

Abstract
Dysregulated immune responses to commensal intestinal bacteria, including Escherichia coli, contribute to the development of inflammatory bowel diseases (IBDs) and experimental colitis. Reciprocally, E. coli responds to chronic intestinal inflammation by upregulating expression of stress response genes, including gadA and gadB. GadAB encode glutamate decarboxylase and protect E. coli from the toxic effects of low pH and fermentation acids, factors present in the intestinal lumen in patients with active IBDs. We hypothesized that E. coli upregulates gadAB during inflammation to enhance its survival and virulence. Using real-time PCR, we determined gadAB expression in luminal E. coli from ex-germfree wild-type (WT) and interleukin-10 (IL-10) knockout (KO) (IL-10−/−) mice selectively colonized with a commensal E. coli isolate (NC101) that causes colitis in KO mice in isolation or in combination with 7 other commensal intestinal bacterial strains. E. coli survival and host inflammatory responses were measured in WT and KO mice colonized with NC101 or a mutant lacking the gadAB genes (NC101ΔgadAB). The susceptibility of NC101 and NC101ΔgadAB to killing by host antimicrobial peptides and their translocation across intestinal epithelial cells were evaluated using bacterial killing assays and transwell experiments, respectively. We show that expression of gadAB in luminal E. coli increases proportionately with intestinal inflammation in KO mice and enhances the susceptibility of NC101 to killing by the host antimicrobial peptide cryptdin-4 but decreases bacterial transmigration across intestinal epithelial cells, colonic inflammation, and mucosal immune responses. Chronic intestinal inflammation upregulates acid tolerance pathways in commensal E. coli isolates, which, contrary to our original hypothesis, limits their survival and colitogenic potential. Further investigation of microbial adaptation to immune-mediated inflammation may provide novel insights into the pathogenesis and treatment of IBDs.
INTRODUCTION
The human gastrointestinal tract is home to a complex community of microbes whose number exceeds the number of host cells by over an order of magnitude (1). In the healthy state, protective host mechanisms, including bacterium-induced regulatory T cells and rapid microbial adaptation, result in a dynamic equilibrium between the intestinal immune system and this large number of microbial symbionts (2, 3). However, mounting evidence suggests that members of the intestinal microbiota, including adherent-invasive strains of Escherichia coli, also induce maladaptive immune responses in patients with inflammatory bowel diseases (IBDs) (4). Currently, the prevailing hypothesis is that a subset of commensal enteric bacteria provides antigenic stimulation that continuously activates pathogenic T cells to cause chronic intestinal injury (5). Similar to human IBDs, the importance of the intestinal microbiota in the pathogenesis of chronic, immune-mediated experimental colitis is evident in gnotobiotic studies using animals with immunoregulatory defects, including the interleukin-10 (IL-10)-deficient (IL-10−/−) mouse. In this model, development of chronic intestinal inflammation requires the presence of commensal microbes (6, 7). Moreover, selective colonization (monoassociation) of IL-10−/− mice with defined commensal bacteria, including adherent-invasive E. coli, causes colitis (7–10).
While the influence of the microbiota on host immune responses has been extensively studied, little is known about the effects of inflamed intestine on bacterial properties that may perpetuate the cycle of mucosal inflammation. Host inflammatory responses enhance the virulence of certain enteric pathogens (11–13), and we recently reported that colitis in E. coli-monoassociated IL-10−/− mice over a long period induces upregulation of stress-response genes, including gadA and gadB, in commensal luminal E. coli (14).
Although the gadA and gadB genes are not located in the same operon, their function appears to be redundant. gadA and gadB (gadAB) encode glutamate decarboxylase proteins that are part of the glutamate-dependent acid resistance system 2 (AR2) in E. coli, which protects the bacteria from acid conditions (pH 2.5 to 4.5), including those encountered during passage through the stomach into the intestine (15, 16). While the roles of E. coli GadAB in bacterial survival during transit through the normal gastrointestinal tract have been described (15, 17), little is known about the effect of these proteins on E. coli viability in the inflamed gut and how they affect host immune responses.
Since active IBDs are variably associated with decreased luminal pH (18) and increased colonic levels of some fermentation acids (19, 20) and because gadAB protect E. coli from the toxic effects of low pH and fermentation acids (16, 21), we hypothesized that E. coli upregulates gadAB in the inflamed colon in order to survive and perpetuate colitis. In the present study, we show that, contrary to our hypothesis, expression of gadAB in luminal commensal E. coli is associated with reduced bacterial survival and attenuated colitis, which may be due in part to increased susceptibility to bacterial killing by host antimicrobial peptides (AMPs) and decreased translocation across intestinal epithelial cells. These results suggest that inflammation-associated changes in commensal bacterial gene expression may paradoxically decrease luminal bacterial survival and attenuate the host inflammatory response.
MATERIALS AND METHODS
Bacterial strains, lysates, and growth curves.
The nonpathogenic murine E. coli strain NC101 was isolated as described previously (8). The Enterococcus faecalis human oral isolate (strain OG1RF) was provided by Mark Huycke. Mutant strain NC101ΔgadAB was constructed using the bacteriophage λ-red recombinase method (22). Mutant strains NC101ΔgadAB attTn7::gadBA and NC101ΔgadAB(pgadB) were generated using standard molecular biology techniques (see the methods in the supplemental material). Bacterial lysates were prepared and bacterial growth curves were generated as described previously (8, 14). In separate experiments, germfree mice were colonized by oral gavage with a mixture of Enterococcus faecalis OG1RF, Bacteroides thetaiotaomicron VPI-5482, Bacteroides vulgatus ATCC 8482, Bifidobacterium longum subsp. infantis ATCC 15697, Lactobacillus rhamnosus GG, Ruminococcus gnavus ATCC 29149, Faecalibacterium prausnitzii A2-165, and either E. coli NC101 or E. coli NC101ΔgadAB that had been cultured anaerobically overnight.
Antimicrobial peptide bacterial killing assays.
Bacteria were grown to mid-exponential phase and washed in 10 mM PIPES buffer, pH 7.3, and the optical density at 600 nm was adjusted to ∼1.0 using PIPES [piperazine-N,N′-bis(2-ethanesulfonic acid)] containing 1% LB. Bacterial suspensions were incubated with either medium control (PIPES plus 1% LB) or recombinant murine cryptdin-4 (a gift from Andrew Oulette) for 1 h, after which viable bacteria were enumerated by plating serial dilutions.
Mice.
Germfree (GF) IL-10−/− and wild-type (WT) controls (both on the 129S6/SvEV background) were originally derived under sterile conditions by hysterectomy at the Gnotobiotic Laboratory (University of Wisconsin, Madison). Mice maintained under GF conditions at the National Gnotobiotic Rodent Resource Center at the University of North Carolina (UNC)—Chapel Hill were monoassociated with bacteria by swabbing the mouth and anus with an overnight bacterial culture of E. coli NC101 or NC101ΔgadAB. The absence of isolator contamination was confirmed by Gram stain and culture of cecal contents on brain heart infusion (BHI) agar plates under aerobic and anaerobic conditions. All animal protocols were approved by the UNC—Chapel Hill Institutional Animal Care and Use Committee.
RNA isolation from cecal contents.
Freshly harvested cecal or midcolonic contents (100 to 300 mg) were snap-frozen in liquid N2 and stored at −80°C. Frozen samples were thawed into 1 ml of Qiagen RNAprotect bacterial reagent while vortexing and incubated at 25°C for 5 min, and bacterial RNA was isolated as described previously (23).
Extraction of DNA from colon tissue.
Bacterial DNA was isolated from snap-frozen cecal tissue using phenol-chloroform extraction, physical disruption, and DNA purification on Qiagen DNeasy blood and tissue columns (see the methods in the supplemental material).
Real-time PCR.
Real-time oligonucleotide primers were designed using either Primer 3 or Applied Biosystems Primer Express (version 3.0) software based on E. coli NC101 genomic DNA sequences published in GenBank (see Table S1 in the supplemental material). First-strand cDNA was synthesized, and quantitative real-time PCRs were performed as described previously (14), except that Bio-Rad 2× iTaq SYBR green PCR Supermix was used. To quantify the number of E. coli 16S rRNA gene copies in cecal tissue, 50 ng of tissue DNA was added to each real-time PCR mixture and the results were normalized to those for murine GAPDH (glyceraldehyde-3-phosphate dehydrogenase) using the ΔCT (where CT is the threshold cycle) method.
Quantification of luminal bacteria.
Luminal E. coli concentrations in monoassociated mice were measured by dilution plating on BHI agar as described previously (14). Cecal E. coli concentrations in animals with bacterial octoassociation (animals colonized with the mixture of the seven commensal strains representative of the normal gut microbiota indicated above plus an E. coli strain) were measured by quantitative PCR of the E. coli 16S rRNA gene as described above, but using bacterial genomic DNA isolated from cecal contents rather than cecal tissue. Absolute quantification of E. coli genome copies per ng of total DNA was performed using a standard curve generated from purified E. coli genomic DNA, assuming that the E. coli NC101 genome is 5.02 Mb and contains 7 copies of the 16S rRNA gene.
Histological scoring.
Composite histological inflammation scores of colon tissue were determined as described previously (14).
Immunofluorescence staining.
To detect E. coli in intestinal tissue, paraffin-embedded intestinal sections were rehydrated in a xylene-alcohol gradient, permeabilized with phosphate-buffered saline (PBS)–0.1% Triton X-100 for 10 min at 25°C, blocked with PBS–5% bovine serum albumin–1% Triton X-100, and incubated in a 1:50 dilution of biotinylated rabbit anti-E. coli polyclonal antibody (Fitzgerald Industries) for 1 h at 25°C, followed by a 1:400 dilution of streptavidin-phycoerythrin (BD Biosciences) for 1 h. Three images per tissue section were acquired from randomly chosen areas using an Olympus IX71 fluorescence microscope. In a blinded fashion, apical epithelial cell-associated bacteria per ×20-magnification field were enumerated in each image, and the numbers from six individual intestinal sections (ileum; 2 sections of the cecum; and the proximal, mid-, and distal colon) were summed for each animal.
Colonic tissue fragment and MLN cultures.
Colonic tissue fragment cultures and IL-12/23p40 enzyme-linked immunosorbent assays (ELISAs) were performed as described previously (7, 14). Multiplex immunoassays for gamma interferon (IFN-γ), IL-1β, IL-6, IL-13, IL-17, monocyte chemoattractant protein 1, and tumor necrosis factor alpha (TNF-α) were performed using a Milliplex system (EMD Millipore) on undiluted colon tissue fragment culture supernatants according to the manufacturer's instructions. For mesenteric lymph node (MLN) cultures, single-cell suspensions of MLNs were stimulated with medium or bacterial lysates, after which the IFN-γ in the culture supernatants was measured by ELISA as described previously (14).
T84 cell cultures and transwell experiments.
The human T84 colorectal cancer cell line was provided by Scott Plevy. T84 cells (2.5 × 105) in high-glucose Dulbecco modified Eagle medium containing 10% fetal bovine serum were cultured in the upper chamber of 12-well transwell plates with a 3-μm pore size (Corning Costar) for 10 to 14 days, at which point tight junctions formed and transepithelial electrical resistance was ∼3,500 Ω for 1 day, as measured with an EVOM2 ohmmeter and an Endohm-12 device. Bacteria (8 × 107) were added in the upper chamber and incubated at either 37°C or 4°C. At the indicated times, viable bacteria in the lower chamber were quantified by plating on LB agar. To enumerate epithelial cell-associated bacteria, monolayers were washed 5 times with PBS and lysed with 1% Triton X-100 for 10 min, and dilutions were plated on LB agar. For confocal imaging, monolayers infected for 5 h were fixed in 4% paraformaldehyde, washed, stained with anti-E. coli antibody as described above but without permeabilization, counterstained with DAPI (4′,6-diamidino-2-phenylindole), and visualized on a Zeiss LSM710 confocal microscope.
Gentamicin protection assays.
T84 cells were cultured and infected with E. coli as described above. Monolayers were washed 3 times with PBS, treated with gentamicin (100 μg/ml) at 37°C for 15 min, followed by 4°C for 2 h, and washed 3 times with PBS. Epithelial cells were lysed as described above, and intracellular bacteria were enumerated by plating dilutions on LB agar.
Statistical analysis.
All data are presented as the mean ± standard error of the mean (SEM). P values were calculated using the unpaired, 2-tailed Student's t test.
RESULTS
Expression of gadA and gadB is upregulated in luminal commensal E. coli during intestinal inflammation.
We have previously shown that E. coli stress response genes, including gadAB, are upregulated in cecal bacteria from IL-10−/− mice with colitis monoassociated with E. coli for 7 weeks compared to their expression in healthy WT controls (14). However, whether increased E. coli gadAB expression is the direct result of IL-10 deficiency or colitis is unknown, nor are the kinetics of gadAB expression during the development of colitis. To test this, we monoassociated germfree (GF) WT and IL-10−/− mice for 5 days to 10 weeks with a commensal murine strain of E. coli (NC101) that has previously been shown to induce colitis in IL-10−/− mice (8). Consistent with previous results, we observed a time-dependent increase in histological colonic inflammation in IL-10−/− but not WT mice (Fig. 1A). Similar to the colitis scores, gadAB expression in cecal NC101 bacteria from IL-10−/− mice increased over time and was significantly higher than that in cecal NC101 bacteria from WT mice at later time points, when colitis was the most severe. Surprisingly, we observed higher expression of gadAB in cecal NC101 bacteria from healthy WT mice than IL-10−/− mice at 5 and 14 days (Fig. 1B and C). These data suggest that multiple factors govern gadAB expression in luminal E. coli in a time-dependent fashion. During early phases of colonization, non-inflammation-dependent factors predominate, whereas in later phases, inflammation-related factors are paramount.
Fig 1.
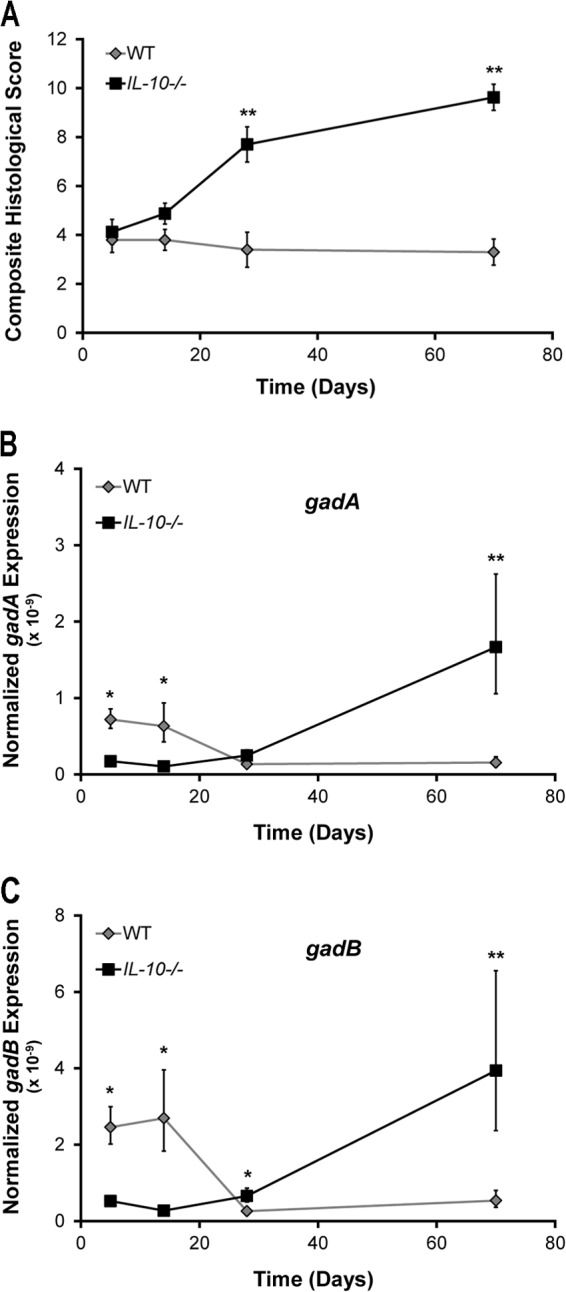
Expression of gadAB is upregulated in luminal E. coli during inflammation. WT and IL-10−/− mice were monoassociated with E. coli NC101 for 5, 14, 35, and 70 days, after which colonic histological inflammation (A) and expression of gadA (B) and gadB (C) normalized to that of the 16S rRNA gene in cecal NC101 were determined. Data are presented as the mean ± SEM (n = 4 to 8 mice per group). *, P < 0.05 relative to WT; **, P < 0.01 relative to WT.
Expression of gadAB enhances susceptibility of E. coli to killing by the host α-defensin cryptdin-4 in vitro and is associated with decreased bacterial survival in vivo.
Based on our hypothesis that E. coli upregulates gadAB as a survival strategy, we predicted that a mutant strain of NC101 that lacks gadAB (NC101ΔgadAB) would be less viable in the inflamed intestine. In contrast, we observed significantly higher concentrations of NC101ΔgadAB bacteria than NC101 bacteria in cecal and colonic luminal contents from both WT and IL-10−/− monoassociated mice (Fig. 2A; see Fig. S1 in the supplemental material). The differences in bacterial viability in the cecum and colon are likely due to the unique environment of these locations, since the growth of NC101ΔgadAB was virtually identical to that of NC101 in vitro (Fig. 2B). The absence of gadAB expression in cecal bacteria from mice monoassociated with NC101ΔgadAB was confirmed by real-time PCR (see Fig. S2 in the supplemental material). These data demonstrate that, contrary to our original hypothesis, expression of gadAB in E. coli impairs luminal bacterial growth in vivo but not in vitro.
Fig 2.
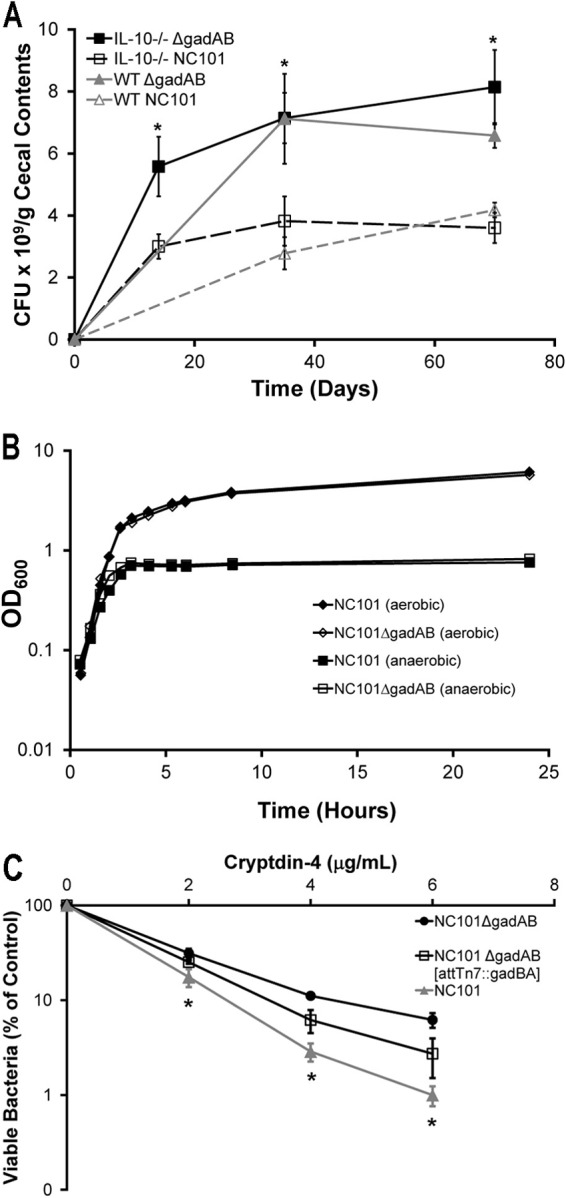
GadAB inhibits luminal E. coli survival and enhances cryptdin-4-mediated killing in vitro. (A) Cecal bacterial concentrations in monoassociated WT and IL-10−/− mice. Data are presented as mean ± SEM (n = 5 mice per group). *, P < 0.05 relative to mice with the same genetic background monoassociated with NC101. (B) NC101 and NC101ΔgadAB growth curves under aerobic or anaerobic conditions. Data are presented as the mean ± SEM (n = 2 separate cultures/strain). OD600, optical density at 600 nm. (C) Killing curves of mid-log-phase NC101, NC101ΔgadAB, and complemented NC101ΔgadAB (NC101ΔgadAB attTn7::gadBA) bacteria treated with recombinant mouse cryptdin-4. Data show the percentage of surviving bacteria relative to the numbers of untreated controls, presented as the mean ± SD of three separate experiments. *, P < 0.05 relative to NC101ΔgadAB.
Host antimicrobial peptides (AMPs), including α-defensins, are secreted into the intestinal lumen, where they limit the growth of certain pathogenic and commensal bacteria. The antimicrobial activity of some AMPs depends in part on a normal proton motive force across the bacterial inner membrane (24–26). Since GadAB help maintain the normal proton gradient in acidic environments (15, 16), we reasoned that expression of gadAB increases the susceptibility of NC101 to killing by AMPs. To test this hypothesis, we treated mid-log-phase NC101 and NC101ΔgadAB with increasing concentrations of the recombinant murine α-defensin cryptdin-4 and found that NC101ΔgadAB was more resistant to cryptdin-4-mediated killing than NC101 at all concentrations of cryptdin-4 tested (Fig. 2C). Complementation of NC101ΔgadAB with chromosomal copies of gadAB partially reversed the phenotype (Fig. 2C). These results indicate that expression of gadAB enhances the susceptibility of NC101 to killing by cryptdin-4 in vitro, but not necessarily in vivo. However, they suggest the possibility that the decreased luminal bacterial concentrations in NC101- versus NC101ΔgadAB-monoassociated mice might be due to differences in AMP susceptibility. Transcription of the cryptdin-4 gene (defa4) and those of other AMPs in ileal tissue from NC101- and NC101ΔgadAB-monoassociated mice was similar, indicating that differences in luminal bacterial concentrations in the two groups of mice are not due to differences in AMP gene expression (see Fig. S3 in the supplemental material).
GadAB in luminal E. coli attenuate the development of experimental colitis.
To determine whether the decreased luminal bacterial concentrations of gadAB-expressing NC101 correlate with decreased colitis, we monoassociated WT and IL-10−/− mice with E. coli NC101 and NC101ΔgadAB for 2, 5, and 10 weeks and quantified colonic inflammation using a validated blinded histological scoring system described previously (8). As expected, IL-10−/− mice colonized with NC101 exhibited increased histological inflammation compared to WT mice at 5 and 10 weeks (Fig. 3A and B). Consistent with the increased luminal concentrations of NC101ΔgadAB compared to those of NC101, histological inflammation scores in NC101ΔgadAB monoassociated IL-10−/− and WT mice were higher than those in NC101-monoassociated mice (Fig. 3A and B).
Fig 3.

The presence of E. coli gadAB is associated with attenuated host proinflammatory immune responses in NC101-monoassociated IL-10−/− and WT mice. (A) Composite blinded histological inflammation scores of colonic segments from IL-10−/− or WT mice monoassociated for the indicated times. (B) Representative photomicrographs of cecum from WT and IL-10−/− mice monoassociated for 5 weeks, as described in panel A. Magnifications, ×20. KO, knockout. (C) Spontaneous secretion of IL-12/23p40 in culture supernatants from proximal colon explants measured by ELISA. (D) Proinflammatory cytokine levels in colon explant cultures from IL-10−/− mice monoassociated with NC101ΔgadAB for 5 weeks relative to those in mice monoassociated with NC101 measured by multiplex immunoassays. MCP-1, monocyte chemoattractant protein 1. (E) IFN-γ secretion by unfractionated MLN cells stimulated ex vivo with E. coli NC101 lysate. Data are presented as the mean ± SEM (n = 5 to 6 mice per group for all panels except panel D, for which 4 mice were used per group). *, P < 0.05 relative to NC101-monoassociated mice of the same genetic background.
The activation of innate immune pathways, including IL-12 and IL-23 secretion, is associated with experimental colitis and human IBDs (8, 27). We therefore tested whether the expression of gadAB in luminal E. coli isolates of monoassociated mice affects the secretion of IL-12/23p40 by colon explants. In accordance with decreased colonic histological inflammation, spontaneous secretion of IL-12/23p40 was lower in colon explants from IL-10−/− mice monoassociated with NC101 for 2 and 5 weeks and WT mice monoassociated with NC101 for 5 and 10 weeks than the corresponding NC101ΔgadAB-monoassociated mice (Fig. 3C). Using multiplex immunoassays, we also detected increased levels of the proinflammatory cytokines IFN-γ, IL-6, IL-17, and TNF-α in cultures of colon explants from IL-10−/− mice monoassociated with NC101ΔgadAB for 5 weeks compared to those in cultures of colon explants from NC101-monoassociated IL-10−/− mice (Fig. 3D).
Because IL-12 and IL-23 promote the clonal expansion of IFN-γ-producing Th1 and IL-17-producing Th17 cells, respectively, both of which play a crucial role in the development of chronic-immune mediated colitis (28), we next aimed to determine whether the increased innate immune responses observed in mice monoassociated with NC101ΔgadAB were associated with enhanced antigen-specific adaptive immune responses. Therefore, we measured IFN-γ secretion by NC101 lysate-stimulated MLN cells isolated from NC101- or NC101ΔgadAB-monoassociated WT and IL-10−/− mice. Consistent with the increased levels of proinflammatory cytokines in cultures of colon explants from NC101ΔgadAB-monoassociated mice, stimulated MLN cells from both WT and IL-10−/− mice colonized with NC101ΔgadAB produced significantly more IFN-γ than MLN cells from mice monoassociated with NC101 (Fig. 3E). MLN cells isolated from NC101- and NC101ΔgadAB-monoassociated mice and stimulated with lysate from the unrelated bacterium Enterococcus faecalis secreted similar and very small amounts of IFN-γ (data not shown).
To confirm that the increased inflammatory responses in NC101ΔgadAB-monoassociated IL-10−/− mice were due to the absence of glutamate decarboxylase function and not an artifact of creating the deletion mutant, we repeated monoassociation studies using a strain of NC101ΔgadAB in which gadB was expressed from a stable episome [NC101ΔgadAB(pgadB)]. Luminal bacterial concentrations, histological scores, and spontaneous IL-12/23p40 secretion by colon explants from IL-10−/− mice monoassociated with NC101ΔgadAB(pgadB) for 2 and 5 weeks were not different from those for NC101-monoassociated IL-10−/− mice (Fig. 4), confirming that glutamate decarboxylase activity in NC101 is responsible for the observed attenuated colitis phenotype. Overall, these data indicate that expression of gadAB in luminal E. coli isolates is associated with reduced colonic inflammation and decreased mucosal innate and adaptive immune responses in monoassociated IL-10−/− mice.
Fig 4.
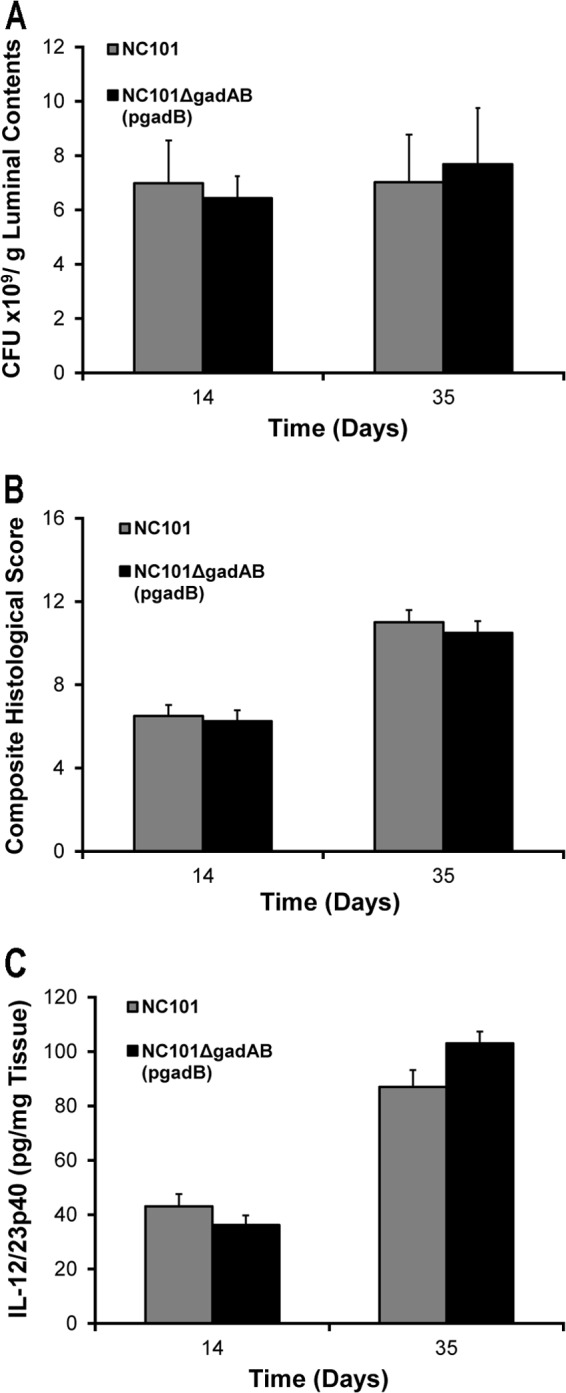
Complementation of NC101ΔgadAB restores the E. coli NC101 phenotype in IL-10−/− monoassociated mice. (A) Luminal bacterial concentrations in the cecum; (B) composite blinded histological inflammation scores of colonic segments; (C) spontaneous secretion of IL-12/23p40 in culture supernatants from proximal colon explants of IL-10−/− mice monoassociated with NC101 or complemented NC101ΔgadAB [NC101ΔgadAB(pgadB)] for the indicated times. Data are presented as the mean ± SEM (n = 6 mice per group).
Since results obtained using monoassociated mice may not accurately represent those observed in mice with a complex intestinal microbiota, we colonized WT and IL-10−/− mice with a mixture of seven commensal strains representative of the normal gut microbiota, together with either E. coli NC101 or NC101ΔgadAB, for 5 and 10 weeks (octoassociation). We found significantly increased expression of gadA and gadB in luminal E. coli isolates from octoassociated IL-10−/− mice at 10 weeks (Fig. 5A and B). Similar to results obtained in monoassociated IL-10−/− mice, luminal E. coli densities, histological inflammation scores, and spontaneous IL-12/23p40 secretion by colon explants from IL-10−/− mice colonized with a complex bacterial mixture that included NC101ΔgadAB were significantly increased compared to those for mice colonized with a complex bacterial mixture that included NC101 at 5 weeks (Fig. 5C to E). These results suggest that expression of gadAB in luminal E. coli NC101 attenuates colitis even in the presence of a complex microbiota.
Fig 5.

Presence of E. coli gadAB is associated with attenuated host proinflammatory immune responses in octoassociated IL-10−/− and WT mice. Expression of gadA (A) and gadB (B) in cecal E. coli from octoassociated mice. (C) Number of E. coli genomes per ng cecal content DNA. (D and E) Composite blinded histological inflammation scores of colonic segments (D) and spontaneous secretion of IL-12/23p40 in culture supernatants from proximal colon explants (E) of WT and IL-10−/− mice octoassociated for 5 weeks with 7 bacteria and either NC101 or NC101ΔgadAB. Data are presented as the mean ± SEM (n = 5 mice per group). *, P < 0.05 relative to WT mice (A and B) or relative to octoassociated and NC101-monoassociated mice of the same genetic background (C to E).
Expression of gadAB decreases translocation of E. coli across intestinal epithelial cells.
Since our initial observations indicated that gadAB expression is associated with reduced colonic inflammation and decreased mucosal immune responses to NC101 and because access to lamina propria antigen-presenting cells is necessary for bacteria to stimulate antigen-specific immune responses, we examined whether decreased colitis in NC101-monoassociated IL-10−/− mice corresponds to a reduced ability of E. coli NC101 to adhere to the intestinal epithelium compared with that for NC101ΔgadAB. We determined the densities of epithelial cell-associated E. coli in intestinal fragments from IL-10−/− mice monoassociated with NC101 and NC101ΔgadAB for 2, 5, and 10 weeks using fluorescence microscopy. Semiquantitative analysis of anti-E. coli antibody-stained intestinal sections revealed significantly increased numbers of luminal bacteria adjacent to the intestinal epithelium in NC101-monoassociated IL-10−/− mice compared to NC101ΔgadAB-monoassociated IL-10−/− mice at 2 weeks but a less pronounced increase at 5 weeks (P = 0.06) (Fig. 6A). To confirm these results, we measured the relative levels of the E. coli 16S rRNA gene copies on extensively washed cecal tissue from monoassociated IL-10−/− mice using quantitative PCR. We detected significantly more E. coli 16S rRNA gene copies on cecal tissue from IL-10−/− mice monoassociated with NC101 than IL-10−/− mice monoassociated with NC101ΔgadAB for 2 and 5 weeks (Fig. 6B). Though the difference was not statistically significant, numerically more E. coli 16S rRNA gene copies and antibody-stained E. coli cells were detected on intestinal tissue from NC101-monoassociated mice than NC101ΔgadAB-monoassociated mice at 10 weeks (Fig. 6A and B). These results indicate that expression of gadAB in NC101 is associated with increased numbers of colonic epithelial cell surface-associated E. coli soon after colonization and are counter to our hypothesis that the attenuated colitis observed in NC101-monoassociated mice compared to that observed in NC101ΔgadAB-monoassociated mice correlates with decreased numbers of epithelial cell-associated bacteria.
Fig 6.
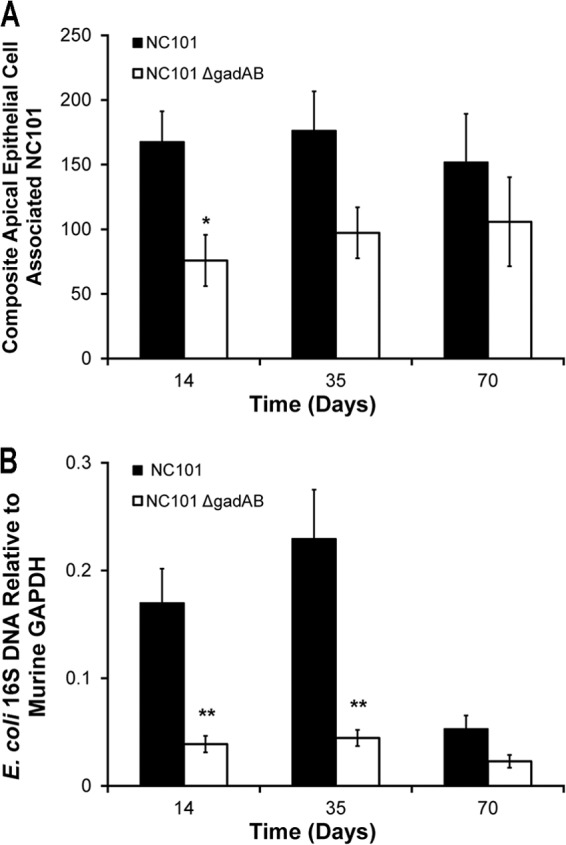
The presence of E. coli gadAB is associated with increased numbers of bacteria at the apical surface of intestinal epithelial cells in vivo. (A) Numbers of apical epithelial cell-associated bacteria in 18 ×20-magnification fields of intestinal tissue per IL-10−/− mouse monoassociated with NC101 or NC101ΔgadAB. (B) Relative numbers of E. coli 16S DNA copies in washed cecal tissue from the same IL-10−/− mice used for the experiment whose results are shown in panel A. Results are normalized to the level of tissue GAPDH expression. Data are presented as the mean ± SEM (n = 5 mice per group). *, P < 0.05 relative to NC101ΔgadAB-monoassociated mice; **, P < 0.01 relative to NC101ΔgadAB-monoassociated mice.
Based on these unexpected results, we postulated that NC101 cells accumulate at the epithelial surface because of decreased bacterial translocation to the lamina propria and therefore cause less colitis than NC101ΔgadAB cells. To test this hypothesis, we infected polarized T84 intestinal epithelial cell monolayers in transwells with NC101 and NC101ΔgadAB under translocation-permissive (37°C) and -nonpermissive (4°C) conditions and determined the numbers of epithelial cell-associated and translocated E. coli cells using culture plating. Similar to the in vivo results, we detected significantly more NC101 bacteria than NC101ΔgadAB bacteria associated with T84 cells after infection at the translocation-permissive temperature of 37°C (Fig. 7A). However, there was no significant difference in the numbers of epithelial cell-associated bacteria between the two strains after infection at the translocation-nonpermissive temperature of 4°C (Fig. 7A). In addition, confocal imaging of infected polarized T84 monolayers in transwells at 37°C also showed higher densities of NC101 bacteria than NC101ΔgadAB bacteria on the epithelial surface (Fig. 7B). These data indicate that NC101ΔgadAB may translocate across epithelial cell monolayers more efficiently than NC101.
Fig 7.

GadAB inhibits translocation of NC101 across intestinal epithelial cells in vitro. (A) Numbers of epithelial cell-associated bacteria in T84 cell monolayers infected at the indicated temperatures. (B) Representative confocal images of T84 intestinal epithelial cells infected for 5 h at 37°C and stained with anti-E. coli antibody (red) and DAPI (blue). (Top) x-y axis; (bottom) z axis. Bar = 20 μm. (C) Numbers of bacteria in the basolateral chamber of monolayers at the indicated times postinfection at 37°C. (D) Intracellular numbers of NC101 and NC101ΔgadAB bacteria in infected intestinal epithelial cell monolayers at 37°C. Data are presented as the mean ± SEM (n = 6 wells per group). *, P < 0.05 relative to NC101; **, P < 0.01 relative to NC101; ***, P < 0.001 relative to NC101.
To confirm that expression of gadAB in NC101 impairs E. coli translocation across epithelial cells in vitro, we directly measured the numbers of NC101 and NC101ΔgadAB bacteria that migrated to the basolateral compartment of polarized T84 cell monolayers at 37°C and found significantly fewer NC101 than NC101ΔgadAB bacteria at all time points investigated (Fig. 7C). Complementation of NC101ΔgadAB with gadAB partially restored the translocation phenotype to that observed for NC101 at 8 and 10 h (see Fig. S4 in the supplemental material). In addition, we detected fewer intracellular bacteria in polarized T84 cells infected with NC101 at 37°C than cells infected with NC101ΔgadAB (Fig. 7D). These data suggest that gadAB expression reduces the ability of NC101 to enter, survive within, and/or exit intestinal epithelial cells in vitro. Together, the results of these in vitro experiments indicate that E. coli gadAB expression inhibits translocation across intestinal epithelial cells, resulting in increased numbers of apical epithelial cell-associated bacteria and may explain how these inflammation-induced bacterial genes attenuate colitis in vivo.
DISCUSSION
While the importance of the commensal enteric microbiota, including E. coli, in the development of IBDs and experimental colitis has previously been established, the mechanisms by which the host inflammatory responses influence the intestinal bacteria are still poorly understood. We reported in a previous study that E. coli bacterial stress response genes, including gadAB, are highly upregulated in IL-10−/− mice with a long-term bacterial monoassociation (14). We show here that the increased expression of gadAB in luminal E. coli NC101 from IL-10−/− mice coincides with advanced histological intestinal inflammation at 10 weeks.
Unexpectedly however, gadAB expression is also elevated in E. coli isolates from WT mice at early time points, suggesting that factors unrelated to histological inflammation also upregulate gadAB. Although the mechanisms involved in the early upregulation of gadAB in WT mice remain to be determined, innate immune responses might be suspected due to the early timing. Alternatively, IL-10 produced by intestinal tissue during early phases of bacterial colonization could directly upregulate E. coli gadAB. However, addition of recombinant IL-10 to E. coli cultures in vitro had no effect on gadAB expression (data not shown). Whatever the cause, it is tempting to speculate that early expression of gadAB in luminal E. coli isolates in WT mice may facilitate intestinal homeostasis, since WT mice monoassociated with NC101ΔgadAB had increased proinflammatory immune responses at later time points (Fig. 3).
The mechanisms responsible for later inflammation-associated expression of gadAB are equally enigmatic. While stationary-phase growth, osmotic shock, norepinephrine, indole, and sodium have all been implicated as potential regulators of gadAB expression (17, 29–31), none of these factors have been reported to be increased in the lumen during colitis. Most of the published data support a role for gadAB in acid tolerance (16). While others have reported decreased luminal pH during colitis (18), we found no significant differences in pH or glutamine or glutamate levels in cecal contents from IL-10−/− mice with colitis versus WT controls (data not shown). Therefore, factors other than low pH and glutamate/glutamine levels are likely stimulating expression of E. coli gadAB in this model system. Interestingly, others have demonstrated that gadB expression is regulated by oxyS, a small regulatory RNA which is important in E. coli stress responses to oxidative environments, such as the oxidative environment found in the inflamed intestine, and which is also upregulated in luminal E. coli isolates from colitic mice (14, 32, 33). Therefore, we speculate that inflammation-associated upregulation of E. coli gadAB may be caused by oxidative stress rather than acidic pH.
Contrary to our initial hypothesis, gadAB expression decreases survival of E. coli NC101 in the inflamed intestine and is associated with reduced innate and adaptive immune responses and attenuated colitis. We provide in vitro evidence that one reason for the decreased luminal concentrations of gadAB-expressing E. coli may be an increased susceptibility to killing by the mouse AMP cryptdin-4. How gadAB expression enhances the susceptibility of E. coli to AMP killing and whether this phenomenon occurs in vivo remain to be determined. However, since gadAB helps maintain a normal proton motive force across the bacterial inner membrane, which may be necessary for cryptdin-4-mediated killing (24–26), we propose that the proton-exporting function of the GadAB system mediates this effect.
Due to the intestinal epithelial barrier that separates potentially proinflammatory microbes from the mucosal immune system, luminal bacterial concentrations do not necessarily correlate with colitis. Translocation of the commensal microbiota or microbial products across the epithelium is required for host immune activation. We show that E. coli GadAB inhibits bacterial translocation in polarized colonic epithelial cell cultures, but the mechanism is unclear. Others have reported that activation of the PhoB regulon is associated with decreased adherence of pathogenic E. coli to intestinal epithelial cells (34). Since PhoB also regulates acid stress responses (35), it is conceivable that GadAB-mediated inhibition of E. coli translocation is also PhoB dependent. However, regulation of gadAB by PhoB has not yet been reported.
We therefore propose a model in which gadAB expression in luminal E. coli increases susceptibility to killing by antimicrobial peptides and decreases migration across the epithelium, which reduces the density of priming microbes in the lamina propria and diminishes host inflammatory responses.
The reason why E. coli would upregulate a gene that ultimately proves to be detrimental to its survival in the colon is still unclear. However, we and others have observed similar findings with different stress response genes (14, 36). It might be more important for commensal E. coli to dampen inflammation triggered by any gut microbe rather than to develop mechanisms to overcome host immune responses. Indeed, others have shown that enterohemorrhagic E. coli suppresses host inflammatory responses to its own toxin (37). In addition, the paradox could be explained by limitations of our experimental system, since selective colonization does not accurately reflect the natural gut environment, where many other commensal microbes are present. These commensals may not only contribute to the inflammatory responses but also potentially interact with E. coli and affect its gene expression. However, we demonstrate that gadAB expression has a detrimental effect on E. coli survival and its colitogenic potential even in the presence of a more complex microbiota (Fig. 5). Moreover, others have also reported that E. coli lacking gadX and gadW genes that regulate expression of gadAB outcompete parental E. coli MG1655 in the streptomycin-treated mouse intestine, which harbors a complex microbiota (38). Interestingly, colitis is not associated with significant upregulation of gadB in cecal E. coli strain LF82 from IL-10−/− mice colonized with a complex defined microbiota relative to its expression in WT controls, suggesting that E. coli transcriptional responses to inflammation are not shared by all E. coli strains (data not shown). Lastly, it is possible that mucosa-associated bacteria that are closer to epithelial and immune cells exhibit gene expression profiles that are different from those of most luminal bacteria. Further studies that dissect the effects of different host cells on bacterial transcription will extend the biological relevance of our findings.
In summary, we have shown that expression of the bacterial stress response genes gadAB is differentially regulated in commensal luminal E. coli isolates from mice with colitis than healthy controls. Furthermore, gadAB expression is associated with decreased luminal bacterial survival and reduced host inflammatory responses in vivo as well as increased sensitivity to the α-defensin cryptdin-4 and an impaired ability to migrate across intestinal epithelial cells. These studies provide evidence that host intestinal inflammation affects commensal microbial function in a manner that reduces inflammation. An enhanced understanding of the mechanisms by which inflammation controls bacterial gene expression and how inflammation-induced bacterial stress responses impact chronic immune-mediated colitis will improve our knowledge of the pathogenesis of IBDs and could lead to the development of novel therapies that target the host-microbe dialogue.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge the Gnotobiotic, Histology, and Immunotechnology Cores at the UNC Center for Gastrointestinal Biology and Disease (supported by NIH grant P30DK34987). This work was funded by NIH grant K08DK087896 (to J.J.H.), a Crohn's and Colitis Foundation of America (CCFA) career development award (to J.J.H.), NIH grant R01DK53347 (to R.B.S.), NIH grant P40OD 010995 (to R.B.S.), and a grant from the CCFA Gnotobiotic Animal Facility (to R.B.S.).
We thank Changsoo Seun for his help with data acquisition.
Footnotes
Published ahead of print 22 July 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.00355-13.
REFERENCES
- 1.Savage DC. 1977. Microbial ecology of the gastrointestinal tract. Annu. Rev. Microbiol. 31:107–133 [DOI] [PubMed] [Google Scholar]
- 2.Ishikawa H, Tanaka K, Maeda Y, Aiba Y, Hata A, Tsuji NM, Koga Y, Matsumoto T. 2008. Effect of intestinal microbiota on the induction of regulatory CD25+ CD4+ T cells. Clin. Exp. Immunol. 153:127–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.O'Mahony C, Scully P, O'Mahony D, Murphy S, O'Brien F, Lyons A, Sherlock G, MacSharry J, Kiely B, Shanahan F, O'Mahony L. 2008. Commensal-induced regulatory T cells mediate protection against pathogen-stimulated NF-kappaB activation. PLoS Pathog. 4:e1000112. 10.1371/journal.ppat.1000112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Darfeuille-Michaud A, Boudeau J, Bulois P, Neut C, Glasser AL, Barnich N, Bringer MA, Swidinski A, Beaugerie L, Colombel JF. 2004. High prevalence of adherent-invasive Escherichia coli associated with ileal mucosa in Crohn's disease. Gastroenterology 127:412–421 [DOI] [PubMed] [Google Scholar]
- 5.Sartor RB. 2008. Microbial influences in inflammatory bowel diseases. Gastroenterology 134:577–594 [DOI] [PubMed] [Google Scholar]
- 6.Hansen JJ, Sartor RB. 2007. Insights from animal models, p 19–55 In The inflammatory bowel disease yearbook, vol 4 Remedica Publishing Ltd, London, United Kingdom [Google Scholar]
- 7.Sellon RK, Tonkonogy S, Schultz M, Dieleman LA, Grenther W, Balish E, Rennick DM, Sartor RB. 1998. Resident enteric bacteria are necessary for development of spontaneous colitis and immune system activation in interleukin-10-deficient mice. Infect. Immun. 66:5224–5231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim SC, Tonkonogy SL, Albright CA, Tsang J, Balish EJ, Braun J, Huycke MM, Sartor RB. 2005. Variable phenotypes of enterocolitis in interleukin 10-deficient mice monoassociated with two different commensal bacteria. Gastroenterology 128:891–906 [DOI] [PubMed] [Google Scholar]
- 9.Mombaerts P, Mizoguchi E, Grusby MJ, Glimcher LH, Bhan AK, Tonegawa S. 1993. Spontaneous development of inflammatory bowel disease in T cell receptor mutant mice. Cell 75:274–282 [DOI] [PubMed] [Google Scholar]
- 10.Rath HC, Herfarth HH, Ikeda JS, Grenther WB, Hamm TE, Jr, Balish E, Taurog JD, Hammer RE, Wilson KH, Sartor RB. 1996. Normal luminal bacteria, especially Bacteroides species, mediate chronic colitis, gastritis, and arthritis in HLA-B27/human beta2 microglobulin transgenic rats. J. Clin. Invest. 98:945–953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Crane JK, Naeher TM, Broome JW, Boedeker EC. 2013. Role of host xanthine oxidase in infection due to enteropathogenic and Shiga-toxigenic Escherichia coli. Infect. Immun. 81:1129–1139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guo L, Lim KB, Gunn JS, Bainbridge B, Darveau RP, Hackett M, Miller SI. 1997. Regulation of lipid A modifications by Salmonella typhimurium virulence genes phoP-phoQ. Science 276:250–253 [DOI] [PubMed] [Google Scholar]
- 13.Winter SE, Thiennimitr P, Winter MG, Butler BP, Huseby DL, Crawford RW, Russell JM, Bevins CL, Adams LG, Tsolis RM, Roth JR, Bäumler AJ. 2010. Gut inflammation provides a respiratory electron acceptor for Salmonella. Nature 467:426–429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Patwa LG, Fan TJ, Tchaptchet S, Liu Y, Lussier YA, Sartor RB, Hansen JJ. 2011. Chronic intestinal inflammation induces stress-response genes in commensal Escherichia coli. Gastroenterology 141:1842–1851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Castanie-Cornet MP, Penfound TA, Smith D, Elliott JF, Foster JW. 1999. Control of acid resistance in Escherichia coli. J. Bacteriol. 181:3525–3535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Foster JW. 2004. Escherichia coli acid resistance: tales of an amateur acidophile. Nat. Rev. Microbiol. 2:898–907 [DOI] [PubMed] [Google Scholar]
- 17.De Biase D, Tramonti A, Bossa F, Visca P. 1999. The response to stationary-phase stress conditions in Escherichia coli: role and regulation of the glutamic acid decarboxylase system. Mol. Microbiol. 32:1198–1211 [DOI] [PubMed] [Google Scholar]
- 18.Fallingborg J, Christensen LA, Jacobsen BA, Rasmussen SN. 1993. Very low intraluminal colonic pH in patients with active ulcerative colitis. Dig. Dis. Sci. 38:1989–1993 [DOI] [PubMed] [Google Scholar]
- 19.Treem WR, Ahsan N, Shoup M, Hyams JS. 1994. Fecal short-chain fatty acids in children with inflammatory bowel disease. J. Pediatr. Gastroenterol. Nutr. 18:159–164 [DOI] [PubMed] [Google Scholar]
- 20.Vernia P, Caprilli R, Latella G, Barbetti F, Magliocca FM, Cittadini M. 1988. Fecal lactate and ulcerative colitis. Gastroenterology 95:1564–1568 [DOI] [PubMed] [Google Scholar]
- 21.Moreau PL. 2007. The lysine decarboxylase CadA protects Escherichia coli starved of phosphate against fermentation acids. J. Bacteriol. 189:2249–2261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U. S. A. 97:6640–6645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rey FE, Faith JJ, Bain J, Muehlbauer MJ, Stevens RD, Newgard CB, Gordon JI. 2010. Dissecting the in vivo metabolic potential of two human gut acetogens. J. Biol. Chem. 285:22082–22090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bonnet M, Rafi MM, Chikindas ML, Montville TJ. 2006. Bioenergetic mechanism for nisin resistance, induced by the acid tolerance response of Listeria monocytogenes. Appl. Environ. Microbiol. 72:2556–2563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kagan BL, Selsted ME, Ganz T, Lehrer RI. 1990. Antimicrobial defensin peptides form voltage-dependent ion-permeable channels in planar lipid bilayer membranes. Proc. Natl. Acad. Sci. U. S. A. 87:210–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lehrer RI, Barton A, Daher KA, Harwig SS, Ganz T, Selsted ME. 1989. Interaction of human defensins with Escherichia coli. Mechanism of bactericidal activity. J. Clin. Invest. 84:553–561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Duerr RH, Taylor KD, Brant SR, Rioux JD, Silverberg MS, Daly MJ, Steinhart AH, Abraham C, Regueiro M, Griffiths A, Dassopoulos T, Bitton A, Yang H, Targan S, Datta LW, Kistner EO, Schumm LP, Lee AT, Gregersen PK, Barmada MM, Rotter JI, Nicolae DL, Cho JH. 2006. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science 314:1461–1463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu B, Tonkonogy SL, Sartor RB. 2011. Antigen-presenting cell production of IL-10 inhibits T-helper 1 and 17 cell responses and suppresses colitis in mice. Gastroenterology 141:653–662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dowd SE. 2007. Escherichia coli O157:H7 gene expression in the presence of catecholamine norepinephrine. FEMS Microbiol. Lett. 273:214–223 [DOI] [PubMed] [Google Scholar]
- 30.Hirakawa H, Hayashi-Nishino M, Yamaguchi A, Nishino K. 2010. Indole enhances acid resistance in Escherichia coli. Microb. Pathog. 49:90–94 [DOI] [PubMed] [Google Scholar]
- 31.Richard H, Foster JW. 2007. Sodium regulates Escherichia coli acid resistance, and influences GadX- and GadW-dependent activation of gadE. Microbiology 153:3154–3161 [DOI] [PubMed] [Google Scholar]
- 32.Altuvia S, Weinstein-Fischer D, Zhang A, Postow L, Storz G. 1997. A small, stable RNA induced by oxidative stress: role as a pleiotropic regulator and antimutator. Cell 90:43–53 [DOI] [PubMed] [Google Scholar]
- 33.Keshavarzian A, Sedghi S, Kanofsky J, List T, Robinson C, Ibrahim C, Winship D. 1992. Excessive production of reactive oxygen metabolites by inflamed colon: analysis by chemiluminescence probe. Gastroenterology 103:177–185 [DOI] [PubMed] [Google Scholar]
- 34.Cheng C, Tennant SM, Azzopardi KI, Bennett-Wood V, Hartland EL, Robins-Browne RM, Tauschek M. 2009. Contribution of the pst-phoU operon to cell adherence by atypical enteropathogenic Escherichia coli and virulence of Citrobacter rodentium. Infect. Immun. 77:1936–1944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lamarche MG, Wanner BL, Crepin S, Harel J. 2008. The phosphate regulon and bacterial virulence: a regulatory network connecting phosphate homeostasis and pathogenesis. FEMS Microbiol. Rev. 32:461–473 [DOI] [PubMed] [Google Scholar]
- 36.Krogfelt KA, Hjulgaard M, Sorensen K, Cohen PS, Givskov M. 2000. rpoS gene function is a disadvantage for Escherichia coli BJ4 during competitive colonization of the mouse large intestine. Infect. Immun. 68:2518–2524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bellmeyer A, Cotton C, Kanteti R, Koutsouris A, Viswanathan VK, Hecht G. 2009. Enterohemorrhagic Escherichia coli suppresses inflammatory response to cytokines and its own toxin. Am. J. Physiol. Gastrointest. Liver Physiol. 297:G576–G581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tucker DL, Tucker N, Ma Z, Foster JW, Miranda RL, Cohen PS, Conway T. 2003. Genes of the GadX-GadW regulon in Escherichia coli. J. Bacteriol. 185:3190–3201 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.