

Abstract
Quorum sensing is a cell-to-cell communication system known to control many bacterial processes. In the present study, the functions of quorum sensing in the pathogenesis of Vibrio vulnificus, a food-borne pathogen, were assessed by evaluating the virulence of a mutant deficient in SmcR, a quorum-sensing regulator and homologue of LuxR. When biofilms were used as an inoculum, the smcR mutant was impaired in virulence and colonization capacity in the infection of mice. The lack of SmcR also resulted in decreased histopathological damage in mouse jejunum tissue. These results indicated that SmcR is essential for V. vulnificus pathogenesis. Moreover, the smcR mutant exhibited significantly reduced biofilm detachment. Upon exposure to INT-407 host cells, the wild type, but not the smcR mutant, revealed accelerated biofilm detachment. The INT-407 cells increased smcR expression by activating the expression of LuxS, an autoinducer-2 synthase, indicating that host cells manipulate the cellular level of SmcR through the quorum-sensing signaling of V. vulnificus. A whole-genome microarray analysis revealed that the genes primarily involved in biofilm detachment and formation are up- and downregulated by SmcR, respectively. Among the SmcR-regulated genes, vvpE encoding an elastolytic protease was the most upregulated, and the purified VvpE appeared to dissolve established biofilms directly in a concentration-dependent manner in vitro. These results suggest that the host cell-induced SmcR enhances the detachment of V. vulnificus biofilms entering the host intestine and thereby may promote the dispersal of the pathogen to new colonization loci, which is crucial for pathogenesis.
INTRODUCTION
Quorum sensing is a bacterial cell-to-cell communication process that involves the sensing of extracellular signal molecules as a method to monitor their cell densities and alter their gene expression in a cell density-dependent manner (for a recent review, see reference 1). The signal molecules, called autoinducers (AI), are produced by members of the community and accumulated in the environments as the bacterial cell density increases. Vibrio harveyi luminescence gene regulation in response to the cell density is frequently used as a model for quorum sensing. Three autoinducer molecules, AI-1, AI-2, and CAI-1, are produced and detected by the periplasmic sensor proteins, LuxN, LuxPQ, and CqsS, respectively, in V. harveyi. At low cell density lacking autoinducers, the sensor proteins possessing both kinase and phosphatase activities are autophosphorylated. Phosphate from all three sensors is transduced to a single phosphate transfer protein, LuxU, and then subsequently to the response regulator LuxO. The phosphorylated LuxO activates the transcription of small regulatory RNAs (sRNAs) that destabilize the luxR mRNA, thereby inhibiting the production of LuxR, a quorum-sensing master regulator. LuxR is required to activate the transcription of the luciferase operon luxCDABE, and thus the bacteria do not express bioluminescence at low cell density. At high cell density, the phosphate flow in the signal transduction pathway is reversed, resulting in a high cellular level of LuxR and expression of bioluminescence (1).
Quorum sensing regulates the production of virulence factors in dozens of pathogens, which consequently affects their pathogenesis (2, 3). Quorum sensing also influences bacterial pathogenesis by regulating the development of biofilms, assemblages of bacteria encased in a matrix (4). Biofilm plays a multifaceted role in the pathogenesis of a wide variety of pathogens and is thought to be a sophisticated, highly evolved cellular process particular to each pathogen, leading to overall success of the pathogens during the infectious cycle (5, 6). In Pseudomonas aeruginosa, which infects and then remains in the host (human) chronically, the accumulation of AI at high cell density ensures biofilm formation to a matured structure (7). In contrast, biofilm formation of Vibrio cholerae, which only temporally occupies the human niche, occurs in the absence of AI at low cell density, and the presence of AI at high cell density represses biofilm formation (8).
It is well documented that V. cholerae forms biofilms on abiotic and biotic surfaces, including those of crustaceans, planktons, and algae in natural environments (9). Furthermore, biofilms are likely a form of pathogenic V. cholerae and an important source for new outbreaks as they provide a means to reach a concentrated infective dose consumed by humans (10). As a LuxR homologue, V. cholerae HapR decreases biofilm formation by repressing the genes encoding VpsT and VpsR, positive regulators of the vps operons required for exopolysaccharide biosynthesis and biofilm formation (11, 12). The hapR mutant has a lower ability to colonize in mouse intestines, suggesting that HapR, by enhancing biofilm detachment, may promote dispersal of V. cholerae cells from biofilms to new loci to colonize (13). It is noteworthy that such influences of quorum-sensing deficiency (hapR mutation) on colonization capacity were observed only when biofilms, not planktonic cells, were used as an inoculum to infect mice (13).
Although little is known about the quorum-sensing regulation of Vibrio vulnificus, a pathogenic marine bacterium, it has been known that the AI-2 (or homologue) produced by LuxS is the quorum-sensing signal molecule of the pathogen (14). SmcR, a LuxR homologue of V. vulnificus, appears to regulate numerous genes involved in the virulence and survival of the pathogen (15–19). However, until now, a definitive analysis of the relationship among quorum sensing, biofilms, and the pathogenesis of V. vulnificus has not been addressed. Accordingly, in order to elucidate the roles of quorum sensing in V. vulnificus pathogenesis, biofilm of the smcR mutant was used to infect mice, and its virulence was compared to that of the parental wild type. Moreover, the influence of host cells on the level of smcR expression and on biofilm development was examined. Finally, the SmcR-regulated genes possibly involved in biofilm development were searched using a microarray. It appeared from the results that host cell-induced SmcR plays an essential role in V. vulnificus pathogenesis, presumably by enhancing the detachment of the pathogen from biofilms and thereby promoting its ability to colonize the intestinal surface.
MATERIALS AND METHODS
Culture conditions and biofilm formation.
The strains and plasmids used in this study are listed in Table 1. Unless otherwise noted, the V. vulnificus strains were grown in Luria-Bertani (LB) medium supplemented with 2.0% (wt/vol) NaCl (LBS) at 30°C. Vibrio fischeri minimal medium containing 32.6 mM glycerol (VFMG) was used for biofilm formation (20). Biofilm was formed using the procedure developed by Zhu and Mekalanos (13) with minor modifications. Briefly, an aliquot of cultures (150 μl) grown to an A600 of 0.8 with LBS broth was used to inoculate 15 ml of VFMG in a polystyrene petri dish (100 by 15 mm; SPL, Seoul, South Korea). Biofilms were formed by incubating these cultures for 12 h at 30°C with very gentle shaking. Planktonic cells were removed by gentle rinsing with phosphate-buffered saline (PBS; pH 7.4), after which the biofilms were collected with a cell scraper (SPL) and diluted in PBS to the desired concentrations. The resulting biofilm suspension was divided into two tubes; one was used as an inoculum to infect mice, and the other was vortexed to disrupt biofilm structures completely and to enumerate the biofilm-derived cells by counting CFU (13). During preliminary studies, multiple independent dilutions were attempted to ensure consistent and accurate numbers of cells in the inoculum. Uniformity of biofilms in the inoculum was confirmed by microscopy, live-dead staining, and quantitating total RNA (see Fig. S1 in the supplemental material).
Table 1.
Strains and plasmids used in this study
| Strain or plasmid | Relevant characteristicsa | Reference or source |
|---|---|---|
| Strains | ||
| V. vulnificus | ||
| MO6-24/O | Clinical isolate; virulent | 54 |
| HS03 | MO6-24/O with smcR::nptI; Kmr | 17 |
| KS12 | MO6-24/O with luxP::nptI; Kmr | This study |
| E. coli | ||
| SM10 λpir | thi thr leu tonA lacY supE recA::RP4-2-Tc::Mu λpir oriT RP4; Kmr; conjugational donor | 27 |
| BL21(DE3) | F− ompT hsdSB (rB− mB−) gal dcm (DE3) | Laboratory collection |
| Plasmids | ||
| pGEM-T Easy | PCR product cloning vector; Apr | Promega |
| pCVD442 | R6K γ ori sacB oriT RP4; Apr | 26 |
| pRK415 | Broad-host-range vector; IncP ori oriT RK2; Tcr | 32 |
| pKS0805 | pGEM-T Easy with the luxP gene; Apr | This study |
| pKS0807 | pCVD442 with luxP::nptI; Apr Kmr | This study |
| pET29a(+) | His6 tag fusion expression vector; Kmr | Novagen |
| pKS1202 | pET29a(+) with 1,827-bp vvpE; Kmr | This study |
| pHS105 | pRK415 with smcR; Tcr | 17 |
Apr, ampicillin resistance; Kmr, kanamycin resistance; Tcr, tetracycline resistance.
Determination of mouse mortality.
Mouse mortalities of the wild type and smcR mutant were compared as described elsewhere (21). Groups of (n = 10) 7-week-old ICR female mice (specific-pathogen-free; Seoul National University), without an iron-dextran pretreatment, were infected intragastrically with 100 μl of the inoculum, representing approximately 108 to 109 biofilm-derived cells of either the wild type or smcR mutant, and mouse mortalities were recorded for 24 h. All manipulations of mice were approved by the Animal Care and Use Committee at Seoul National University.
Competition assay.
Colonization activities of each strain were determined by competition assays as previously described (22). Briefly, four ICR female mice were intragastrically given drinking water containing polymyxin B (100 U/ml) for 24 h and then 100 μl of the inoculum, prepared by combining biofilm-derived cells of the wild type and smcR mutant at a 1:1 ratio, representing approximately 108 CFU of each strain. The mice were sacrificed after a 1- to 24-h infection, and their intestines were collected, washed, and homogenized. Equal amounts of the homogenates were spread on LBS agar containing either polymyxin B (100 U/ml) alone to enumerate the sum of the wild-type and smcR mutant cells or polymyxin B and kanamycin (100 μg/ml) to specifically count the smcR mutant cells. The ratio of CFU recovered from the intestines to the number of CFU inoculated is defined as the colonization index (22).
Histopathological examination.
Five female mice were infected as described above for competition assays, except that 100 μl of the inoculum, representing approximately 108 biofilm-derived cells of either the wild type or smcR mutant, was used. At 18 h postinfection, the mice were sacrificed, and segments taken from jejunum were sectioned transversely and fixed overnight in 10% neutral buffered formalin. The fixed jejunum was processed in paraffin, cut into five microsections, and stained with hematoxylin-eosin, and images of the stained jejuna were photographed using a microscope (Fluoview 300 fluorescence microscope; Olympus, Tokyo, Japan).
Biofilm formation and detachment assay.
Biofilms were formed by incubating the polystyrene microtiter plates (Nunc, Roskilde, Denmark) containing 200 μl of VFMG culture for 12 h at 30°C as described above. Once the planktonic cells were removed, the biofilm cells on the wall were washed with PBS and then stained with 1% (wt/vol) crystal violet (CV) solution for 15 min at room temperature. Biofilms were quantitated by measuring the amount of CV eluted from the biofilms as the absorbance at 570 nm (A570) (23).
To determine the effects of host cells on biofilm detachment, INT-407 (ATCC CCL-6) human intestine epithelial cells were prepared and suspended in a cell culture medium (minimum essential medium with 1% [vol/vol] fetal bovine serum [MEMF]) (Gibco-BRL, Gaithersburg, MD) to the desired concentrations as previously described (24). The INT-407 cells in 200 μl of MEMF were added to the biofilms preformed on the microtiter plate walls, after which the detached planktonic cells and remaining biofilms were quantitated by determining CFU counts and staining with CV, respectively, as described above. In a similar way, the effects of purified VvpE, an elastolytic protease, on biofilm detachment were analyzed by staining residual biofilms with CV and visualized using a digital imaging system (UTA-1100; UMAX Technologies, Fremont, CA).
Cloning of V. vulnificus luxP and generation of the luxP mutant.
The entire luxP coding region was amplified from the genomic DNA of V. vulnificus MO6-24/O by PCR using the oligonucleotide primers 5′-CGATACTCTTTACACGGTCAGGCTA-3′ and 5′-CTGTTCGTTTGTCATGGCTGATC-3′. The amplified 1.1 kb of DNA was ligated into pGEM-T Easy (Promega, Madison, WI) resulting in pKS0805 (Table 1). To inactivate luxP in pKS0805 in vitro, a 1.2 kb of nptI DNA conferring resistance to kanamycin (25) was inserted into a unique BamHI site present within the luxP coding region, and the resulting luxP::nptI cartridge was ligated with SphI-SacI-digested pCVD422 (26) to form pKS0807 (Table 1). Escherichia coli SM10 λpir tra (containing pKS0807) (27) was used as a conjugal donor to V. vulnificus MO6-24/O. The conjugation and isolation of the transconjugants were conducted as previously described (28), and a resulting luxP::nptI mutant chosen for further analysis was named KS12 (Table 1).
qRT-PCR and Western blot analysis.
For quantitative real-time PCR (qRT-PCR), total RNAs of biofilms were isolated using an RNeasy Mini Kit (Qiagen, Valencia, CA), and cDNA was synthesized by using an iScript cDNA Synthesis Kit (Bio-Rad Laboratories, Hercules, CA). Real-time PCR amplification of the cDNA was performed by using a Chromo 4 real-time PCR detection system (Bio-Rad Laboratories) with a pair of specific primers, listed in Table 2, as described previously (28). Relative expression levels of the specific transcripts were calculated using the 16S rRNA expression level as the internal reference for normalization. When necessary, biofilms exposed to INT-407 cells were harvested and used to isolate total RNAs.
Table 2.
Oligonucleotides used for quantitative real-time PCR
| Locus taga | Oligonucleotide sequence (5′–3′)b |
|
|---|---|---|
| Forward | Reverse | |
| VVMO6_00535 | CACACACTTCACCACGCTCAATG | AACATCGCCAACATCACCAACG |
| VVMO6_00513 | CGATGGAAGATGTGTTGAAAGT | TCCTGCTGCCAGAATGTT |
| VVMO6_04125 | GAAAAAGATATTACTAACTTGTTTA | TTCCTGACCACCTCAGACAG |
| VVMO6_04124 | TACGGGAATCGCTATGGCT | TCATGGCTGATCTGAATATCTAAAC |
| VVMO6_02022 | GCGGATGCACTGTGTCATT | TCAGAAGCCGTTTGCCTG |
| VVMO6_02023 | CCGCCTCCTTTGAACCAG | CGGAATGTTACCATCACACG |
| VVMO6_03339 | GCTAGATATCGAGCTAACCAGAC | CAATCTTGCGGAAAATATTATAAA |
| VVMO6_00926 | GACGTTTGAGTTTGGTTTACGTAGC | CCACAACTGAAGGTGATCGGG |
| VVMO6_03635 | GGTTGGCTTATCCATTGATGA | TTGTTGATGCTTGTAGTTAGTGA |
| VVMO6_03732 | ATGGCATTGAGTGTATCCCTGTATT | GCTTTAGTGTCCAGCGCAGTAATAC |
| VVMO6_04095 | ATCGTCGTGGTTCTGGAA | TGTCTTCGTCCGTGTCAT |
| VVMO6_02139 | TGCACATTTTGGAAAACAGTCATC | TGCTGACCATTTCCACCATGT |
| VVMO6_00361 | TTTGAAAGATTTGGGTGTGGATTAC | CTTGTCCACCATGTATGCCTAGCTC |
| VVMO6_04435 | TTTTGGTGAGTCAGAGCGTTGC | CTGAAATGCCCGTAACGTGGT |
| VVMO6_04367 | CAATATTGCAGCTTTAACGTCACAC | AACCGACCACAAGCTCTTGGG |
Locus tags are based on the database of the V. vulnificus MO6-24/O genome (GenBank accession numbers CP002469.1 and CP002470.1) as presented in Fig. 7 (except VVMO6_00535 for smcR, VVMO6_00513 for luxS, VVMO6_04125 for luxP, VVMO6_04124 for luxQ, VVMO6_02022 for luxU, and VVMO6_02023 for luxO, which were used in Fig. 6).
The oligonucleotides were designed using the genomic sequence of V. vulnificus MO6-24/O (55).
For Western blot analysis, biofilms exposed to INT-407 cells were harvested and lysed using complete lysis B buffer (Roche, Mannheim, Germany) for 1 min, and residual cell debris was removed by centrifugation. Protein samples from the cell lysates, equivalent to 10 μg of total protein, were resolved using 12% SDS-PAGE (29). Western immunoblotting of SmcR was performed using a rat anti-SmcR antiserum as described previously (17).
Transcriptome analysis.
The V. vulnificus Whole Genome Twin-Chip manufactured by Ebiogen (Seoul, South Korea) was used. Total cellular RNAs were isolated from biofilms, and aminoallyl-cDNA was synthesized using an Amino Allyl cDNA Labeling Kit (Ambion, Austin, TX) according to the manufacturer's procedures. The aminoallyl cDNAs from the wild type and smcR mutant were labeled with Cy3 and Cy5 (Amersham Pharmacia, Uppsala, Sweden), respectively, and equal amounts of the labeled cDNA were combined to hybridize the microarray slides at 42°C for 16 h. The arrays were washed, dried, scanned, and analyzed by GenePix Pro, version 3.0, software (Axon Instruments, Union City, CA), and the open reading frame (ORF) spots that showed 1.58-fold or greater difference in expression with a P value of ≤0.05 were considered to be regulated by SmcR.
Overexpression and purification of the elastolytic protease VvpE.
The coding region of vvpE encoding an elastolytic protease was amplified by PCR using the primers VVPE-F (5′-AAACATATGAAACACAATCAACGTCATCG-3′) and VVPE-R (5′-AAAACTCGAGATATTGCAGCTTTAACGTC-3′) and then subcloned into a His6 tag expression vector, pET29a(+) (Novagen, Madison, WI), resulting in pKS1202. The His-tagged VvpE protein was overexpressed in E. coli BL21(DE3) and then purified by affinity chromatography according to the manufacturer's procedure (Qiagen). One unit of the VvpE activity was defined as an increase in the A495 of 0.01 per hour when the purified VvpE was added to 1 ml of solution containing 20 mg of elastin-Congo red in 10 mM sodium phosphate (pH 7.0), as described previously (24).
Data analyses.
Averages and standard errors of the mean (SEM) were calculated from at least three independent experiments. Mouse mortality was evaluated using a log rank test program (http://bioinf.wehi.edu.au/software/russell/logrank/). All other data were analyzed by Student's t tests with SAS software (SAS Institute, Inc.). Significance of differences between experimental groups was accepted at a P value of <0.05.
Microarray data accession number.
All primary microarray data were deposited in the Gene Expression Omnibus (GEO [http://www.ncbi.nlm.nih.gov/projects/geo/]) database under accession number GSE32270.
RESULTS
SmcR is important for virulence in mice.
In order to determine the role of quorum sensing in V. vulnificus pathogenesis, mouse mortality of a mutant with disruption of the smcR gene was evaluated. Mice were infected intragastrically with the wild-type or smcR mutant biofilms, and the numbers of dead mice were then counted for 24 h. As shown in Fig. 1A and B, the deaths of mice infected with the smcR mutant biofilm were consistently and significantly delayed (P < 0.05, log rank test) compared to those of mice infected with the parental wild-type biofilm. The results suggested that SmcR is important for the virulence of V. vulnificus. Neither defective nor advantageous growth was observed for both strains in VFMG shaking cultures (data not shown). Moreover, acid resistance levels of the wild-type and smcR mutant biofilms did not significantly differ from each other, indicating that their abilities to survive the passage through the acidic environment of the mouse stomach are likely to be similar (see Fig. S1 in the supplemental material) (30). Therefore, it was apparent that the reduced virulence observed in the smcR mutant biofilm was not due to altered growth and survival in vivo.
Fig 1.
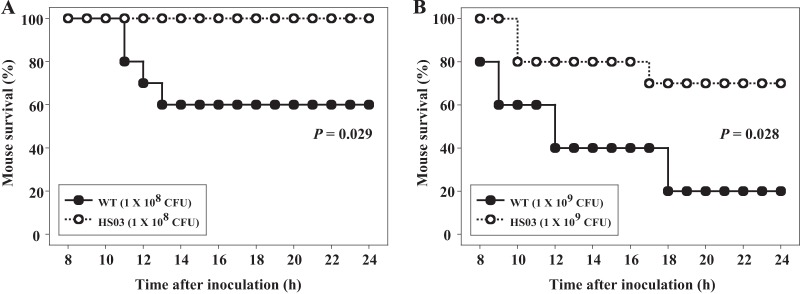
Mortality of the V. vulnificus strains for mice. Seven-week-old specific-pathogen-free female ICR mice (n = 10) were intragastrically infected using different numbers of biofilm-derived cells of the wild type or smcR mutant as indicated. Mouse survival was monitored for 24 h. The data were analyzed by using a log rank test. WT, wild type; HS03, smcR mutant.
SmcR is required for intestinal colonization in vivo.
Colonization of the small intestine has been known to be an essential step in the pathogenesis of many enteropathogenic bacteria (31). Therefore, we hypothesized that the different levels of virulence observed in the wild-type and smcR mutant biofilms could be the result of the difference in their abilities to colonize the mouse small intestine. To examine this hypothesis, mice were coinoculated intragastrically with wild-type and smcR mutant biofilms, and both strains colonized on the intestine were recovered and enumerated (Fig. 2). After infection, both strains appeared to colonize the mouse small intestine as early as 1 h, and the colonization index increased as the period of postinoculation was prolonged, reaching a maximum level at 18 h. The colonization index of the wild type ranged from 10−3 to 10−1, and was consistently and significantly (about 10-fold) higher than that of the smcR mutant. The results demonstrated that the wild type clearly out-competed the smcR mutant in the small intestine and indicated that SmcR is required for the increased colonization observed in the wild-type biofilm.
Fig 2.
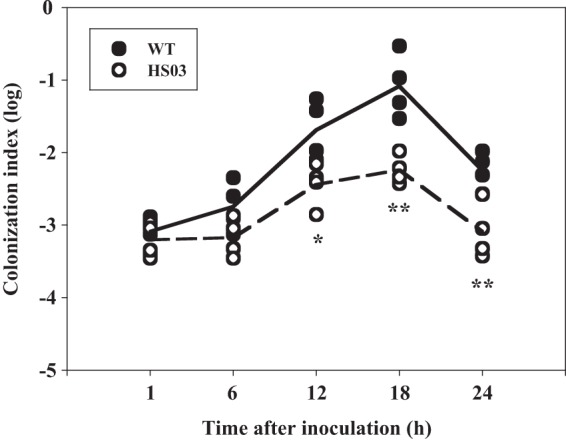
Colonization activities of the V. vulnificus strains. Four mice were infected intragastrically with an inoculum prepared by mixing equal numbers of biofilm-derived cells of the wild type and the smcR mutant, and then the bacterial cells colonized on the mouse intestine were enumerated (as CFU) at each time point. Each circle corresponds to the colonization index for an individual mouse. The median values are displayed as a solid line (wild type) or dashed line (mutant) on the graph. *, P < 0.05; **, P < 0.01, relative to the wild type at the indicated time. WT, wild type; HS03, smcR mutant.
Histopathology of mice infected with V. vulnificus.
Histological examinations of the jejunum were performed to address whether any pathological changes occurred in the mouse intestine during infection (Fig. 3). There were no histopathological changes in the jejunum of mice that received PBS alone as a control (Fig. 3A). In comparison to the PBS-injected mice, the mice infected with the wild-type biofilm exhibited increased dilation of the jejunum along with a markedly thinner wall (Fig. 3B, top). Moreover, marked damage of the jejunum villi as evidenced by their irregular morphology and shorter lengths was observed in the mice infected with the wild-type biofilm (Fig. 3B, bottom). In contrast, the mice infected with the smcR mutant biofilm exhibited dilation and wall thicknesses of the jejunum comparable to those of the control (Fig. 3C, top). The regularity and length of the villi of the mice infected with the smcR mutant biofilm were intermediate between those of the controls and the mice infected with the wild-type biofilm (Fig. 3C, bottom). Consequently, the combined results of the mouse infection studies led us to conclude that the quorum-sensing regulator SmcR is essential for the virulence of the bacteria when biofilm is used as an inoculum.
Fig 3.
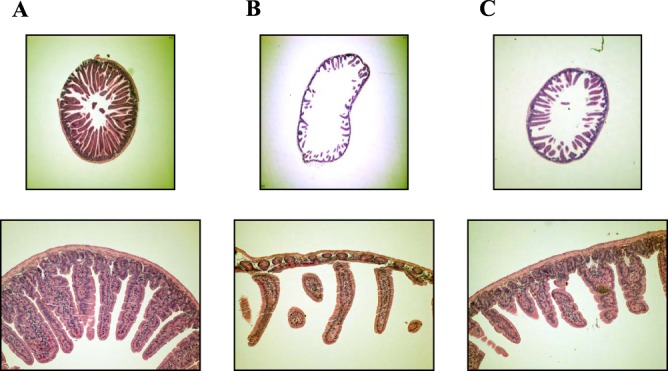
Histopathology of mouse jejunum after infection with the V. vulnificus strains. Groups (n = 5) of mice were infected intragastrically with equal numbers of biofilm-derived cells of the wild type and the smcR mutant. Jejuna of the mice infected with PBS (A) or with the wild-type (B) or the smcR mutant (C) biofilm were histopathologically examined as described in Materials and Methods. Photographs representing tissues with the median histopathological changes observed in mice of each group are presented. For all panels, the top images are cross-sections (magnification, ×40) and bottom images are villi (magnification, ×100) of the jejuna.
Effects of smcR mutation on biofilm development.
In contrast to biofilms, planktonic cells of the wild type and smcR mutant failed to show a difference in mouse mortality rates (data not shown), indicating that SmcR plays little to no role in the virulence of planktonic cells. This difference in the role of SmcR in biofilms and planktonic cells suggests that SmcR may affect the virulence of V. vulnificus by a mechanism that involves a process or processes specific only to biofilms. To test this possibility, biofilm development of the wild type and smcR mutant was assessed using an assay based on CV staining at different time points (Fig. 4). The results revealed that biofilm formation by the wild type initiated at 4 h and reached a maximum level at 12 h. In contrast, the smcR mutant arrived at the maximum earlier and formed much thicker biofilms than the wild type (Fig. 4). Both biofilms of the wild type and smcR mutant steadily decreased from the maximum level during extended periods of incubation. However, when the decrease rate (slope) of biofilms was analyzed by using SigmaPlot, version 10.0 (Systat Software Inc., San Jose, CA), the decrease rate (slope) of biofilms of the wild type was twice as high as that of the smcR mutant (wild type, −0.031 ± 0.0008; smcR mutant, −0.015 ± 0.0012). This result indicated that the smcR mutation decreased detachment of cells from the biofilm and that SmcR could affect virulence by controlling biofilm development of V. vulnificus.
Fig 4.
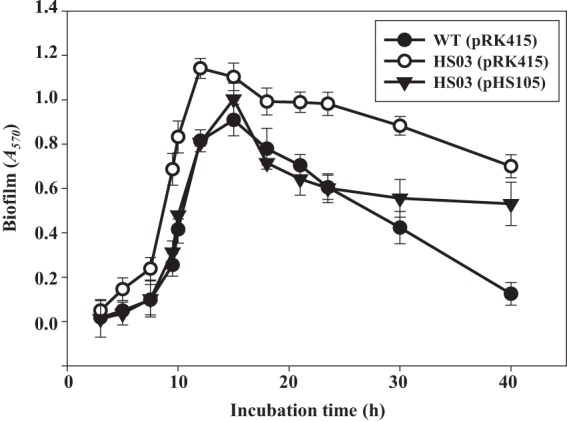
Kinetics of biofilm formation and detachment. The wild-type and smcR mutant biofilms were grown on the microtiter plate walls and quantitated using CV staining (Materials and Methods) at the indicated time points over 40 h of incubation at 30°C. Error bars represent the SEM. Mean and SEM values were calculated from three independent experiments, and the P value for the slope of the regression was less than 0.0001. WT(pRK415), wild type; HS03(pRK415), smcR mutant; HS03(pHS105), complemented strain.
It was examined whether the reintroduction of recombinant smcR could complement the decreased biofilm detachment of the smcR mutant. For this purpose, plasmid pHS105 constructed by subcloning the smcR coding region into the broad-host-range vector pRK415 (32) carrying a tetracycline (Tc) resistance gene (17) (Table 1) was used. Biofilm development of the complemented strain (the smcR mutant containing pHS105) was analyzed in the absence of Tc to rule out the effect of Tc on biofilm development. The complemented strain delayed biofilm formation, as observed in the wild type, and restored the decreased biofilm detachment of the smcR mutant during the first 24 h of biofilm development. The resulting biofilm formation and detachment activities were comparable to those of the wild type (Fig. 4). The results indicated that the decreased biofilm detachment activity of the smcR mutant results from the inactivation of functional smcR rather than from any polar effects on genes downstream of smcR. However, the introduction of pHS105 into HS03 caused a partial complementation of biofilm detachment after 24 h of biofilm development (Fig. 4). One possible explanation for this incomplete complementation is that plasmid pHS105 was not well maintained in biofilm cells after 24 h in the absence of Tc. Consistent with this, biofilm of HS03(pHS105) developed for 12 h in the absence of Tc was not able to restore the reduced virulence of HS03, as determined by measuring mouse mortality, the colonization index, and histopathological lesions (data not shown).
Effects of host cells on biofilm detachment.
Increasing numbers of INT-407 host cells (ranging from 104 to 106 cells in MEMF) were added to the biofilms preformed on microtiter plate walls and incubated for 10 h. The wild-type biofilm decreased when exposed to the INT-407 cells, and the rate of decrease was accelerated along with the increase in the numbers of the host cells (Fig. 5A). In contrast, the smcR mutant biofilm did not significantly decrease when exposed to INT-407 cells (Fig. 5A). Similarly, when 105 INT-407 cells were added to and incubated with the preformed biofilms, the wild-type biofilm decreased in an incubation time-dependent manner (Fig. 5B). In contrast, the decrease of the smcR mutant biofilm was not apparent, regardless of the length of incubation with INT-407 cells (Fig. 5B). This accelerated decrease in the wild-type biofilm after exposure to INT-407 cells could be the result of either an increased detachment of cells from the biofilm or increased death of biofilm cells.
Fig 5.

Effects of INT-407 cells on biofilm detachment. (A and C) Various numbers of INT-407 cells were added to the wild-type and smcR mutant biofilms preformed on microtiter plate walls and then incubated for 10 h. (B and D) A total of 105 INT-407 cells were added to the biofilms and incubated at various times. The biofilms on the microtiter plate wall were quantitated using CV staining (Materials and Methods), and the results are shown in panels A and B. The cells detached from the biofilms were enumerated by counting CFU, and results are presented in panels C and D as percentages of the total number of cells (sum of the cells detached from and present in the biofilm). Error bars represent the SEM. Mean and SEM values were calculated from at least three independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001, relative to the smcR mutant. WT(pRK415), wild type; HS03(pRK415), smcR mutant; HS03(pHS105), complemented strain.
To distinguish these two possibilities, changes in the number of planktonic cells detached from established biofilms were determined after different numbers of INT-407 cells were added to and incubated with the biofilms for 10 h (Fig. 5C). The number of planktonic cells from the wild-type biofilm, enumerated by CFU counts ml−1, increased when more numbers of INT-407 cells were added (Fig. 5C). Similarly, detachment of the wild-type biofilms incubated with 105 INT-407 cells was increased by extending the length of incubation (Fig. 5D). In contrast, however, the smcR mutant detached from the biofilm to a much lower extent than the wild type, and the rate of detachment was not significantly affected by the INT-407 cells (Fig. 5C and D). Similar to results from the biofilm development tests, complementation of the smcR mutation with a functional smcR (pHS105) restored the decreased biofilm detachment to levels similar, if not equivalent, to those of the wild type (Fig. 5A and B). The results suggested that the biofilm decrease which was accelerated in the presence of INT-407 host cells was attributed to the concomitant increase in detachment rather than to the death of biofilm cells and that the increased detachment was mediated by the activity of SmcR.
Effects of host cells on SmcR expression.
To confirm that SmcR indeed mediates the host cell acceleration of biofilm detachment, we examined smcR expression in the presence of INT-407 cells by qRT-PCR and Western blot analyses (Fig. 6). The qRT-PCR analyses were performed with RNA isolated from the wild-type biofilms exposed to different numbers of INT-407 cells. The results revealed that the smcR transcript increased along with the increase in the numbers of INT-407 cells (Fig. 6A). The level of smcR expression was almost 2-fold greater in the biofilms exposed to 106 INT-407 cells than in the biofilms exposed to MEMF alone (control). The cellular level of the SmcR protein was also higher in the biofilms exposed to INT-407 cells (Fig. 6B). The results implied that the biofilm detachment accelerated by the host cells might result from increased smcR expression.
Fig 6.
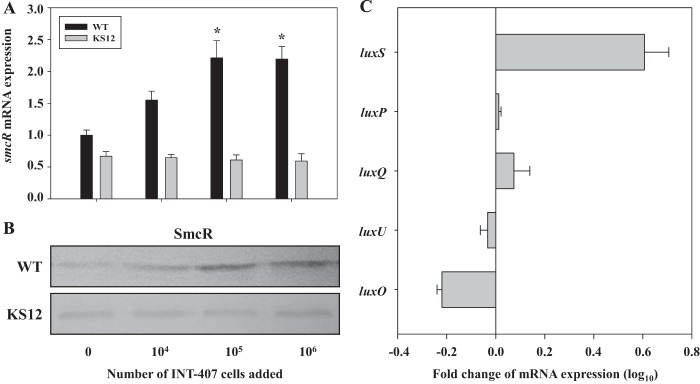
Effects of INT-407 cells on smcR expression. (A and B) Total RNAs and proteins were isolated from the wild-type and luxP mutant biofilms exposed to various numbers of INT-407 cells as indicated. The relative levels of smcR expression (A) were determined by qRT-PCR analyses, and the expression level of smcR in the biofilm exposed to MEMF (control) was set as 1. The smcR expression levels are the means ± SEM of results from three independent experiments. *, P < 0.05, relative to the biofilms exposed to MEMF alone (control). Protein samples were resolved by SDS-PAGE, and SmcR was detected by Western blotting (B) using a rat anti-SmcR antiserum. WT, wild type; KS12, luxP mutant. (C) Biofilms of the wild type exposed to either 105 INT-407 cells or MEMF for 10 h were harvested and used to isolate total RNAs. The expression levels of the genes listed on the left were determined by qRT-PCR analyses. Each column represents the mRNA level of each gene expressed in the biofilms exposed to INT-407 cells relative to that in the biofilms exposed to MEMF. Error bars represent the SEM. The relative levels of expression of each gene are the means ± SEM of results from three independent experiments.
To investigate the involvement of quorum-sensing signaling in the host cell induction of SmcR, the expression of smcR in the luxP mutant biofilm was assessed (Fig. 6A and B). The level of smcR expression in the luxP mutant biofilm was lower than that of the wild type and not induced by exposure to INT-407 cells, indicating that the quorum-sensing signaling of V. vulnificus is involved in the host cell induction of smcR. To further understand the effects of host cells on the quorum sensing of V. vulnificus, the expression levels of AI-2 quorum-sensing components such as luxS, luxP, luxQ, luxU, and luxO were compared in the wild-type biofilms exposed to either INT-407 cells or MEMF (control) (Fig. 6C). Exposure to INT-407 cells resulted in about a 4-fold (log10 0.6) increase in luxS expression, indicating that the host cell induces smcR expression through the quorum-sensing signaling of V. vulnificus by increasing the cellular level of LuxS. The marginal reduction of luxO expression (about 1.5-fold; log10 0.2) can be explained by the previous observation that LuxR represses luxO expression in V. harveyi (33). Taken together, the results also led us to postulate that this host cell-induced and SmcR-dependent detachment of V. vulnificus from biofilms might confer enhanced colonization potential, thereby leading to an increase in virulence.
SmcR-regulated genes involved in biofilm development.
A whole-genome microarray analysis was used to compare the transcriptional profiles of the wild-type and smcR mutant biofilms. The microarray analysis predicted 180 genes potentially regulated by SmcR, 74 of which were upregulated and 106 of which were downregulated (data not shown). Among them, nine genes that presumably participate in biofilm development (34) were selected. Two genes encoding VpsT and a GGDEF domain protein, known to induce biofilm formation (11, 12, 35), were downregulated. In contrast, seven genes known to contribute to biofilm detachment, such as the EAL domain protein VieA (36), a DNase (37), the nitrate reductase component NapD (38), two methyl-accepting chemotaxis transducer BdlA-like proteins (39, 40), the GGDEF and EAL domain protein MshH (41), and the elastolytic protease VvpE (17) were upregulated (Fig. 7). Since many of the genes that we predicted were not previously reported to be SmcR regulated, SmcR regulation of the selected nine genes was experimentally verified. qRT-PCR revealed that SmcR indeed regulates transcription of all of the genes predicted, of which VVMO6_04367 encoding VvpE was the most greatly upregulated (approximately 10-fold) by SmcR (Fig. 7).
Fig 7.
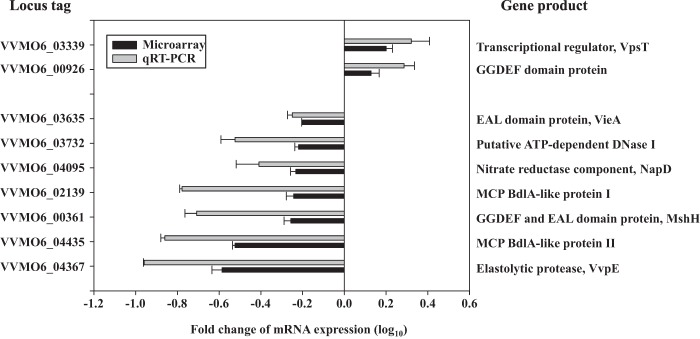
SmcR-regulated genes possibly involved in biofilm development. Nine genes possibly involved in biofilm development were chosen from the pool of the SmcR regulon members predicted by a microarray analysis. The ORF spots that showed 1.58-fold or greater difference in expression levels with a P value of ≤0.05 were considered to be regulated by SmcR. Regulation of their transcription by SmcR was confirmed by qRT-PCR. Each column represents the mRNA expression level in biofilm cells of the smcR mutant relative to that of the isogenic wild type. Locus tags are based on the database of the V. vulnificus MO6-24/O genome, which was retrieved from GenBank (accession numbers CP002469.1 and CP002470.1), and the products of the nine genes are listed on the right. The expression levels of the genes are the means ± SEM of results from three independent experiments. MCP, methyl-accepting chemotaxis protein.
Effects of VvpE on biofilm detachment.
To examine the effects of elastolytic protease on biofilm detachment, the smcR mutant biofilm preformed on the microtiter plate walls was treated with recombinant VvpE for 4 h (Fig. 8). VvpE reduced the biofilm mass in a concentration-dependent manner, as visualized using a digital imaging system (Fig. 8A). When treated with 50 units of VvpE, the residual mass of the biofilm was almost 2-fold less than that of the biofilm treated with a buffer (control) (Fig. 8B). Addition of 50 units of VvpE to the culture with VFMG did not inhibit bacterial growth (data not shown). The results demonstrated that VvpE, a gene product of the SmcR regulon, directly dissolves the matrix of the V. vulnificus biofilm and also indicated that the biopolymers of the matrix consist of materials, at least in part, of protein nature.
Fig 8.
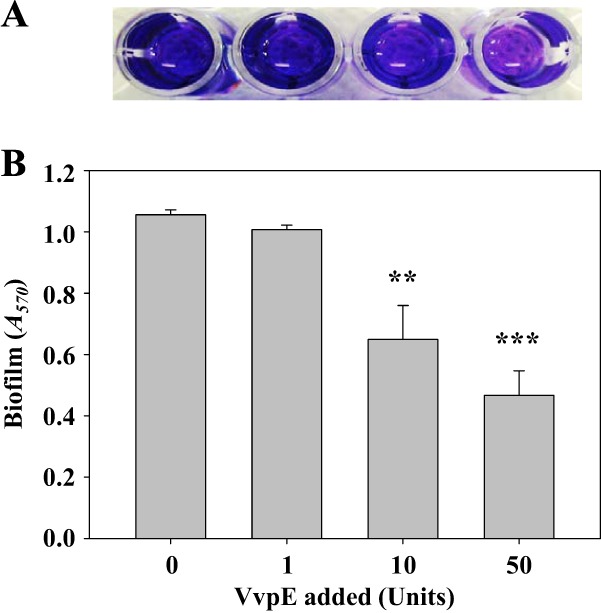
Effects of VvpE on biofilm detachment. Various amounts of purified VvpE were added exogenously to biofilms of the smcR mutant preformed on microtiter plate walls and then incubated for 4 h as described in Materials and Methods. (A) The remaining biofilms were stained with CV and photographed using a digital imaging system. (B) The amount of CV eluted from the biofilms was quantitated as the absorbance at 570 nm (A570). Error bars represent the SEM. Mean and SEM values were calculated from three independent experiments. **, P < 0.01; ***, P < 0.001, relative to the biofilm treated with a buffer (control).
DISCUSSION
In most natural ecosystems, bacteria reside predominantly in sessile communities known as biofilms rather than as free-living planktonic cells (42, 43). Biofilm formation provides bacteria with protection from antimicrobial agents and host immune defense systems during infection as well as from a variety of stresses in the environment (4, 44). Therefore, biofilms of pathogenic bacteria are considered one of the most important causes of new outbreaks and account for 65% of bacterial infections in humans (45). This is highlighted by the observation that filtration of drinking water through sari cloth to remove V. cholerae attached to particulates, such as diverse planktons greater than 20 μm in size, reduced cholera incidence by 48% in Bangladesh (10). There are several lines of evidence supporting the idea that V. vulnificus bacteria embed themselves in oyster tissues and form biofilms to colonize and to persist in oysters as a primary infection route of the pathogen (46, 47). Recently, scanning electron microscopy analysis revealed V. vulnificus biofilm structures formed on oyster shells (48). Therefore, the data obtained in this study in which biofilms rather than planktonic cells were used in mouse infection experiments could be more realistic and reliable (Fig. 1, 2, and 3). The data emphasized that SmcR plays a role in the pathogenesis of V. vulnificus by enhancing biofilm detachment (dispersal) and thereby promoting its ability to colonize the intestinal surface. Consistent with this, a previous study in which planktonic cells were used to inject mice intraperitoneally demonstrated that SmcR is not required for the virulence of V. vulnificus (16).
Biofilms are specialized and highly differentiated architectures of bacteria encased in an extracellular polymeric substance (EPS) matrix that forms the framework for three-dimensional communities (5, 49). The major components of the EPS are polysaccharides, proteins, and DNA, which are distributed between the cells in a nonhomogeneous pattern (49). Biofilm development of bacteria can be divided into several stages. Motile planktonic cells attach to a surface, and the sessile cells aggregate and subsequently develop into matured biofilms. During these biofilm formation stages, the bacteria multiply and concomitantly produce the EPS, which holds the bacterial cells together in a mass. The final stage of biofilm development is the detachment of cells from the mature biofilms into planktonic cells that migrate and disperse to new places to initiate the new life cycle of biofilms (6, 34). This stage facilitates the dissemination of pathogens in new environments and dispersal of infections within a single host (6).
Exopolysaccharides are the most prevalent component of the Vibrio EPS, and the structural and regulatory genes involved in the V. cholerae exopolysaccharide (VPS) production and, in turn, biofilm formation have been extensively studied (for a recent review, see reference 12). The vps structural genes required for VPS production are organized into two operons (vps I and vps II), and their expression is positively regulated by the transcriptional regulator VpsT (50, 51). Expression of vpsT is repressed by quorum sensing (HapR) (8, 13) and induced by the second messenger, cyclic di-GMP (c-di-GMP), which is synthesized by GGDEF domain proteins (guanylate cyclases) (11, 35, 52). A previous study characterized V. vulnificus biofilms formed in different media and identified three gene clusters involved in exopolysaccharide biosynthesis (23). However, the effects of diverse regulatory components such as c-di-GMP and quorum sensing on the expression of the exopolysaccharide genes remain unclear. This study demonstrated that SmcR represses vpsT and a gene encoding a GGDEF domain protein (Fig. 7), suggesting that quorum sensing also represses V. vulnificus biofilm formation by limiting the cellular level of VpsT and guanylate cyclase activities.
In addition, seven genes encoding a variety of proteins, most likely involved in biofilm detachment directly or indirectly, were found to be regulated by SmcR (Fig. 7 and 8). An EAL domain protein (cyclic diguanylate phosphodiesterase), VieA, is known to repress vps operons and consequently the biofilm formation of V. cholerae, presumably by decreasing the cellular level of c-di-GMP (36). MshH is a GGDEF and EAL domain protein known to repress the biofilm formation of V. cholerae (41). BdlA, a methyl-accepting chemotaxis transducer is known to control the biofilm detachment of P. aeruginosa that is triggered by nitric oxide (NO) (39, 40). NapD is a component of nitrate reductase that is required for the production of the signaling molecule NO (38). VvpE is a homologue of the hemagglutinin (HA)/protease which has been suggested to release V. cholerae from the host intestinal epithelium (17, 53), and DNase I is known to dissolve established biofilms of P. aeruginosa directly (37). Among these genes involved in biofilm detachment, genes encoding VieA, MshH, BdlA-like proteins, NapD, and the DNase I are newly identified as an SmcR regulon in this study. Therefore, SmcR inversely regulates two distinct groups of genes that are important for biofilm formation and detachment, and the net effects of such regulation appeared to enhance the detachment of V. vulnificus from biofilms.
In summary, this study revealed that the quorum-sensing regulator SmcR is essential for the virulence of V. vulnificus when biofilm is used as an inoculum. Our data presented here extended our understanding of the role of quorum sensing in V. vulnificus pathogenesis by demonstrating that host cells increase smcR expression, and, in turn, accelerate biofilm detachment. The accelerated detachment may permit V. vulnificus entering the host in the form of biofilms to leave the biofilm structure in order to colonize the intestinal surface. It is noteworthy that smcR expression increased through quorum-sensing signaling of V. vulnificus but in response to the increasing density of host cells rather than to V. vulnificus cells present in biofilms (Fig. 6). Whereas integration of the host information by SmcR into biofilm development and its implication in the pathogenesis of V. vulnificus have yet to be fully explored, the overall success of the organism during pathogenesis would be enhanced through this ability to sense and combine information about its own community and the surrounding host. Additional work is needed to identify the host cell signals affecting smcR expression and to clarify how SmcR inversely regulates the genes contributing to the distinct stages of biofilm development. However, the results of this study suggest that a small molecule inhibiting the activity of SmcR could successfully inhibit the dispersal of the consumed V. vulnificus biofilms and thereby the pathogenicity of the pathogen in the host.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants to S.H.C from the Mid-Career Researcher Program (2012R1A2A1A03009679) and the Public Welfare and Safety Research Program (2012M3A2A1051679) through the National Research Foundation funded by the Ministry of Science, ICT, and Future Planning and by the R&D Convergence Center Support Program of the Ministry of Agriculture, Food and Rural Affairs, Republic of Korea.
Footnotes
Published ahead of print 29 July 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.00561-13.
REFERENCES
- 1.Ng WL, Bassler BL. 2009. Bacterial quorum-sensing network architectures. Annu. Rev. Genet. 43:197–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Antunes LC, Ferreira RB, Buckner MM, Finlay BB. 2010. Quorum sensing in bacterial virulence. Microbiology 156:2271–2282 [DOI] [PubMed] [Google Scholar]
- 3.Rutherford ST, Bassler BL. 2012. Bacterial quorum sensing: its role in virulence and possibilities for its control. Cold Spring Harb. Perspect. Med. 2:a012427. 10.1101/cshperspect.a012427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Karatan E, Watnick P. 2009. Signals, regulatory networks, and materials that build and break bacterial biofilms. Microbiol. Mol. Biol. Rev. 73:310–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hall-Stoodley L, Costerton JW, Stoodley P. 2004. Bacterial biofilms: from the natural environment to infectious diseases. Nat. Rev. Microbiol. 2:95–108 [DOI] [PubMed] [Google Scholar]
- 6.Kaplan JB. 2010. Biofilm dispersal: mechanisms, clinical implications, and potential therapeutic uses. J. Dent. Res. 89:205–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Davies DG, Parsek MR, Pearson JP, Iglewski BH, Costerton JW, Greenberg EP. 1998. The involvement of cell-to-cell signals in the development of a bacterial biofilm. Science 280:295–298 [DOI] [PubMed] [Google Scholar]
- 8.Hammer BK, Bassler BL. 2003. Quorum sensing controls biofilm formation in Vibrio cholerae. Mol. Microbiol. 50:101–104 [DOI] [PubMed] [Google Scholar]
- 9.Faruque SM, Albert MJ, Mekalanos JJ. 1998. Epidemiology, genetics, and ecology of toxigenic Vibrio cholerae. Microbiol. Mol. Biol. Rev. 62:1301–1314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Colwell RR, Huq A, Islam MS, Aziz KMA, Yunus M, Khan NH, Mahmud A, Sack RB, Nair GB, Chakraborty J, Sack DA, Russek-Cohen E. 2003. Reduction of cholera in Bangladeshi villages by simple filtration. Proc. Natl. Acad. Sci. U. S. A. 100:1051–1055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Waters CM, Lu W, Rabinowitz JD, Bassler BL. 2008. Quorum sensing controls biofilm formation in Vibrio cholerae through modulation of cyclic di-GMP levels and repression of vpsT. J. Bacteriol. 190:2527–2536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yildiz FH, Visick KL. 2009. Vibrio biofilms: so much the same yet so different. Trends Microbiol. 17:109–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhu J, Mekalanos JJ. 2003. Quorum sensing-dependent biofilms enhance colonization in Vibrio cholerae. Dev. Cell 5:647–656 [DOI] [PubMed] [Google Scholar]
- 14.Kim SY, Lee SE, Kim YR, Kim CM, Ryu PY, Choy HE, Chung SS, Rhee JH. 2003. Regulation of Vibrio vulnificus virulence by the LuxS quorum-sensing system. Mol. Microbiol. 48:1647–1664 [DOI] [PubMed] [Google Scholar]
- 15.McDougald D, Rice SA, Kjelleberg S. 2001. SmcR-dependent regulation of adaptive phenotypes in Vibrio vulnificus. J. Bacteriol. 183:758–762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shao CP, Hor LI. 2001. Regulation of metalloprotease gene expression in Vibrio vulnificus by a Vibrio harveyi LuxR homologue. J. Bacteriol. 183:1369–1375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jeong HS, Lee MH, Lee KH, Park SJ, Choi SH. 2003. SmcR and cyclic AMP receptor protein coactivate Vibrio vulnificus vvpE encoding elastase through the RpoS-dependent promoter in a synergistic manner. J. Biol. Chem. 278:45072–45081 [DOI] [PubMed] [Google Scholar]
- 18.Lee JH, Rhee JE, Park UR, Ju HM, Lee BC, Kim TS, Jeong HS, Choi SH. 2007. Identification and functional analysis of Vibrio vulnificus smcR a novel global regulator. J. Microbiol. Biotechnol. 17:325–334 [PubMed] [Google Scholar]
- 19.Shao CP, Lo HR, Lin JH, Hor LI. 2011. Regulation of cytotoxicity by quorum-sensing signaling in Vibrio vulnificus is mediated by SmcR, a repressor of hlyU. J. Bacteriol. 193:2557–2565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cao X, Studer SV, Wassarman K, Zhang Y, Ruby EG, Miyashiro T. 2012. The novel sigma factor-like regulator RpoQ controls luminescence, chitinase activity, and motility in Vibrio fischeri. mBio 3(1):e00285–11. 10.1128/mBio.00285-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lim MS, Kim JA, Lim JG, Kim BS, Jeong KC, Lee KH, Choi SH. 2011. Identification and characterization of a novel serine protease, VvpS, that contains two functional domains and is essential for autolysis of Vibrio vulnificus. J. Bacteriol. 193:3722–3732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jeong HG, Oh MH, Kim BS, Lee MY, Han HJ, Choi SH. 2009. The capability of catabolic utilization of N-acetylneuraminic acid, a sialic acid, is essential for Vibrio vulnificus pathogenesis. Infect. Immun. 77:3209–3217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim HS, Park SJ, Lee KH. 2009. Role of NtrC-regulated exopolysaccharides in the biofilm formation and pathogenic interaction of Vibrio vulnificus. Mol. Microbiol. 74:436–453 [DOI] [PubMed] [Google Scholar]
- 24.Jeong KC, Jeong HS, Rhee JH, Lee SE, Chung SS, Starks AM, Escudero GM, Gulig PA, Choi SH. 2000. Construction and phenotypic evaluation of a Vibrio vulnificus vvpE mutant for elastolytic protease. Infect. Immun. 68:5096–5106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Oka A, Sugisaki H, Takanami M. 1981. Nucleotide sequence of the kanamycin resistance transposon Tn903. J. Mol. Biol. 147:217–226 [DOI] [PubMed] [Google Scholar]
- 26.Donnenberg MS, Kaper JB. 1991. Construction of an eae deletion mutant of enteropathogenic Escherichia coli by using a positive-selection suicide vector. Infect. Immun. 59:4310–4317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miller VL, Mekalanos JJ. 1988. A novel suicide vector and its use in construction of insertion mutations: osmoregulation of outer membrane proteins and virulence determinants in Vibrio cholerae requires toxR. J. Bacteriol. 170:2575–2583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim SM, Lee DH, Choi SH. 2012. Evidence that the Vibrio vulnificus flagellar regulator FlhF is regulated by a quorum sensing master regulator SmcR. Microbiology 158:2017–2025 [DOI] [PubMed] [Google Scholar]
- 29.Sambrook J, Russell DW. 2001. Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 30.McConnell EL, Basit AW, Murdan S. 2008. Measurements of rat and mouse gastrointestinal pH, fluid and lymphoid tissue, and implications for in-vivo experiments. J. Pharm. Pharmacol. 60:63–70 [DOI] [PubMed] [Google Scholar]
- 31.Falkow S, Isberg RR, Portnoy DA. 1992. The interaction of bacteria with mammalian cells. Annu. Rev. Cell Biol. 8:333–363 [DOI] [PubMed] [Google Scholar]
- 32.Keen NT, Tamaki S, Kobayashi D, Trollinger D. 1988. Improved broad-host-range plasmids for DNA cloning in gram-negative bacteria. Gene 70:191–197 [DOI] [PubMed] [Google Scholar]
- 33.Tu KC, Long T, Svenningsen SL, Wingreen NS, Bassler BL. 2010. Negative feedback loops involving small regulatory RNAs precisely control the Vibrio harveyi quorum-sensing response. Mol. Cell 37:567–579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McDougald D, Rice SA, Barraud N, Steinberg PD, Kjelleberg S. 2012. Should we stay or should we go: mechanisms and ecological consequences for biofilm dispersal. Nat. Rev. Microbiol. 10:39–50 [DOI] [PubMed] [Google Scholar]
- 35.Beyhan S, Bilecen K, Salama SR, Casper-Lindley C, Yildiz FH. 2007. Regulation of rugosity and biofilm formation in Vibrio cholerae: comparison of VpsT and VpsR regulons and epistasis analysis of vpsT, vpsR, and hapR. J. Bacteriol. 189:388–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tamayo R, Tischler AD, Camilli A. 2005. The EAL domain protein VieA is a cyclic diguanylate phosphodiesterase. J. Biol. Chem. 280:33324–33330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Whitchurch CB, Tolker-Nielsen T, Ragas PC, Mattick JS. 2002. Extracellular DNA required for bacterial biofilm formation. Science 295:1487. 10.1126/science.295.5559.1487 [DOI] [PubMed] [Google Scholar]
- 38.Barraud N, Hassett DJ, Hwang SH, Rice SA, Kjelleberg S, Webb JS. 2006. Involvement of nitric oxide in biofilm dispersal of Pseudomonas aeruginosa. J. Bacteriol. 188:7344–7353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Barraud N, Schleheck D, Klebensberger J, Webb JS, Hassett DJ, Rice SA, Kjelleberg S. 2009. Nitric oxide signaling in Pseudomonas aeruginosa biofilms mediates phosphodiesterase activity, decreased cyclic di-GMP levels, and enhanced dispersal. J. Bacteriol. 191:7333–7342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Morgan R, Kohn S, Hwang SH, Hassett DJ, Sauer K. 2006. BdlA, a chemotaxis regulator essential for biofilm dispersion in Pseudomonas aeruginosa. J. Bacteriol. 188:7335–7343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pickering BS, Smith DR, Watnick PI. 2012. Glucose-specific enzyme IIA has unique binding partners in the Vibrio cholerae biofilm. mBio 3(6):e00228–12. 10.1128/mBio.00228-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.September SM, Els FA, Venter SN, Brozel VS. 2007. Prevalence of bacterial pathogens in biofilms of drinking water distribution systems. J. Water Health 5:219–227 [PubMed] [Google Scholar]
- 43.Wimpenny J, Manz W, Szewzyk U. 2000. Heterogeneity in biofilms. FEMS Microbiol. Rev. 24:661–671 [DOI] [PubMed] [Google Scholar]
- 44.Hall-Stoodley L, Stoodley P. 2005. Biofilm formation and dispersal and the transmission of human pathogens. Trends Microbiol. 13:7–10 [DOI] [PubMed] [Google Scholar]
- 45.Costerton JW. 2001. Cystic fibrosis pathogenesis and the role of biofilms in persistent infection. Trends Microbiol. 9:50–52 [DOI] [PubMed] [Google Scholar]
- 46.Paranjpye RN, Johnson AB, Baxter AE, Strom MS. 2007. Role of type IV pilins in persistence of Vibrio vulnificus in Crassostrea virginica oysters. Appl. Environ. Microbiol. 73:5041–5044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Froelich B, Oliver JD. 2013. The interactions of Vibrio vulnificus and the oyster Crassostrea virginica. Microb. Ecol. 65:807–816. 10.1007/s00248-012-0162-3 [DOI] [PubMed] [Google Scholar]
- 48.Guo Y, Rowe-Magnus DA. 2010. Identification of a c-di-GMP-regulated polysaccharide locus governing stress resistance and biofilm and rugose colony formation in Vibrio vulnificus. Infect. Immun. 78:1390–1402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Flemming HC, Wingender J. 2010. The biofilm matrix. Nat. Rev. Microbiol. 8:623–633 [DOI] [PubMed] [Google Scholar]
- 50.Casper-Lindley C, Yildiz FH. 2004. VpsT is a transcriptional regulator required for expression of vps biosynthesis genes and the development of rugose colonial morphology in Vibrio cholerae O1 El Tor. J. Bacteriol. 186:1574–1578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yildiz FH, Dolganov NA, Schoolnik GK. 2001. VpsR, a member of the response regulators of the two-component regulatory systems, is required for expression of vps biosynthesis genes and EPSETr-associated phenotypes in Vibrio cholerae O1 El Tor. J. Bacteriol. 183:1716–1726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Srivastava D, Harris RC, Waters CM. 2011. Integration of cyclic di-GMP and quorum sensing in the control of vpsT and aphA in Vibrio cholerae. J. Bacteriol. 193:6331–6341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Finkelstein RA, Boesman-Finkelstein M, Chang Y, Hase CC. 1992. Vibrio cholerae hemagglutinin/protease, colonial variation, virulence, and detachment. Infect. Immun. 60:472–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wright AC, Simpson LM, Oliver JD, Morris JG., Jr 1990. Phenotypic evaluation of acapsular transposon mutants of Vibrio vulnificus. Infect. Immun. 58:1769–1773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Park JH, Cho YJ, Chun J, Seok YJ, Lee JK, Kim KS, Lee KH, Park SJ, Choi SH. 2011. Complete genome sequence of Vibrio vulnificus MO6-24/O. J. Bacteriol. 193:2062–2063 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.