

Abstract
Natural killer (NK) cells directly recognize and kill fungi, such as the pathogenic fungus Cryptococcus neoformans, via cytolytic mechanisms. However, the precise signaling pathways governing this NK cell microbicidal activity and the implications for fungal recognition are still unknown. Previously, it was reported that NK cell anticryptococcal activity is mediated through a conserved phosphatidylinositol 3-kinase–extracellular signal-regulated kinase 1/2 (PI3K-ERK1/2) pathway. Using YT (a human NK-like cell line) and primary human NK cells, we sought to identify the upstream, receptor-proximal signaling elements that led to fungal cytolysis. We demonstrate that Src family kinases were activated in response to C. neoformans. Furthermore, pharmacologic inhibition with an Src kinase inhibitor blocked C. neoformans-induced downstream activation of PI3K and ERK1/2 and abrogated cryptococcal killing. At the same time, the inhibitor disrupted the polarization of perforin-containing granules toward the NK cell-cryptococcal synapse but had no effect on conjugate formation between the organism and the NK cell. Finally, small interfering RNA (siRNA) double (but not single) knockdown of two Src family kinases, Fyn and Lyn, blocked cryptococcal killing. Together these data demonstrate a mechanism whereby the Src family kinases, Fyn and Lyn, redundantly mediate anticryptococcal activity through the activation of PI3K and ERK1/2, which in turn facilitates killing by inducing the polarization of perforin-containing granules to the NK cell-cryptococcal synapse.
INTRODUCTION
Natural killer (NK) cells possess an enormous capacity for immune recognition and cytotoxicity, extending far beyond just tumor and virus-infected cells (1, 2). Numerous studies have now demonstrated that NK cells also possess the ability to directly recognize and kill bacteria (3, 4), parasites (5), and fungi (6, 7). In particular, NK cells have been shown to kill Cryptococcus neoformans (8–11). This devastating fungal pathogen, often acquired through the inhalation of C. neoformans yeast cells or spores, is associated with tremendous morbidity and mortality in immunocompromised individuals such as those living with AIDS (12). Uncontrolled growth of C. neoformans in the lungs of these individuals leads to the development of pneumonia, and in more severe cases, yeast cells disseminate from the lungs to the brain, resulting in meningoencephalitis or even death (12).
It has become increasingly evident that NK cells mediate anticryptococcal activity through contact-dependent cytotoxicity (8, 13). Microscopic studies have revealed direct contact and conjugate formation between NK cells and C. neoformans, and the magnitude of conjugate formation correlated with anticryptococcal activity (8–10). Other studies have suggested that NK cell microbicidal activity against C. neoformans is likely mediated through a receptor-ligand interaction (14). It follows that receptor ligation converges on signaling cascades that facilitate microbicidal activity. In the context of C. neoformans, the pathways underlying NK cell anticryptococcal activity have recently begun to be defined. It is clear that NK cell anticryptococcal activity is mediated through phosphatidylinositol-3-kinase (PI3K)-dependent extracellular signal-regulated kinases 1 and 2 (ERK1/2) signaling (PI3K→ERK1/2), which stimulates the polarization and release of perforin (15). However, the upstream (receptor-proximal) signaling elements that lead to the activation of PI3K and perforin-dependent anticryptococcal activity are unknown.
Some receptors utilize Src (v-src sarcoma [Schmidt-Ruppin A-2] viral oncogene homolog) family kinases, while others do not. For example, NK cells express receptors that are involved in microbial recognition, including Toll-like receptors, C-type lectin receptors, complement, and scavenger receptors. While some of these receptors employ PI3K (16–18), the canonical pathways do not use Src family kinases (19–21), which are a diverse group of protein tyrosine kinases (Src, Fyn, Hck, Lck, Lyn, Yes, Fgr, Blk, and Yrk) involved in numerous cellular processes ranging from growth and differentiation to survival (22, 23). In contrast, many of the NK cell cytotoxicity-activating receptors involved in tumor cell killing, such as killer cell immunoglobulin-like receptors (KIR) and natural cytotoxicity receptors (NCR), mediate activation of PI3K→ERK1/2 through Src family kinases. Thus, the requirement for Src family kinases will provide an important distinction between fundamentally different receptor pathways in the recognition and killing of Cryptococcus neoformans by NK cells.
Among the many Src family kinases, Fyn (FYN oncogene related to SRC, FGR, and YES) and Lck (lymphocyte-specific protein tyrosine kinase) have emerged as the principal members involved in NK cell cytotoxicity against tumor cells. These kinases have been shown to physically associate with, and mediate phosphorylation of, the immunoreceptor tyrosine-based activation motif (ITAM)-containing adaptor DAP12 (24). Similarly, cross-linking of NCR was found to induce activation of Fyn and Lck (25). Activation through the signaling lymphocyte activation molecule (SLAM)-family receptor 2B4 has also been shown to proceed through Fyn (26, 27–29). Finally, in addition to its role in natural cytotoxicity, Lck has also been found to mediate antibody-dependent cell-mediated cytotoxicity (ADCC) through the Fc receptor CD16 (30–33). Some studies have also suggested the possibility of other Src family members being involved in cytotoxicity. For instance, the kinase Lyn (v-yes-1 Yamaguchi sarcoma virus-related oncogene homolog) has been found to associate with the NK cell-activating receptors CD94 and NKR-P1 (34). Furthermore, cross-linking of IgG-CD16 complexes has been found to result in Lyn activation (35). Thus, while Fyn and Lck have classically been associated with NK cell cytotoxicity, these findings raise the potential for Lyn to be the key player in microbicidal activity against C. neoformans. Alternatively, given the possibility of redundancy among members of the Src family (24, 36–38), anticryptococcal activity may be mediated by not one, but several, of these kinases.
Herein we investigate the upstream (receptor-proximal) signaling events that facilitate PI3K→ERK1/2 and anticryptococcal activity, focusing on the role of Src family kinases. To determine whether Src family kinases were activated and required for anticryptococcal activity, a human NK cell line and primary human NK cells were stimulated with C. neoformans, and dual infrared detection of proteins by immunoblotting was performed. Meanwhile, CFU assays were performed under pharmacologic inhibition of Src family kinases to gauge function. Immunoblotting was performed to assess activation of PI3K→ERK1/2 in the presence of an Src family kinase inhibitor. Furthermore, digital deconvolution of stacked microscopy images was used to visualize the effect of inhibiting Src family kinases on perforin polarization to the NK cell-cryptococcal synapse. Finally, single or double knockdown of individual Src family kinases using small interfering RNA (siRNA) was performed to determine the specific member(s) involved in anticryptococcal activity.
MATERIALS AND METHODS
Cell preparation and culture.
The human NK-like leukemic cell line (YT) was a generous gift from C. Clayberger (National Cancer Institute, Bethesda MD). YT cells were cultured at 37°C and 5% CO2 in complete RPMI medium consisting of RPMI 1640 medium, 10% heat-inactivated fetal bovine serum, 1% nonessential amino acids, 1% sodium pyruvate, 1% penicillin, and 1% streptomycin (all from Invitrogen, Carlsbad, CA).
For isolation of primary human NK cells, human peripheral blood was obtained by venipuncture from healthy volunteers (in compliance with the University of Calgary Conjoint Health Research Ethics Board of the University of Calgary, Protocol 23363) and anticoagulated with heparin (10 U/ml blood). Peripheral blood mononuclear cells were purified by centrifugation on a Ficoll-Hypaque (GE Healthcare, Mississauga, ON, Canada) density gradient and washed three times in Hanks balanced salt solution (Invitrogen, Carlsbad, CA). NK cells were magnetically separated through LS columns using a MACS NK cell isolation kit (Miltenyi Biotec, Auburn, CA) as per the manufacturer's instructions. NK cells collected in the negative fraction were labeled with anti-human CD3-phycoerythrin (PE) (BD Biosciences, San Jose CA) and CD56-fluorescein isothiocyanate (FITC) (Biolegend, San Diego CA) and analyzed using a Guava EasyCyte 96-well flow cytometer (Guava Technologies, Hayward, CA) to assess purity.
C. neoformans strain B3501 was obtained from the ATCC (catalog number 34873). C. neoformans was grown to log phase in Sabouraud dextrose broth (Difco) at 32°C with gentle shaking and stored at 4°C.
Antibodies.
Rabbit anti-human Hck, Blk, Fgr, Yes, and phospho-ERK1/2 (p-ERK1/2) and mouse anti-human ERK1/2 and Src were purchased from Cell Signaling Technology (Danvers, MA). Mouse anti-human CD3-phycoerythrin (PE), perforin (clone δG9), Fyn, and Lck antibodies were purchased from BD Biosciences (San Jose, CA). Rabbit anti-human p-Akt1, -2, and -3 (p-Akt1/2/3) and mouse anti-human Akt1 and Lyn antibodies were purchased from Santa Cruz Biotechnologies (Santa Cruz, CA). Anti-phosphotyrosine clone 4G10 and rabbit anti-Fyn antiserum were purchased from Millipore (Billerica, MA). Alexa-350-conjugated phalloidin, rabbit anti-human p-Src family (pY418), and Alexa-555-conjugated goat anti-mouse IgG were purchased from Invitrogen (Carlsbad, CA). Mouse anti-human beta-actin was purchased from Sigma-Aldrich (St. Louis, MO). Goat anti-human Fyn was purchased from AbD Serotec (Raleigh, NC). Goat anti-rabbit IgG infrared (IR) dye 700DX, goat anti-mouse IgG IR dye 800, and donkey anti-goat IgG IR dye 700DX were all purchased from Rockland (Gilbertsville, PA).
NK cell anticryptococcal activity.
Anticryptococcal activity was determined as previously described (39). Briefly, C. neoformans (targets) was grown to log phase in Sabouraud dextrose broth at 32°C with gentle shaking and incubated with YT cells (effectors) in a round-bottom 96-well plate at 37°C. Unless otherwise indicated, a starting effector-to-target (E/T) ratio of 200:1 was used (Cryptococcus alone increases approximately 100-fold during the course of the assay). CFU counts were determined at 0 (starting inoculum), 24, and in some instances 48 h. Primary human NK cell anticryptococcal activity was determined similarly, using a starting E/T ratio of 1,000:1 unless otherwise indicated. For some experiments, cells were pretreated with dimethyl sulfoxide (DMSO) or dasatinib (100 nM) (a generous gift from May Ho, University of Calgary, Calgary, AB, Canada) for 1 h at 37°C prior to incubation with C. neoformans. In other experiments, cells were washed in serum-free RPMI medium and incubated with water (vehicle control) or methyl-beta-cyclodextrin (MBCD) (2 mM) (Sigma-Aldrich, St. Louis, MO) for 30 min at 37°C prior to incubation with C. neoformans.
Stimulation and immunoblotting.
YT and primary human NK cells were incubated with C. neoformans in serum-free RPMI medium at 37°C. An E/T ratio of 1:100 was used unless otherwise indicated. For some experiments, cells were pretreated with DMSO, dasatinib (100 nM), or LY294002 (50 μM) (Calbiochem) for 1 h at 37°C. As a positive control, cells were incubated at 37°C for 10 min with pervanadate, which was prepared as previously described (40). Immediately after stimulation, cells were centrifuged at 3,000 × g for 30s and lysed on ice in NP-40 lysis buffer (Invitrogen) supplemented with phosphatase and protease inhibitor cocktails (both from Roche Applied Science). Lysates were diluted in NuPAGE lithium dodecyl sulfate (LDS) sample buffer and reducing agent (both from Invitrogen) and boiled.
Samples were resolved on 4 to 12% NuPAGE Bis-Tris gels (Invitrogen) and transferred onto nitrocellulose membrane (Bio-Rad). Immunoblots were probed and visualized using an Odyssey infrared imaging system (Li-Cor). Analysis was performed on Odyssey application software. Densitometry was performed using ImageJ, version 1.44o (National Institutes of Health, Bethesda MD). For immunoprecipitation, whole-cell lysates were clarified at 13,000 rpm for 15 min at 4°C. Supernatants were mixed with rabbit anti-Fyn antiserum and rotated overnight at 4°C. Samples were then mixed with protein A and G-agarose beads (Sigma-Aldrich) and rotated for several hours at 4°C. After several washes in NP-40 lysis buffer, samples were resuspended in NuPAGE LDS sample buffer containing 1.5% β-mercaptoethanol and then boiled at 100°C.
Gene silencing with siRNA.
For each protein listed, four individual small interfering RNA (siRNA) sequences were ordered at a time and arbitrarily numbered. Fyn siRNA sequences two (CGCAUGAAUUAUAUCCAUA), three (CAGGAAUGGCUUACAUCGA), and four (CUGUGAAGCAUUCGAGACA) and Lyn siRNA sequences one (UGGCAUACAUCGAGCGGAA), two (AAGCUAAAAUAACCGGAUA), and three (AGAUUGGAGAAGGCUUGUA) and a nontargeting control siRNA were all purchased from Dharmacon RNAi Technologies (Lafayette, CO). For transfection, YT cells were washed in serum-free RPMI medium and resuspended in Nucleofector solution (kit V) (Amaxa, Walkersville, MD). These cells (3 × 106) were then premixed with 0.2 nmol of nontargeting control siRNA (0.4 nmol for double knockdowns) or 0.2 nmol of a single sequence of target-specific siRNA in 100-μl volumes. They were then transferred into cuvettes and nucleofected using an Amaxa Nucleofector II device (program O-017). Immediately after, cells were diluted in prewarmed complete RPMI medium and incubated at 37°C and 5% CO2.
Immunofluorescence microscopy.
YT cells were left untreated or pretreated with DMSO or dasatinib (100 nM) for 30 min at 37°C. YT cells were then incubated alone or with C. neoformans for 4 h at an E/T ratio of 1:5. Cells were fixed with 3.7% formaldehyde, mounted on slides with Prolong Gold (Molecular Probes), and visualized by differential interference contrast (DIC) using a DeltaVision IX70 microscope (Applied Precision, Issaquah, WA) with a PlanApo 60× objective (1.46 numerical aperture [NA]). This wide-angle Olympus microscope is equipped with stacking capabilities. Samples were assessed blinded to sample type, and counts were taken of contiguous fields in a serpentine pattern along the slide. Conjugates were scored if there was a clear contact site with a concave interface in YT cells formed by the C. neoformans conjugate. Results were graphed as percentages of YT cells that formed conjugates with C. neoformans.
For some experiments, cells were fixed with formaldehyde and permeabilized with Perm/Wash (BD). They were then labeled with mouse anti-human perforin, followed by Alexa-555-conjugated goat anti-mouse IgG and phalloidin-Alexa-350 for staining F-actin. Cells were washed, mounted on slides, and visualized by DIC microscopy using a DeltaVision microscope. DIC and fluorescent images represent one deconvolved Z-stack obtained using the digital deconvolution program SoftWoRx (Applied Precision, Issaquat, WA). Cells with F-actin polymerization at the site of C. neoformans contact with YT cells were considered to have formed a stable conjugate. Movement of perforin to the synapse/polymerized F-actin was quantified by assigning a score from 0 to 3 as follows. A score of 0 was assigned when perforin granules were predominately in the half of the cell distal to the synapse or randomly throughout the cell. A score of 1 was assigned when granules were predominantly distributed in the half of the cell proximal to the synapse. A score of 2 was assigned when granules were in loose approximation to the synapse. A score of 3 was assigned when granules were in intimate association with the synapse.
Statistics.
Data expressed are the means ± standard errors of the means (SEM). Statistical significance was determined by performing an analysis of variance (ANOVA), followed by a Bonferroni multiple comparison test. In some instances (only two groups present), Student's t test was performed. Analyses were performed using GraphPad Prism software.
RESULTS
Anticryptococcal activity is lipid raft dependent and accompanied by early tyrosine phosphorylation of a 60-kDa protein.
Previous studies have elucidated several critical elements in the anticryptococcal pathway, including PI3K, ERK1/2, and perforin (13, 15). However, the more receptor-proximal elements (i.e., those upstream of PI3K) have not yet been identified. Several upstream mediators involved in lymphocyte activation and tumor cell killing have been shown to associate with lipid rafts (41). Others have been found to operate independently of lipid rafts (42). Thus, the contribution of lipid rafts was investigated to narrow the range of signaling molecules that could potentially be involved in PI3K activation and cryptococcal killing. This was done using methyl-beta-cyclodextrin (MBCD), a cholesterol-depleting agent known for its ability to disrupt lipid rafts (43). Thus, YT cells were pretreated with an equal volume of H2O (vehicle control, 5%) or 2 mM MBCD and then incubated with C. neoformans for 24 h in serum-free medium. Cultures containing cells treated with MBCD had significantly more CFU than those containing untreated or H2O-treated cells (Fig. 1A). No appreciable effects on viability were observed from MBCD treatment in these experiments (71.3% viability in MBCD treated cells versus 70.9% in control cells). These results suggested that upstream signaling during anticryptococcal activity requires lipid rafts.
Fig 1.

YT cell anticryptococcal activity is lipid raft dependent and accompanied by early tyrosine phosphorylation of a 60-kDa protein. (A) YT cells were left untreated or pretreated with H2O (vehicle control, 5% [vol/vol]) or 2 mM MBCD for 30 min. They were then washed several times in serum-free medium and incubated with C. neoformans (Crypto) for 24 h. CFU counts were determined at 0 and 24 h. **, P < 0.01; ***, P < 0.001. Data are representative of three experiments. (B) YT cells were left unstimulated or stimulated with C. neoformans for 1 to 30 min. Immunoblotting was performed with anti-phospho-Tyr (p-Tyr; clone 4G10). Data are representative of three experiments.
To further elucidate the upstream signaling molecules that could potentially mediate cryptococcal killing, we assessed tyrosine phosphorylation in response to C. neoformans. Thus, YT cells were stimulated with C. neoformans for 1 to 30 min and immunoblotted with the anti-phospho-Tyr (p-Tyr) clone 4G10. Bands were detected at several molecular masses including, approximately, 104 kDa, 99 kDa, 72 kDa, 60 kDa, and 49 kDa (Fig. 1B). A few of these corresponded to the molecular masses of well-known signaling proteins. In particular, the band at approximately 60 kDa corresponded to the molecular mass of several members of the Src family. As Src family kinases are known to signal through lipid rafts (44, 45), these data indicated that Src family kinases might be involved in anticryptococcal activity.
Src family kinases are activated and required for cryptococcal killing.
To determine whether Src family kinases are activated during cryptococcal killing, YT cells were left unstimulated or were stimulated with C. neoformans for 1 to 15 min, and immunoblotting was performed for phosphorylation of ERK1/2 and the conserved activating tyrosine, Y418, of Src family kinases (p-Src family). Consistent with previous studies (15), C. neoformans was found to effectively induce the phosphorylation of ERK1/2 in YT cells (Fig. 2A and B). At the same time, Src family kinase phosphorylation was detected as early as 1 min poststimulation and returned to baseline by 15 min. Primary human NK cells (purity of >90%) stimulated with C. neoformans were also found to signal through Src family kinases, with phosphorylation occurring maximally at 5 min and returning to baseline by 30 min (Fig. 2D and E).
Fig 2.

Src family kinases are activated and necessary for killing of C. neoformans. (A) YT cells were left unstimulated or were stimulated with C. neoformans for 1 to 15 min. Immunoblotting was performed for p-Src family (pY418) and p-ERK1/2. Data are representative of at least three experiments. (B) Corresponding densitometry to the experiment shown in panel A. (C) YT cells were left untreated or were pretreated with DMSO (vehicle control) or 100 nM dasatinib for 1 h. Cells were then incubated with C. neoformans for 24 h. CFU counts were determined at time (T) 0 and 24 h. *, P < 0.05; **, P < 0.01. Data are representative of three experiments. (D) Primary human NK cells (purity of >90%) were left unstimulated or were stimulated with C. neoformans for 1 to 30 min at an E/T ratio of 1:50. Stimulation with pervanadate (PV) served as a positive control. Immunoblotting was performed for the p-Src family (pY418). Data are representative of three experiments. (E) Corresponding densitometry to the experiment shown in panel D. (F) Primary human NK cells were left untreated or were pretreated with DMSO or 100 nM dasatinib for 1 h. They were then incubated with C. neoformans for 24 h. CFU counts were determined at 0 and 24 h. *, P < 0.05. Data are representative of three experiments.
To address whether Src family kinases are also required for cryptococcal killing, cells were treated with a pharmacologic Src family kinase inhibitor, dasatinib. Dasatinib's primary targets are the Src family kinases and Abelson tyrosine kinase (46). It binds reversibly to the Src family kinase ATP site, thereby competitively inhibiting tyrosine kinase function (47). Dasatinib was chosen due to its broad Src family kinase coverage, relatively high potency, and lack of toxicity toward NK cells and C. neoformans. Furthermore, it has previously been shown to effectively block NK cell killing of tumor cells (48). Dasatinib was similarly found to be effective at inhibiting cryptococcal killing. Cultures containing YT or primary human NK cells pretreated with dasatinib were found to have significantly more CFU after 24 h than those containing untreated cells or cells pretreated with the vehicle control DMSO alone (Fig. 2C and F). Dasatinib was not found to exert any adverse effects on viability at the concentration used in these experiments (data not shown).
Src family kinases mediate activation of PI3K→ERK1/2 during anticryptococcal activity.
Given that the activation of PI3K→ERK1/2 has previously been shown to be required for killing of C. neoformans (15), we questioned whether Src family kinases might mediate activation of PI3K→ERK1/2. Thus, YT cells were left untreated or were pretreated with DMSO or dasatinib and then stimulated with C. neoformans. Immunoblotting was then performed to assess phosphorylation of Src family kinases, Akt (v-akt murine thymoma viral oncogene homolog) (indicating PI3K activation), and ERK1/2. While untreated and DMSO-treated cells produced Akt and ERK1/2 phospho-signals in response to C. neoformans, cells treated with dasatinib failed to demonstrate increased phosphorylation of these signaling proteins (Fig. 3A and B). The pharmacologic PI3K inhibitor LY294002 was similarly able to block the phosphorylation of Akt and ERK1/2, as demonstrated previously (15), but had no effect on the phosphorylation of Src family kinases. These results demonstrate that C. neoformans-induced activation of PI3K→ERK is dependent on upstream Src family kinase signals.
Fig 3.

Activation of PI3K and ERK in response to C. neoformans is dependent on upstream Src family kinase signals. (A) YT cells were left untreated or were pretreated with DMSO (vehicle control), 100 nM dasatinib, or 50 μM LY294002 for 1 h. These cells were then left unstimulated or were stimulated with C. neoformans for 1 min. Stimulation with pervanadate served as a positive control. Immunoblotting was performed for p-Src family (pY418), p-Akt1/2/3 (indicating PI3K activation), and p-ERK1/2. The arrow indicates Akt 2 (lower band). Data are representative of three experiments. (B) Corresponding densitometry is shown.
Src family kinases mediate polarization of perforin-containing granules to the YT cell-cryptococcal synapse during killing.
Several possible mechanisms may account for the dependence of anticryptococcal activity on Src family signals. For instance, Src family kinases may regulate the formation of conjugates between NK cells and C. neoformans. Indeed, some studies have reported a requirement for Src family kinases in conjugate formation with tumor cells (49, 50). Thus, the inability of dasatinib-treated YT cells to kill C. neoformans could be due to a lack of conjugate formation altogether. To investigate this possibility, microscopy was used to visualize the number of conjugates formed with DMSO-treated cells compared to cells treated with dasatinib. No significant difference was found between the two groups (Fig. 4A), suggesting that Src family kinases are not required for the formation of NK cell-cryptococcal conjugates.
Fig 4.

Src family kinases mediate polarization of perforin-containing granules to the site of contact with C. neoformans during killing. (A) YT cells were left untreated or were pretreated with DMSO or 100 nM dasatinib for 30 min. YT cells were then incubated alone or with C. neoformans for 4 h at an E/T ratio of 1:5. Cells were fixed and mounted on slides and visualized using a DeltaVision microscope (a single Z section). Conjugates were scored if there was a clear contact site accompanied by a concave depression in the YT cell in contact with C. neoformans. Samples were assessed blinded to sample type, and counts were taken of contiguous fields in a contiguous serpentine pattern across the slide. Data are representative of 250 cells from one of two independent experiments. NS, not significant. (B) Cells were fixed, permeabilized, and labeled with mouse anti-human perforin followed by Alexa-555-conjugated goat anti-mouse IgG (red) and phalloidin–Alexa-350 for staining F-actin (blue). Cells were washed, mounted on slides, and visualized using a DeltaVision microscope. YT cells with F-actin polymerization at the site of contact with C. neoformans were considered to have formed a stable conjugate. Two different representative images are indicated by Crypto #1 and Crypto #2. (C) Movement of perforin to the synapse/polymerized F-actin was quantified by assigning a score from 0 to 3 as follows. A score of 0 was assigned when perforin granules were predominately in the half of the cell distal to the synapse. A score of 1 was assigned when granules were predominantly distributed in the half of the cell proximal to the synapse. A score of 2 was assigned when granules were in loose approximation to the synapse. A score of 3 was assigned when granules were in intimate association with the synapse. ***, P < 0.001. Data are representative of 16 established conjugates from one of two independent experiments.
Another possibility may be that Src family kinases influence the movement of perforin-containing granules to the NK cell-cryptococcal synapse. This is suggested by our finding that Src family kinases mediate the activation of PI3K→ERK, which has previously been shown to induce the polarization of perforin-containing granules to the NK cell-cryptococcal synapse (15). To test this hypothesis, YT cells were pretreated with DMSO or dasatinib and then incubated with C. neoformans for 4 h. Cells were then stained for perforin and F-actin and visualized by microscopy. In cells treated with DMSO there was both F-actin polymerization and perforin trafficking to the YT cell-cryptococcal synapse (Fig. 4B). On the other hand, cells treated with dasatinib showed little perforin movement to the site of contact, despite having formed a stable polymerized F-actin plate. Quantitative analysis revealed a statistically significant difference between the polarizations of perforin in DMSO- and dasatinib-treated cells (Fig. 4C). Thus, the inability of perforin-containing granules to traffic to the YT cell-cryptococcal synapse in the presence of dasatinib provided evidence that Src family kinases are required for the granule polarization that is necessary for anticryptococcal activity.
Fyn is not solely required for YT cell anticryptococcal activity.
To begin determining the specific Src family kinase(s) involved in anticryptococcal activity, YT and primary human NK cells were screened for Src family kinase expression. Based on immunoblotting, both YT and primary human NK cells were found to express several Src family members, including lithium dodecyl sulfate (Fig. 5A). Given the major role of Fyn in NK cell cytotoxicity against tumor cells (24, 25, 28), we wondered whether it might similarly mediate microbicidal activity against C. neoformans. Thus, Fyn was immunoprecipitated from YT cells stimulated with C. neoformans for 1 to 15 min and then subjected to immunoblotting for p-Tyr using 4G10. Phosphorylation of Fyn was detected within 1 min of stimulation and returned to resting levels by 15 min (Fig. 5B and C). These results demonstrate that Fyn is activated in response to C. neoformans. To determine whether Fyn is also required for cryptococcal killing, YT cells were transfected with nonspecific siRNA or two different sequences of Fyn siRNA to minimize the possibility of off-target effects. Forty-eight hours later (determined to be the time of maximal knockdown), immunoblotting was performed to confirm knockdown (Fig. 5D), and cells were incubated with C. neoformans for an additional 24 h to assess anticryptococcal activity. Surprisingly, neither Fyn siRNA sequence had any effect on CFU counts (Fig. 5E) despite achieving >90% knockdown. Thus, the ability of NK cells to kill C. neoformans is not entirely dependent on Fyn.
Fig 5.

Fyn is not solely required for YT cell killing of C. neoformans. (A) Immunoblotting was performed for the indicated Src family kinases in YT and primary human NK cells. Mixed primary human T and B cells (T&B) served as a positive control. (B) YT cells were left unstimulated or were stimulated with C. neoformans for 1 to 15 min. Stimulation with pervanadate (PV) served as a positive control. Whole-cell lysates were immunoprecipitated (IP) for Fyn using anti-Fyn antiserum. Immunoblotting (IB) was performed for p-Tyr (4G10) and Fyn. Data are representative of two experiments. (C) Corresponding densitometry for the experiment shown in panel B. (D) YT cells were mock transfected or transfected with nonspecific (NS) siRNA or Fyn siRNA (sequence [seq] 3 or 4). At 48 h posttransfection, immunoblotting was performed for Fyn to assess knockdown. (E) Mock- and siRNA-transfected YT cells were incubated with C. neoformans for an additional 24 h. CFU counts were determined at 72 h posttransfection. T = 0, starting inoculum; NS, not significant. Data are representative of four experiments.
Fyn and Lyn are redundant in mediating anticryptococcal activity.
Despite becoming activated in response to C. neoformans, Fyn was not essential for anticryptococcal activity. One explanation for these results may be the phenomenon of redundancy: that is, some members of the Src family are able to assume the function of other members in their absence. Given that Src family kinases share a conserved domain structure, redundancy is certainly possible and has been suggested by several studies. In NK and T cells in particular, there is evidence to support redundancy between the kinases Fyn and Lck (24, 36, 38). Meanwhile, the possibility of redundancy among other Src family kinases such as Fyn and Lyn in NK and T cells has yet to be explored. In any case, the redundant Src family kinase(s) would also have to be knocked down to elucidate a response. Among the three remaining Src family members expressed by YT cells, it was felt that Lyn would be the most likely to substitute for Fyn in mediating anticryptococcal activity when Fyn is absent (51). Thus, YT cells were transfected with nonspecific siRNA, Fyn siRNA alone, Lyn siRNA alone, or both Fyn and Lyn siRNAs together. Forty-eight hours posttransfection, immunoblotting was performed to assess knockdown (Fig. 6A), and cells were incubated with C. neoformans for an additional 48 h to assess cryptococcal killing. Fyn or Lyn knockdown individually had no significant effect on CFU counts (Fig. 6B). However, when these kinases were knocked down together, there was a significant increase in CFU counts, reflecting impaired anticryptococcal activity compared to both the nonspecific siRNA and mock-transfected groups. No adverse effects on viability were observed from the double knockdown itself. To minimize the possibility of off-target effects, replicate experiments were performed with different Fyn and Lyn sequences (data not shown). Thus, a redundancy exists between Fyn and Lyn such that anticryptococcal activity is spared in the absence of one or the other.
Fig 6.
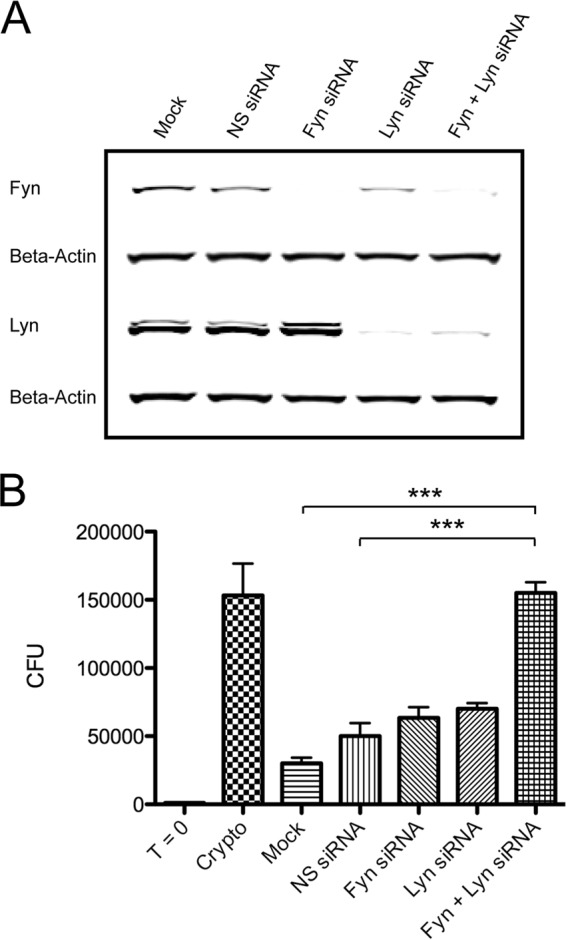
Fyn and Lyn are redundant in mediating killing of C. neoformans. (A) YT cells were mock transfected or transfected with nonspecific (NS) siRNA, Fyn siRNA alone, Lyn siRNA alone, or both Fyn and Lyn siRNA. At 48 h posttransfection, immunoblotting was performed for Fyn and Lyn to assess knockdown. (B) YT cells were incubated with C. neoformans for an additional 48 h. CFU counts were determined at 96 h posttransfection. T = 0, starting inoculum; ***, P < 0.001. Data are representative of three experiments.
DISCUSSION
In this study, we have identified the following key aspects of the NK cell signaling response to C. neoformans. (i) Src family kinases are activated and necessary for NK cell killing of C. neoformans. (ii) Src family kinases mediate activation of the previously described PI3K→ERK1/2 pathway during anticryptococcal activity. (iii) Unlike conjugate formation, the polarization of perforin-containing granules to the NK cell-cryptococcal synapse during killing is dependent on Src family kinase signals. (iv) Neither Fyn (despite becoming activated in response to C. neoformans) nor Lyn is solely required for cryptococcal killing. (v) The Src family kinases Fyn and Lyn redundantly mediate anticryptococcal activity.
Previous studies have indicated that Src family kinases play a pivotal role in upstream signaling during NK cell killing of tumor cells. Tumor cells stimulate NK cell receptors that associate with immunoreceptor tyrosine-based activation motif (ITAM)-containing (52) transmembrane adaptors, resulting in the phosphorylation of ITAMs by Src family kinases (53). These are then able to bind spleen tyrosine kinase (Syk) (54, 55) and a zeta-chain-associated protein of 70 kDa (ZAP-70) (56), which may in turn facilitate recruitment of downstream signaling molecules such as PI3K (57). Similarly, the YINM sequence of NKG2D and the adaptors (SAP and EAT-2) of the SLAM family of receptors require Src family kinase-dependent phosphorylation (26, 27, 29, 58–60). While it is evident that NK cells also mediate killing of C. neoformans, very little is known regarding the signaling mechanisms underpinning this microbicidal activity. Building on previous work that described some of the major downstream signaling elements involved in the NK cell response to C. neoformans, our data demonstrated an essential role for Src family kinases in anticryptococcal activity. Thus, although tumor cells and C. neoformans are vastly different targets, NK cells appear to utilize a conserved family of upstream signaling molecules (Src family kinases) for cytotoxicity.
Our results demonstrate that Src family kinases are required for NK cell anticryptococcal activity. Indeed, dasatinib treatment impaired NK cell killing of C. neoformans. As with all pharmacologic inhibitors, however, the possibility of nonspecific effects was considered, which prompted confirmation with knockdown experiments (to be discussed later). Furthermore, our results show that the requirement of Src family kinases for anticryptococcal activity is rooted in their ability to mediate activation of PI3K→ERK1/2. Indeed, inhibition of Src family kinase signaling with dasatinib was found to impair activation of PI3K and ERK1/2 in response to C. neoformans. Interestingly, Src family kinase activation was found to noticeably outlast that of ERK1/2 following stimulation with C. neoformans. This finding may reflect the fact that deactivation of ERK1/2 through phosphatase activity proceeds at a faster pace than that of Src family kinases following stimulation with C. neoformans.
Prior studies have shown that the polarization of perforin-containing granules toward the site of tumor contact is dependent on the activation of PI3K→ERK1/2 (61), which is dependent on Src family kinases (62). These data were consistent with our observation that the polarization of perforin-containing granules toward the site of contact with C. neoformans is dependent on PI3K→ERK1/2 (15) and Src family kinases (present data). Thus, our data established a link between upstream Src family kinase signals and perforin-dependent cryptococcal killing. Interestingly, unlike the trafficking of perforin-containing granules, actin polymerization at the NK cell-cryptococcal synapse was still able to occur in the presence of dasatinib, suggesting that this process is regulated independently of Src family kinases. Studies with tumor cells have shown that actin polymerization during NK cell cytotoxicity is mediated to a large extent by Rho family GTPases, which in turn are activated by the Vav family of guanine nucleotide exchange factors (GEF) (53). Thus, one explanation may be that Vav family members become activated independently of Src family kinases in response to NK cell stimulation. In any case, it seems that both Src family kinase-dependent and -independent signaling pathways occurring in parallel govern NK cell anticryptococcal activity.
It is evident from tumor cell studies that Fyn and Lck are the predominant Src family kinases involved in NK cell cytotoxic pathways. For instance, cross-linking of the murine receptor Ly49D (24) and natural cytotoxicity receptors (NKp30, NKp44, and NKp46) (25), which mediate killing through the ITAM-dependent pathway, was found to induce Fyn and Lck activation. Similarly, Lck has been found to mediate ADCC through the Fc receptor CD16 (30–33) and may also play an important role in the NKG2D/DNAX-activating protein of 10 kDa (DAP-10)-dependent pathway (58). Finally, the SLAM family receptor 2B4 has been shown to induce cytotoxicity through activation of Fyn (26, 29). Given these reports, it was anticipated that Fyn would be required for anticryptococcal activity. Thus, it came as a surprise when Fyn knockdown was not sufficient to abrogate killing of C. neoformans. One explanation for these results may be that another Src family kinase(s) was redundant with Fyn, thus masking the effect of the knockdown. Such a relationship among Src family members has been recognized and suggested by several studies (24, 36, 37) although it has not been firmly established which Src family kinases are redundant. Among the remaining kinases expressed by YT cells, we felt that Lyn would be the most likely to be redundant with Fyn for several reasons. First, in addition to Lck and Fyn, Lyn has been found to associate with several NK cell cytotoxic receptors, including β1 integrins (63), CD16 (35), NKR-P1, and CD94 (34). Second, Fyn and Lck (but not Src) have been shown to compensate for Lyn deficiency in B cell receptor signaling, which provides some evidence that redundancy may exist between Fyn and Lyn (51). Indeed, our work demonstrates that Fyn and Lyn are redundant in mediating anticryptococcal activity. This conclusion was derived from the observation that neither Fyn nor Lyn knockdown individually was sufficient to disrupt cryptococcal killing, whereas concomitant knockdown of both kinases abrogated killing. While we acknowledge that these data are obtained from YT cells, these data provide the first direct evidence that Fyn and Lyn have the capacity for redundancy in NK cells. In contrast, previous studies with NK and T cells have primarily suggested redundancy between Fyn and Lck (24, 36, 38). Our data also expand on the previously underrecognized role of Lyn in NK cell cytotoxicity. Indeed, while Lyn has been shown to associate with NK cell cytotoxic receptors and become phosphorylated (34, 35, 63), it has never been shown to be required for NK cell cytolytic function. Thus, while Fyn and Lck have classically been shown to be the dominant Src family kinases involved in NK cell lysis of tumor targets, other Src family kinases, such as Lyn, might have a more important role in NK cell cytotoxicity than once thought, particularly in the context of direct microbicidal activity.
We have established that the Src family kinases Fyn and Lyn are involved in the activation of PI3K→ERK1/2 and killing of C. neoformans. However, as other Src family members were also expressed, our data do not preclude the possibility that knockdown of another combination of Src family kinases might also have an inhibitory effect on anticryptococcal activity. Nevertheless, knockdown of two Src family kinases, as opposed to three or more, was sufficient to disrupt anticryptococcal activity. Thus, while a “critical mass” effect may occur with even greater redundancy, our data suggest that there is a limit to the extent to which redundancy among Src family kinases may compensate for loss of some of the Src family members (e.g., Fyn and Lyn) and still preserve function. It is also still unclear which signaling molecule(s) couples Src family kinases to PI3K. In this regard, there are several possibilities. (i) Cryptococcal killing may be mediated by one of the major NK cell cytotoxic pathways such as the ITAM, DAP-10, or immunoreceptor tyrosine-based switch motif (ITSM) or adhesion-protein dependent pathways (53). Previous studies have demonstrated that anticryptococcal activity proceeds independently of LFA-1 (64), but there have been no published data excluding the involvement of ITAMs, DAP-10, SLAM family receptors, or adhesion proteins such as other β1-integrins, DNAM-1, CRTAM, etc. (ii) Rather than requiring an intermediate step, Src family kinases may directly associate with and activate PI3K during anticryptococcal activity. Indeed, several studies have demonstrated the ability of the p85 subunit of PI3K (specifically two N-terminal proline-rich regions) to bind the SH3 domain of the Src family kinases Lck, Fyn, and Lyn, which resulted in increased PI3K activity (65, 66). Such a mechanism of PI3K activation was found to occur in T cells upon cross-linking of CD3 (67) and may thus also take place in NK cells. (iii) Cryptococcal killing may be mediated altogether through a unique, not yet described pathway. It follows that PI3K activation may be coupled to Src family kinases through signaling molecule(s) not classically associated with NK cell cytotoxicity. In this case, further investigation of the anticryptococcal pathway will no doubt uncover these molecule(s).
In conclusion, we have demonstrated an essential role for Src family kinases in perforin-dependent cryptococcal killing by NK cells. In doing so, we have further uncovered the pathway responsible for the remarkable ability of NK cells to directly recognize and kill microbial targets.
ACKNOWLEDGMENTS
We thank J. P. Deans and M. Ho for their labs' technical assistance and helpful discussions.
This work was supported by a grant from the Canadian Institutes for Health Research (C.H.M., 247301) and the Jessie Bowden Lloyd Professorship in Immunology (C.H.M.) and by an equipment and infrastructure grant from the Canadian Foundation for Innovation and the Alberta Science and Research Authority.
Immunofluorescence microscopy was performed in the Live Cell Imaging Facility of the Snyder Institute for Chronic Disease with valuable assistance of Pina Colarusso.
We have no conflicts of interest.
Footnotes
Published ahead of print 5 August 2013
REFERENCES
- 1.Lanier LL. 2008. Evolutionary struggles between NK cells and viruses. Nat. Rev. Immunol. 8:259–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Long EO. 2002. Tumor cell recognition by natural killer cells. Semin. Cancer Biol. 12:57–61 [DOI] [PubMed] [Google Scholar]
- 3.Nencioni L, Villa L, Boraschi D, Berti B, Tagliabue A. 1983. Natural and antibody-dependent cell-mediated activity against Salmonella typhimurium by peripheral and intestinal lymphoid cells in mice. J. Immunol. 130:903–907 [PubMed] [Google Scholar]
- 4.Esin S, Batoni G, Counoupas C, Stringaro A, Brancatisano FL, Colone M, Maisetta G, Florio W, Arancia G, Campa M. 2008. Direct binding of human NK cell natural cytotoxicity receptor NKp44 to the surfaces of mycobacteria and other bacteria. Infect. Immun. 76:1719–1727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roland J, Soulard V, Sellier C, Drapier AM, Di Santo JP, Cazenave PA, Pied S. 2006. NK cell responses to Plasmodium infection and control of intrahepatic parasite development. J. Immunol. 177:1229–1239 [DOI] [PubMed] [Google Scholar]
- 6.Petkus AF, Baum LL. 1987. Natural killer cell inhibition of young spherules and endospores of Coccidioides immitis. J. Immunol. 139:3107–3111 [PubMed] [Google Scholar]
- 7.Jimenez BE, Murphy JW. 1984. In vitro effects of natural killer cells against Paracoccidioides brasiliensis yeast phase. Infect. Immun. 46:552–558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Murphy JW, Hidore MR, Nabavi N. 1991. Binding interactions of murine natural killer cells with the fungal target Cryptococcus neoformans. Infect. Immun. 59:1476–1488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nabavi N, Murphy JW. 1985. In vitro binding of natural killer cells to Cryptococcus neoformans targets. Infect. Immun. 50:50–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Murphy JW, Hidore MR, Wong SC. 1993. Direct interactions of human lymphocytes with the yeast-like organism, Cryptococcus neoformans. J. Clin. Invest. 91:1553–1566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hidore MR, Nabavi N, Sonleitner F, Murphy JW. 1991. Murine natural killer cells are fungicidal to Cryptococcus neoformans. Infect. Immun. 59:1747–1754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Park BJ, Wannemuehler KA, Marston BJ, Govender N, Pappas PG, Chiller TM. 2009. Estimation of the current global burden of cryptococcal meningitis among persons living with HIV/AIDS. AIDS 23:525–530 [DOI] [PubMed] [Google Scholar]
- 13.Ma LL, Wang CL, Neely GG, Epelman S, Krensky AM, Mody CH. 2004. NK cells use perforin rather than granulysin for anticryptococcal activity. J. Immunol. 173:3357–3365 [DOI] [PubMed] [Google Scholar]
- 14.Hidore MR, Murphy JW. 1989. Murine natural killer cell interactions with a fungal target, Cryptococcus neoformans. Infect. Immun. 57:1990–1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wiseman JC, Ma LL, Marr KJ, Jones GJ, Mody CH. 2007. Perforin-dependent cryptococcal microbicidal activity in NK cells requires PI3K-dependent ERK1/2 signaling. J. Immunol. 178:6456–6464 [DOI] [PubMed] [Google Scholar]
- 16.Laird MH, Rhee SH, Perkins DJ, Medvedev AE, Piao W, Fenton MJ, Vogel SN. 2009. TLR4/MyD88/PI3K interactions regulate TLR4 signaling. J. Leukoc. Biol. 85:966–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Caparros E, Munoz P, Sierra-Filardi E, Serrano-Gomez D, Puig-Kroger A, Rodriguez-Fernandez JL, Mellado M, Sancho J, Zubiaur M, Corbi AL. 2006. DC-SIGN ligation on dendritic cells results in ERK and PI3K activation and modulates cytokine production. Blood 107:3950–3958 [DOI] [PubMed] [Google Scholar]
- 18.Li B, Allendorf DJ, Hansen R, Marroquin J, Ding C, Cramer DE, Yan J. 2006. Yeast beta-glucan amplifies phagocyte killing of iC3b-opsonized tumor cells via complement receptor 3-Syk-phosphatidylinositol 3-kinase pathway. J. Immunol. 177:1661–1669 [DOI] [PubMed] [Google Scholar]
- 19.Chalifour A, Jeannin P, Gauchat JF, Blaecke A, Malissard M, N′Guyen T, Thieblemont N, Delneste Y. 2004. Direct bacterial protein PAMP recognition by human NK cells involves TLRs and triggers alpha-defensin production. Blood 104:1778–1783 [DOI] [PubMed] [Google Scholar]
- 20.Meng F, Lowell CA. 1997. Lipopolysaccharide (LPS)-induced macrophage activation and signal transduction in the absence of Src-family kinases Hck, Fgr, and Lyn. J. Exp. Med. 185:1661–1670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Keck S, Freudenberg M, Huber M. 2010. Activation of murine macrophages via TLR2 and TLR4 is negatively regulated by a Lyn/PI3K module and promoted by SHIP1. J. Immunol. 184:5809–5818 [DOI] [PubMed] [Google Scholar]
- 22.Parsons SJ, Parsons JT. 2004. Src family kinases, key regulators of signal transduction. Oncogene 23:7906–7909 [DOI] [PubMed] [Google Scholar]
- 23.Boggon TJ, Eck MJ. 2004. Structure and regulation of Src family kinases. Oncogene 23:7918–7927 [DOI] [PubMed] [Google Scholar]
- 24.Mason LH, Willette-Brown J, Taylor LS, McVicar DW. 2006. Regulation of Ly49D/DAP12 signal transduction by Src-family kinases and CD45. J. Immunol. 176:6615–6623 [DOI] [PubMed] [Google Scholar]
- 25.Augugliaro R, Parolini S, Castriconi R, Marcenaro E, Cantoni C, Nanni M, Moretta L, Moretta A, Bottino C. 2003. Selective cross-talk among natural cytotoxicity receptors in human natural killer cells. Eur. J. Immunol. 33:1235–1241 [DOI] [PubMed] [Google Scholar]
- 26.Bloch-Queyrat C, Fondaneche MC, Chen R, Yin L, Relouzat F, Veillette A, Fischer A, Latour S. 2005. Regulation of natural cytotoxicity by the adaptor SAP and the Src-related kinase Fyn. J. Exp. Med. 202:181–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Latour S, Roncagalli R, Chen R, Bakinowski M, Shi X, Schwartzberg PL, Davidson D, Veillette A. 2003. Binding of SAP SH2 domain to FynT SH3 domain reveals a novel mechanism of receptor signalling in immune regulation. Nat. Cell Biol. 5:149–154 [DOI] [PubMed] [Google Scholar]
- 28.Chan B, Lanyi A, Song HK, Griesbach J, Simarro-Grande M, Poy F, Howie D, Sumegi J, Terhorst C, Eck MJ. 2003. SAP couples Fyn to SLAM immune receptors. Nat. Cell Biol. 5:155–160 [DOI] [PubMed] [Google Scholar]
- 29.Chen R, Relouzat F, Roncagalli R, Aoukaty A, Tan R, Latour S, Veillette A. 2004. Molecular dissection of 2B4 signaling: implications for signal transduction by SLAM-related receptors. Mol. Cell. Biol. 24:5144–5156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cone JC, Lu Y, Trevillyan JM, Bjorndahl JM, Phillips CA. 1993. Association of the p56lck protein tyrosine kinase with the Fc gamma RIIIA/CD16 complex in human natural killer cells. Eur. J. Immunol. 23:2488–2497 [DOI] [PubMed] [Google Scholar]
- 31.Pignata C, Prasad KV, Robertson MJ, Levine H, Rudd CE, Ritz J. 1993. Fc gamma RIIIA-mediated signaling involves src-family Lck in human natural killer cells. J. Immunol. 151:6794–6800 [PubMed] [Google Scholar]
- 32.Salcedo TW, Kurosaki T, Kanakaraj P, Ravetch JV, Perussia B. 1993. Physical and functional association of p56lck with FcγRIIIA (CD16) in natural killer cells. J. Exp. Med. 177:1475–1480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ting AT, Dick CJ, Schoon RA, Karnitz LM, Abraham RT, Leibson PJ. 1995. Interaction between Lck and Syk family tyrosine kinases in Fcγ receptor-initiated activation of natural killer cells. J. Biol. Chem. 270:16415–16421 [DOI] [PubMed] [Google Scholar]
- 34.Cerny J, Fiserova A, Horvath O, Bezouska K, Pospisil M, Horejsi V. 1997. Association of human NK cell surface receptors NKR-P1 and CD94 with Src-family protein kinases. Immunogenetics 46:231–236 [DOI] [PubMed] [Google Scholar]
- 35.Mota G, Moldovan I, Calugaru A, Hirt M, Kozma E, Galatiuc C, Brasoveanu L, Boltz-Nitulescu G. 2004. Interaction of human immunoglobulin G with CD16 on natural killer cells: ligand clearance, FcγRIIIA turnover and effects of metalloproteinases on FcγRIIIA-mediated binding, signal transduction and killing. Scand. J. Immunol. 59:278–284 [DOI] [PubMed] [Google Scholar]
- 36.Groves T, Smiley P, Cooke MP, Forbush K, Perlmutter RM, Guidos CJ. 1996. Fyn can partially substitute for Lck in T lymphocyte development. Immunity 5:417–428 [DOI] [PubMed] [Google Scholar]
- 37.Sillman AL, Monroe JG. 1994. Surface IgM-stimulated proliferation, inositol phospholipid hydrolysis, Ca2+ flux, and tyrosine phosphorylation are not altered in B cells from p59fyn−/− mice. J. Leukoc. Biol. 56:812–816 [DOI] [PubMed] [Google Scholar]
- 38.van Oers NS, Lowin-Kropf B, Finlay D, Connolly K, Weiss A. 1996. alpha beta T cell development is abolished in mice lacking both Lck and Fyn protein tyrosine kinases. Immunity 5:429–436 [DOI] [PubMed] [Google Scholar]
- 39.Levitz SM, Dupont MP, Smail EH. 1994. Direct activity of human T lymphocytes and natural killer cells against Cryptococcus neoformans. Infect. Immun. 62:194–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Evans GA, Garcia GG, Erwin R, Howard OM, Farrar WL. 1994. Pervanadate simulates the effects of interleukin-2 (IL-2) in human T cells and provides evidence for the activation of two distinct tyrosine kinase pathways by IL-2. J. Biol. Chem. 269:23407–23412 [PubMed] [Google Scholar]
- 41.Watzl C, Long EO. 2003. Natural killer cell inhibitory receptors block actin cytoskeleton-dependent recruitment of 2B4 (CD244) to lipid rafts. J. Exp. Med. 197:77–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hundt M, Harada Y, De Giorgio L, Tanimura N, Zhang W, Altman A. 2009. Palmitoylation-dependent plasma membrane transport but lipid raft-independent signaling by linker for activation of T cells. J. Immunol. 183:1685–1694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Simons K, Toomre D. 2000. Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol. 1:31–39 [DOI] [PubMed] [Google Scholar]
- 44.Filipp D, Zhang J, Leung BL, Shaw A, Levin SD, Veillette A, Julius M. 2003. Regulation of Fyn through translocation of activated Lck into lipid rafts. J. Exp. Med. 197:1221–1227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Arcaro A, Aubert M, Espinosa del Hierro ME, Khanzada UK, Angelidou S, Tetley TD, Bittermann AG, Frame MC, Seckl MJ. 2007. Critical role for lipid raft-associated Src kinases in activation of PI3K-Akt signalling. Cell Signal. 19:1081–1092 [DOI] [PubMed] [Google Scholar]
- 46.Montero JC, Seoane S, Ocana A, Pandiella A. 2011. Inhibition of SRC family kinases and receptor tyrosine kinases by dasatinib: possible combinations in solid tumors. Clin. Cancer Res. 17:5546–5552 [DOI] [PubMed] [Google Scholar]
- 47.Gratacap MP, Martin V, Valera MC, Allart S, Garcia C, Sie P, Recher C, Payrastre B. 2009. The new tyrosine-kinase inhibitor and anticancer drug dasatinib reversibly affects platelet activation in vitro and in vivo. Blood 114:1884–1892 [DOI] [PubMed] [Google Scholar]
- 48.Blake SJ, Bruce Lyons A, Fraser CK, Hayball JD, Hughes TP. 2008. Dasatinib suppresses in vitro natural killer cell cytotoxicity. Blood 111:4415–4416 [DOI] [PubMed] [Google Scholar]
- 49.Burshtyn DN, Shin J, Stebbins C, Long EO. 2000. Adhesion to target cells is disrupted by the killer cell inhibitory receptor. Curr. Biol. 10:777–780 [DOI] [PubMed] [Google Scholar]
- 50.Aucher A, Magdeleine E, Joly E, Hudrisier D. 2008. Capture of plasma membrane fragments from target cells by trogocytosis requires signaling in T cells but not in B cells. Blood 111:5621–5628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Takata M, Sabe H, Hata A, Inazu T, Homma Y, Nukada T, Yamamura H, Kurosaki T. 1994. Tyrosine kinases Lyn and Syk regulate B cell receptor-coupled Ca2+ mobilization through distinct pathways. EMBO J. 13:1341–1349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shibuya A, Campbell D, Hannum C, Yssel H, Franz-Bacon K, McClanahan T, Kitamura T, Nicholl J, Sutherland GR, Lanier LL, Phillips JH. 1996. DNAM-1, a novel adhesion molecule involved in the cytolytic function of T lymphocytes. Immunity 4:573–581 [DOI] [PubMed] [Google Scholar]
- 53.Tassi I, Klesney-Tait J, Colonna M. 2006. Dissecting natural killer cell activation pathways through analysis of genetic mutations in human and mouse. Immunol. Rev. 214:92–105 [DOI] [PubMed] [Google Scholar]
- 54.Brumbaugh KM, Binstadt BA, Billadeau DD, Schoon RA, Dick CJ, Ten RM, Leibson PJ. 1997. Functional role for Syk tyrosine kinase in natural killer cell-mediated natural cytotoxicity. J. Exp. Med. 186:1965–1974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jiang K, Zhong B, Gilvary DL, Corliss BC, Vivier E, Hong-Geller E, Wei S, Djeu JY. 2002. Syk regulation of phosphoinositide 3-kinase-dependent NK cell function. J. Immunol. 168:3155–3164 [DOI] [PubMed] [Google Scholar]
- 56.Vivier E, da Silva AJ, Ackerly M, Levine H, Rudd CE, Anderson P. 1993. Association of a 70-kDa tyrosine phosphoprotein with the CD16:ζ:γ complex expressed in human natural killer cells. Eur. J. Immunol. 23:1872–1876 [DOI] [PubMed] [Google Scholar]
- 57.Shim EK, Jung SH, Lee JR. 2011. Role of two adaptor molecules SLP-76 and LAT in the PI3K signaling pathway in activated T cells. J. Immunol. 186:2926–2935 [DOI] [PubMed] [Google Scholar]
- 58.Billadeau DD, Upshaw JL, Schoon RA, Dick CJ, Leibson PJ. 2003. NKG2D-DAP10 triggers human NK cell-mediated killing via a Syk-independent regulatory pathway. Nat. Immunol. 4:557–564 [DOI] [PubMed] [Google Scholar]
- 59.Tassi I, Colonna M. 2005. The cytotoxicity receptor CRACC (CS-1) recruits EAT-2 and activates the PI3K and phospholipase Cγ signaling pathways in human NK cells. J. Immunol. 175:7996–8002 [DOI] [PubMed] [Google Scholar]
- 60.Eissmann P, Beauchamp L, Wooters J, Tilton JC, Long EO, Watzl C. 2005. Molecular basis for positive and negative signaling by the natural killer cell receptor 2B4 (CD244). Blood 105:4722–4729 [DOI] [PubMed] [Google Scholar]
- 61.Jiang K, Zhong B, Gilvary DL, Corliss BC, Hong-Geller E, Wei S, Djeu JY. 2000. Pivotal role of phosphoinositide-3 kinase in regulation of cytotoxicity in natural killer cells. Nat. Immunol. 1:419–425 [DOI] [PubMed] [Google Scholar]
- 62.James AM, Hsu HT, Dongre P, Uzel G, Mace EM, Banerjee PP, Orange JS. 2013. Rapid activation receptor- or IL-2-induced lytic granule convergence in human natural killer cells requires Src, but not downstream signaling. Blood 121:2627–2637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rabinowich H, Manciulea M, Herberman RB, Whiteside TL. 1996. Beta1 integrin-mediated activation of focal adhesion kinase and its association with Fyn and Zap-70 in human NK cells. J. Immunol. 157:3860–3868 [PubMed] [Google Scholar]
- 64.Jones GJ, Wiseman JC, Marr KJ, Wei S, Djeu JY, Mody CH. 2009. In contrast to anti-tumor activity, YT cell and primary NK cell cytotoxicity for Cryptococcus neoformans bypasses LFA-1. Int. Immunol. 21:423–432 [DOI] [PubMed] [Google Scholar]
- 65.Kapeller R, Prasad KV, Janssen O, Hou W, Schaffhausen BS, Rudd CE, Cantley LC. 1994. Identification of two SH3-binding motifs in the regulatory subunit of phosphatidylinositol 3-kinase. J. Biol. Chem. 269:1927–1933 [PubMed] [Google Scholar]
- 66.Pleiman CM, Hertz WM, Cambier JC. 1994. Activation of phosphatidylinositol-3′ kinase by Src-family kinase SH3 binding to the p85 subunit. Science 263:1609–1612 [DOI] [PubMed] [Google Scholar]
- 67.Prasad KV, Janssen O, Kapeller R, Raab M, Cantley LC, Rudd CE. 1993. Src-homology 3 domain of protein kinase p59fyn mediates binding to phosphatidylinositol 3-kinase in T cells. Proc. Natl. Acad. Sci. U. S. A. 90:7366–7370 [DOI] [PMC free article] [PubMed] [Google Scholar]