

Abstract
Infection with Helicobacter pylori is associated with development of ulcer disease and gastrointestinal adenocarcinoma. The infection leads to a large infiltration of immune cells and the formation of organized lymphoid follicles in the human gastric mucosa. Still, the immune system fails to eradicate the bacteria, and the substantial regulatory T cell (Treg) response elicited is probably a major factor permitting bacterial persistence. Dendritic cells (DCs) are professional antigen-presenting cells that can activate naive T cells, and maturation of DCs is crucial for the initiation of primary immune responses. The aim of this study was to investigate the presence and localization of mature human DCs in H. pylori-infected gastric mucosa. Gastric antral biopsy specimens were collected from patients with H. pylori-associated gastritis and healthy volunteers, and antrum tissue was collected from patients undergoing gastric resection. Immunohistochemistry and flow cytometry showed that DCs expressing the maturation marker dendritic cell lysosome-associated membrane glycoprotein (DC-LAMP; CD208) are enriched in the H. pylori-infected gastric mucosa and that these DCs are specifically localized within or close to lymphoid follicles. Gastric DC-LAMP-positive (DC-LAMP+) DCs express CD11c and high levels of HLA-DR but little CD80, CD83, and CD86. Furthermore, immunofluorescence analyses demonstrated that DC-LAMP+ DCs are in the same location as FoxP3-positive putative Tregs in the follicles. In conclusion, we show that DC-LAMP+ DCs with low costimulatory capacity accumulate in the lymphoid follicles in human H. pylori-infected gastric tissue, and our results suggest that Treg-DC interactions may promote chronic infection by rendering gastric DCs tolerogenic.
INTRODUCTION
Infection with the Gram-negative bacterium Helicobacter pylori is widespread throughout the world and is associated with development of gastric and duodenal ulcer disease as well as gastric adenocarcinoma and mucosa-associated lymphoid tissue lymphoma (1–3). The infection generally leads to a large infiltration of immune cells, such as neutrophils, macrophages, and T and B lymphocytes, into the gastric mucosa (4, 5). Lymphocytes are found scattered in the lamina propria, but they also form organized lymphoid follicles, which are not present in the uninfected gastric mucosa (6, 7). Even though the H. pylori infection induces an inflammatory response and induction of specific B and T cell immunity, the immune response fails to eradicate the bacteria and the infection becomes chronic. T lymphocytes in particular play an essential role in the pathogenesis of H. pylori-induced gastritis, and prominent induction of both Th1 and Th17 responses has been demonstrated (8–11). One important aspect of T cell immunity in H. pylori infection is the substantial immune suppression exerted by H. pylori-specific regulatory T cells (Tregs) present in both the circulation and gastric mucosa of H. pylori-infected individuals (12–14). However, the inductive sites for the adaptive immune response to H. pylori, as well as the mechanisms for Treg priming, have not yet been clearly identified.
Dendritic cells (DCs) are professional antigen-presenting cells that can activate naive T cells, and they drive the host immune reaction by modulating the functions or interactions of effector T and B cells. In addition, DCs are well suited to recognize pathogens and to facilitate their uptake. The migration of DCs from the site of antigen capture to the draining lymph nodes and their simultaneous maturation are crucial for both the initiation and amplification of primary immune responses (15–17). During maturation, DCs upregulate membrane expression of molecules involved in migration to lymph nodes, especially CCR7. In addition, molecules important for activation and costimulation of T cells, such as major histocompatibility complex (MHC) classes I and II, CD80, CD83, and CD86, are induced. The interaction of DCs with H. pylori in vitro induces maturation of the DCs and secretion of proinflammatory cytokines, such as inteleukin-6 (IL-6), IL-8, IL-12, and IL-23 (18–25), and recent studies demonstrated that RIG-1- and MyD88-dependent Toll-like receptor signaling is crucial for the DC maturation induced by H. pylori (26, 27). However, H. pylori phase variation in lipopolysaccharide glycosylation influences DC production of stimulating and immunomodulating cytokines, thereby contributing to shaping the resulting T cell response (28, 29). Furthermore, a mature DC phenotype does not necessarily correlate with a functional immunogenic stage of the DCs but can in fact be related to DCs that induce tolerance (30). Along these lines, DCs generated in vitro in the continued presence of H. pylori have an exhausted phenotype, which in turn may lead to defective antigen presentation and Th1 responses (31).
Our recent studies have shown substantial accumulation of dendritic cell lysosome-associated membrane glycoprotein-positive (DC-SIGN+) DCs in the gastric mucosa of H. pylori-infected individuals but also that these gastric DCs have a semimature phenotype with high levels of expression of MHC class II antigens but low levels of or absent expression of CD80, CD83, and CD86 (32). Gastric macrophages and DCs in H. pylori-infected mice also fail to upregulate costimulatory molecules, even after 6 months of infection (33). Indeed, several recent studies have demonstrated that murine DCs exposed to H. pylori in vivo or in vitro adopt a tolerogenic state and drive induction of Tregs (32, 34, 35) and that gastric tissue factors may act in synergy to keep gastric DCs in a tolerogenic state (36). The induction of regulatory mechanisms by H. pylori infection may even reduce disease symptoms in experimental colitis and inhibit allergic reactions in the airways (32, 37–39). These findings, combined with the observation that mature DCs are present in the gastric mucosa in humans and mice with autoimmune gastritis (40), suggest that the gastric mucosa may have potential for antigen presentation by mature DCs and prompted us to investigate if mature DCs may be retained in the H. pylori-infected gastric mucosa. We used the maturation marker DC-LAMP (CD208) to define mature DCs, as it is expressed on all mature DC subtypes but not on immature DCs or other immune cells (41, 42). Using tissue material from patients with H. pylori-associated gastritis and from healthy volunteers, we showed that there is an accumulation of DCs expressing DC-LAMP close to and in lymphoid follicles in the infected gastric mucosa. The DC-LAMP+ DCs expressed high levels of MHC class II molecules but little CD80, CD83, and CD86 and were in the same location as FoxP3-positive (FoxP3+) T cells in the follicles.
MATERIALS AND METHODS
Volunteers, patients, and collection of specimens.
This study was approved by the Regional Research Ethics Committee of West Sweden, and informed consent was obtained from all participants. Participating donors were recruited from among blood donors at Sahlgrenska University Hospital after serologic analysis, and H. pylori infection was subsequently confirmed or excluded by culture on Scirrow plates (43). A subject was considered to be H. pylori infected if he or she was positive by both serology and culture and uninfected if negative by both tests. One biopsy specimen from each volunteer was fixed in formalin, paraffin embedded, and examined by an experienced histopathologist for the grade of gastritis and the presence of Helicobacter-like organisms (HLOs) using the updated Sydney system (44). Chronic and acute gastritis, as well as HLOs, were scored independently by an experienced pathologist on a scale ranging from 0 to 3 (0 = none, 1 = mild, 2 = moderate, and 3 = severe).
Gastric antrum biopsy specimens were collected from 18 H. pylori-infected (12 males and 6 females; age range, 25 to 67 years) and 20 uninfected volunteers (9 males and 11 females; age range, 23 to 63 years) by endoscopy and used for immunohistochemical staining, RNA preparation, and protein extraction. In order to obtain sufficiently large tissue samples from the antrum for flow cytometric analysis of DCs, biopsy specimens from four H. pylori-infected patients (2 males and 2 females; age range, 64 to 81 years) and three uninfected patients (3 females; age range, 68 to 75 years) undergoing surgery for gastric adenocarcinoma (n = 2), pancreatic cancer (n = 4), or chronic pancreatitis (n = 1) at the Sahlgrenska University Hospital were also included in the study. Tissue was collected from the antrum, and in the gastric cancer patients, tissue was removed at least 5 cm distant from the tumor. None of the patients had undergone radiotherapy or chemotherapy prior to surgery. H. pylori infection was determined on the basis of serology, culture, and pathology reports.
Immunohistochemical staining.
The presence of mature DC-LAMP+ DCs and CD303+ plasmacytoid DCs (pDCs) was determined by immunohistochemical staining of frozen sections of gastric mucosa from both H. pylori-infected and uninfected volunteers. Cryo-cut tissue sections (8 μm thick) were fixed for 10 min in ice-cold acetone, and endogenous peroxidase was blocked with glucose-oxidase for 20 min. Thereafter, the slides were incubated with primary mouse monoclonal antibodies to DC-LAMP (CD208; clone 104.G4; Immunotech a.s., Prague, Czech Republic) or CD303 (clone AC144; Miltenyi Biotec, Bergisch Gladbach, Germany) in phosphate-buffered saline (PBS) containing 5% human and rabbit serum at room temperature for 30 min, followed by a horseradish peroxidase-conjugated rabbit antibody to mouse immunoglobulins (DakoCytomation, Glostrup, Denmark). Isotype control antibodies were always run in parallel. The area stained with the respective antibodies was calculated relative to the total area of each tissue section, using Biopix image analysis software.
Immunofluorescence staining.
The potential interaction between DC-LAMP+ DCs and FoxP3+ cells was assessed by immunofluorescence staining of frozen sections from H. pylori-infected volunteers. The slides were fixed in paraformaldehyde and blocked with 1% goat and donkey serum, glucose-oxidase, and an avidin-biotin blocking kit (Vector Laboratories, Peterborough, United Kingdom) before staining with mouse anti-human FoxP3 (clone 236A/E7; eBioscience, Hatfield, United Kingdom), followed by biotinylated goat antimouse antibodies (Caltag, Burlingame, CA) in PBS containing 0.1% of saponin. The signal was amplified by a TSA kit with Alexa Fluor 488-conjugated tyramide (Invitrogen, Stockholm, Sweden). Thereafter, remaining antimouse binding sites were blocked with Fab fragments of goat anti-mouse IgG antibodies (Jackson ImmunoResearch, West Grove, PA), and the slides were stained with anti-DC-LAMP and Alexa Fluor 594-conjugated donkey antibody to mouse immunoglobulins (Invitrogen). Finally, slides were mounted using a DAPI (4′,6-diamidino-2-phenylindole)-containing mounting medium.
To detect potential expression of DC-LAMP and CD68 on the same cells, antibodies to CD68 (clone EBM11; Dako Cytomation) were first preincubated with the secondary antibody, goat anti-mouse IgG conjugated to Alexa Fluor 488. Thereafter, the remaining binding sites were blocked by adding 5% mouse serum. The sections were blocked with 5% human serum and stained with DC-LAMP, as described above, followed by the preincubated CD68 antibody for 1 h. Isotype control antibodies were always run in parallel.
Isolation of gastric lamina propria leukocytes.
Unaffected antral tissue from gastrectomy patients was stripped of muscle and fat layers and cut into 5-mm pieces. Epithelial cells were removed by incubating the tissue for 15 min at 37°C with 2 mM EDTA and 1 mM dithiothreitol in Hanks' balanced salt solution (HBSS) supplemented with 2% fetal calf serum a total of three times. Lamina propria leukocytes were then released by incubating the remaining tissue for 1 h at 37°C with 100 U/ml collagenase type VIII (Sigma-Aldrich) and 0.1 mg/ml DNase (Sigma-Aldrich) in RPMI 1640 containing 10% fetal calf serum.
Flow cytometry analyses.
Lamina propria leukocytes (2 × 106/sample) were stained with the following antibodies: anti-CD80 (clone L307.4), CD86 (2331), CD83 (HB15e), CD11c (S-HCL-3), CCR7 (3D12), DC-LAMP (I10-1112), and HLA-DR (L243) (all from BD Biosciences) and CD14 (M5E2; Biolegend, San Diego, CA). Appropriate isotype controls were used. 7-Aminoactinomycin D (7AAD; Sigma-Aldrich) was used to exclude dead cells. To detect DC-LAMP, cells were fixed in BD Cell Fix (BD Biosciences) and permeabilized with 0.5% saponin (Sigma-Aldrich) in HBSS. Samples were acquired on an LSR-II flow cytometer (BD Biosciences) and analyzed using FlowJo software (Tree Star Inc., Ashland, OR).
Real-time PCR analyses.
Total RNA from antral biopsy specimens collected by endoscopy was purified by use of an RNeasy minikit (Qiagen, Hilden, Germany). cDNA was synthesized from 500 ng total RNA and oligo(dT) primers using an Omniscript reverse transcription-PCR kit (Qiagen). Real-time PCR was performed with CCL19, CCL21, and hypoxanthine phosphoribosyltransferase (HPRT) primers using standard procedures for 40 cycles (Applied Biosystems, Foster City, CA). The relative levels of CCL19 and CCL21 mRNA were calculated using the ΔΔCT method, with HPRT used as an internal standard and a sample from a noninfected volunteer with a low and stable threshold cycle (CT) value used as a calibrator.
ELISA analyses.
Proteins were extracted from gastric tissue using saponin, as previously described (45). Briefly, frozen tissue was thawed in a solution containing 2% saponin, 0.1% bovine serum albumin, and protease inhibitors at 4°C overnight. The samples were then centrifuged and the supernatants were used for detection of CCL19 by enzyme-linked immunosorbent assay (ELISA; Duoset; R&D Systems, Abingdon, United Kingdom). Chemokine concentrations were related to the total protein concentration in the respective samples, which was determined by a protein assay kit (Pierce, Rockford, IL). Before protein analysis, the samples were passed through desalting columns (Pierce) to remove detergents remaining from the extraction procedure.
Statistical analyses.
Differences between H. pylori-infected and uninfected individuals were evaluated using the nonparametric two-sided Mann-Whitney test, and P values of less than 0.05 were considered significant.
RESULTS
Inflammation and bacterial load.
To investigate DC subpopulations in gastric tissues, antral biopsy samples were collected from both H. pylori-infected and uninfected individuals. Tissues from 16 uninfected subjects were histologically normal without inflammation or HLOs, while biopsy specimens from 4 uninfected subjects had mild chronic gastritis but no HLOs. In contrast, active chronic inflammation and HLOs were observed in biopsy samples from the antrum of all 18 H. pylori-infected subjects, except for 1 individual, who had a negative score for HLOs but was serology and culture positive. The H. pylori-infected individuals had a chronic inflammation score of 2.08 ± 0.55 (mean ± standard deviation); the active inflammation score was 1.42 ± 0.73, and the HLO score was 1.83 ± 0.92. Atrophy and metaplasia were seen in two of the H. pylori-infected subjects, and metaplasia alone was seen in one of the H. pylori-infected subjects.
DC-LAMP+ DCs are located in close association with lymphoid follicles in H. pylori-infected gastric mucosa.
DC-LAMP is a type I transmembrane glycoprotein that is absent on immature DCs but is rapidly upregulated upon DC maturation (41, 42). It is expressed in the endosomal/lysosomal compartment and may be involved in MHC class II processing. We used immunohistochemistry to detect DC-LAMP+ DCs in biopsy specimens from human antrum and could show that the frequencies of DC-LAMP+ DCs were significantly higher (P < 0.01) in H. pylori-infected patients than uninfected volunteers (Fig. 1A). The increased numbers of DC-LAMP+ DCs in patients with H. pylori-associated gastritis were also confirmed by flow cytometry analyses, showing severalfold higher frequencies of DC-LAMP+ cells in H. pylori-infected than uninfected individuals (Fig. 2A and C). In immunohistochemistry, DC-LAMP+ cells were sometimes seen scattered in the lamina propria but were mainly associated with lymphoid follicles.
Fig 1.

DC-LAMP+ DCs in the human antrum mucosa. Biopsy specimens were collected from 8 H. pylori-infected (Hp+) and 10 uninfected (Hp−) individuals, and expression of DC-LAMP was determined by immunoperoxidase staining. (A) Frequencies of DC-LAMP+ DCs, expressed as a percentage of the stained area in the gastric mucosa. Symbols represent individual values, and lines indicate the median. (B to E) Representative staining of DC-LAMP from uninfected (B) and H. pylori-infected (C to E) individuals. (F) Isotype control for staining in panel E. **, P < 0.01. Magnifications, ×20.
Fig 2.

Phenotype of gastric DC-LAMP+ DCs from H. pylori-infected subjects. Gastric DCs from uninfected (Hp−) and H. pylori-infected (Hp+) individuals were analyzed by flow cytometry for expression of HLA-DR and DC-LAMP (A) or CD11c and DC-LAMP (B). The lower dot plots show staining with an isotype control. Numbers by the gates indicate cell frequencies among all live (7AAD−) cells. (C) Percentage of DC-LAMP+ DCs, gated as described for panels A and B, in the gastric lamina propria of uninfected and H. pylori-infected subjects. Symbols represent individual values, which were calculated from several stainings in each individual, and horizontal lines indicate the median. All staining with the isotype control was subtracted from the DC-LAMP staining. (D) Flow cytometric analysis of the phenotype of gastric DC-LAMP+ HLA-DRhigh cells identified as described for panels A and B. Thick lines, staining with the indicated antibody; dotted lines, staining with isotype controls. The data shown are from two H. pylori-infected individuals.
Lymphoid follicles were not found in any of the uninfected volunteers but were present in the sections from six out of the eight H. pylori-infected individuals analyzed. In five of these individuals, DC-LAMP+ DCs were found to accumulate close to or inside the follicles, and only a few were seen in other parts of the lamina propria (Fig. 1C to E). Since the DC-LAMP+ DCs were associated with lymphoid follicles in H. pylori-infected individuals, where Tregs have also previously been shown to accumulate (46, 47), we used immunofluorescence to evaluate if DC-LAMP+ DCs would have the possibility to interact with FoxP3+ putative Tregs. Indeed, these experiments showed that DC-LAMP+ DCs are located in the same part of the lymphoid follicles as the FoxP3+ T cells, suggesting that these DCs may be able to interact directly with Tregs (Fig. 3A). Unfortunately, we did not perform quantitative H. pylori cultures to correlate bacterial counts with DC-LAMP+ DC frequencies. However, there was no correlation between HLO scores and the densities of DC-LAMP+ cells.
Fig 3.

Immunofluorescence staining of DCs in the antrum mucosa. Biopsy specimens were collected from H. pylori-infected individuals, and expression of DC-LAMP, FoxP3, and CD68 was determined by immunofluorescence. (A) Representative double staining of DC-LAMP (red) and FoxP3 (green) in two lymphoid follicles from H. pylori-infected individuals. Magnification, ×20. Nuclei are stained blue with DAPI, and follicles are encircled. (B) Representative double staining with DC-LAMP (red) and CD68 (green) close to a lymphoid follicle in an H. pylori-infected individual.
pDCs in H. pylori-infected gastric mucosa.
pDCs are a subtype of dendritic cells, found in blood and lymphoid organs, with the capacity to mount both protective and tolerogenic immune responses (48). Previously, subsets of pDCs were also shown to induce Treg differentiation (49). Here, pDCs were assessed by expression of CD303 (BDCA-2) (50), and immunohistochemical staining for CD303 showed that gastric pDCs are present in very low numbers in both uninfected and infected individuals. The proportion of the tissue area stained ranged from 0 to 0.0097% (median, 0.0020%) in uninfected volunteers and from 0 and 0.014% (median, 0.0040%) in H. pylori-infected individuals, and there was no significant difference between the groups with regard to CD303 expression.
Phenotypic characterization of gastric DCs.
To exclude the possibility that DC-LAMP was expressed by mucosal macrophages, double staining with antibodies to DC-LAMP and CD68, a surface molecule preferentially expressed on monocytes and macrophages, was performed. However, DC-LAMP and CD68 were not coexpressed; in fact, the markers were expressed by cells in different areas of the lamina propria (Fig. 3B).
To further characterize the DC-LAMP+ DCs in the gastric mucosa, flow cytometry of cells isolated from patients undergoing gastric resection was used to analyze the expression of selected surface markers on DC-LAMP+ DCs. These DCs were identified among live cells (7AAD negative [7AAD−]) expressing high levels of HLA-DR together with DC-LAMP (Fig. 2A). Most DC-LAMP+ DCs coexpressed the DC marker CD11c (Fig. 2B) but lacked expression of the pattern recognition receptor CD14 (data not shown). HLA-DRhigh CD11chigh cells typically make up 0.5 to 2.5% of all cells isolated from H. pylori-infected human lamina propria, and among these, 0.1 to 10% were DC-LAMP+. We could also conclude that the DC-LAMP+ DCs represent a DC subset other than the DC-SIGN+ cells that we have recently reported in increased numbers during H. pylori infection (32), as the two populations are located in distinct tissue compartments and since the DC-SIGN+ DCs coexpress CD14, which the DC-LAMP+ DCs lack.
To determine their capacity to present antigens to T cells, we also analyzed the expression of costimulatory molecules by the DC-LAMP+ DCs. Surprisingly, since DC-LAMP is generally considered a maturation marker, the DC-LAMP+ DCs had low to negligible expression of CD86 and lacked expression of CD80 and CD83 (Fig. 2D). When analyzing the few DC-LAMP+ DCs in the uninfected mucosa, we found no obvious differences in the phenotype of DC-LAMP+ DCs in H. pylori-infected and uninfected patients (data not shown).
CCL19 but not CCL21 levels are increased in H. pylori-infected gastric mucosa.
DC maturation results in upregulation of the chemokine receptor CCR7 and subsequent migration to draining lymph nodes. To evaluate possible mechanisms for the accumulation of DC-LAMP+ DCs in the H. pylori-infected gastric mucosa, we investigated the expression of the CCR7 ligands CCL19 and CCL21. Real-time PCR analyses showed that the relative expression of CCL19 mRNA, but not CCL21 mRNA, was significantly higher (P < 0.001) in H. pylori-infected individuals than in uninfected volunteers (Fig. 4A and B). Furthermore, there were significantly higher (P < 0.01) concentrations of CCL19 protein in biopsy specimens from H. pylori-infected individuals than in those from uninfected individuals (Fig. 4C). These results suggested that increased levels of CCL19 in the gastric mucosa may result in retention of maturing DCs in the gastric mucosa. However, we could not detect expression of CCR7 on DC-LAMP+ DCs isolated either from H. pylori-infected gastric mucosa (Fig. 2D) or from uninfected tissues (data not shown), suggesting that CCL19-mediated signals may not be the primary cause of DC recruitment to the lymphoid follicles.
Fig 4.
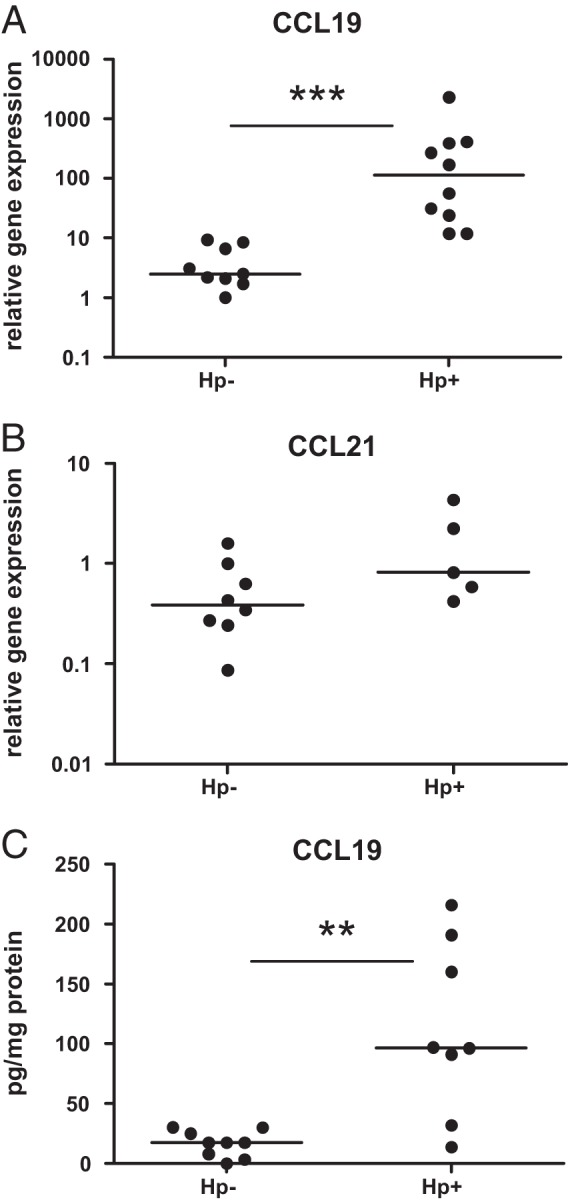
Expression of CCL19 and CCL21 in human antrum mucosa. Biopsy specimens were collected from 9 uninfected (Hp−) and 10 H. pylori-infected (Hp+) individuals and used for mRNA and protein purification. Relative expression of CCL19 (A) and CCL21 (B) mRNA was determined by real-time PCR. (C) The CCL19 protein concentration in relation to the total amount of protein was determined by ELISA. Symbols represent individual values, and lines indicate the median. **, P < 0.01; ***, P < 0.001.
DISCUSSION
Infection with H. pylori gives rise to a chronic inflammation in the gastric mucosa, which is characterized by infiltration of neutrophils, macrophages, and lymphocytes. In this study, we show that the frequencies of DC-LAMP+ putative mature DCs are also increased in human H. pylori-associated gastritis and that the DC-LAMP+ DCs are localized in close association with lymphoid follicles in the mucosa of H. pylori-infected stomach tissue. H. pylori is generally held to be noninvasive and to rarely invade beyond the gastric epithelium. However, recent reports have suggested that mucosal DCs may sample antigen in the lumen by opening tight junctions between epithelial cells, a feature also recently described for gastric DCs, which may thus get in contact with live H. pylori bacteria (35, 51, 52).
In animal models, infection with H. pylori results in recruitment of DCs to the gastric mucosa (53, 54), and a recent study in humans also showed increased frequencies of HLA-DR-positive (HLA-DR+) putative DCs in H. pylori-infected individuals (25). In addition, we have recently shown that semimature (i.e., HLA-DRhigh but CD80− and CD86−) DC-SIGN+ DCs accumulate in the gastric mucosa of H. pylori-infected individuals (32). Bimczok et al. also showed that a majority of gastric HLA-DR+ cells do not express CD80 or CD86 (25, 36). In this study, we used DC-LAMP to identify and investigate mature DCs, to focus on DCs with a potential ability to present antigens to local T cells (41, 42). Our study shows that DC-LAMP is expressed on a small population of CD11chigh gastric cells devoid of the macrophage marker CD68 and of CD14 in H. pylori-infected individuals, while it is more or less absent in the tissue from healthy subjects. Furthermore, the DC-LAMP+ DCs have a high level of expression of MHC class II molecules but express only low levels of the costimulatory molecule CD86 and no CD80 and CD83 at all. Thus, our findings extend previous studies of human gastric DCs by providing details on a carefully identified subpopulation of mature DCs. The DC-LAMP+ DCs are a small population compared to all putative antigen-presenting cells present in the gastric lamina propria, but their distinct localization in and around lymphoid follicles indicates that they may have important functions in relation to T cell priming and illustrates the importance of examining local immune responses in order not to miss small but significant cell populations that are not present in blood or other easily accessible tissues. Our findings also indicate that the lymphoid follicles forming in the setting of chronic H. pylori infection have a specialized function and may be involved in shaping the local T cell response. Unfortunately, the low frequencies of DC-LAMP+ DCs make it impossible to isolate enough cells to perform functional studies, such as studies to determine the production of tolerance-promoting cytokines and enzymes or their effect on T cell priming. It is interesting to note that DC-LAMP+ DCs were localized to the same tissue area as FoxP3+ putative Tregs, which were recently demonstrated to be enriched in the T cell zone of lymphoid follicles in H. pylori-induced gastritis (46, 47). Taken together, our observations suggest that a subpopulation of gastric DCs is able to present antigen to T cells locally in the gastric lamina propria, but the relative lack of costimulatory molecules would suggest that these DC-LAMP+ DCs may, rather, be involved in inducing tolerance to local antigens. Interactions between DC-LAMP+ DCs and Tregs in the lymphoid follicles may have rendered the DCs tolerogenic through Treg-derived signals (55). Treg-mediated suppression of conventional T cells leads to increased bacterial numbers, at least in murine Helicobacter infection (56, 57), but may still be beneficial to the host, as it reduces the inflammation that would otherwise be more harmful and cause ulcers or premalignant epithelial lesions in some individuals. In animal experiments, Treg induction by H. pylori infection, presumably by interactions with tolerogenic DCs, may even reduce experimental colitis and allergy, demonstrating the anti-inflammatory potential of such H. pylori-induced Tregs (32, 37–39). It is essential to understand the potential tolerization mechanisms induced by H. pylori infection also in humans, as they appear to protect against exaggerated inflammatory responses. A better insight into these mechanisms may provide therapeutic options to treat inflammatory conditions, such as childhood asthma and inflammatory bowel disease (58, 59).
In intestinal tissues, a specialized population of CD103+ DCs with migrating potential has been shown to migrate to the draining lymph nodes and promote differentiation of the subsequently activated T cells to Tregs, which in turn home back to the intestinal mucosa (60). As very few of the CD11chigh putative DCs in the gastric mucosa express CD103 (data not shown), the many Tregs present in gastric mucosa (13, 46, 47) may have been induced somewhere other than the draining lymph nodes, maybe the Peyer's patches, and may then have migrated to the gastric mucosa, as previously demonstrated for IgA-secreting cells induced by intestinal antigen delivery (61).
Having established the presence of DC-LAMP+ DCs with a low costimulatory ability in human H. pylori-associated gastritis, we sought to determine the mechanisms of their retention. As we have previously shown that DCs stimulated with live H. pylori cells increase their expression of CCR7 and migrate in response to CCL19 (21), we determined the presence of the CCR7 ligands CCL19 and CCL21 in the gastric mucosa. We could demonstrate substantially increased CCL19 mRNA and protein levels in the H. pylori-infected gastric mucosa, whereas CCL21 levels were similar in H. pylori-infected and uninfected tissues. Lymphoid follicles that form in tissues during autoimmune or infectious disease have been shown to express CL19 and CCL21 (62). However, the expression of CCL19 and CCL21 in gastric lymphoid follicles has not been evaluated, and to our knowledge, this is the first study to demonstrate increased levels of CCL19 in the H. pylori-infected gastric mucosa. Thus, there would be a possibility that mature DCs expressing CCR7 are retained in the gastric mucosa due to local production of CCL19. However, flow cytometry showed that DC-LAMP+ DCs in the gastric mucosa lacked expression of CCR7. Similarly, the majority of DC-LAMP+ DCs in inflamed skin and lymph nodes do not express CCR7 (63). The lack of surface CCR7 could be due to receptor internalization upon chemokine binding, a phenomenon observed in vitro in response to CCL19 (64–66). However, as CCR7 recirculates to the cell surface, at least in vitro, and since DCs migrating to lymph nodes in response to CCL19 retain their CCR7 expression (67), the lack of CCR7 on DC-LAMP+ DCs may reflect a more immature phenotype of the DCs. On the basis of our results, it is reasonable to propose that a subpopulation of maturing DCs remains in the gastric mucosa and localizes to lymphoid follicles, instead of migrating to draining gastric lymph nodes, but at this stage, the process appears to be independent of CCL19-CCR7 interactions.
In conclusion, we have demonstrated that DC-LAMP+ DCs expressing high levels of MHC class II molecules but little CD80, CD83, and CD86 accumulate in the lymphoid follicles in H. pylori-infected human gastric mucosa and that these DCs are located close to follicular FoxP3+ T cells. Furthermore, the level of the chemokine CCL19 was significantly increased in the gastric tissue of H. pylori-infected individuals. Based on these observations, we propose that a subset of maturing DCs does not migrate to the draining lymph nodes in human H. pylori-induced gastritis but is instead recruited to gastric lymphoid follicles, where they may interact with Tregs and then contribute to T cell tolerization by antigen presentation in the context of suboptimal costimulation.
ACKNOWLEDGMENTS
This work was supported by grants from the Swedish Science Council, Sahlgrenska University Hospital, the Foundation for Strategic Research through the MIVAC Center of Excellence, the Inga-Britt and Arne Lundberg Foundation, and the Professor Nanna Svartz Foundation.
We thank all volunteers that participated in the study and Eva Peras, My Engström, and Diana Lustgarten for help with collection of gastric tissues. We also acknowledge Anna Holmberg, Johan Mattsson, and the core facility for cellular imaging at the Sahlgrenska Academy for help with confocal microscopy.
We have no competing interests to declare.
Footnotes
Published ahead of print 22 July 2013
REFERENCES
- 1.Fischbach W, Chan AO, Wong BC. 2005. Helicobacter pylori and gastric malignancy. Helicobacter 10(Suppl 1):34–39 [DOI] [PubMed] [Google Scholar]
- 2.Huang JQ, Sridhar S, Hunt RH. 2002. Role of Helicobacter pylori infection and non-steroidal anti-inflammatory drugs in peptic-ulcer disease: a meta-analysis. Lancet 359:14–22 [DOI] [PubMed] [Google Scholar]
- 3.Makola D, Peura DA, Crowe SE. 2007. Helicobacter pylori infection and related gastrointestinal diseases. J. Clin. Gastroenterol. 41:548–558 [DOI] [PubMed] [Google Scholar]
- 4.Figueiredo C, Machado JC, Yamaoka Y. 2005. Pathogenesis of Helicobacter pylori infection. Helicobacter 10(Suppl 1):14–20 [DOI] [PubMed] [Google Scholar]
- 5.Del Giudice G, Michetti P. 2004. Inflammation, immunity and vaccines for Helicobacter pylori. Helicobacter 9(Suppl 1):23–28 [DOI] [PubMed] [Google Scholar]
- 6.Genta RM, Hamner HW, Graham DY. 1993. Gastric lymphoid follicles in Helicobacter pylori infection: frequency, distribution, and response to triple therapy. Hum. Pathol. 24:577–583 [DOI] [PubMed] [Google Scholar]
- 7.Houghton J, Wang TC. 2005. Helicobacter pylori and gastric cancer: a new paradigm for inflammation-associated epithelial cancers. Gastroenterology 128:1567–1578 [DOI] [PubMed] [Google Scholar]
- 8.Bamford KB, Fan X, Crowe SE, Leary JF, Gourley WK, Luthra GK, Brooks EG, Graham DY, Reyes VE, Ernst PB. 1998. Lymphocytes in the human gastric mucosa during Helicobacter pylori infection have a T helper cell 1 phenotype. Gastroenterology 114:482–492 [DOI] [PubMed] [Google Scholar]
- 9.Luzza F, Parrello T, Monteleone G, Sebkova L, Romano M, Zarrilli R, Imeneo M, Pallone F. 2000. Up-regulation of IL-17 is associated with bioactive IL-8 expression in Helicobacter pylori-infected human gastric mucosa. J. Immunol. 165:5332–5337 [DOI] [PubMed] [Google Scholar]
- 10.Garhart CA, Redline RW, Nedrud JG, Czinn SJ. 2002. Clearance of Helicobacter pylori infection and resolution of postimmunization gastritis in a kinetic study of prophylactically immunized mice. Infect. Immun. 70:3529–3538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sayi A, Kohler E, Hitzler I, Arnold I, Schwendener R, Rehrauer H, Muller A. 2009. The CD4+ T cell-mediated IFN-gamma response to Helicobacter infection is essential for clearance and determines gastric cancer risk. J. Immunol. 182:7085–7101 [DOI] [PubMed] [Google Scholar]
- 12.Lundgren A, Suri-Payer E, Enarsson K, Svennerholm AM, Lundin BS. 2003. Helicobacter pylori-specific CD4+ CD25high regulatory T cells suppress memory T-cell responses to H. pylori in infected individuals. Infect. Immun. 71:1755–1762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Enarsson K, Lundgren A, Kindlund B, Hermansson M, Roncador G, Banham AH, Lundin BS, Quiding-Jarbrink M. 2006. Function and recruitment of mucosal regulatory T cells in human chronic Helicobacter pylori infection and gastric adenocarcinoma. Clin. Immunol. 121:358–368 [DOI] [PubMed] [Google Scholar]
- 14.Lundgren A, Stromberg E, Sjoling A, Lindholm C, Enarsson K, Edebo A, Johnsson E, Suri-Payer E, Larsson P, Rudin A, Svennerholm AM, Lundin BS. 2005. Mucosal FOXP3-expressing CD4+ CD25high regulatory T cells in Helicobacter pylori-infected patients. Infect. Immun. 73:523–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Banchereau J, Briere F, Caux C, Davoust J, Lebecque S, Liu YJ, Pulendran B, Palucka K. 2000. Immunobiology of dendritic cells. Annu. Rev. Immunol. 18:767–811 [DOI] [PubMed] [Google Scholar]
- 16.Schuurhuis DH, Fu N, Ossendorp F, Melief CJ. 2006. Ins and outs of dendritic cells. Int. Arch. Allergy Immunol. 140:53–72 [DOI] [PubMed] [Google Scholar]
- 17.Bar-On L, Jung S. 2010. Defining dendritic cells by conditional and constitutive cell ablation. Immunol. Rev. 234:76–89 [DOI] [PubMed] [Google Scholar]
- 18.Kranzer K, Eckhardt A, Aigner M, Knoll G, Deml L, Speth C, Lehn N, Rehli M, Schneider-Brachert W. 2004. Induction of maturation and cytokine release of human dendritic cells by Helicobacter pylori. Infect. Immun. 72:4416–4423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hafsi N, Voland P, Schwendy S, Rad R, Reindl W, Gerhard M, Prinz C. 2004. Human dendritic cells respond to Helicobacter pylori, promoting NK cell and Th1-effector responses in vitro. J. Immunol. 173:1249–1257 [DOI] [PubMed] [Google Scholar]
- 20.Guiney DG, Hasegawa P, Cole SP. 2003. Helicobacter pylori preferentially induces interleukin 12 (IL-12) rather than IL-6 or IL-10 in human dendritic cells. Infect. Immun. 71:4163–4166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hansson M, Lundgren A, Elgbratt K, Quiding-Jarbrink M, Svennerholm AM, Johansson EL. 2006. Dendritic cells express CCR7 and migrate in response to CCL19 (MIP-3beta) after exposure to Helicobacter pylori. Microbes Infect. 8:841–850 [DOI] [PubMed] [Google Scholar]
- 22.Khamri W, Walker MM, Clark P, Atherton JC, Thursz MR, Bamford KB, Lechler RI, Lombardi G. 2010. Helicobacter pylori stimulates dendritic cells to induce interleukin-17 expression from CD4+ T lymphocytes. Infect. Immun. 78:845–853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Andres S, Schmidt HM, Mitchell H, Rhen M, Maeurer M, Engstrand L. 2011. Helicobacter pylori defines local immune response through interaction with dendritic cells. FEMS Immunol. Med. Microbiol. 61:168–178 [DOI] [PubMed] [Google Scholar]
- 24.Fehlings M, Drobbe L, Moos V, Renner Viveros P, Hagen J, Beigier-Bompadre M, Pang E, Belogolova E, Churin Y, Schneider T, Meyer TF, Aebischer T, Ignatius R. 2012. Comparative analysis of the interaction of Helicobacter pylori with human dendritic cells, macrophages, and monocytes. Infect. Immun. 80:2724–2734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bimczok D, Clements RH, Waites KB, Novak L, Eckhoff DE, Mannon PJ, Smith PD, Smythies LE. 2010. Human primary gastric dendritic cells induce a Th1 response to H. pylori. Mucosal Immunol. 3:260–269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rad R, Ballhorn W, Voland P, Eisenacher K, Mages J, Rad L, Ferstl R, Lang R, Wagner H, Schmid RM, Bauer S, Prinz C, Kirschning CJ, Krug A. 2009. Extracellular and intracellular pattern recognition receptors cooperate in the recognition of Helicobacter pylori. Gastroenterology 136:2247–2257 [DOI] [PubMed] [Google Scholar]
- 27.Rad R, Brenner L, Krug A, Voland P, Mages J, Lang R, Schwendy S, Reindl W, Dossumbekova A, Ballhorn W, Wagner H, Schmid RM, Bauer S, Prinz C. 2007. Toll-like receptor-dependent activation of antigen-presenting cells affects adaptive immunity to Helicobacter pylori. Gastroenterology 133:150–163.e3 [DOI] [PubMed] [Google Scholar]
- 28.Bergman MP, Engering A, Smits HH, van Vliet SJ, van Bodegraven AA, Wirth HP, Kapsenberg ML, Vandenbroucke-Grauls CM, van Kooyk Y, Appelmelk BJ. 2004. Helicobacter pylori modulates the T helper cell 1/T helper cell 2 balance through phase-variable interaction between lipopolysaccharide and DC-SIGN. J. Exp. Med. 200:979–990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gringhuis SI, den Dunnen J, Litjens M, van der Vlist M, Geijtenbeek TB. 2009. Carbohydrate-specific signaling through the DC-SIGN signalosome tailors immunity to Mycobacterium tuberculosis, HIV-1 and Helicobacter pylori. Nat. Immunol. 10:1081–1088 [DOI] [PubMed] [Google Scholar]
- 30.Reis e Sousa C. 2006. Dendritic cells in a mature age. Nat. Rev. Immunol. 6:476–483 [DOI] [PubMed] [Google Scholar]
- 31.Mitchell P, Germain C, Fiori PL, Kahmri W, Forster GR, Ghosh S, Lechler RI, Bamford KB, Lombardi G. 2007. Chronic exposure to Helicobacter pylori impairs dendritic cell function. Infect. Immun. 75:810–819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Oertli M, Sundquist M, Hitzler I, Engler DB, Arnold IC, Reuter S, Maxeiner J, Hansson M, Taube C, Quiding-Jarbrink M, Muller A. 2012. DC-derived IL-18 drives Treg differentiation, murine Helicobacter pylori-specific immune tolerance, and asthma protection. J. Clin. Invest. 122:1082–1096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Quiding-Jarbrink M, Raghavan S, Sundquist M. 2010. Enhanced M1 macrophage polarization in human Helicobacter pylori-associated atrophic gastritis and in vaccinated mice. PLoS One 5:e15018. 10.1371/journal.pone.0015018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hitzler I, Oertli M, Becher B, Agger EM, Muller A. 2011. Dendritic cells prevent rather than promote immunity conferred by a Helicobacter vaccine using a mycobacterial adjuvant. Gastroenterology 141:186–196 [DOI] [PubMed] [Google Scholar]
- 35.Kao JY, Zhang M, Miller MJ, Mills JC, Wang B, Liu M, Eaton KA, Zou W, Berndt BE, Cole TS, Takeuchi T, Owyang SY, Luther J. 2010. Helicobacter pylori immune escape is mediated by dendritic cell-induced Treg skewing and Th17 suppression in mice. Gastroenterology 138:1046–1054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bimczok D, Grams JM, Stahl RD, Waites KB, Smythies LE, Smith PD. 2011. Stromal regulation of human gastric dendritic cells restricts the Th1 response to Helicobacter pylori. Gastroenterology 141:929–938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Luther J, Owyang SY, Takeuchi T, Cole TS, Zhang M, Liu M, Erb-Downward J, Rubenstein JH, Chen CC, Pierzchala AV, Paul JA, Kao JY. 2011. Helicobacter pylori DNA decreases pro-inflammatory cytokine production by dendritic cells and attenuates dextran sodium sulphate-induced colitis. Gut 60:1479–1486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Arnold IC, Dehzad N, Reuter S, Martin H, Becher B, Taube C, Muller A. 2011. Helicobacter pylori infection prevents allergic asthma in mouse models through the induction of regulatory T cells. J. Clin. Invest. 121:3088–3093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Higgins PD, Johnson LA, Luther J, Zhang M, Sauder KL, Blanco LP, Kao JY. 2011. Prior Helicobacter pylori infection ameliorates Salmonella typhimurium-induced colitis: mucosal crosstalk between stomach and distal intestine. Inflamm. Bowel Dis. 17:1398–1408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ninomiya T, Matsui H, Akbar SM, Murakami H, Onji M. 2000. Localization and characterization of antigen-presenting dendritic cells in the gastric mucosa of murine and human autoimmune gastritis. Eur. J. Clin. Invest. 30:350–358 [DOI] [PubMed] [Google Scholar]
- 41.Karthaus N, Torensma R, Tel J. 2012. Deciphering the message broadcast by tumor-infiltrating dendritic cells. Am. J. Pathol. 181:733–742 [DOI] [PubMed] [Google Scholar]
- 42.de Saint-Vis B, Vincent J, Vandenabeele S, Vanbervliet B, Pin JJ, Ait-Yahia S, Patel S, Mattei MG, Banchereau J, Zurawski S, Davoust J, Caux C, Lebecque S. 1998. A novel lysosome-associated membrane glycoprotein, DC-LAMP, induced upon DC maturation, is transiently expressed in MHC class II compartment. Immunity 9:325–336 [DOI] [PubMed] [Google Scholar]
- 43.Hamlet A, Thoreson AC, Nilsson O, Svennerholm AM, Olbe L. 1999. Duodenal Helicobacter pylori infection differs in cagA genotype between asymptomatic subjects and patients with duodenal ulcers. Gastroenterology 116:259–268 [DOI] [PubMed] [Google Scholar]
- 44.Dixon MF, Genta RM, Yardley JH, Correa P. 1997. Histological classification of gastritis and Helicobacter pylori infection: an agreement at last? The International Workshop on the Histopathology of Gastritis. Helicobacter 2(Suppl 1):S17–S24 [DOI] [PubMed] [Google Scholar]
- 45.Bergquist C, Mattsson-Rydberg A, Lonroth H, Svennerholm A. 2000. Development of a new method for the determination of immune responses in the human stomach. J. Immunol. Methods 234:51–59 [DOI] [PubMed] [Google Scholar]
- 46.Hansson M, Lundgren A, Elgbratt K, Quiding-Jarbrink M, Svennerholm AM, Johansson EL. 2006. Dendritic cells express CCR7 and migrate in response to CCL19 (MIP-3beta) after exposure to Helicobacter pylori. Microbes Infect. 8:841–850 [DOI] [PubMed] [Google Scholar]
- 47.Kandulski A, Wex T, Kuester D, Peitz U, Gebert I, Roessner A, Malfertheiner P. 2008. Naturally occurring regulatory T cells (CD4+, CD25high, FOXP3+) in the antrum and cardia are associated with higher H. pylori colonization and increased gene expression of TGF-beta1. Helicobacter 13:295–303 [DOI] [PubMed] [Google Scholar]
- 48.Colonna M, Trinchieri G, Liu YJ. 2004. Plasmacytoid dendritic cells in immunity. Nat. Immunol. 5:1219–1226 [DOI] [PubMed] [Google Scholar]
- 49.Hadeiba H, Sato T, Habtezion A, Oderup C, Pan J, Butcher EC. 2008. CCR9 expression defines tolerogenic plasmacytoid dendritic cells able to suppress acute graft-versus-host disease. Nat. Immunol. 9:1253–1260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dzionek A, Fuchs A, Schmidt P, Cremer S, Zysk M, Miltenyi S, Buck DW, Schmitz J. 2000. BDCA-2, BDCA-3, and BDCA-4: three markers for distinct subsets of dendritic cells in human peripheral blood. J. Immunol. 165:6037–6046 [DOI] [PubMed] [Google Scholar]
- 51.Rescigno M, Urbano M, Valzasina B, Francolini M, Rotta G, Bonasio R, Granucci F, Kraehenbuhl JP, Ricciardi-Castagnoli P. 2001. Dendritic cells express tight junction proteins and penetrate gut epithelial monolayers to sample bacteria. Nat. Immunol. 2:361–367 [DOI] [PubMed] [Google Scholar]
- 52.Necchi V, Manca R, Ricci V, Solcia E. 2009. Evidence for transepithelial dendritic cells in human H. pylori active gastritis. Helicobacter 14:208–222 [DOI] [PubMed] [Google Scholar]
- 53.Nishi T, Okazaki K, Kawasaki K, Fukui T, Tamaki H, Matsuura M, Asada M, Watanabe T, Uchida K, Watanabe N, Nakase H, Ohana M, Hiai H, Chiba T. 2003. Involvement of myeloid dendritic cells in the development of gastric secondary lymphoid follicles in Helicobacter pylori-infected neonatally thymectomized BALB/c mice. Infect. Immun. 71:2153–2162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kao JY, Rathinavelu S, Eaton KA, Bai L, Zavros Y, Takami M, Pierzchala A, Merchant JL. 2006. Helicobacter pylori-secreted factors inhibit dendritic cell IL-12 secretion: a mechanism of ineffective host defense. Am. J. Physiol. Gastrointest. Liver Physiol. 291:G73–G81 [DOI] [PubMed] [Google Scholar]
- 55.Sakaguchi S, Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T. 2009. Regulatory T cells: how do they suppress immune responses? Int. Immunol. 21:1105–1111 [DOI] [PubMed] [Google Scholar]
- 56.Raghavan S, Fredriksson M, Svennerholm AM, Holmgren J, Suri-Payer E. 2003. Absence of CD4+CD25+ regulatory T cells is associated with a loss of regulation leading to increased pathology in Helicobacter pylori-infected mice. Clin. Exp. Immunol. 132:393–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rad R, Brenner L, Bauer S, Schwendy S, Layland L, da Costa CP, Reindl W, Dossumbekova A, Friedrich M, Saur D, Wagner H, Schmid RM, Prinz C. 2006. CD25+/Foxp3+ T cells regulate gastric inflammation and Helicobacter pylori colonization in vivo. Gastroenterology 131:525–537 [DOI] [PubMed] [Google Scholar]
- 58.Chen Y, Blaser MJ. 2008. Helicobacter pylori colonization is inversely associated with childhood asthma. J. Infect. Dis. 198:553–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Luther J, Dave M, Higgins PD, Kao JY. 2010. Association between Helicobacter pylori infection and inflammatory bowel disease: a meta-analysis and systematic review of the literature. Inflamm. Bowel Dis. 16:1077–1084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Agace WW, Persson EK. 2012. How vitamin A metabolizing dendritic cells are generated in the gut mucosa. Trends Immunol. 33:42–48 [DOI] [PubMed] [Google Scholar]
- 61.Mattsson A, Lonroth H, Quiding-Jarbrink M, Svennerholm AM. 1998. Induction of B cell responses in the stomach of Helicobacter pylori-infected subjects after oral cholera vaccination. J. Clin. Invest. 102:51–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Drayton DL, Liao S, Mounzer RH, Ruddle NH. 2006. Lymphoid organ development: from ontogeny to neogenesis. Nat. Immunol. 7:344–353 [DOI] [PubMed] [Google Scholar]
- 63.Katou F, Ohtani H, Nakayama T, Nagura H, Yoshie O, Motegi K. 2003. Differential expression of CCL19 by DC-Lamp+ mature dendritic cells in human lymph node versus chronically inflamed skin. J. Pathol. 199:98–106 [DOI] [PubMed] [Google Scholar]
- 64.Bardi G, Lipp M, Baggiolini M, Loetscher P. 2001. The T cell chemokine receptor CCR7 is internalized on stimulation with ELC, but not with SLC. Eur. J. Immunol. 31:3291–3297 [DOI] [PubMed] [Google Scholar]
- 65.Otero C, Groettrup M, Legler DF. 2006. Opposite fate of endocytosed CCR7 and its ligands: recycling versus degradation. J. Immunol. 177:2314–2323 [DOI] [PubMed] [Google Scholar]
- 66.Byers MA, Calloway PA, Shannon L, Cunningham HD, Smith S, Li F, Fassold BC, Vines CM. 2008. Arrestin 3 mediates endocytosis of CCR7 following ligation of CCL19 but not CCL21. J. Immunol. 181:4723–4732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ohl L, Mohaupt M, Czeloth N, Hintzen G, Kiafard Z, Zwirner J, Blankenstein T, Henning G, Forster R. 2004. CCR7 governs skin dendritic cell migration under inflammatory and steady-state conditions. Immunity 21:279–288 [DOI] [PubMed] [Google Scholar]