Manual of clinical microbiology, 10th ed. ASM Press, Washington, DC [Google Scholar]
- 25.Garcia LS. (ed). 2010. Clinical microbiology procedures handbook, 3rd ed. ASM Press, Washington, DC [Google Scholar]
- 26.Jiang Y, Hofstadler SA. 2003. A highly efficient and automated method of purifying and desalting PCR products for analysis by electrospray ionization mass spectrometry. Anal. Biochem. 316:50–57 [DOI] [PubMed] [Google Scholar]
- 27.Sampath R, Russell KL, Massire C, Eshoo MW, Harpin V, Blyn LB, Melton R, Ivy C, Pennella T, Li F, Levene H, Hall TA, Libby B, Fan N, Walcott DJ, Ranken R, Pear M, Schink A, Gutierrez J, Drader J, Moore D, Metzgar D, Addington L, Rothman R, Gaydos CA, Yang S, St George K, Fuschino ME, Dean AB, Stallknecht DE, Goekjian G, Yingst S, Monteville M, Saad MD, Whitehouse CA, Baldwin C, Rudnick KH, Hofstadler SA, Lemon SM, Ecker DJ. 2007. Global surveillance of emerging influenza virus genotypes by mass spectrometry. PLoS One 2:e489. 10.1371/journal.pone.0000489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fernandez-Sabe N, Roson B, Carratala J, Dorca J, Manresa F, Gudiol F. 2003. Clinical diagnosis of Legionella pneumonia revisited: evaluation of the Community-Based Pneumonia Incidence Study Group scoring system. Clin. Infect. Dis. 37:483–489 [DOI] [PubMed] [Google Scholar]
- 29.Johansson N, Kalin M, Tiveljung-Lindell A, Giske CG, Hedlund J. 2010. Etiology of community-acquired pneumonia: increased microbiological yield with new diagnostic methods. Clin. Infect. Dis. 50:202–209 [DOI] [PMC free article] [PubMed] [Google Scholar]