

Abstract
Following Anopheles mosquito-mediated introduction into a human host, Plasmodium parasites infect hepatocytes and undergo intensive replication. Accumulating evidence indicates that CD8+ T cells induced by immunization with attenuated Plasmodium sporozoites can confer sterile immunity at the liver stage of infection; however, the mechanisms underlying this protection are not clearly understood. To address this, we generated recombinant Plasmodium berghei ANKA expressing a fusion protein of an ovalbumin epitope and green fluorescent protein in the cytoplasm of the parasite. We have shown that the ovalbumin epitope is presented by infected liver cells in a manner dependent on a transporter associated with antigen processing and becomes a target of specific CD8+ T cells from the T cell receptor transgenic mouse line OT-I, leading to protection at the liver stage of Plasmodium infection. We visualized the interaction between OT-I cells and infected hepatocytes by intravital imaging using two-photon microscopy. OT-I cells formed clusters around infected hepatocytes, leading to the elimination of the intrahepatic parasites and subsequent formation of large clusters of OT-I cells in the liver. Gamma interferon expressed in CD8+ T cells was dispensable for this protective response. Additionally, we found that polyclonal ovalbumin-specific memory CD8+ T cells induced by de novo immunization were able to confer sterile protection, although the threshold frequency of the protection was relatively high. These studies revealed a novel mechanism of specific CD8+ T cell-mediated protective immunity and demonstrated that proteins expressed in the cytoplasm of Plasmodium parasites can become targets of specific CD8+ T cells during liver-stage infection.
INTRODUCTION
Plasmodium sporozoites are transmitted by the bites of Anopheles mosquitoes under the skin and are transported via the bloodstream to the liver, where they infect hepatocytes. Immunization with irradiated sporozoites can induce sterile protection at preerythrocytic stages of infection in both mice and humans (1–3). Similarly, sterile protective immunity is induced by Plasmodium parasites that have been genetically attenuated by a gene deletion and which arrest at the hepatic stage (4, 5). Recent studies have shown that the infection of mice under a chloroquine shield induces a protective immune response at the hepatic stage of infection (6). Immunization by these methods induces multiple different mechanisms of protection involving CD8+ T cells, CD4+ T cells, B cells, and NK cells (7, 8). Among the major effector cells are CD8+ T cells, which recognize malaria antigen in association with major histocompatibility complex class I (MHC-1) during liver-stage infection (9).
Targets for protective immunity against malaria were identified using antibodies obtained from mice immunized with irradiated sporozoites, including circumsporozoite protein (CSP), which was extensively investigated (10, 11). CSP is expressed on the surface of sporozoites and liver-stage malaria parasites and is the most advanced target antigen of liver-stage vaccine development. The major liver-stage effector cells specific for CSP are CD8+ T cells, as shown by the depletion of CD8+ T cells with the antibody abrogating protection and by the resistance to subsequent challenge infection conferred by cloned specific T cells. Further studies using CSP transgenic mice indicated that additional protective antigens are present, although CSP is the major antigen that can induce protection against preerythrocytic forms of malaria in BALB/c mice (12). Additional candidate antigens at the liver stage of infection include sporozoite surface protein 2 (SSP), which was identified using an antibody produced by BALB/c mice after immunization with irradiated sporozoites and which induces protection that is mediated by CD8+ T cells, CD4+ T cells, and antibodies (13–15). Protective immunity via immunization is much more difficult to establish in C57BL/6 (B6) mice than in BALB/c mice, partly because the H-2b-restricted cytotoxic T lymphocyte (CTL) epitope is not present in CSP (16). However, protection is induced in B6 mice by immunization with attenuated Plasmodium parasites or infection under a chloroquine shield. This protective immunity is also mediated by CD8+ T cells, whose target antigen is not CSP. The latter studies suggest the existence of unknown target antigens recognized by CD8+ T cells in infected hepatocytes, in addition to CSP and SSP2.
Research efforts are in progress to identify novel malaria antigen targets expressed at the liver stage. Genome-wide expression profiling studies have indicated that many malaria proteins are expressed during liver-stage infection (17, 18). However, the criteria that would frame the search for target malaria antigens have not yet been established. Several studies have suggested that the localization of antigen within microbial pathogens is important for the generation of specific T cells and the resulting protection. It is generally thought that secreted antigens are more accessible to antigen presentation pathways and induce strong T cell immune responses (19). For example, intracellular bacteria such as Mycobacterium tuberculosis remain in the phagosome, where they survive and replicate. The secreted form of the antigens expressed in these bacteria can be presented via the MHC-I pathway, through a process that appears to be facilitated by an increase in permeation of the endosomal membrane by the microbe (20, 21). In an infection model using recombinant Trypanosoma cruzi expressing an ovalbumin (OVA) epitope, it was shown that host cells were able to present OVA via the MHC-I pathway when the antigen was produced in secretory form but not the cytoplasmic or transmembrane form (22). It has also been proposed that CSP is released from the surface of sporozoites directly into the cytoplasm of host hepatocytes, where it binds to RNA-associated host cell targets (23, 24). Furthermore, CSP is released from the surface of sporozoites when they travel through hepatocytes before reaching the final infected hepatocyte and appears to be presented by these traversed hepatocytes to specific T cells (25). Therefore, the search for candidate malaria antigens for liver-stage infection is generally focused on molecules expressed on the surface of parasites. However, it is not clear whether intracytoplasmic molecules are able to become targets of the protective immune responses during liver-stage infection.
In this study, we generated recombinant parasites that exhibited cytoplasmic expression of an OVA epitope presented by MHC-I. We examined whether this epitope was presented by infected hepatocytes and whether it became a target of specific OT-I CD8+ T cells leading to protection at the liver stage of infection. We also examined the mechanisms underlying the presentation of this antigen and visualized the interaction of OT-I cells with infected hepatocytes by intravital imaging using two-photon microscopy (TPM). The results of these experiments suggest that CD8+ T cells can recognize cytoplasmic malaria antigens, form clusters around infected hepatocytes, and protect against parasites.
MATERIALS AND METHODS
Parasites.
Recombinant Plasmodium berghei ANKA expressing class II and class I OVA epitopes fused to the N and C termini of a P. yoelii hsp70 fragment (PbA-hsOVA), respectively, and P. berghei ANKA expressing an OVA class I epitope fused to the C terminus of green fluorescent protein (GFP) (PbA-gfpOVA) were constructed as previously described (26) (Fig. 1A). PbA-hsOVA expresses a recombinant fusion protein containing the N-terminal sequence (amino acids [aa] 1 to 5) of P. berghei hsp70, an OVA MHC-II epitope from positions 323 to 339 (OVA323-339), a truncated sequence (aa 201 to 398) of P. yoelii hsp70, and an OVA257-264 MHC-I epitope. PbA-gfpOVA expresses a protein containing an OVA257-264 MHC-I epitope fused to the C terminus of GFP. After transfection, mice were infected and were maintained under the presence of the antimalaria drug pyrimethamine. PbA-gfpOVA was enriched by sorting of GFP-positive erythrocytes using a FACSAria cell sorter (BD Biosciences, San Jose, CA). The stable transfectant was cloned by limiting dilution in mice and was maintained by alternating passage between Anopheles stephensi and BALB/c mice. Sporozoites were prepared from the salivary glands of A. stephensi mosquitoes after 18 to 24 days of infection with PbA-hsOVA or PbA-gfpOVA.
Fig 1.

Expression of a model antigen in the cytoplasm of recombinant P. berghei ANKA. (A) Schematic representation of the transgenic P. berghei ANKA constructs used in this study. (B) PbA-gfpOVA sporozoites, HepG2 cells infected with PbA-gfpOVA sporozoites in vitro, and infected RBCs (iRBC) were stained with BODIPY-TR-C5-ceramide and DRAQ5, which mark membrane structure and nuclei, respectively. Images were obtained using confocal microscopy. Arrowheads, margin of the RBC. Bars, 5 μm.
Animals.
OT-I and OT-II transgenic mice expressing the T cell receptor (TCR) specific for OVA257-264/Kb and OVA323-339/IAb, respectively, were provided by H. Kosaka (Osaka University, Osaka, Japan) (27, 28). TAP knockout (TAP−/−) mice (B6 background) were provided by H. Watanabe (Ryukyu University, Okinawa, Japan) (29). B6.SJL and OT-I or OT-II mice were interbred, and the offspring were intercrossed to obtain CD45.1+ OT-I or OT-II mice. DsRed transgenic, gamma interferon knockout (IFN-γ−/−), and perforin knockout (perforin−/−) mice were purchased from The Jackson Laboratory (Bar Harbor, ME). DsRed transgenic mice and OT-I mice were crossed to produce DsRed/OT-I mice. OT-I and IFN-γ−/− or perforin−/− mice were bred to produce IFN-γ−/− OT-I mice, perforin−/− OT-I mice, and IFN-γ−/− perforin−/− OT-I mice. B6 and BALB/c mice were purchased from SLC (Shizuoka, Japan). Mice were maintained in the Laboratory Animal Center for Animal Research at Nagasaki University and were used at the age of 8 to 14 weeks. To generate bone marrow chimeras, B6 or TAP−/− mice were lethally irradiated (900 rads) and received bone marrow cells (1.0 × 107; prepared from TAP−/− or B6 mice) intravenously on the following day. Mice were left for at least 2 months before infection to allow reconstitution of the lymphoid system. The animal experiments reported herein were approved by the Institutional Animal Care and Use Committee of Nagasaki University and were conducted according to the guidelines for Animal Experimentation at Nagasaki University.
Adoptive transfer and P. berghei ANKA infection.
To prepare activated OT-I cells, pooled cells from the spleen and inguinal lymph nodes of OT-I mice were prepared and cultured in the presence of OVA257-264 peptide (2 μg/ml) for 3 days. OT-II cells were purified from spleen and inguinal lymph node cells of OT-II mice using anti-CD4 IMag (BD Biosciences). Dendritic cells were prepared from B6 splenocytes using CD11c-coated microbeads and an AutoMACS cell separator (Myltenyi Biotec, Bergisch Gladbach, Germany). OT-II cells (6 × 106/ml) and dendritic cells (1 × 105/ml) were cocultured in the presence of OVA323-339 peptide (3 μg/ml) for 5 days. Mice received OT-I (1 × 105 to 100 × 105) or OT-II (3 × 107) cells through the tail vein and were challenged with 300 to 500 infectious sporozoites 2 days later. The proportion of OT-I (CD45.1) cells in the total CD8+ T cell population was determined by staining peripheral blood lymphocytes (PBLs) with allophycocyanin–anti-CD8 and phycoerythrin (PE)–Cy7–anti-CD45.1 monoclonal antibodies (MAbs). For the experiments involving de novo priming of CD8+ T cells (see Fig. 6) and determination of the parasite burden in the liver (see Fig. 3), mice were challenged with 1,000 and 5,000 sporozoites, respectively. Mice were monitored for parasitemia daily (starting 4 days after infection) by microscopic examination of standard blood films. The parasite burden was determined by real-time PCR using liver RNA and is expressed as the ratio of the cDNA of Plasmodium 18S rRNA to the cDNA of mouse glyceraldehyde-3-phosphate dehydrogenase (G3PDH), as described previously (30).
Fig 6.
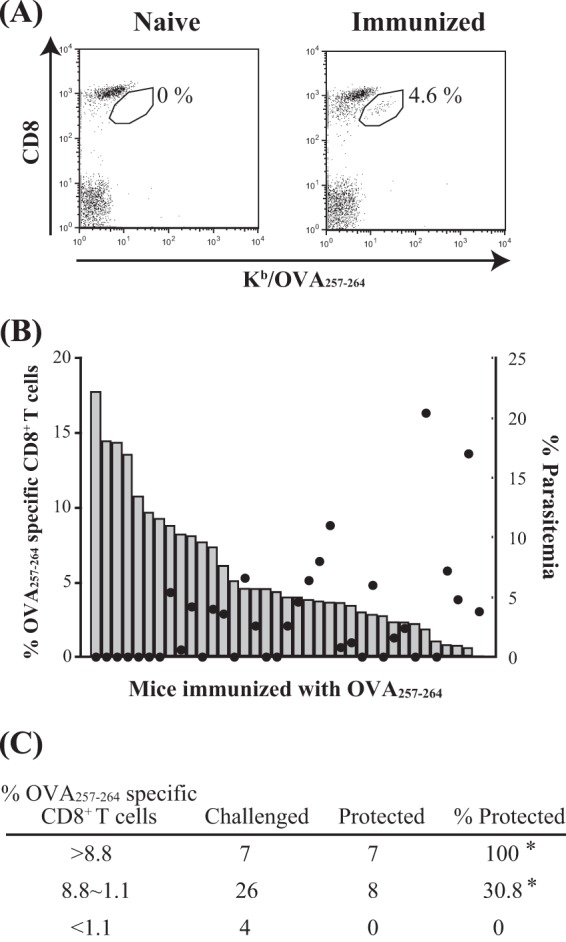
OVA-specific memory CD8+ T cells were protective against infection with PbA-hsOVA. B6 mice were immunized with OVA257-264 as described in Materials and Methods. Two months later, the proportion of OVA-specific memory CD8+ T cells was determined by staining PBLs with anti-CD8 MAb and OVA257-264/Kb tetramer. (A) Representative flow cytometry profiles of PBLs from naive and immunized mice. (B) Each bar in the graph shows the proportion of OVA-specific memory CD8+ T cells in the total CD8+ T cells for an individual mouse (left axis). The data are arranged from left to right in order of high to low specific CD8+ T cell ratios. These mice were challenged by intravenous injection of PbA-hsOVA sporozoites (1,000/mouse). Parasitemia was assessed 8 days after challenge; each dot shows the level of parasitemia in an individual mouse (right axis). (C) Data from 37 mice are summarized. *, P < 0.05%. The experiments were performed 3 times; pooled data are shown.
Fig 3.

TAP-dependent presentation of MHC-I epitope by infected host cells. (A and B) B6 (A and B) or TAP−/− (B) mice were inoculated (or not inoculated, for controls) with activated OT-I CD8+ T cells and infected with sporozoites (5 × 103) of PbA-hsOVA or P. berghei ANKA (PbA). (A) The numbers in parentheses indicate the proportion of OT-I cells in the total CD8+ T cell population in PBLs at the time of infection. (C) Bone marrow (BM) chimeras were generated between B6 and TAP−/− mice (as described in Materials and Methods), and the mice were inoculated with OT-I cells and infected with PbA-hsOVA sporozoites. Two days after infection, RNA was extracted from the livers of the infected mice and the parasite burden was determined by real-time PCR. The experiments were performed twice (A) or 3 times (B, C); representative data are shown. ns, not significant; *, P < 0.05; **, P < 0.01.
Confocal and two-photon microscopy.
PbA-gfpOVA sporozoites were obtained from the salivary glands of infected A. stephensi mosquitoes. To prepare P. berghei ANKA-infected hepatocytes, HepG2 cells (1 × 104) were cultured in HepG2 medium (500 μl; Dulbecco modified Eagle medium containing 10% fetal calf serum, 1% penicillin-streptomycin, and 1% nonessential amino acids) using a Fluorodish cell culture dish (World Precision Instruments, Sarasota, FL) for 3 days, as described previously (31). PbA-gfpOVA sporozoites (1 × 104) were added to the culture and incubated for 3 h, followed by the addition of invasion medium (500 μl; HepG2 medium supplemented with 3 mg/ml of glucose). The medium was replaced 12 h later, and the culture was maintained in the invasion medium for a total of 24 h, after which cells were stained. PbA-gfpOVA-infected red blood cells (RBCs) were collected from the tail vein of the infected mice. Sporozoites, infected HepG2 cells, and RBCs were incubated in the presence of boron-dipyrromethene (BODIPY)-TR-C5-ceramide (5 μM; Invitrogen, Carlsbad, CA) for 15 min at 37°C, washed 3 times with phosphate-buffered saline, and stained with DRAQ5 (1.25 μM; Biostatus, Leicestershire, United Kingdom) for 30 min at 37°C. Images were acquired with an inverted TCS SP5 MP confocal microscope with a ×63 glycerol immersion lens (Leica Microsystems, Wetzlar, Germany).
For intravital imaging, spleen cells and lymph node cells from DsRed/OT-I mice were cultured in the presence of OVA257-264 for 3 days. Activated DsRed/OT-I cells (3 × 106 to 10 × 106) were adoptively transferred into B6 mice. Two days later, the mice were infected (or not infected, for controls) with PbA-gfpOVA sporozoites (1 × 104). At 40 to 48 h postinfection, mice were anesthetized with isoflurane. The abdomen was then shaved, a midline incision was made through the dermis and peritoneum, and the liver was carefully exteriorized. Mice were placed on a platform with a centrally located hole, where a cover glass was attached. An O ring with a 9.8-mm inner diameter was placed on the cover glass to prevent movement of the liver during imaging. Images were acquired with an inverted TCS SP5 TPM equipped with an OPO laser (Leica Microsystems) and with a ×25 (numerical aperture, 0.95) water immersion objective. During observation with fluorescence microscopy (DMI6000B; Leica Microsystems), the numbers of GFP-positive (GFP+) infected hepatocytes and OT-I clusters were determined by counting manually within the field inside the O ring (∼75 mm2). The number of OT-I cells in each cluster was determined using Imaris software (Bitplane, Zurich, Switzerland), after acquiring a 3-dimensional image of each cluster with TPM.
Generation of OVA-specific memory CD8+ T cells.
Specific memory CD8+ T cells were induced in mice as described previously (32), with slight modifications. B6 mice were immunized intravenously with bone marrow-derived dendritic cells (2.5 × 105) pulsed with OVA257-264 peptide (1 mM). Seven to 9 days later, these mice were boosted by infection with Listeria monocytogenes expressing OVA (LM-OVA; 1 × 106 to 10 × 106 CFU) (33). After 2 months, PBLs from these mice were stained with fluorescein isothiocyanate–anti-CD8 MAb and PE–OVA257-264/H-2Kb tetramer (MBL, Nagoya, Japan), and the proportion of OVA-specific CD8+ T cells was determined using a FACSCanto II flow cytometer (BD Biosciences).
Statistical analysis.
Data are expressed as means ± standard deviations (SDs). Statistical analysis was performed using the Mann-Whitney U test for the comparison of two experimental groups, and the data were analyzed using GraphPad Prism software. Differences with P values of <0.05 were considered significant.
RESULTS
Cytoplasmic expression of OVA-GFP fusion proteins in recombinant P. berghei ANKA.
To investigate the mechanisms of protection against liver-stage malaria, we generated two recombinant P. berghei ANKA constructs (Fig. 1A). The first construct expresses a fusion protein of the OVA257-264 epitope fused to the C terminus of GFP (PbA-gfpOVA); the second expresses a fusion protein of the OVA323-339 MHC-II epitope, a portion of P. yoelii hsp70, and the OVA257-264 MHC-I epitope (PbA-hsOVA). The sequence of P. yoelii hsp70 was used because an antigen fused to this portion of hsp70 was shown to promote priming of specific T cell responses (34, 35). Since the fusion protein constructs did not contain a signal sequence, their expression was expected to be limited to the cytoplasm of the parasite. To confirm the localization of the expressed protein, confocal microscopy was used to examine the expression of the fusion protein in sporozoites and infected cells after staining with the membrane marker BODIPY-TR-C5-ceramide and nuclear marker DRAQ5 (36) (Fig. 1B). The GFP-fused protein was localized in the cytoplasm of PbA-gfpOVA sporozoites. At 24 h postinfection with sporozoites, GFP was detected within the parasitophorous membrane of the infected HepG2 cells but was not observed in the host cytoplasm. We also examined the expression of GFP in the infected RBCs and observed that GFP was also localized within the parasitophorous membrane in these cells.
OT-I cell-mediated protection against liver-stage infection with P. berghei ANKA.
We examined whether CD8+ T cells from OT-I mice are protective against liver-stage infection with PbA-hsOVA and PbA-gfpOVA. OT-I cells were activated prior to transfer, since previous studies indicated that the activation of specific CD8+ T cells was required for protection against sporozoite infection at the liver stage (37). B6 mice were inoculated with different doses of preactivated OT-I cells and then infected with PbA-hsOVA or wild-type P. berghei ANKA sporozoites, and the levels of parasitemia were monitored daily (Fig. 2A). Transferred OT-I cells were identified as CD45.1+ CD8low T cells (38). Mice that received 1 × 107 OT-I cells were completely protected from challenge infection with PbA-hsOVA but not with P. berghei ANKA, indicating that the protective effect was specific to the OVA-expressing parasites. We also observed that the protection was OT-I dose dependent and that mice receiving less than 1 × 106 OT-I cells developed parasitemia (Fig. 2A). OT-I cells constituted 42.1% and 3.4% of the CD8+ T cell population in PBLs from mice receiving 1 × 107 and 1 × 106 OT-I cells, respectively, indicating that high levels of OT-I cells were required for sterile protection at the liver stage of infection. Similarly, sterile protection was observed when mice receiving OT-I cells were infected with PbA-gfpOVA sporozoites (Fig. 2B). We also examined whether CD4+ T cells from OT-II mice were protective against the liver-stage infection with PbA-hsOVA (Fig. 2C). Although parasitemia appeared 5 days after infection in both mice to which OT-II was transferred and mice to which OT-II was not transferred, the levels of parasitemia were lower in mice to which OT-II was transferred, suggesting that OT-II cells have protective roles against infection with PbA-hsOVA. However, sterile immunity was never achieved at the liver stage by inoculation with OT-II cells, although the proportion of OT-II cells in the CD4+ T cell population was as high as 43.8%.
Fig 2.

OT-I cells protect against infection with sporozoites of OVA-expressing P. berghei ANKA. B6 mice were inoculated with activated OT-I CD8+ T cells (0 to 107) (A, B) and infected with sporozoites (300/mouse) of PbA-hsOVA or wild-type P. berghei ANKA (PbA) (A) or PbA-gfpOVA (B) 2 days later. (C) Alternatively, after transfer with OT-II CD4+ T cells (3 × 107 or 0), mice were infected with PbA-hsOVA sporozoites (500/mouse). (A to C) Representative flow cytometry profiles of PBLs from mice to which OT-I cells (1× 107) (A, B) or OT-II cells (3 × 107) (C) were transferred are also shown. The proportions of OT-I or OT-II cells within the total CD8+ or CD4+ T cell populations are indicated. Note that the levels of CD8 expression on activated T cells are reduced, as reported previously (38). The number in the upper left of each graph indicates the number of transferred cells; the number in parentheses indicates the proportion of OT-I cells in the total CD8+ T cell population in PBLs at the time of infection (A and B) or the proportion of OT-II cells in the total CD4+ T cells in PBL (C). Parasitemia was monitored daily starting at 4 days after infection. Values significantly different (P < 0.05) from those for mice not receiving T cells are indicated (*). The experiments were performed twice (B, C) or 3 times (A); representative data are shown.
To confirm that the observed decrease in parasitemia was due to the inhibition of parasite growth at the liver stage, the parasite burden in the liver was examined by real-time PCR of parasite rRNA (Fig. 3A). OT-I cells were found to significantly inhibit the parasite burden in the liver of mice infected with PbA-hsOVA (90.1% reduction) but not in those infected with P. berghei ANKA, indicating that the protection was specific to the OVA-expressing P. berghei ANKA. We next wanted to examine whether the OVA antigen-presenting pathway utilizes the classical MHC class I pathway. To this end, B6 and TAP−/− mice were inoculated with OT-I cells, infected with PbA-hsOVA, and examined for parasite burden in the liver. OT-I cells significantly inhibited the parasite burden in B6 mice (99.8% reduction) but not in TAP−/− mice after challenge infection with PbA-hsOVA sporozoites, indicating that the antigen presentation pathway did utilize the classical TAP-dependent pathway (Fig. 3B). Furthermore, we generated bone marrow chimeras between B6 and TAP−/− mice to examine whether the TAP expressed in hematopoietic cells or hepatocytes is critical for the protection. After inoculation with OT-I and infection with PbA-hsOVA, the parasite burden in the liver was significantly reduced in bone marrow chimeras when B6 mice were used as recipients. The reductions were 98.2% in the B6 → B6 chimera compared to the B6 → TAP−/− chimera and 98.1% in the TAP−/− → B6 chimera compared to the TAP−/− → TAP−/− chimera, indicating that TAP expression in the radioresistant host is critical for the protection against challenge infection with PbA-hsOVA (Fig. 3C). These results strongly suggest that hepatocytes infected with PbA-hsOVA sporozoites process and present the OVA epitope via the classical MHC class I pathway, which is consistent with the findings of a previous study using P. berghei expressing a mutant CS protein containing an OVA epitope (39).
In vivo imaging of the interaction between OT-I cells and infected hepatocytes.
After observing the protective effect of OT-I cells, we aimed to directly visualize the interaction of infected hepatocytes with the effector OT-I cells using TPM. For this purpose, mice were inoculated (or not inoculated, for controls) with preactivated DsRed/OT-I cells and infected with PbA-gfpOVA sporozoites. Two days later, the livers of the infected mice were surgically exposed and imaging was performed. In mice infected with PbA-gfpOVA sporozoites, GFP+ cells were clearly visible after 24 h and the quantity of GFP continued to increase for 24 to 48 h after infection (data not shown). We observed a defined surface area (75 mm2) of the liver using TPM at 40 to 48 h after sporozoite infection. When a low dose (3 × 106) of OT-I cells was inoculated into the mice, we observed numerous OT-I clusters formed around GFP+ cells (Fig. 4A). The number of OT-I cells in each of these clusters was relatively small (mean, 34.2; range, 10 to 71) (Fig. 4D, GFP +). Using time-lapse imaging, we were able to observe the disappearance of GFP+ cells while in contact with OT-I cells, suggesting that the OT-I cells are directly involved in the elimination of intrahepatic parasites (Fig. 4A, right; see Movie S1 in the supplemental material). When the number of inoculated OT-I cells was increased to the dose sufficient for sterile protection (1 × 107), fewer GFP+ cells remained in the liver (Fig. 4B and C, left), and the number of OT-I clusters increased (Fig. 4B and C, right). The number of OT-I clusters in the liver of the OT-I-inoculated, P. berghei ANKA-infected mice was similar to the number of GFP+ cells in the P. berghei ANKA-infected mice not inoculated with OT-I, suggesting that the clusters were formed following elimination of infected hepatocytes by OT-I cells (compare the left and right panels of Fig. 4E). Additionally, we determined that the number of OT-I cells in clusters containing GFP+ cells (mean, 28.4; range, 14 to 48) was much lower than that in clusters that did not contain GFP+ cells (mean, 293.8; range, 15 to 1,415) in mice inoculated with 1 × 107 OT-I cells (Fig. 4D). The OT-I clusters were barely detectable in OT-I-inoculated mice without P. berghei ANKA infection and, if present, were formed by small numbers of OT-I cells (Fig. 4E, right, and F).
Fig 4.

Clustering of OT-I cells around GFP+ infected hepatocytes during liver-stage infection with PbA-gfpOVA. Activated DsRed/OT-I CD8+ T cells were transferred into B6 mice at a dose of 3 × 106 (A, C, D, F) or 1 × 107 (B, C to F), and the mice were infected with PbA-gfpOVA sporozoites (1 × 104). (E, F) Some mice did not receive DsRed/OT-I or were not infected with PbA-gfpOVA as controls. At 48 h after infection, the liver was imaged with TPM. (A, B) The 2-dimensional projections of 3-dimensional imaging volumes are shown. Bars, 10 μm. A still image of GFP+ cell disappearance while in contact with OT-I cells is shown (A, right; a time-lapse image is shown in Movie S1 in the supplemental material). (C, E) The numbers of GFP+ cells and T cell clusters within a surface area of 75 mm2 were counted using fluorescence microscopy. (D, F) GFP+ cells and T cell clusters were imaged in 3 dimensions using TPM, and the number of OT-I cells within each cluster was determined using Imaris software. (D) The number of OT-I cells was determined separately for clusters containing and not containing GFP+ cells. Bars indicate averages. *, P < 0.05; **, P < 0.01; ***, P < 0.0001.
Effector function of OT-I cells.
The clustering of OT-I cells around infected hepatocytes suggests that the effector mechanisms of CD8+ T cells in liver-stage malaria might be different from the classical CTL killing mechanisms. Thus, we evaluated the effector function of CD8+ T cells during protection at the liver stage of infection with PbA-hsOVA or PbA-gfpOVA. CD8+ T cells were prepared from OT-I, IFN-γ−/− OT-I, perforin−/− OT-I, or IFN-γ−/− perforin−/− OT-I mice, activated in vitro, and transferred into B6 mice, which were infected with sporozoites of PbA-hsOVA or PbA-gfpOVA and examined for parasitemia (Fig. 5). After infection with PbA-hsOVA, no parasitemia was detected in mice receiving IFN-γ−/− OT-I, perforin−/− OT-I, or IFN-γ−/− perforin−/− OT-I cells, indicating that the expression of IFN-γ and perforin in CD8+ T cells was dispensable for the protection against liver-stage infection (Fig. 5A). When the mice were infected with PbA-gfpOVA, a delayed onset of parasitemia was detected in 2/5 infected mice to which IFN-γ−/− perforin−/− OT-I cells were transferred and 1/5 mice to which perforin−/− OT-I cells were transferred (Fig. 5B). These results suggest that IFN-γ and perforin are partially involved in the protective effects of OT-I cells, although these molecules are not essential for protection. The difference in the results of infection with PbA-hsOVA and PbA-gfpOVA may be due to the differences in the efficiency of antigen presentation; the OVA epitope may be more efficiently presented to OT-I cells for PbA-hsOVA infection than for PbA-gfpOVA infection.
Fig 5.
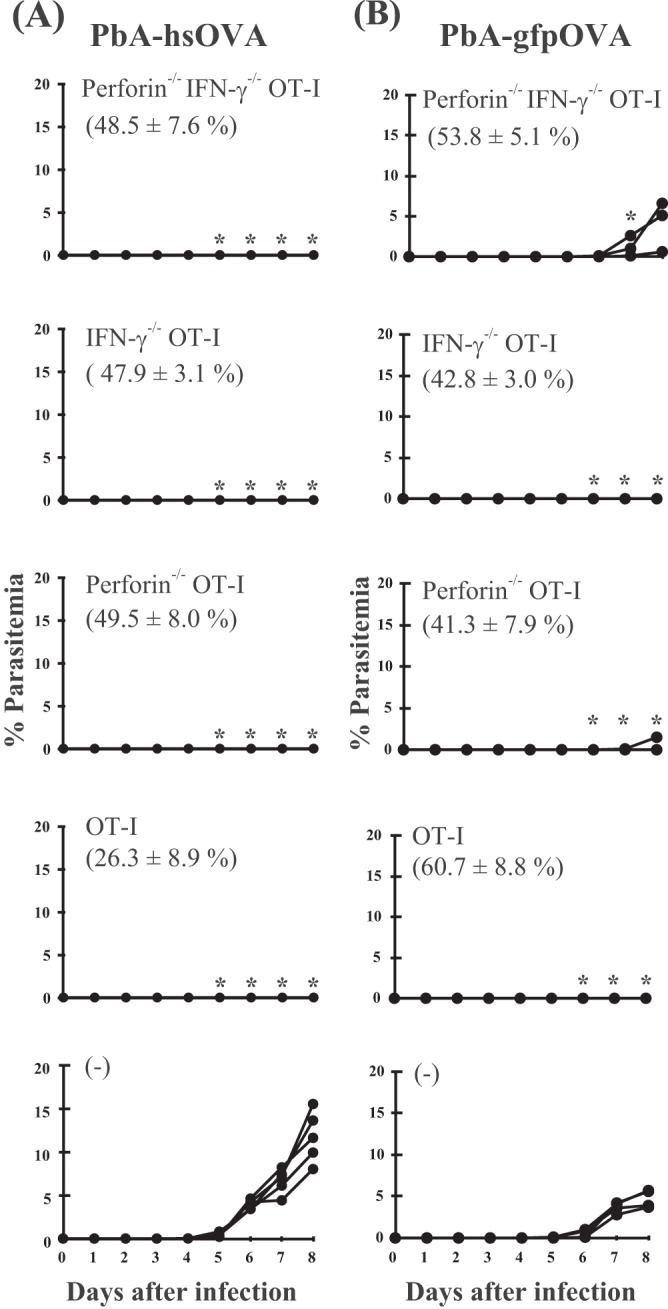
Perforin and IFN-γ expressed in CD8+ T cells are dispensable for sterile immunity at the liver stage of infection. Activated CD8+ T cells from perforin−/− IFN-γ−/− OT-I, IFN-γ−/− OT-I, perforin−/− OT-I, or wild-type OT-I mice or no cells [(−)] were adoptively transferred into B6 mice, which were then infected with sporozoites (300/mouse) of PbA-hsOVA (A) or PbA-gfpOVA (B), and the levels of parasitemia were monitored. Values significantly different (P < 0.05) from those for mice not receiving OT-I cells are indicated (*). In each graph, the number in parentheses indicates the proportion of OT-I cells in the total CD8+ T cell population in PBLs at the time of infection. The experiments were performed 3 times; representative data are shown.
Finally, we examined whether OVA-specific polyclonal memory CD8+ T cells were protective against infection with PbA-hsOVA sporozoites following a previously described protocol (40). Mice were primed with OVA257–264-pulsed dendritic cells and boosted with LM-OVA infection. Two months later, we examined the proportion of OVA-specific CD8+ T cells in PBLs by staining with OVA257-264/Kb tetramer. These mice were infected with PbA-hsOVA sporozoites, and the levels of parasitemia in peripheral blood were determined 8 days after infection (Fig. 6). Comparison of the number of tetramer-positive cells with the occurrence of parasitemia showed that mice bearing OVA-specific CD8+ T cells at levels more than 9.3% of the total amount of CD8+ T cells were completely protected from the sporozoite challenge, while both protected and unprotected mice were included among those bearing specific CD8+ T cells in the range of 1.1 to 8.8%.
DISCUSSION
In this study, we established a novel system to investigate the cellular and molecular mechanisms underlying the protective immune response against liver-stage infection with malaria parasites using a model malaria antigen, OVA. Unlike the CSP model, which utilizes BALB/c mice, our model can be applied in B6 mice. Cockburn et al. generated a model in which CSP containing an OVA epitope was expressed on the surface of sporozoites and used B6 mice for the study of protective immunity at the liver stage of infection (39). Our model is distinct from this model, in that the antigen is expressed in the cytoplasm of malaria parasites and can become a target of specific CD8+ T cells during the liver stage of Plasmodium infection, leading to sterile protection. Protection was achieved both by the inoculation of activated OT-I cells and by the induction of polyclonal OVA-specific memory CD8+ T cells. Since protection by OT-I cells was dependent on TAP molecule expression in nonhematopoietic host cells, consistent with the previous study (39), it is reasonable to speculate that OVA expressed in the cytoplasm of the parasite is somehow transported into the cytoplasm of hepatocytes for antigen processing. However, we did not detect any leakage of GFP into the cytoplasm of the infected hepatocytes by confocal imaging. A possible explanation for this is that cytoplasmic malaria antigens are processed to smaller peptides prior to transfer into the host cells. Alternatively, the amount of the protein transported to the cytoplasm may have been too low for visualization by our methods. Whatever the molecular mechanisms, these results imply that malaria proteins expressed in the cytoplasm of malaria parasites can be targets of protective immune responses and should not be excluded from the pool of candidate malaria vaccine targets.
In our experimental model, we employed intravital imaging to visualize the interaction between P. berghei ANKA-infected hepatocytes and specific CD8+ T cells. In the absence of inoculation with OT-I cells, infected hepatocytes were observed as isolated GFP+ cells, as shown previously by others (41–43). When we used a lower number of OT-I cells for inoculation (3 × 106), clustering of OT-I cells around the infected hepatocytes was observed, suggesting that OT-I cells recognize the MHC/OVA epitope expressed on the surface of hepatocytes and make direct contacts with them. Using time-lapse imaging, we were able to observe the disappearance of GFP+ intrahepatic parasites during their interaction with OT-I cells, implying that the OT-I clusters are directly involved in the elimination of the parasites in the liver. When the number of inoculated OT-I cells was increased to a level sufficient for sterile immunity (1 × 107), the number of GFP+ cells was dramatically reduced. Furthermore, we observed OT-I clusters that did not contain GFP+ hepatocytes, and some OT-I clusters were large (containing more than 1,000 OT-I cells), suggesting that the accumulation of OT-I cells in the cluster continued after the elimination of GFP+ hepatocytes. After submission of the manuscript, Cockburn et al. (44) published an imaging study of CSP-specific CD8+ T cells eliminating liver-stage malaria parasites and showed that CD8+ T cells form clusters around infected hepatocytes, similar to the findings of our study. Thus, cluster formation is not limited to our model system but occurs in Plasmodium-specific CD8+ T cells eliminating malaria parasites during liver-stage infection.
The effector mechanisms of CD8+ T cell-mediated elimination of intrahepatic parasites are complex. An earlier study suggested that perforin- or Fas-mediated killing is not the main pathway of parasite elimination during the hepatic stage of the infection (45). Additionally, a recent study using CSP-specific transgenic T cells suggested that IFN-γ is not essential for the protection of mice against infection with P. yoelii sporozoites (46). However, IFN-γ and tumor necrosis factor alpha have been reported to be important for protection against liver-stage infection with P. berghei as well as P. yoelii, while perforin is important for protection against infection with P. yoelii but not P. berghei (47, 48). In our study, IFN-γ expressed in CD8+ T cells was dispensable for the elimination of infected hepatocytes during infection with PbA-gfpOVA, whereas perforin was partially involved in this process. Therefore, unlike the elimination of virus-infected or transformed cells (49), perforin/granzyme-mediated killing is not the essential pathway for the elimination of malaria parasites in the liver. Effector CD8+ T cells were shown herein to form clusters around infected hepatocytes, leading to the elimination of the intrahepatic parasites. These features suggest that a novel mechanism might be involved in the protective immune responses of CD8+ T cells against intrahepatic parasites. It is intriguing to speculate that other hepatic immune cells, such as dendritic cells, Kupffer cells, and liver sinusoidal endothelial cells (43), are involved in parasite elimination.
Schmidt et al. showed that the proportion of CSP-specific memory CD8+ T cells correlated with sterilizing immunity at the liver stage, with protective effects being observed when more than 1% of CD8+ T cells in PBLs were CSP specific (40). In our model, the threshold frequency of OVA-specific memory CD8+ T cells was much higher and more than 8% OVA-specific CD8+ T cells were required to achieve sterile immunity in 100% of mice. The probability of sterile immunity was reduced to 28.6% (8/27) when OVA-specific CD8+ T cells constituted 1.1 to 8.8% of PBLs. Therefore, the threshold frequency of memory CD8+ T cells required for sterile immunity in our OVA system was higher than that required in the CSP system. The localization of antigen expression may influence the efficacy and timing of antigen presentation by hepatocytes. CSP is expressed on the surface of the parasite; thus, it may be readily accessible to the cytoplasm of the infected hepatocytes soon after infection. Further, CSP might be transferred to sinusoidal endothelial cells when sporozoites migrate through hepatic sinusoids prior to infection and these cells cross-present CSP to specific CD8+ T cells in a manner similar to that for hepatocyte-infecting viruses (50). In contrast, proteins expressed in the cytoplasm of parasites might be transferred to host cells relatively late after infection and thus may have a narrower window for sterile protection. Alternatively, the outcome of the individual studies may be affected by differences in the mouse strain used (BALB/c for the CSP study and B6 in our OVA study) or the levels of antigen expressed. A recent transcriptome approach revealed that approximately 2,000 genes are active during liver-stage infection (14). It is possible that many of the proteins encoded by these genes are expressed in the cytoplasm of parasites and that combined polyclonal CD8+ T cell responses against different sets of these antigens might achieve sterile protection against malaria parasites in the liver.
Our study showed that malaria proteins expressed in the cytoplasm of parasites can be targets of the protective immune responses by CD8+ T cells. We also visualized the interaction between the infected hepatocytes and specific effector CD8+ T cells which led to the elimination of the parasites in the liver and revealed a novel aspect of the effector mechanisms of protective immunity in liver-stage infection. These findings enhance our understanding of the cellular and molecular mechanisms underlying the protective immune responses during the liver stage of malaria infection and identify novel candidates for malaria vaccine targets.
Supplementary Material
ACKNOWLEDGMENTS
We thank H. Kosaka and H. Watanabe for providing mice, Y. Yoshikai and H. Shen for providing LM-OVA, M. Ishii (Osaka University, Osaka, Japan) and T. Okada (Riken Center for Integrative Medical Sciences, Yokohama, Japan) for help in setting up two-photon microscopy, and N. Kawamoto for technical assistance.
This work was supported by Japan Society for the Promotion of Science (JSPS) KAKENHI grant numbers 23113514 and 25113717 and by the Global COE Program, Nagasaki University.
Footnotes
Published ahead of print 29 July 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.00570-13.
REFERENCES
- 1.Nussenzweig RS, Vanderberg J, Most H, Orton C. 1967. Protective immunity produced by the injection of X-irradiated sporozoites of Plasmodium berghei. Nature 216:160–162 [DOI] [PubMed] [Google Scholar]
- 2.Hoffman SL, Doolan DL. 2000. Malaria vaccines-targeting infected hepatocytes. Nat. Med. 6:1218–1219 [DOI] [PubMed] [Google Scholar]
- 3.Overstreet MG, Cockburn IA, Chen YC, Zavala F. 2008. Protective CD8 T cells against Plasmodium liver stages: immunobiology of an ‘unnatural' immune response. Immunol. Rev. 225:272–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mueller AK, Labaied M, Kappe SH, Matuschewski K. 2005. Genetically modified Plasmodium parasites as a protective experimental malaria vaccine. Nature 433:164–167 [DOI] [PubMed] [Google Scholar]
- 5.Butler NS, Schmidt NW, Vaughan AM, Aly AS, Kappe SH, Harty JT. 2011. Superior antimalarial immunity after vaccination with late liver stage-arresting genetically attenuated parasites. Cell Host Microbe 9:451–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Belnoue E, Voza T, Costa FT, Gruner AC, Mauduit M, Rosa DS, Depinay N, Kayibanda M, Vigario AM, Mazier D, Snounou G, Sinnis P, Renia L. 2008. Vaccination with live Plasmodium yoelii blood stage parasites under chloroquine cover induces cross-stage immunity against malaria liver stage. J. Immunol. 181:8552–8558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Doolan DL, Hoffman SL. 2000. The complexity of protective immunity against liver-stage malaria. J. Immunol. 165:1453–1462 [DOI] [PubMed] [Google Scholar]
- 8.Rodrigues M, Nussenzweig RS, Zavala F. 1993. The relative contribution of antibodies, CD4+ and CD8+ T cells to sporozoite-induced protection against malaria. Immunology 80:1–5 [PMC free article] [PubMed] [Google Scholar]
- 9.Good MF, Doolan DL. 2010. Malaria vaccine design: immunological considerations. Immunity 33:555–566 [DOI] [PubMed] [Google Scholar]
- 10.Romero P, Maryanski JL, Corradin G, Nussenzweig RS, Nussenzweig V, Zavala F. 1989. Cloned cytotoxic T cells recognize an epitope in the circumsporozoite protein and protect against malaria. Nature 341:323–326 [DOI] [PubMed] [Google Scholar]
- 11.Hafalla JC, Silvie O, Matuschewski K. 2011. Cell biology and immunology of malaria. Immunol. Rev. 240:297–316 [DOI] [PubMed] [Google Scholar]
- 12.Kumar KA, Sano G, Boscardin S, Nussenzweig RS, Nussenzweig MC, Zavala F, Nussenzweig V. 2006. The circumsporozoite protein is an immunodominant protective antigen in irradiated sporozoites. Nature 444:937–940 [DOI] [PubMed] [Google Scholar]
- 13.Khusmith S, Charoenvit Y, Kumar S, Sedegah M, Beaudoin RL, Hoffman SL. 1991. Protection against malaria by vaccination with sporozoite surface protein 2 plus CS protein. Science 252:715–718 [DOI] [PubMed] [Google Scholar]
- 14.Rogers WO, Malik A, Mellouk S, Nakamura K, Rogers MD, Szarfman A, Gordon DM, Nussler AK, Aikawa M, Hoffman SL. 1992. Characterization of Plasmodium falciparum sporozoite surface protein 2. Proc. Natl. Acad. Sci. U. S. A. 89:9176–9180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang R, Charoenvit Y, Corradin G, De La Vega P, Franke ED, Hoffman SL. 1996. Protection against malaria by Plasmodium yoelii sporozoite surface protein 2 linear peptide induction of CD4+ T cell- and IFN-γ-dependent elimination of infected hepatocytes. J. Immunol. 157:4061–4067 [PubMed] [Google Scholar]
- 16.Weiss WR, Good MF, Hollingdale MR, Miller LH, Berzofsky JA. 1989. Genetic control of immunity to Plasmodium yoelii sporozoites. J. Immunol. 143:4263–4266 [PubMed] [Google Scholar]
- 17.Tarun AS, Peng X, Dumpit RF, Ogata Y, Silva-Rivera H, Camargo N, Daly TM, Bergman LW, Kappe SH. 2008. A combined transcriptome and proteome survey of malaria parasite liver stages. Proc. Natl. Acad. Sci. U. S. A. 105:305–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Speake C, Duffy PE. 2009. Antigens for pre-erythrocytic malaria vaccines: building on success. Parasite Immunol. 31:539–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kaufmann SH, Hess J. 1999. Impact of intracellular location of and antigen display by intracellular bacteria: implications for vaccine development. Immunol. Lett. 65:81–84 [DOI] [PubMed] [Google Scholar]
- 20.Mazzaccaro RJ, Gedde M, Jensen ER, van Santen HM, Ploegh HL, Rock KL, Bloom BR. 1996. Major histocompatibility class I presentation of soluble antigen facilitated by Mycobacterium tuberculosis infection. Proc. Natl. Acad. Sci. U. S. A. 93:11786–11791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Teitelbaum R, Cammer M, Maitland ML, Freitag NE, Condeelis J, Bloom BR. 1999. Mycobacterial infection of macrophages results in membrane-permeable phagosomes. Proc. Natl. Acad. Sci. U. S. A. 96:15190–15195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Garg N, Nunes MP, Tarleton RL. 1997. Delivery by Trypanosoma cruzi of proteins into the MHC class I antigen processing and presentation pathway. J. Immunol. 158:3293–3302 [PubMed] [Google Scholar]
- 23.Khan ZM, Ng C, Vanderberg JP. 1992. Early hepatic stages of Plasmodium berghei: release of circumsporozoite protein and host cellular inflammatory response. Infect. Immun. 60:264–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hugel FU, Pradel G, Frevert U. 1996. Release of malaria circumsporozoite protein into the host cell cytoplasm and interaction with ribosomes. Mol. Biochem. Parasitol. 81:151–170 [DOI] [PubMed] [Google Scholar]
- 25.Bongfen SE, Torgler R, Romero JF, Renia L, Corradin G. 2007. Plasmodium berghei-infected primary hepatocytes process and present the circumsporozoite protein to specific CD8+ T cells in vitro. J. Immunol. 178:7054–7063 [DOI] [PubMed] [Google Scholar]
- 26.Miyakoda M, Kimura D, Yuda M, Chinzei Y, Shibata Y, Honma K, Yui K. 2008. Malaria-specific and nonspecific activation of CD8+ T cells during blood stage of Plasmodium berghei infection. J. Immunol. 181:1420–1428 [DOI] [PubMed] [Google Scholar]
- 27.Hogquist KA, Jameson SC, Heath WR, Howard JL, Bevan MJ, Carbone FR. 1994. T cell receptor antagonist peptides induce positive selection. Cell 76:17–27 [DOI] [PubMed] [Google Scholar]
- 28.Barnden MJ, Allison J, Heath WR, Carbone FR. 1998. Defective TCR expression in transgenic mice constructed using cDNA-based α- and β-chain genes under the control of heterologous regulatory elements. Immunol. Cell Biol. 76:34–40 [DOI] [PubMed] [Google Scholar]
- 29.Van Kaer L, Ashton-Rickardt PG, Ploegh HL, Tonegawa S. 1992. TAP1 mutant mice are deficient in antigen presentation, surface class I molecules, and CD4−8+ T cells. Cell 71:1205–1214 [DOI] [PubMed] [Google Scholar]
- 30.Kawabata Y, Udono H, Honma K, Ueda M, Mukae H, Kadota J, Kohno S, Yui K. 2002. Merozoite surface protein 1-specific immune response is protective against exoerythrocytic forms of Plasmodium yoelii. Infect. Immun. 70:6075–6082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ishino T, Yano K, Chinzei Y, Yuda M. 2004. Cell-passage activity is required for the malarial parasite to cross the liver sinusoidal cell layer. PLoS Biol. 2:e4. 10.1371/journal.pbio.0020004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Badovinac VP, Messingham KA, Jabbari A, Haring JS, Harty JT. 2005. Accelerated CD8+ T-cell memory and prime-boost response after dendritic-cell vaccination. Nat. Med. 11:748–756 [DOI] [PubMed] [Google Scholar]
- 33.Dudani R, Chapdelaine Y, van Faassen H, Smith DK, Shen H, Krishnan L, Sad S. 2002. Multiple mechanisms compensate to enhance tumor-protective CD8+ T cell response in the long-term despite poor CD8+ T cell priming initially: comparison between an acute versus a chronic intracellular bacterium expressing a model antigen. J. Immunol. 168:5737–5745 [DOI] [PubMed] [Google Scholar]
- 34.Udono H, Yamano T, Kawabata Y, Ueda M, Yui K. 2001. Generation of cytotoxic T lymphocytes by MHC class I ligands fused to heat shock cognate protein 70. Int. Immunol. 13:1233–1242 [DOI] [PubMed] [Google Scholar]
- 35.Huang Q, Richmond JF, Suzue K, Eisen HN, Young RA. 2000. In vivo cytotoxic T lymphocyte elicitation by mycobacterial heat shock protein 70 fusion proteins maps to a discrete domain and is CD4+ T cell independent. J. Exp. Med. 191:403–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gruring C, Heiber A, Kruse F, Ungefehr J, Gilberger TW, Spielmann T. 2011. Development and host cell modifications of Plasmodium falciparum blood stages in four dimensions. Nat. Commun. 2:165. [DOI] [PubMed] [Google Scholar]
- 37.Sano G, Hafalla JC, Morrot A, Abe R, Lafaille JJ, Zavala F. 2001. Swift development of protective effector functions in naive CD8+ T cells against malaria liver stages. J. Exp. Med. 194:173–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rai D, Pham NL, Harty JT, Badovinac VP. 2009. Tracking the total CD8 T cell response to infection reveals substantial discordance in magnitude and kinetics between inbred and outbred hosts. J. Immunol. 183:7672–7681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cockburn IA, Tse SW, Radtke AJ, Srinivasan P, Chen YC, Sinnis P, Zavala F. 2011. Dendritic cells and hepatocytes use distinct pathways to process protective antigen from plasmodium in vivo. PLoS Pathog. 7:e1001318. 10.1371/journal.ppat.1001318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schmidt NW, Podyminogin RL, Butler NS, Badovinac VP, Tucker BJ, Bahjat KS, Lauer P, Reyes-Sandoval A, Hutchings CL, Moore AC, Gilbert SC, Hill AV, Bartholomay LC, Harty JT. 2008. Memory CD8 T cell responses exceeding a large but definable threshold provide long-term immunity to malaria. Proc. Natl. Acad. Sci. U. S. A. 105:14017–14022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sturm A, Amino R, van de Sand C, Regen T, Retzlaff S, Rennenberg A, Krueger A, Pollok JM, Menard R, Heussler VT. 2006. Manipulation of host hepatocytes by the malaria parasite for delivery into liver sinusoids. Science 313:1287–1290 [DOI] [PubMed] [Google Scholar]
- 42.Amino R, Thiberge S, Martin B, Celli S, Shorte S, Frischknecht F, Menard R. 2006. Quantitative imaging of Plasmodium transmission from mosquito to mammal. Nat. Med. 12:220–224 [DOI] [PubMed] [Google Scholar]
- 43.Frevert U, Nardin E. 2008. Cellular effector mechanisms against Plasmodium liver stages. Cell. Microbiol. 10:1956–1967 [DOI] [PubMed] [Google Scholar]
- 44.Cockburn IA, Amino R, Kelemen RK, Kuo SC, Tse SW, Radtke A, Mac-Daniel L, Ganusov VV, Zavala F, Menard R. 2013. In vivo imaging of CD8+ T cell-mediated elimination of malaria liver stages. Proc. Natl. Acad. Sci. U. S. A. 110:9090–9095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Renggli J, Hahne M, Matile H, Betschart B, Tschopp J, Corradin G. 1997. Elimination of P. berghei liver stages is independent of Fas (CD95/Apo-I) or perforin-mediated cytotoxicity. Parasite Immunol. 19:145–148 [DOI] [PubMed] [Google Scholar]
- 46.Chakravarty S, Baldeviano GC, Overstreet MG, Zavala F. 2008. Effector CD8+ T lymphocytes against liver stages of Plasmodium yoelii do not require γ interferon for antiparasite activity. Infect. Immun. 76:3628–3631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Butler NS, Schmidt NW, Harty JT. 2010. Differential effector pathways regulate memory CD8 T cell immunity against Plasmodium berghei versus P. yoelii sporozoites. J. Immunol. 184:2528–2538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Trimnell A, Takagi A, Gupta M, Richie TL, Kappe SH, Wang R. 2009. Genetically attenuated parasite vaccines induce contact-dependent CD8+ T cell killing of Plasmodium yoelii liver stage-infected hepatocytes. J. Immunol. 183:5870–5878 [DOI] [PubMed] [Google Scholar]
- 49.Trapani JA, Smyth MJ. 2002. Functional significance of the perforin/granzyme cell death pathway. Nat. Rev. Immunol. 2:735–747 [DOI] [PubMed] [Google Scholar]
- 50.Wohlleber D, Kashkar H, Gartner K, Frings MK, Odenthal M, Hegenbarth S, Borner C, Arnold B, Hammerling G, Nieswandt B, van Rooijen N, Limmer A, Cederbrant K, Heikenwalder M, Pasparakis M, Protzer U, Dienes HP, Kurts C, Kronke M, Knolle PA. 2012. TNF-induced target cell killing by CTL activated through cross-presentation. Cell Rep. 2:478–487 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.