

TEXT
Chromatin is organized into domains that are permissive or repressive for transcription, depending largely on regional patterns of covalent histone modifications. These epigenetic marks recruit nuclear factors that regulate transcription either directly or via their actions to alter chromatin accessibility. For example, dimethylation of histone-3 at lysine-9 (H3K9Me2) recruits repressive chromatin modifiers and, accordingly, inhibits gene transcription. Deposition of the H3K9me2 modification is mediated by the histone methyltransferases (HMT) G9a (or KMT1C) and GLP (or KMT1D) (1). These enzymes act predominantly in euchromatin to coordinate repressive changes in gene expression programs during cell fate decisions (2).
The importance of G9a/GLP in human health is underscored by diseases that are linked to perturbations in this HMT complex. For example, G9a activity participates in the resolution of inflammatory responses via its repression of proinflammatory cytokines (3). Amplification of G9a is observed in diverse classes of tumors, and in hepatocellular carcinomas, G9a knockdown inhibits their continued growth (4). As such, HMT enzymes, including G9a/GLP, are attractive targets for the design of small-molecule inhibitors that could reverse gene repression linked to pathogenesis. In this regard, two groups have reported inhibitors, BRD4770 and UNC0636, which have nanomolar affinity and exquisite specificity for G9a/GLP, rendering them promising compounds for future in vivo studies (5, 6).
In this issue of Molecular and Cellular Biology, Artal-Martinez de Narvajas et al. (7) report that treatment of tumor cell lines with G9a inhibitors leads to the formation of autophagosome-like structures, suggesting that G9a plays a role in regulating gene expression changes governing this cellular process. Macroautophagy is used by nearly all cells to remove damaged organelles and extraneous proteins. Normally, cells in homeostasis inhibit autophagy by the action of mammalian target of rapamycin complex 1 (mTORC1). However, upon experiencing starvation or other types of stress, the same cells suppress mTORC1 kinase activity and rapidly form autophagosomes (8). Initiation of macroautophagy is thought to be transcription independent, because the mTORC1 kinase directly represses the function of proteins involved in autophagosome formation (Fig. 1, left). However, recent studies have hinted that transcription-dependent steps may drive macroautophagy following mTORC1-mediated initiation (9).
Fig 1.
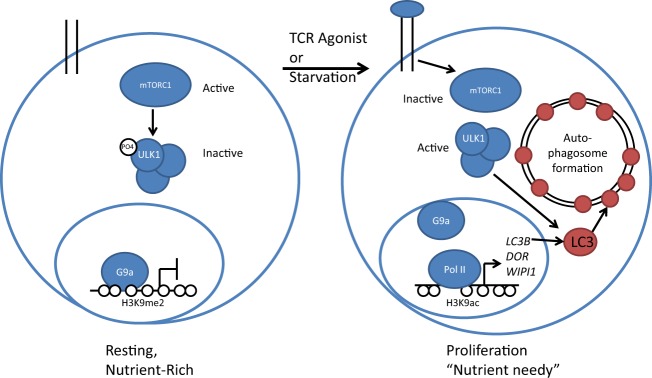
Repression of autophagosome formation by G9a. Under resting, nutrient-rich conditions (left), G9a represses autophagosome genes LC3B, DOR, and WIPI1 by modifying the chromatin around gene promoters with the repressive H3K9me2 mark. In the cytoplasm, mTORC1 kinase phosphorylates and inactivates complexes containing ULK1 that drive autophagosome formation. Upon starvation or during T cell proliferation (right), G9a and H3K9me2 are removed from relevant promoters, leading to expression of LCB3, DOR, and WIPI1, which are essential components of autophagosomes. This transcription-dependent process synergizes with inactivation of mTORC1, which releases restrictions on ULK1 complexes and guides autophagic flux to its completion.
The study by Artal-Martinez de Narvajas et al. (7) now shows that full engagement of the autophagy pathway is indeed transcription dependent and requires relief of G9a repression at several relevant genes. Specifically, the authors demonstrate that in unstressed cells, G9a binds directly to the promoters of LC3B, WIPI1, and DOR, whose products are required for the formation of autophagosomes (10). In keeping with its role as a repressor, RNA interference (RNAi)-mediated depletion of G9a enhances H3K9 acetylation, polymerase II (Pol II) binding, and transcription of these three genes while attenuating levels of the H3K9me2 repressive mark. Direct regulation of LC3B and WIPI1 is dependent on the HMT function of G9a, since deletion of its enzymatic SET domain reverses repression of these autophagy-associated genes. Importantly, while G9a inhibition is sufficient to induce autophagosome formation, it does not lead to their fusion with lysosomes, which is required to complete the process known as autophagic flux (11). Instead, inhibition of both G9a and mTORC1 kinase activity is required to drive autophagy to completion.
Artal-Martinez de Narvajas et al. extend their findings to show that G9a also restricts autophagy in primary cells, namely, human CD4+ T lymphocytes (7). Prior studies have shown that autophagosomes form in these cells during T cell receptor (TCR)-mediated proliferation (12). Indeed, mice rendered incapable of inducing autophagy in this lineage exhibit impaired T cell proliferation and survival (13). In their study, Artal-Martinez de Narvajas et al. used TCR agonists and starvation each to induce autophagy in CD4+ cells and demonstrated that sustained expression of LC3B, DOR, and WIPI1 again requires expulsion of G9a from their promoters (7). While prior studies have shown that G9a is dispensable for lymphocyte development, this HMT is necessary for differentiation of mature T cells into distinct functional lineages (14). However, the connections between proliferation-induced autophagy and differentiation into functional T cell subsets during an immune response remain to be established.
Overall, this study establishes a new regulatory pathway that represses the initial stages of autophagy (7). Reversal of G9a-mediated transcriptional repression, in conjunction with mTORC1 inhibition, drives autophagy, recovering nutrients that maintain viability during cellular stress and proliferative bursts. In addition, Artal-Martinez de Narvajas et al. (7) provide numerous avenues for future studies. For example, the mechanisms by which G9a is removed or displaced from promoters at autophagosome genes as well its relationship to other chromatin modifiers, such as the autophagy-associated histone acetyltransferase hMOF (15), remain to be determined. To date, at least two transcription factors, TFEB and JunB, have been implicated in the regulation of autophagy (9, 16). As Atral-Martinez de Narvajas et al. suggest the leading candidate for links to G9a repression of autophagy in T lymphocytes is JunB, which is a component of signaling pathways downstream of TCR activation. Another interesting possibility for future exploration is a role for G9a in autophagy that is independent of its function in histone modification. Perhaps the G9a/GLP complex methylates directly lysines on proteins involved in autophagosome formation.
Finally, the study described here (7) should stimulate further exploration of the therapeutic potential for this G9a-mediated repression pathway. Indeed, perturbations of autophagy have been implicated in cancer, neurodegeneration, autoimmunity, and the development of diabetes (8). The therapeutic potential of G9a inhibitors for reversal of these pathogenic processes was bolstered by a recent study showing that BRD4770 can synergize with other inducers of autophagy to block the growth of pancreatic cancer cells in vitro (17). Together, G9a, mTORC1, and autophagy stand at the crossroads of molecular targets for inhibitors that could ultimately find clinical applications for the treatment of multiple pathologies.
ACKNOWLEDGMENTS
This work was supported by NIH grants AI079732 (E.M.O.) and AI007163 (P.L.C.).
Footnotes
Published ahead of print 26 August 2013
The views expressed in this Commentary do not necessarily reflect the views of the journal or of ASM.
REFERENCES
- 1.Tachibana M, Ueda J, Fukuda M, Takeda N, Ohta T, Iwanari H, Sakihama T, Kodama T, Hamakubo T, Shinkai Y. 2005. Histone methyltransferases G9a and GLP form heteromeric complexes and are both crucial for methylation of euchromatin at H3-K9. Genes Dev. 9:815–826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tachibana M, Sugimoto K, Nozaki M, Ueda J, Ohta T, Ohki M, Fukuda M, Takeda N, Niida H, Kato H, Shinkai Y. 2002. G9a histone methyltransferase plays a dominant role in euchromatic histone H3 lysine 9 methylation and is essential for early embryogenesis. Genes Dev. 16:1779–1791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen X, El Gazzar M, Yoza BK, McCall CE. 2009. The NF-κB factor RelB and histone H3 lysine methyltransferase G9a directly interact to generate epigenetic silencing in endotoxin tolerance. J. Biol. Chem. 284:27857–27865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kondo Y, Shen L, Suzuki S, Kurokawa T, Masuko K, Tanaka Y, Kato H, Mizuno Y, Yokoe M, Sugauchi F, Hirashima N, Orito E, Osada H, Ueda R, Guo Y, Chen X, Issa J-PJ, Sekido Y. 2007. Alterations of DNA methylation and histone modifications contribute to gene silencing in hepatocellular carcinomas. Hepatol. Res. 37:974–983 [DOI] [PubMed] [Google Scholar]
- 5.Yuan Y, Wang Q, Paulk J, Kubicek S, Kemp MM, Adams DJ, Shamji AF, Wagner BK, Schreiber SL. 2012. A small-molecule probe of the histone methyltransferase G9a induces cellular senescence in pancreatic adenocarcinoma. ACS Chem. Biol. 7:1152–1157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vedadi M, Barsyte-Lovejoy D, Liu F, Rival-Gervier S, Allali-Hassani A, Labrie V, Wigle TJ, Dimaggio PA, Wasney GA, Siarheyeva A, Dong A, Tempel W, Wang S-C, Chen X, Chau I, Mangano TJ, Huang X-P, Simpson CD, Pattenden SG, Norris JL, Kireev DB, Tripathy A, Edwards A, Roth BL, Janzen WP, Garcia BA, Petronis A, Ellis J, Brown PJ, Frye SV, Arrowsmith CH, Jin J. 2011. A chemical probe selectively inhibits G9a and GLP methyltransferase activity in cells. Nat. Chem. Biol. 7:648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Artal-Martinez de Narvajas A, Gomez TS, Zhang J-S, Mann AO, Taoda Y, Gorman JA, Herreros-Villanueva M, Gress TM, Ellenrieder V, Bujanda L, Kim D-H, Kozikowski AP, Koenig A, Billadeau DD. 2013. Epigenetic regulation of autophagy by the methyltransferase G9a. Mol. Cell. Biol. 33:3983–3993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Levine B, Kroemer G. 2008. Autophagy in the pathogenesis of disease. Cell 132:27–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roczniak-Ferguson A, Petit CS, Froehlich F, Qian S, Ky J, Angarola B, Walther TC, Ferguson SM. 2012. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci. Signal. 5:ra42. 10.1126/scisignal.2002790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu J, Dang Y, Su W, Liu C, Ma H, Shan Y, Pei Y, Wan B, Guo J, Yu L. 2006. Molecular cloning and characterization of rat LC3A and LC3B—two novel markers of autophagosome. Biochem. Biophys. Res. Commun. 339:437–442 [DOI] [PubMed] [Google Scholar]
- 11.Hansen TE, Johansen T. 2011. Following autophagy step by step. BMC Biol. 9:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li C, Capan E, Zhao Y, Zhao J, Stolz D, Watkins SC, Jin S, Lu B. 2006. Autophagy is induced in CD4+ T cells and important for the growth factor-withdrawal cell death. J. Immunol. 177:5163–5168 [DOI] [PubMed] [Google Scholar]
- 13.Pua HH, Dzhagalov I, Chuck M, Mizushima N, He Y-W. 2007. A critical role for the autophagy gene Atg5 in T cell survival and proliferation. J. Exp. Med. 204:25–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thomas LR, Miyashita H, Cobb RM, Pierce S, Tachibana M, Hobeika E, Reth M, Shinkai Y, Oltz EM. 2008. Functional analysis of histone methyltransferase g9a in B and T lymphocytes. J. Immunol. 181:485–493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Füllgrabe J, Lynch-Day MA, Heldring N, Li W, Struijk RB, Ma Q, Hermanson O, Rosenfeld MG, Klionsky DJ, Joseph B. 2013. The histone H4 lysine 16 acetyltransferase hMOF regulates the outcome of autophagy. Nature 10.1038/nature12313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yogev O, Goldberg R, Anzi S, Yogev O, Shaulian E. 2010. Jun proteins are starvation-regulated inhibitors of autophagy. Cancer Res. 70:2318–2327 [DOI] [PubMed] [Google Scholar]
- 17.Yuan Y, Tang AJ, Castoreno AB, Kuo S, Wang Q, Kuballa P, Xavier R, Shamji AF, Schreiber SL, Wagner BK. 2013. Gossypol and an HMT G9a inhibitor act in synergy to induce cell death in pancreatic cancer cells. Cell Death Dis. 4:e690. 10.1038/cddis.2013.191 [DOI] [PMC free article] [PubMed] [Google Scholar]