

Abstract
A vaccine candidate that elicits humoral and cellular responses to multiple sporozoite and liver-stage antigens may be able to confer protection against Plasmodium falciparum malaria; however, a technology for formulating and delivering such a vaccine has remained elusive. Here, we report the preclinical assessment of an optimized DNA vaccine approach that targets four P. falciparum antigens: circumsporozoite protein (CSP), liver stage antigen 1 (LSA1), thrombospondin-related anonymous protein (TRAP), and cell-traversal protein for ookinetes and sporozoites (CelTOS). Synthetic DNA sequences were designed for each antigen with modifications to improve expression and were delivered using in vivo electroporation (EP). Immunogenicity was evaluated in mice and nonhuman primates (NHPs) and assessed by enzyme-linked immunosorbent assay (ELISA), gamma interferon (IFN-γ) enzyme-linked immunosorbent spot (ELISpot) assay, and flow cytometry. In mice, DNA with EP delivery induced antigen-specific IFN-γ production, as measured by ELISpot assay and IgG seroconversion against all antigens. Sustained production of IFN-γ, interleukin-2, and tumor necrosis factor alpha was elicited in both the CD4+ and CD8+ T cell compartments. Furthermore, hepatic CD8+ lymphocytes produced LSA1-specific IFN-γ. The immune responses conferred to mice by this approach translated to the NHP model, which showed cellular responses by ELISpot assay and intracellular cytokine staining. Notably, antigen-specific CD8+ granzyme B+ T cells were observed in NHPs. Collectively, the data demonstrate that delivery of gene sequences by DNA/EP encoding malaria parasite antigens is immunogenic in animal models and can harness both the humoral and cellular arms of the immune system.
INTRODUCTION
Malaria is a mosquito-borne disease caused by Plasmodium parasites that poses a significant global health burden. The World Health Organization estimated that in 2010 there were approximately 216 million cases of malaria and 655,000 deaths due to malaria parasite infection, the majority of which are in young children in Africa (1). There are multiple species of Plasmodium but only five that can cause malaria in humans. Of these five, Plasmodium falciparum is the predominant pathogenic species for severe disease and death. Preventive measures and treatment options can reduce the risk and severity of infection. However, the increasing resistance to antimalarial drugs by Plasmodium species further complicates successful treatment of malaria. Thus, the development of a vaccine to prevent malaria infection and subsequent clinical disease remains an important global goal.
The form of the Plasmodium parasite that is transmitted to humans, the sporozoite, is delivered to the skin by the bite of an infected female Anopheles mosquito. The sporozoites that do not remain in the skin can enter the bloodstream and migrate to the liver. In the liver, they invade hepatocytes, undergo replication, and are released as merozoites that then invade red blood cells (RBCs). Many current malaria vaccine strategies target sporozoite and/or liver stages (preerythrocytic stage [PE]) of infection in an effort to prevent progression to the blood stages, which are associated with the clinical manifestation of the disease and continued transmission.
High levels of protection from parasite infection in humans has been achieved through repeated bites of P. falciparum-infected mosquitoes while undergoing a prophylactic regimen of chloroquine (2, 3) and by vaccination with RTS,S, an adjuvanted recombinant protein. RTS,S targets the circumsporozoite protein (CSP). In phase III RTS,S clinical trials, 1-year follow-up data from children who were 5 to 17 months old at first vaccination reported 55.8% efficacy (97.5% confidence interval [CI], 50.6 to 60.4) against clinical malaria and 47.3% efficacy (95% CI, 22.4 to 64.2) against severe malaria incidence compared to the control group (4). Sporozoite-based approaches have provided a gold standard for malaria vaccine development, because they confer protection in animal models and humans and have served as an important tool to gain insight into the immune mechanisms underlying protection (3). Unfortunately, widespread use of attenuated and irradiated sporozoites may be hindered by practical and logistical considerations. For example, a recent clinical trial investigated the protective efficacy of cryopreserved, irradiated sporozoites delivered via intradermal or subcutaneous injection (5). This approach demonstrated less than 5% efficacy in malaria naive adults.
In general, targeting multiple antigens to broaden cellular and humoral anti-P. falciparum immune responses appears to be important in the development of malaria vaccines (6). Protection conferred by sporozoite-based approaches is thought to be primarily T-cell mediated and dependent on multiple proteins expressed during the early stages of invasion of the liver (7, 8). Naturally acquired immunity, which residents of areas in which malaria is endemic acquire after repeated infection, has been associated with antibodies to several different P. falciparum proteins (9). Thus, there is evidence that both arms of the immune system contribute to protection. DNA vaccines are an attractive approach for targeting multiple antigens in a single formulation and can generate both humoral and cellular responses, including cytotoxic T lymphocytes (CTLs) (10, 11). DNA vaccines offer several significant advantages over viral vector-based vaccines, including long-term stability, the potential for fewer cold chain requirements than conventional vaccines (10, 12), and no concern for vector serology inhibiting immune boosting with subsequent applications of the same vaccine. Early DNA-based vaccine studies failed to elicit reliable or robust immune responses in humans (13, 14) but were safe and well tolerated. Since these early studies, significant technological advancement has been made to enhance the immune potency of the DNA platform (15). Among these advancements are improved physical methods of delivery, such as in vivo electroporation (EP), which increases the uptake of the vaccine plasmids by cells, and optimization of vaccine vectors and encoded antigens. In addition to augmenting DNA vaccine immunogenicity in multiple animal models, including nonhuman primates (NHPs) (16–18), delivery of DNA plasmids with EP has also been employed in clinical trials (11).
Here, we describe immune responses induced by an optimized DNA-based multiple immunogen approach, delivered by EP, targeting four P. falciparum PE antigens: CSP, thrombospondin-related anonymous protein (TRAP), cell-traversal protein for ookinetes and sporozoites (CelTOS), and liver-stage antigen 1 (LSA1). Of the four antigens incorporated into the multivalent vaccine approach described here, CSP has been the most extensively studied in the clinic as the antigen targeted by RTS,S (19). TRAP and LSA1 have also been evaluated in clinical trials. The viral-vectored ME-TRAP, a string of multiple PE antigen epitopes fused to the full-length TRAP antigen, demonstrated partial protection in efficacy trials (20, 21), while an adjuvanted recombinant LSA1 vaccine (FMP-011) did not protect against P. falciparum challenge (22). However, the latter did not drive CD8+ T cell responses. CelTOS, which is currently undergoing evaluation in the clinic (NCT01540474), has shown promise in preclinical studies. In mice, an adjuvanted P. falciparum CelTOS recombinant protein vaccine protected animals challenged with Plasmodium berghei (23).
In this study, the constructs conferred antigen-specific IgG seroconversion when delivered individually or as multiantigen cocktails. Further, all antigens induced cellular immune responses, as evidenced by production of antigen-specific gamma interferon (IFN-γ) by enzyme-linked immunosorbent spot (ELISpot) assay and IFN-γ, tumor necrosis factor alpha (TNF-α), and interleukin-2 (IL-2) production in both the CD4+ and CD8+ T cell compartments by flow cytometry. In addition, we demonstrate that immunization in the periphery drives antigen-specific IFN-γ production by liver-resident CD8+ T cells in mice. The immunogenicity of this approach was also evaluated in rhesus macaques. In NHPs, the multivalent immunogens elicited high antibody titers and cellular immune responses in both the CD4+ and CD8+ T cell compartments. Furthermore, a large portion of antigen-specific CD8+ T cells also produced granzyme B (GrzB), indicating the potential of this population to act as CTLs. Collectively, the data show that this approach is highly immunogenic in animal models and can harness both the humoral and cellular arms of the immune system. Further study of these immunogens in future malaria vaccine studies may be informative.
MATERIALS AND METHODS
Plasmids.
TRAP and CSP consensus immunogens were designed from the available full-length sequences in the GenBank database with previously described modifications (24). The LSA1 antigen was designed based on the P. falciparum NF54 strain (accession number CAA39663) (http://www.ncbi.nlm.nih.gov/protein) and contains 8 of the 86.5 central repeats. The P. falciparum 3D7 sequence was used for the CelTOS antigen (accession number BAD97684.1) (http://www.ncbi.nlm.nih.gov/protein). A human influenza virus hemagglutinin (HA) tag was included at the C terminus of all antigen sequences for detection of expression. The antigen sequences were optimized for mRNA stability and codon usage in humans. All gene sequences were synthesized by GenScript and were subcloned into the BamHI and XhoI sites of a modified pVAX1 vector.
Western blotting.
RD cells (human muscle) (CCL-136; ATCC) were transfected with the pVAX1 (negative control), CSP, LSA1, TRAP, or CelTOS plasmid using Lipofectamine (Invitrogen) according to the manufacturer's instructions. SDS-PAGE of whole-cell lysate was completed, and the antigens were visualized with an anti-HA polyclonal antibody (Covance) followed by anti-rabbit horseradish peroxidase (HRP)-conjugated IgG (Cell Signaling). Bands were visualized using ECL Western blotting substrate (Pierce).
Immunofluorescence.
RD cells were transfected with 1 μg of the plasmid DNA (pDNA) constructs using TurboFect (Thermo Scientific) according to the manufacturer's instructions. The cells were fixed with 2% paraformaldehyde 48 h after transfection and then permeabilized. Antigen expression was detected with a polyclonal rabbit anti-HA antibody (Invitrogen) and DyLight-488-labeled anti-rabbit conjugated secondary antibody (Thermo Scientific). 4′,6-Diamidino-2-phenylindole (DAPI) staining was used to visualize the nucleus.
Animals. (i) Mice.
Female 4- to 6-week-old BALB/c mice were purchased from Jackson Laboratories. All animals were housed in a temperature-controlled, light-cycled facility at the University of Pennsylvania. Animal care was carried out according to the guidelines of the National Institutes of Health and the University of Pennsylvania Institutional Care and Use Committee.
(ii) Nonhuman primates.
Five Indian-origin rhesus macaques were housed at BIOQUAL (Rockville, MD) according to the standards of the Institutional Animal Care and Use Committee and the American Association for Accreditation of Laboratory Animal Care.
Immunizations. (i) Mice.
Mice were immunized with 30 μg plasmid (120 μg total DNA for the multiantigen formulation) by intramuscular (i.m.) injection into the tibialis anterior muscle followed by in vivo electroporation using the CELLECTRA adaptive constant current electroporation device (Inovio Pharmaceuticals). Square-wave pulses at a 0.1-A constant current were delivered as previously described (25). Each pulse was 52 ms in length with a 1-s delay between pulses. The mice received a total of 3 immunizations that were administered 3 weeks apart.
(ii) NHPs.
A 1.0-mg volume of each plasmid (4.0 mg DNA total) DNA was delivered i.m. to a single site in the quadriceps, followed by in vivo electroporation with the CELLECTRA adaptive constant current electroporation device. Three square-wave pulses at a 0.5-A constant current were delivered as previously described (17). Each pulse was 52 ms in length with a 1-s delay between pulses.
Immunohistochemistry.
Proteins were overexpressed in the liver by hydrodynamic tail vein injection of 50 μg of each plasmid (200 μg total DNA) in 2.0 ml PBS. Mice were sacrificed 24 h postinjection, livers were collected and fixed in 10% buffered formalin solution for 24 h, and the samples were stored in 70% ethanol prior to embedding, sectioning, and staining with sera (1:500) from mice immunized with the individual pDNAs.
ELISA.
Enzyme-linked immunosorbent assay (ELISA) was used to determine antigen-specific serum IgG titers. Ninety-six-well Nunc-Immuno MaxiSorp plates (Fisher Scientific) were coated overnight at 4°C with 1 to 5 μg/ml of protein. Full-length P. falciparum CSP was kindly provided by the PATH Malaria Vaccine Initiative (Washington, DC) and Gennova Biopharmaceuticals Ltd. (Pune, India). Full-length LSA1 was kindly provided by David Lanar (WRAIR, Silver Spring, MD). The TRAP and CelTOS proteins were synthesized by GenScript using baculovirus and bacterial systems, respectively. Plates were washed with PBS, 0.05% Tween 20 (PBST), blocked for 1 h at room temperature with 10% bovine serum albumin (BSA)-PBST, and incubated with serial dilutions of serum, which were 3-fold dilutions beginning with 1:50, from immunized or naive animals for 1 h at room temperature. Plates were then washed 3 times with PBST. For mouse assays, HRP-conjugated goat anti-mouse IgG (Santa Cruz Biotechnologies) was added at a dilution of 1:5,000 in PBST. For monkey assays, HRP-conjugated mouse anti-monkey IgG (Southern Biotech) was added at a 1:5,000 dilution in PBST. Bound enzyme was detected by SigmaFAST O-phenylenediamine dihydrochloride (OPD; Sigma-Aldrich), and the optical density was determined at 450 nm (OD450). Titers are reported as the reciprocal dilution at which the OD450 was 1.0. A nonlinear regression sigmoidal dose-response curve was used to estimate the values where OD was 1.0 (Prism 5; GraphPad Software). Any detectable background levels at baseline (week 0) were subtracted from IgG levels detected after immunizations.
IFA.
The sporozoite slides were prepared from NF54 mosquitoes. The mosquitoes were dissected by cutting the thorax at the scutum. The heads and the anterior portions of the scutum were kept, while the remaining portions of the thorax and abdomen were discarded. The sporozoites were isolated using Osaki tubes and then purified using a DEAE column (Whatman). Sporozoites were counted using a hemocytometer, and a sporozoite suspension of 5,000 sporozoites/well was added to each well. Slides were dried overnight and then wrapped in aluminum foil and placed in a desiccator at −20°C. For the immunofluorescence assay (IFA), the negative control was obtained from a pool of normal rhesus sera and was diluted to 1:50 in PBS plus 1% BSA (Fisher Scientific). The positive control was obtained from a subject immunized with R32NS181 and also diluted to 1:50 in PBS plus 1% BSA. Slides were thawed for 20 min at room temperature and then blocked with 16 μl of PBS plus 1% BSA. Fifteen μl of each sample then was added to each well and incubated for 1 h in a humidity chamber at room temperature. The slides were washed 3 times for 2 min each in 1× PBS. Sixteen μl of the secondary antibody, goat anti-rhesus IgG (H+L) fluorescein isothiocyanate (FITC; SouthernBiotech) diluted to 1:2,000 in PBS plus 1% BSA, was added to each well. The slides were incubated for 1 h at room temperature in the dark and then were washed 3 times for 2 min in 1× PBS. Finally, 1 μl of Vectashield with DAPI (Vector laboratories) was added to each well, and the slide was covered with a coverslip. The slide was covered with aluminum foil until read. Slides were read using an Olympus Provis UV microscope with a 40× objective, exposure time of 1/3.5 s, an isotropic signal of 400, and a resolution of 1,360 by 1,024. The well was first scanned, and then a representative photograph was taken. Images then were analyzed using Image-Pro Plus (Media Cybernetics) utilizing a software macro designed such that sporozoites were identified based on size and dimensions while ignoring luminous artifacts. Sporozoites were manually selected until all sporozoites were analyzed (a minimum of three). The luminosity for each parasite was automatically averaged and reported, maintaining a percent coefficient of variance of less than 20%. Titer cutoffs were determined based on the last dilution that gave a positive luminosity, defined as the mean luminosity 2 standard deviations above the mean of that of parasites incubated with the negative control.
Lymphocyte isolation. (i) Splenocyte isolation.
Splenocytes were aseptically isolated and placed in 5 ml of R10 media (RPMI medium 1640 supplemented with 10% fetal bovine serum, 1% antibiotic-antimycotic, and 0.1% 2-mercaptoethanol). Splenocytes were isolated by mechanical disruption of the spleen using a Stomacher machine (Seward Laboratory Systems Inc.), and the resulting product was filtered using a 40-μm cell strainer (BD Falcon). The resulting product was centrifuged and the pellet was treated for 5 min with ACK lysis buffer (Lonza) for lysis of RBCs. The splenocytes were centrifuged, washed in PBS, and then resuspended in R10 media.
(ii) Hepatic lymphocyte isolation.
The livers of mice were perfused in vivo with 5 ml cold PBS through the right ventricle of the heart. The portal vein was then cut and the liver perfused again with 5 ml cold PBS. The gallbladder was removed and discarded. The liver was then removed and placed into 5 ml of R10 media. The lymphocytes were isolated by mechanical disruption using a Stomacher machine (Seward Laboratory Systems Inc.), and the resulting product was filtered using a 60-μm mesh screen (Sigma-Aldrich). Lymphocytes were resuspended in 8 ml of 40% Percoll (GE Healthcare) containing 100 U/ml heparin (Fisher) and layered onto 3 ml 70% Percoll. The lymphocyte layer was isolated after centrifugation, washed in PBS, and treated with ACK lysis buffer for 2 to 3 min. The lymphocytes were then centrifuged, washed in PBS, and resuspended in R10 media.
(iii) PBMC isolation.
Twenty ml of blood was collected in K2-EDTA tubes at the indicated time points. Peripheral blood mononuclear cells (PBMCs) were isolated using standard Ficoll-Hypaque (GE Healthcare) procedures with Accuspin tubes (Sigma-Aldrich). PBMCs were treated with ACK buffer for 2 to 3 min, washed in PBS, and resuspended in R10 media.
IFN-γ ELISpot assay.
Splenocytes (mice) or PBMCs (NHPs) were used at a concentration of 2 × 105 cells/well. Antigen-specific peptides were 15-mer peptides spanning the entire length of the consensus immunogen, not including the HA tag or leader sequence, overlapping by 11 amino acids and were synthesized by GenScript. The final concentration of each peptide used in the ELISpot assays was approximately 2.5 μg/ml. ELISpot data are expressed in spot-forming units (SFU), which is the number of antigen-specific T cells producing IFN-γ per 106 splenocytes (mice) or 106 PBMCs (NHPs).
(i) Mice.
Mouse IFN-γ capture antibody (R&D Systems) was used to coat flat-bottom Immobilon-P plates (Millipore) overnight at 4°C. Splenocytes were stimulated overnight (approximately 18 h) at 37°C, 5% CO2, in the presence of R10 (negative control), concanavalin A (positive control) (Sigma-Aldrich), or antigen-specific peptide pools. Plates were then washed in PBS, and mouse IFN-γ detection antibody (R&D Systems) was added to the plates overnight at 4°C. Streptavidin-alkaline phosphatase (ALP) (MabTech) was added to the plates for 2 h, and antigen-specific spots were visualized with 5-bromo-4-chloro-3-indolylphosphate-nitroblue tetrazolium (BCIP-NBT) substrate (MabTech).
NHPs.
IFN-γ ELISpot assay was carried out with the ELISpotPRO for monkey IFN-γ (MabTech) according to the manufacturer's instructions. PBMCs were stimulated overnight (approximately 18 h) at 37°C, 5% CO2 in the presence of R10 (negative control), anti-CD3 monoclonal antibody (positive control), or antigen-specific peptide pools. Plates were washed and spots detected using ALP-conjugated antibody and BCIP-NBT substrate.
Intracellular cytokine staining.
Intracellular cytokine staining was completed using the CytoFix/CytoPerm kit per the manufacturer's instructions (BD Biosciences). Mouse splenocytes, mouse hepatic lymphocytes, or NHP PBMCs were washed with PBS and resuspended in R10 and counted. Cells (1 × 106 to 2 × 106) were seeded in 96-well round-bottom plates in a volume of R10 media (negative control), media containing antigen-specific peptides pools at the same concentration used for ELISpot assay, or media containing 10 ng/ml phorbol myristate acetate (Sigma-Aldrich). Ionomycin (250 ng/ml; Sigma-Aldrich) (positive control) was added, and plates were incubated at 37°C, 5% CO2 for 6 h (mice) or overnight (8 to 10 h; NHPs). All stimulation media contained 1 μg/μl each of GolgiPlug (BD Biosciences) and GolgiStop (BD Biosciences). At the end of the incubation period, plates were spun down and washed twice with PBS. Cells were stained as described below, given a final wash with PBS, and resuspended in 1% paraformaldehyde (Tousimis). Cells were analyzed on a modified BD LSR II, and data were analyzed with FlowJo 9.2.5 (Tree Star, Inc.). For the analysis of antigen-specific responses, CD4+ CD8− and CD8+ CD4− cells were identified within the live CD3+ population.
Mice.
Cells were stained externally with a violet dye for viability (LIVE/DEAD violet viability dye; Invitrogen), CD4-peridinin chlorophyll protein (PerCP)-Cy5.5 (BD Biosciences), and CD8-antigen-presenting cells (APC) (BD Biosciences) at 4°C for 30 min, washed twice in PBS, and then fixed and permeabilized for 30 min at 4°C in Cytofix/Cytoperm solution. Cells were washed twice in Perm/Wash buffer and then stained intracellularly in Perm/Wash buffer with the following antibodies: CD3-phycoerythrin (PE)-Cy5 (BD Biosciences), IL-2-PE (BD Biosciences), IFN-γ-Alexa Fluor-700 (BD Biosciences), and TNF-α-FITC (BD Biosciences) for 30 min at 4°C.
NHPs.
Cells were stained externally with a violet dye for viability (LIVE/DEAD violet viability dye; Invitrogen) for 5 min at room temperature and surface stained for 30 min at room temperature with the following antibodies: CD4-PE-Cy5.5 (Biolegend), CD14-Pacific Blue (BD Biosciences), CD16-Pacific Blue (BD Biosciences), CD45RA-Q605 (Invitrogen), and CD28-PE-Cy5 (Beckman Coulter). Cells were washed twice with PBS, resuspended in Cytofix/Cytoperm solution, and fixed for 15 min at room temperature. Cells were washed twice in Perm/Wash buffer and then stained intracellularly in Perm/Wash buffer for 1 h at room temperature with CD3-APC-Cy7 (BD Biosciences), IFN-γ-PE-Cy7 (BD Biosciences), IL-2-PE (BD Biosciences), TNF-α-Alexa Fluor-700 (BD Biosciences), and granzyme B-PE-Texas Red (Invitrogen).
Statistics.
Statistical analysis was performed using PASWS Statistics 18 (IBM Corporation). Analysis between mouse groups was performed using a Student's t test or analysis of variance (ANOVA) as appropriate. Analyses of NHP data were carried out using a related-samples Wilcoxon signed rank test. All data are expressed as the means ± standard errors of the means (SEM) unless otherwise noted. A P value of less than 0.05 was considered statistically significant.
RESULTS
Design of plasmids and in vitro antigen expression.
The CSP and TRAP consensus immunogens were designed as previously described (24), with modifications based on all available full-length sequences in the GenBank database (67 and 28 sequences, respectively). Because CelTOS is highly conserved among Plasmodium species (23, 26), the sequence for the 3D7 strain of P. falciparum was used as the antigen sequence. The LSA1 antigen was designed based on the P. falciparum NF54 strain and contains 8 of the 86.5 central repeats. Synthetic engineering of the antigen-coding sequences also allowed for the inclusion of other optimization approaches, such as incorporation of a highly efficient leader sequence, improved mRNA stability, and codon optimization in the context of a highly efficient promoter. Together, these enhance transcription and translation, thereby improving expression for greater antigen production (27, 28). The antigens were inserted into a modified pVAX1 backbone using the BamHI and XhoI restriction enzyme sites. Prior to in vivo immunogenicity studies, Western blotting and immunofluorescence were used to confirm expression of the antigens by the vectors. Protein bands of the expected molecular sizes were detected by Western blotting (Fig. 1A), and all antigens were visualized by immunofluorescence (Fig. 1B).
Fig 1.
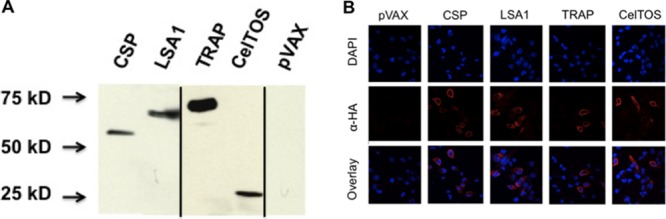
In vitro expression of antigens. Antigen constructs were transfected into RD cells to confirm expression of the antigen proteins. (A) Visualization of the antigen proteins by Western blotting. Bands of the expected protein sizes were detected by Western blotting: CSP, 43 kDa; LSA1, 66 kDa; TRAP, 65 kDa; and CelTOS, 24 kDa. Expression of CSP and LSA1 was analyzed on a separate blot from expression of LSA1 and CelTOS. Vertical black lines indicate splicing of gel images. (B) All antigen proteins detected via immunofluorescence by probing for the C-terminal HA tag.
Humoral immunogenicity in mice.
To evaluate if these antigens were eliciting humoral responses, mice were immunized with the individual CSP, LSA1, TRAP, or CelTOS pDNA (n = 5/group) or the multiantigen formulation containing all 4 constructs (MAV4). To formulate MAV4, the same dose of each plasmid used to evaluate immune responses to the individual antigens was combined and delivered in a single injection site. Antibody levels in the sera were evaluated both 1 and 10 weeks after the last immunization. CSP and TRAP IgG levels (Fig. 2B and D) were not significantly different when the pDNAs were given individually compared to combined delivery. Combined delivery did decrease LSA1- and CelTOS-specific IgG levels (P < 0.05) (Fig. 2C and E). Antibody responses were still detectable 10 weeks after mice received the last immunization with MAV4 but were lower than the levels found 1 week after the 3rd immunization (P < 0.05 for all antigens) (Fig. 2F).
Fig 2.

Humoral responses in mice. Antibody titers were measured by ELISA 1 week following the 3rd immunization and are expressed as the reciprocal dilution of the titer where the OD was 1.0. (A) Titration curves for the individual vaccines. (B) CSP, (C) LSA1, (D) TRAP, and (E) CelTOS IgG endpoint titers induced by the immunogens alone or combined in a single dose 1 week following the last immunization (n = 5/group) are shown. (F) Memory antibody responses induced by the multiple immunogen vaccine 10 weeks following the 3rd immunization. (G) Antigen proteins were overexpressed in the liver by a single hydrodynamic tail vein injection. Livers were harvested and sections stained with sera (1:500) from mice vaccinated with the individual immunogens. Images are representative of 2 independent experiments. *, P < 0.05.
The data shown in Fig. 2A to F demonstrate that the antibodies elicited by these immunogens bind to linear epitopes. It was next examined if the antibodies can also recognize contextual antigen in vivo. The proteins were overexpressed in the liver of mice via a single hydrodynamic tail vein injection containing all four antigen constructs. The antigen proteins were detected by immunohistochemistry (Fig. 2G), and all antibodies effectively recognized their respective antigens in the liver.
Cellular immunogenicity in mice.
Cellular immune responses play a pivotal role in conferring protection from P. falciparum infection at the PE stage (29–33). Prior to combining the CSP, LSA1, TRAP, and CelTOS plasmids into the single-dose MAV4 formulation, cellular immunogenicity induced by the individual pDNAs was assessed by IFN-γ ELISpot assay (Fig. 3A). Responses were evaluated 1 week after the last immunization and demonstrated that the constructs elicited antigen-specific cellular responses in the spleen. Specifically, average numbers were the following: CSP, 1,607 SFU; LSA1, 1,908 SFU; TRAP, 929 SFU; and CelTOS, 477 SFU.
Fig 3.

Antigen-specific IFN-γ production in mice. Antigen-specific IFN-γ responses induced by the immunogens individually (A) and when combined by IFN-γ ELISpot assay (B). For the multivalent immunogen vaccine, antigen-specific IFN-γ production was measured at 1 (n = 8) and 10 (n = 9) weeks after the 3rd immunization. It was evaluated if a change in the ratio of CSP-specific Th1/Th2 responses contributed to the decrease in CSP-specific responses with MAV4. CSP-specific IgG2a (C) and IgG1 (D) titers were used as surrogate markers of Th1 and Th2 responses, respectively.
After confirming induction of cellular immune responses individually, the immunogenicity of MAV4 was evaluated by IFN-γ ELISpot assay and flow cytometry. Antigen-specific IFN-γ production was determined by ELISpot assay at both peak (1 week after the last immunization) (n = 8) and memory (10 weeks after the last immunization) (n = 9) time points (Fig. 3B). Following the third immunization, high levels of antigen-specific IFN-γ responses were detected (3,172 SFU). LSA1-specific IFN-γ production (2,188 SFU) comprised 69% of the total IFN-γ response. CSP-, TRAP-, and CelTOS-specific IFN-γ production comprised 11% (336 SFU), 16% (502 SFU), and 5% (145 SFU), respectively. IFN-γ production 10 weeks after the 3rd immunization was still robust (1,280 SFU) but decreased significantly (P = 0.01) from the response observed at the peak time point. The relative contribution of each antigen response to the total IFN-γ response was similar at both time points.
The individual CSP pDNA (Fig. 3A) induced higher levels of IFN-γ production than MAV4 (Fig. 3B). Others have previously reported that combining a CSP pDNA construct with pDNAs encoding different P. falciparum antigens decreased CSP-specific antibody titers and IFN-γ production (34, 35). In this study, CSP titers did not decrease with MAV4 but IFN-γ production did decrease. We sought to determine if the decrease in the CSP-specific IFN-γ elicited by MAV4 resulted from Th2 skewing. In mice, the IgG1 isotype is associated with Th2 responses while IgG2a is associated with Th1 responses (36). Levels of IgG2a (Fig. 3C) and IgG1 (Fig. 3D) induced by CSP alone or MAV4 were similar, indicating the change in IFN-γ production levels was not due to immune polarization toward Th2 responses.
Production of antigen-specific IFN-γ, IL-2, and TNF-α in both the CD4+ and CD8+ T cell compartments was evaluated in mice at the peak and memory time points by flow cytometry. CSP- and LSA1-specific CD4+ T cells comprised the majority of the peak antigen-specific CD4+ T cell response, 0.56 and 0.44%, respectively (Fig. 4A). The magnitude of the CD4+ T cell response decreased slightly from the peak (1.52%) to the memory (1.01%) time point, but this decrease was not statistically significant (P = 0.346). Interestingly, the relative contribution of each antigen to the total CD4+ T cell response was more balanced at the memory time point. The majority of the peak CD4+ T cell response was composed of monofunctional cells producing either IL-2 or TNF-α (Fig. 4B). Interestingly, at the memory time point the levels of single cytokine-producing cells decreased concurrently with a trend toward increased levels of polyfunctional CD4+ T cells (Fig. 4C).
Fig 4.

Characterization of CD4+ and CD8+ T cell responses in mice. Antigen-specific CD4+ (A to C) and CD8+ (D to F) T cell responses elicited by the multivalent immunogen were characterized in splenocytes by flow cytometry 1 (n = 8) and 10 (n = 9) weeks after the 3rd immunization. (A) The total magnitude of the CD4+ T cell response. Polyfunctionality of the CD4+ T cell response 1 (B) and 10 (C) weeks after the 3rd immunization. (D) The magnitude of the CD8+ T cell response. Polyfunctionality of the CD8+ T cell response 1 (E) and 10 (F) weeks after the 3rd immunization.
Peak CD8+ T cell responses were predominantly directed toward the CSP (0.97%) and LSA1 (1.23%) antigens. The magnitude of the CD8+ T cell response did not significantly decrease (P = 0.646) from the peak (2.86%) to the memory (2.51%) time point (Fig. 4D). The peak CD8+ T cell response was predominantly composed of single cytokine-producing cells (Fig. 4E). Similar to the CD4+ T cell response, there was an increase in the percentage of polyfunctional CD8+ T cells at the memory time point (Fig. 4F). Notably, the population of CD8+ IFN-γ+ TNF-α+ cells increased from 0.24 to 0.85%, a phenotype that has been associated with protection from malaria and other parasitic diseases in the mouse model (37–39).
The liver stage of the P. falciparum life cycle, which lasts 5 to 7 days, is the time period when the parasite is most vulnerable to immune recognition and elimination by CD8+ T cells. Liver resident effector memory CD8+ T cells likely are essential for rapid elimination of liver-stage parasites because of this short window of vulnerability (29). LSA1 was used as a model antigen to evaluate hepatic antigen-specific responses, because expression of LSA1 is specific to the liver stage of infection and its levels increase as the stage progresses (40). Accordingly, LSA1-specific hepatic CD8+ T cell responses were evaluated by flow cytometry 1 and 10 weeks after the 3rd immunization with MAV4. The majority of the LSA1-specific response was composed of CD8+ IFN-γ+ T cells at both time points (Fig. 5B and C). Importantly, the magnitude of the LSA1-specific CD8+ T cell response did not significantly decrease (P = 0.285) for at least 10 weeks following the last immunization (Fig. 5A).
Fig 5.

LSA1-specific CD8+ T cell responses in the liver. Hepatic lymphocytes were isolated from mice vaccinated with the multivalent immunogens 1 (n = 8) or 10 (n = 9) weeks after the 3rd vaccination. (A) Magnitude of the LSA1-specific CD8+ T cell immune response at the acute and memory time points. Polyfunctionality of the CD8+ T cell responses at 1 (B) and 10 (C) weeks after the 3rd immunization.
Immunogenicity in nonhuman primates.
The immunogenicity of MAV4 was further explored in the NHP model. Indian-origin rhesus macaques (n = 5) received 4 immunizations with 1.0 mg of each antigen construct (4.0 mg total plasmid) at weeks 0, 6, 12, and 25. pDNA-induced antigen-specific responses were demonstrated to continuously boost for at least 4 immunizations in previous studies in our laboratory (41). For this reason, a similar study design was used to evaluate the immunogenicity of MAV4 in NHPs. Antibody responses were evaluated by IFA and ELISA. IFA titers were determined at baseline, 2 weeks after the 3rd and 4th immunizations (weeks 14 and 27, respectively), and 8 weeks after the final immunization (week 33) (Fig. 6A). The IFA method determined that MAV4 induced antibodies that recognize and bind sporozoite coat proteins but does not allow for identification of antigen-specific titers. Antigen-specific antibody titers were determined by ELISA at week 0 and 2 weeks following each immunization (weeks 2, 8, 14, and 27). Memory antibody responses were assessed 8 weeks following the 3rd immunization (week 20) as well as 8 (week 33) and 12 (week 37) weeks after the 4th immunization (Fig. 6B to E). Humoral responses to LSA1, TRAP, and CelTOS were similar through week 27, while CSP-specific IgG levels remained lower throughout the study. Overall, antibody responses against all antigens were boostable and were detectable for at least 12 weeks after the 4th immunization.
Fig 6.

Humoral responses in NHPs. (A) IFA titers at weeks 0, 14, 27, and 33. CSP (B)-, LSA1 (C)-, TRAP (D)-, and CelTOS (E)-specific seroconversion in NHPs was measured by ELISA at the indicated time points. Arrows indicate vaccination time points at weeks 0, 6, 12, and 25. Seroconversion levels are expressed as the dilution of sera at which an OD value of 1 was obtained. Arrows indicate immunization time points (n = 5).
Cellular immunogenicity was evaluated by IFN-γ ELISpot assay and flow cytometry in PBMCs at the time points indicated in Fig. 7. The magnitude of IFN-γ production was boosted by each immunization, and in general, IFN-γ was produced in response to the individual immunogens and boosted with each immunization (Fig. 7A). Antigen-specific CD4+ T cells were detected 2 weeks following the 2nd immunization (week 8) and increased with the 3rd immunization (week 14). While the overall percentage of antigen-specific cytokine-producing CD4+ T cells did not increase with the 4th immunization (week 27) (Fig. 7B), the phenotype of the response shifted to include a higher percentage of IFN-γ+ cells and fewer IL-2+ cells (Fig. 7C). At all time points, with the exception of week 14, the majority of the CD4+ T cell response was attributed to IFN-γ+ or TNF-α+ cells.
Fig 7.

Cellular immune responses in NHPs. Antigen-specific T cell responses were assessed by IFN-γ ELISpot assay (A) and flow cytometry (B to F). Arrows in panel A indicate immunization time points. IFN-γ responses were boosted with each immunization. (B) Total percentage of CD4+ T cell responses to each antigen. (C) The majority of antigen-specific CD4+ T cells produced IFN-γ, IL-2, or TNF-α. (D) Total percentage of CD8+ T cell responses to each antigen. (E) The majority of antigen-specific CD8+ T cells produced IFN-γ, IL-2, or TNF-α, but an IFN-γ+ and IL-2+ or TNF-α+ population was observed following the 2nd immunization and was maintained for at least 12 weeks following the 4th immunization. (F) CD8+ T cell antigen-specific production of granzyme B was observed following the 2nd immunization, and the percentage of CD8+ GrB+ T cells increased with the 4th immunization. (n = 5). *, P < 0.05.
Cytokine production in the CD8+ T cell compartment was detectable 2 weeks following the 2nd immunization (week 8) (Fig. 7D). The percentage of antigen-specific CD8+ T cells did not increase following the 3rd immunization but did significantly increase following the 4th immunization (P = 0.043). The increase in the response after the 4th immunization was attributed primarily to an increase in IFN-γ production (Fig. 7E). The percentage of antigen-specific CD8+ T cells did not significantly decrease for at least 12 weeks after the 4th immunization (P = 0.225). We next examined assays relevant for gaining insight into killing function. Antigen-specific CD8+ GrzB+ T cells were detected after the 2nd immunization (week 8) (Fig. 7F). Similar to the overall percentage of CD8+ T cells, the CD8+ GrzB+ population increased from week 8 to week 27 (P = 0.042) and did not decrease for at least 12 weeks after the 4th immunization (P = 0.345) (Fig. 7F). Notably, the majority of antigen-specific CD8+ T cells were also GrzB+ at all time points, suggesting the potential for cytotoxic function (Fig. 7E and F).
DISCUSSION
Previous studies investigating DNA-based vaccine approaches targeting P. falciparum failed to induce potent immune responses in animal models and humans (13, 20, 34, 35, 42). For example, in a study conducted by Sedegah et al. (34) in the mouse model, IFN-γ production against P. falciparum CSP, TRAP, and LSA1 did not exceed 200 SFU when pDNAs were given individually. In a separate study (35), inclusion of the molecular adjuvant granulocyte-macrophage colony-stimulating factor (GM-CSF) in a five-antigen DNA-based approach did significantly increase CSP (266 SFU)-, LSA1 (459 SFU)-, and TRAP (216 SFU)-specific IFN-γ responses. While the responses described above cannot be compared directly to the data obtained in the current study, it is noteworthy that the IFN-γ responses induced by the GM-CSF-adjuvanted formulation (35) did not exceed those of unadjuvanted MAV4, which delivered 3.5-fold less DNA. Specifically, CSP-, LSA1-, and TRAP-specific IFN-γ responses in the current study induced by MAV4 were 365, 2,188, and 502 SFU, respectively (Fig. 3B).
The ability of this DNA-based approach to confer immune responses to multiple P. falciparum antigens most likely is due to the substantial optimization of the antigen coding sequences as well as EP delivery (11, 43, 44). Synthetic engineering of the antigen-coding sequences allowed for the inclusion of optimization approaches, such as incorporation of a highly efficient leader sequence, improved mRNA stability, and codon optimization. Delivery with EP increases the uptake of DNA plasmids by cells in the target tissue, resulting in an increase in antigen production (45) and immunogenicity (16–18). Thus, the optimization of antigens and antigen delivery greatly improved the immune responses induced by the DNA platform.
There have been previous reports that mixing of pDNA encoding different P. falciparum antigens decreases the immune responses to some of the individual antigens in the mixture. Specifically, these studies reported lower LSA1- and CSP-specific antibody titers and CSP-specific IFN-γ production as a result of combining plasmids into a multiantigen formulation (34, 35). In this study, using DNA immunogens delivered with EP, there was not a significant difference in CSP antibody titers (Fig. 2) or LSA1-specific IFN-γ production (Fig. 3) conferred by MAV4 compared to responses elicited by the immunogens individually. In agreement with prior studies (34, 35), the magnitude of CSP-specific IFN-γ production was lower when the CSP antigen was combined with the LSA1, TRAP, and CelTOS antigens (Fig. 3A and B). The decreased magnitude of CSP-specific IFN-γ production was not due to MAV4 skewing CSP responses toward Th2 (Fig. 3C and D). Studies investigating the mechanism underlying reduced responses to CSP when it is codelivered with other pDNAs and exploring adjuvants to enhance CSP immune responses would be of value for moving multiple-antigen approaches forward.
A malaria vaccine that elicits high-titer and high-affinity antibody responses could be important to reduce the number of sporozoites that successfully infect hepatocytes and undergo replication, thereby lessening or eliminating progression to the blood stage of P. falciparum infection, which is associated with clinical malaria symptoms. Previous studies in both animals and humans demonstrated the ability of antibodies to confer protection at the PE stages of P. falciparum infection (46–49). Thus, successful induction of high antibody titers to relevant target antigens is likely a key component of a highly efficacious vaccine. In this study, in both mice and NHPs, MAV4 induced antibodies to all antigens (Fig. 2 and 7). The IFA approach determined that MAV4 induces antibodies that recognize and bind to proteins on the sporozoite surface (Fig. 6A). Of the four antigens evaluated in this study, roles for CSP and CelTOS antibodies in protection from malaria have been studied.
There is clear evidence that antibodies targeting CSP can confer protection from malaria infection in humans. Higher levels of CSP-specific antibodies are likely to inhibit the migration of sporozoites from the skin to the liver (50) and have been correlated with protection in RTS,S phase II clinical trials (51). Shott et al. compared immune responses induced by full-length CSP delivered via the Ad5, Ad35 vectors, and RTS,S/AS01B in the mouse model (52). Mean CSP IgG titers were reported (at a geometric mean titer [GMT] OD of 1) of 933, 1,105, and 2,529 induced by Ad5, Ad35, and RTS,S/AS01B, respectively. Mean CSP titers generated by the single antigen (12,191) and MAV4 (6,456) exceeded those reported for the viral platforms and RTS,S in mice. In NHPs, RTS,S/AS01B CSP titers (at a GMT OD of 1) peaked at approximately 1,500 (53), which exceeded that of MAV4, which peaked at 587 at week 27 (Fig. 6B). In both the mouse and NHP models, assay differences do not allow for a direct comparison of MAV4 CSP titers. In this study, full-length CSP was used to quantitate IgG binding titers, while the other studies used peptides encoding the central repeat region. Future studies could investigate a MAV4 CSP protein prime-boost approach to increase CSP titers.
There is also evidence for anti-CelTOS antibody-mediated protection in mice, but studies have not been completed in higher animal species or humans. The sera from mice immunized with recombinant P. falciparum CelTOS protein adjuvanted with Montanide ISA 720 contained CelTOS-specific IgG that was found to bind sporozoites and inhibit the invasion of hepatocytes in vitro. Immunization also protected approximately 60% of mice against a heterologous challenge with Plasmodium berghei sporozoites. Minimal cellular immune responses were reported (IFN-γ SFU of <100), suggesting that the observed CelTOS-mediated protection is primarily antibody mediated. In this study, pDNA CelTOS drives antibody responses in both the mouse (Fig. 2E and F) and NHP (Fig. 6E) model. The mouse anti-CelTOS IgG titers reported here are not directly comparable to those reported by Bergmann-Leitner et al. because of assay differences (23). This is the first report of CelTOS-specific IgG in NHPs. It would be interesting to explore if pDNA CelTOS and MAV4 can confer protection from heterologous challenge with Plasmodium berghei sporozoites in the mouse model. The protective efficacy of CelTOS has not yet been confirmed in humans, and the role of anti-CelTOS antibodies in protection still needs to be clarified.
Cellular immune responses, including CD4+ and CD8+ T cells, are important for conferring protection from P. falciparum infection at the PE stage. Of the four antigens investigated in this study, the role of CSP-specific CD4+ T cells has been the best characterized. CSP-specific CD4+ T cells are important for both naturally acquired (54) and immunization-mediated protection (51). In this study, in mice, approximately one-third of the peak CD4+ T cell response was CSP specific (0.60%) (Fig. 4A to C). In NHPs, following the 2nd and 3rd vaccinations, the CD4+ T cell response was primarily composed of TNF-α+- and IL-2+-producing cells, which are immune responses believed to be associated with protection from P. falciparum infection (Fig. 7B and C). Specifically, in phase IIa RTS,S trials, central memory and effector CD4+ T cells producing IL-2 and TNF-α were associated with a higher rate of protection from P. falciparum challenge (39).
The protective effects of CD8+ T cell-mediated immunity has been established in animal models (32, 33, 39, 46, 55), but the vaccine that has shown the most efficacy in humans, RTS,S, does not confer appreciable levels of CD8+ T cell responses (53, 56). In addition to immune responses directed at the whole sporozoite, CD8+ T cells targeting CSP can mediate protection in mice (57, 58). Thus, there remains a need for the development of a vaccine approach that can confer CD8+ T cell immune responses to P. falciparum antigens, particularly directed at CSP. MAV4 elicited CD8+ T cell responses to all antigens in both mice and NHPs. CD8+ T cells produced high levels of IFN-γ and TNF-α to all antigens, including CSP, which mimics the immune responses believed to be required for mediated protection (32, 33, 39, 46). Importantly, sustained IFN-γ production by hepatic CD8+ T cells, which has been correlated with long-term protection (55), was detected in the mouse model at both peak and memory time points (Fig. 5). In NHPs, the majority of the antigen-specific CD8+ T cells were also GrzB+, indicating this population has the potential to function as cytotoxic T cells. Further studies will be required to determine if CD8+ GrzB+ T cells can effectively target and kill P. falciparum-infected hepatocytes.
In general, polyfunctional lymphocytes are thought to be optimized for effector function. An increase in the level of multifunctional T cells has been associated with protection from some diseases, including malaria. Specifically, some data suggest induction of polyfunctional CD8+ T cells to PE-stage antigens decreases progression to clinical disease in preclinical (39) and clinical models (2, 3). Increased CD8+ T cell production of IFN-γ alone or concurrently with TNF-α (IFN-γ+ TNF-α+) correlated with increased protection from Plasmodium berghei challenge in mice. Also, in a model of malaria infection in humans, which received immunizations of live sporozoites while undergoing prophylactic chloroquine, it was demonstrated that sporozoite vaccination induces and maintains polyfunctional peak memory T cell responses (IFN-γ+ IL-2+) (3). MAV4 effectively induced polyfunctional T cells in the CD4+ and CD8+ compartments in both mice (Fig. 4) and NHPs (Fig. 7). In mice, the higher levels of polyfunctional T cells were observed at the memory time point (Fig. 4C and F). There was a notable increase in CD8+ IFN-γ+ TNF-α+ T cells, a phenotype that was demonstrated to correlate with protection from P. berghei and other parasitic organisms (37–39). Importantly, the phenotype of the cellular immune responses observed in mice translated to the NHP model (Fig. 7) (23).
In summary, MAV4, an optimized DNA-based multiantigen P. falciparum vaccine approach, can effectively drive robust humoral and cellular immune responses. The responses induced mimic the immune responses believed to be associated with protection from P. falciparum infection in both the mouse (39) and NHP (59) models, as well as in humans (3). Importantly, in mice, administering MAV4 in the periphery effectively drove CD8+ T cell responses in the liver. Further studies of this multiple-immunogen approach will explore heterologous prime-boost approaches and incorporation of cytokine adjuvants to further enhance immune responses.
ACKNOWLEDGMENTS
This work was supported by funding from the PATH Malaria Vaccine Initiative and the National Institutes of Health (NIH grant T32AI070099).
D.B.W. and his laboratory have several commercial relationships. He has received consulting fees and stock ownership for Advisory Board/Review Board Service, speaking support, and research support from commercial entities, including Inovio, BMS, Pfizer, VGXi, Virxsys, J & J, Merck, Sanofi Pasteur, Althea, Novo Nordisk, SSI, Aldevron, Novartis, Incyte, and possibly others. No writing assistance was utilized in the production of the manuscript. N.Y.S., M.P.M., A.S.B., A.S.K., J.Y., and M.Y. are employees of Inovio Pharmaceuticals, Inc., and as such receive a salary and own stock/stock options in the company. The other authors declare no conflict of interest.
A.K.K.K. is a military service member of the United States Government. This work was prepared as part of his official duties. Title 17 U.S.C. §105 provides that “Copyright protection under this title is not available for any work of the United States Government.” Title 17 U.S.C. §101 defines a U.S. Government work as a work prepared by a military service member or employee of the U.S. Government as part of that person's official duties.
The views expressed in this article are those of the authors and do not necessarily reflect the official policy or position of the Department of the Army, the Department of Defense, or the U.S. Government.
Footnotes
Published ahead of print 29 July 2013
REFERENCES
- 1.World Health Organization 2011. World malaria report. World Health Organization, Geneva, Switzerland [Google Scholar]
- 2.Roestenberg M, McCall M, Hopman J, Wiersma J, Luty AJ, Van Gemert GJ, Van De Vegte-Bolmer M, Van Schaijk B, Teelen K, Arens T, Spaarman L, De Mast Q, Roeffen W, Snounou G, Renia L, Van Der Ven A, Hermsen CC, Sauerwein R. 2009. Protection against a malaria challenge by sporozoite inoculation. N. Engl. J. Med. 361:468–477 [DOI] [PubMed] [Google Scholar]
- 3.Teirlinck AC, Mccall MB, Roestenberg M, Scholzen A, Woestenenk R, De Mast Q, Van Der Ven AJ, Hermsen CC, Luty AJ, Sauerwein RW. 2011. Longevity and composition of cellular immune responses following experimental Plasmodium falciparum malaria infection in humans. PLoS Pathog. 7:e1002389. 10.1371/journal.ppat.1002389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Agnandji ST, Lell B, Soulanoudjingar SS, Fernandes JF, Abossolo BP, Conzelmann C, Methogo BGNO, Doucka Y, Flamen A, Mordmüller B, Issifou S, Kremsner PG, Sacarlal J, Aide P, Lanaspa M, Aponte JJ, Nhamuave A, Quelhas D, Bassat Q, Mandjate S, Macete E, Alonso P, Abdulla S, Salim N, Juma O, Shomari M, Shubis K, Machera F, Hamad AS, Minja R, Mtoro A, Sykes A, Ahmed S, Urassa AM, Ali AM, Mwangoka G, Tanner M, Tinto H, Dapos-Alessandro U, Sorgho H, Valea I, Tahita MC, Kaboré W, Ouédraogo S, Sandrine Y, Guiguemdé RT, Ouédraogo JB, Hamel MJ, Kariuki S, Odero C, Oneko M, Otieno K, Awino N, Omoto J, Williamson J, Muturi-Kioi V, Laserson KF, Slutsker L, Otieno W, Otieno L, Nekoye O, Gondi S, Otieno A, Ogutu B, Wasuna R, Owira V, Jones D, Onyango AA, Njuguna P, Chilengi R, Akoo P, Kerubo C, Gitaka J, Maingi C, Lang T, Olotu A, Tsofa B, Bejon P, Peshu N, Marsh K, Owusu-Agyei S, Asante KP, Osei-Kwakye K, Boahen O, Ayamba S, Kayan K, Owusu-Ofori R, Dosoo D, Asante I, Adjei G, Adjei G, Chandramohan D, Greenwood B, Lusingu J, Gesase S, Malabeja A, Abdul O, Kilavo H, Mahende C, Liheluka E, Lemnge M, Theander T, Drakeley C, Ansong D, Agbenyega T, Adjei S, Boateng HO, Rettig T, Bawa J, Sylverken J, Sambian D, Agyekum A, Owusu L, Martinson F, Hoffman I, Mvalo T, Kamthunzi P, Nkomo R, Msika A, Jumbe A, Chome N, Nyakuipa D, Chintedza J, Ballou WR, Bruls M, Cohen J, Guerra Y, Jongert E, Lapierre D, Leach A, Lievens M, Ofori-Anyinam O, Vekemans J, Carter T, Leboulleux D, Loucq C, Radford A, Savarese B, Schellenberg D, Sillman M, Vansadia P. 2011. First results of phase 3 trial of RTS,S/AS01 malaria vaccine in African children. N. Engl. J. Med. 365:1863–1875 [DOI] [PubMed] [Google Scholar]
- 5.Epstein JE, Tewari K, Lyke KE, Sim BKL, Billingsley PF, Laurens MB, Gunasekera A, Chakravarty S, James ER, Sedegah M, Richman A, Velmurugan S, Reyes S, Li M, Tucker K, Ahumada A, Ruben AJ, Li T, Stafford R, Eappen AG, Tamminga C, Bennett JW, Ockenhouse CF, Murphy JR, Komisar J, Thomas N, Loyevsky M, Birkett A, Plowe CV, Loucq C, Edelman R, Richie TL, Seder RA, Hoffman SL. 2011. Live attenuated malaria vaccine designed to protect through hepatic CD8+ T cell immunity. Science 334:475–480 [DOI] [PubMed] [Google Scholar]
- 6.Good MF, Doolan DL. 2010. Malaria vaccine design: immunological considerations. Immunity 33:555–566 [DOI] [PubMed] [Google Scholar]
- 7.Good MF, Doolan DL. 1999. Immune effector mechanisms in malaria. Curr. Opin. Immunol. 11:412–419 [DOI] [PubMed] [Google Scholar]
- 8.Kappe SH, Buscaglia CA, Nussenzweig V. 2004. Plasmodium sporozoite molecular cell biology. Annu. Rev. Cell Dev. Biol. 20:29–59 [DOI] [PubMed] [Google Scholar]
- 9.Doolan DL, Dobano C, Baird JK. 2009. Acquired immunity to malaria. Clin. Microbiol. Rev. 22:13–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu MA. 2011. DNA vaccines: an historical perspective and view to the future. Immunol. Rev. 239:62–84 [DOI] [PubMed] [Google Scholar]
- 11.Bagarazzi ML, Yan J, Morrow MP, Shen X, Parker RL, Lee JC, Giffear M, Pankhong P, Khan AS, Broderick KE, Knott C, Lin F, Boyer JD, Draghia-Akli R, White CJ, Kim JJ, Weiner DB, Sardesai NY. 2012. Immunotherapy against HPV16/18 generates potent TH1 and cytotoxic cellular immune responses. Sci. Transl. Med. 4:155ra138. 10.1126/scitranslmed.3004414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kutzler MA, Weiner DB. 2008. DNA vaccines: ready for prime time? Nat. Rev. Genet. 9:776–788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Epstein JE, Gorak EJ, Charoenvit Y, Wang R, Freydberg N, Osinowo O, Richie TL, Stoltz EL, Trespalacios F, Nerges J, Ng J, Fallarme-Majam V, Abot E, Goh L, Parker S, Kumar S, Hedstrom RC, Norman J, Stout R, Hoffman SL. 2002. Safety, tolerability, and lack of antibody responses after administration of a PfCSP DNA malaria vaccine via needle or needle-free jet injection, and comparison of intramuscular and combination intramuscular/intradermal routes. Hum. Gene Ther. 13:1551–1560 [DOI] [PubMed] [Google Scholar]
- 14.MacGregor RR, Boyer JD, Ugen KE, Lacy KE, Gluckman SJ, Bagarazzi ML, Chattergoon MA, Baine Y, Higgins TJ, Ciccarelli RB, Coney LR, Ginsberg RS, Weiner DB. 1998. First human trial of a DNA-based vaccine for treatment of human immunodeficiency virus type 1 infection: safety and host response. J. Infect. Dis. 178:92–100 [DOI] [PubMed] [Google Scholar]
- 15.Ferraro B, Morrow MP, Hutnick NA, Shin TH, Lucke CE, Weiner DB. 2011. Clinical applications of DNA vaccines: current progress. Clin. Infect. Dis. 53:296–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rosati M, Valentin A, Jalah R, Patel V, Von Gegerfelt A, Bergamaschi C, Alicea C, Weiss D, Treece J, Pal R, Markham PD, Marques ET, August JT, Khan A, Draghia-Akli R, Felber BK, Pavlakis GN. 2008. Increased immune responses in rhesus macaques by DNA vaccination combined with electroporation. Vaccine 26:5223–5229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hirao LA, Wu L, Khan AS, Hokey DA, Yan J, Dai A, Betts MR, Draghia-Akli R, Weiner DB. 2008. Combined effects of IL-12 and electroporation enhances the potency of DNA vaccination in macaques. Vaccine 26:3112–3120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hirao LA, Wu L, Khan AS, Satishchandran A, Draghia-Akli R, Weiner DB. 2008. Intradermal/subcutaneous immunization by electroporation improves plasmid vaccine delivery and potency in pigs and rhesus macaques. Vaccine 26:440–448 [DOI] [PubMed] [Google Scholar]
- 19.Vekemans J, Leach A, Cohen J. 2009. Development of the RTS,S/AS malaria candidate vaccine. Vaccine 27:G67–G71 [DOI] [PubMed] [Google Scholar]
- 20.McConkey S, Reece W, Moorthy V. 2003. Enhanced T-cell immunogenicity of plasmid DNA vaccines boosted by recombinant modified vaccinia virus Ankara in humans. Nat. Med. 9:729–735 [DOI] [PubMed] [Google Scholar]
- 21.Dunachie SJ, Walther M, Vuola JM, Webster DP, Keating SM, Berthoud T, Andrews L, Bejon P, Poulton I, Butcher G. 2006. A clinical trial of prime-boost immunisation with the candidate malaria vaccines RTS,S/AS02A and MVA-CS. Vaccine 24:2850–2859 [DOI] [PubMed] [Google Scholar]
- 22.Cummings JF, Spring MD, Schwenk RJ, Ockenhouse CF, Kester KE, Polhemus ME, Walsh DS, Yoon I-K, Prosperi C, Juompan LY, Lanar DE, Krzych U, Hall BT, Ware LA, Stewart VA, Williams J, Dowler M, Nielsen RK, Hillier CJ, Giersing BK, Dubovsky F, Malkin E, Tucker K, Dubois M-C, Cohen JD, Ballou WR, Heppner DG., Jr 2010. Recombinant liver stage antigen-1 (LSA-1) formulated with AS01 or AS02 is safe, elicits high titer antibody and induces IFN-Î3/IL-2 CD4+ T cells but does not protect against experimental Plasmodium falciparum infection. Vaccine 28:5135–5144 [DOI] [PubMed] [Google Scholar]
- 23.Bergmann-Leitner ES, Mease RM, De La Vega P, Savranskaya T, Polhemus M, Ockenhouse C, Angov E. 2010. Immunization with pre-erythrocytic antigen CelTOS from Plasmodium falciparum elicits cross-species protection against heterologous challenge with Plasmodium berghei. PLoS One 5:e12294. 10.1371/journal.pone.0012294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yan J, Hokey DA, Morrow MP, Corbitt N, Harris K, Harris D, Weiner DB. 2009. Novel SIVmac DNA vaccines encoding Env, Pol and Gag consensus proteins elicit strong cellular immune responses in cynomolgus macaques. Vaccine 27:3260–3266 [DOI] [PubMed] [Google Scholar]
- 25.Hirao LA, Draghia-Akli R, Prigge JT, Yang M, Satishchandran A, Wu L, Hammarlund E, Khan AS, Babas T, Rhodes L, Silvera P, Slifka M, Sardesai NY, Weiner DB. 2011. Multivalent smallpox DNA vaccine delivered by intradermal electroporation drives protective immunity in nonhuman primates against lethal monkeypox challenge. J. Infect. Dis. 203:95–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kariu T, Ishino T, Yano K, Chinzei Y, Yuda M. 2006. CelTOS, a novel malarial protein that mediates transmission to mosquito and vertebrate hosts. Mol. Microbiol. 59:1369–1379 [DOI] [PubMed] [Google Scholar]
- 27.Cheung YK, Cheng SC, Sin FW, Xie Y. 2004. Plasmid encoding papillomavirus type 16 (HPV16) DNA constructed with codon optimization improved the immunogenicity against HPV infection. Vaccine 23:629–638 [DOI] [PubMed] [Google Scholar]
- 28.Yan J, Yoon H, Kumar S, Ramanathan MP, Corbitt N, Kutzler M, Dai A, Boyer JD, Weiner DB. 2007. Enhanced cellular immune responses elicited by an engineered HIV-1 subtype B consensus-based envelope DNA vaccine. Mol. Ther. 15:411–421 [DOI] [PubMed] [Google Scholar]
- 29.Morrot A, Hafalla JC, Cockburn IA, Carvalho LH, Zavala F. 2005. IL-4 receptor expression on CD8+ T cells is required for the development of protective memory responses against liver stages of malaria parasites. J. Exp. Med. 202:551–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reyes-Sandoval A, Wyllie DH, Bauza K, Milicic A, Forbes EK, Rollier CS, Hill A. 2011. CD8+ T effector memory cells protect against liver-stage malaria. J. Immunol. 187:1347–1357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schofield L, Ferreira A, Altszuler R, Nussenzweig V, Nussenzweig RS. 1987. Interferon-gamma inhibits the intrahepatocytic development of malaria parasites in vitro. J. Immunol. 139:2020–2025 [PubMed] [Google Scholar]
- 32.Butler NS, Schmidt NW, Harty JT. 2010. Differential effector pathways regulate memory CD8 T cell immunity against Plasmodium berghei versus P. yoelii sporozoites. J. Immunol. 184:2528–2538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ferreira A, Schofield L, Enea V, Schellekens H, Van Der Meide P, Collins WE, Nussenzweig RS, Nussenzweig V. 1986. Inhibition of development of exoerythrocytic forms of malaria parasites by gamma-interferon. Science 232:881–884 [DOI] [PubMed] [Google Scholar]
- 34.Sedegah M, Charoenvit Y, Minh L, Belmonte M, Majam VF, Abot S, Ganeshan H, Kumar S, Bacon DJ, Stowers A, Narum DL, Carucci DJ, Rogers WO. 2004. Reduced immunogenicity of DNA vaccine plasmids in mixtures. Gene Ther. 11:448–456 [DOI] [PubMed] [Google Scholar]
- 35.Sedegah M, Charoenvit Y, Aguiar J, Sacci J, Hedstrom R, Kumar S, Belmonte A, Lanar DE, Jones TR, Abot E, Druilhe P, Corradin G, Epstein JE, Richie TL, Carucci DJ, Hoffman SL. 2004. Effect on antibody and T-cell responses of mixing five GMP-produced DNA plasmids and administration with plasmid expressing GM-CSF. Genes Immun. 5:553–561 [DOI] [PubMed] [Google Scholar]
- 36.Dekruyff R, Rizzo L, Umetsu D. 1993. Induction of immunoglobulin synthesis by CD4+ T cell clones. Semin. Immunol. 5:421–430 [DOI] [PubMed] [Google Scholar]
- 37.Bogdan C, Moll H, Solbach W, Rollinghoff M. 1990. Tumor necrosis factor-alpha in combination with interferon-gamma, but not with interleukin 4 activates murine macrophages for elimination of Leishmania major amastigotes. Eur. J. Immunol. 20:1131–1135 [DOI] [PubMed] [Google Scholar]
- 38.Liew FY, Millott S, Parkinson C, Palmer RM, Moncada S. 1990. Macrophage killing of Leishmania parasite in vivo is mediated by nitric oxide from L-arginine. J. Immunol. 144:4794–4797 [PubMed] [Google Scholar]
- 39.Reyes-Sandoval A, Berthoud T, Alder N, Siani L, Gilbert SC, Nicosia A, Colloca S, Cortese R, Hill AV. 2010. Prime-boost immunization with adenoviral and modified vaccinia virus Ankara vectors enhances the durability and polyfunctionality of protective malaria CD8+ T-cell responses. Infect. Immun. 78:145–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fidock DA, Gras-Masse H, Lepers JP, Brahimi K, Benmohamed L, Mellouk S, Guerin-Marchand C, Londono A, Raharimalala L, Meis JF. 1994. Plasmodium falciparum liver stage antigen-1 is well conserved and contains potent B and T cell determinants. J. Immunol. 153:190–204 [PubMed] [Google Scholar]
- 41.Hirao LA, Wu L, Satishchandran A, Khan AS, Draghia-Akli R, Finnefrock AC, Bett AJ, Betts MR, Casimiro DR, Sardesai NY, Kim JJ, Shiver JW, Weiner DB. 2010. Comparative analysis of immune responses induced by vaccination with SIV antigens by recombinant Ad5 vector or plasmid DNA in rhesus macaques. Mol. Ther. 18:1568–1576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sedegah M, Rogers WO, Belmonte M, Belmonte A, Banania G, Patterson NB, Rusalov D, Smith L, Ferrari M, Richie TL, Doolan DL. 2009. Vaxfectin enhances both antibody and in vitro T cell responses to each component of a 5-gene Plasmodium falciparum plasmid DNA vaccine mixture administered at low doses. Vaccine 28:1–11 [DOI] [PubMed] [Google Scholar]
- 43.Shedlock DJ, Talbott KT, Wu SJ, Wilson CM, Muthumani K, Boyer JD, Sardesai NY, Awasthi S, Weiner DB. 2012. Vaccination with synthetic constructs expressing cytomegalovirus immunogens is highly T cell immunogenic in mice. Hum. Vaccin. Immunother. 8:1668–1681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dobano C, Sedegah M, Rogers WO, Kumar S, Zheng H, Hoffman SL, Doolan DL. 2009. Plasmodium: mammalian codon optimization of malaria plasmid DNA vaccines enhances antibody responses but not T cell responses nor protective immunity. Exp. Parasitol. 122:112–123 [DOI] [PubMed] [Google Scholar]
- 45.Titomirov AV, Sukharev S, Kistanova E. 1991. In vivo electroporation and stable transformation of skin cells of newborn mice by plasmid DNA. Biochim. Biophys. Acta 1088:131–134 [DOI] [PubMed] [Google Scholar]
- 46.Schofield L, Villaquiran J, Ferreira A, Schellekens H, Nussenzweig R, Nussenzweig V. 1987. Gamma interferon, CD8+ T cells and antibodies required for immunity to malaria sporozoites. Nature 330:664–666 [DOI] [PubMed] [Google Scholar]
- 47.Potocnjak P, Yoshida N, Nussenzweig RS, Nussenzweig V. 1980. Monovalent fragments (Fab) of monoclonal antibodies to a sporozoite surface antigen (Pb44) protect mice against malarial infection. J. Exp. Med. 151:1504–1513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Charoenvit Y, Mellouk S, Cole C, Bechara R, Leef MF, Sedegah M, Yuan LF, Robey FA, Beaudoin RL, Hoffman SL. 1991. Monoclonal, but not polyclonal, antibodies protect against Plasmodium yoelii sporozoites. J. Immunol. 146:1020–1025 [PubMed] [Google Scholar]
- 49.Hollingdale MR, Nardin EH, Tharavanij S, Schwartz AL, Nussenzweig RS. 1984. Inhibition of entry of Plasmodium falciparum and P. vivax sporozoites into cultured cells; an in vitro assay of protective antibodies. J. Immunol. 132:909–913 [PubMed] [Google Scholar]
- 50.Mishra S, Nussenzweig RS, Nussenzweig V. 2012. Antibodies to Plasmodium circumsporozoite protein (CSP) inhibit sporozoite's cell traversal activity. J. Immunol. Methods 377:47–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kester KE, Cummings JF, Ofori-Anyinam O, Ockenhouse CF, Krzych U, Moris P, Schwenk R, Nielsen RA, Debebe Z, Pinelis E, Juompan L, Williams J, Dowler M, Stewart VA, Wirtz RA, Dubois M-C, Lievens M, Cohen J, Ballou WR, Heppner DG. 2009. Randomized, double-blind, phase 2a trial of falciparum malaria vaccines RTS,S/AS01B and RTS,S/AS02A in malaria-naive adults: safety, efficacy, and immunologic associates of protection. J. Infect. Dis. 200:337–346 [DOI] [PubMed] [Google Scholar]
- 52.Shott JP, Mcgrath SM, Pau MG, Custers JHV, Ophorst O, Demoitié M-A, Dubois M-C, Komisar J, Cobb M, Kester KE, Dubois P, Cohen J, Goudsmit J, Heppner DG, Stewart VA. 2008. Adenovirus 5 and 35 vectors expressing Plasmodium falciparum circumsporozoite surface protein elicit potent antigen-specific cellular IFN-γ and antibody responses in mice. Vaccine 26:2818–2823 [DOI] [PubMed] [Google Scholar]
- 53.Mettens P, Dubois PM, Demoitié M-A, Bayat B, Donner M-N, Bourguignon P, Stewart VA, Heppner DG, Jr, Garçon DGN, Cohen J. 2008. Improved T cell responses to Plasmodium falciparum circumsporozoite protein in mice and monkeys induced by a novel formulation of RTS,S vaccine antigen. Vaccine 26:1072–1082 [DOI] [PubMed] [Google Scholar]
- 54.Reece WHH, Pinder M, Gothard PK, Milligan P, Bojang K, Doherty T, Plebanski M, Akinwunmi P, Everaere S, Watkins KR, Voss G, Tornieporth N, Alloueche A, Greenwood BM, Kester KE, McAdam KPWJ, Cohen J, Hill AVS. 2004. A CD4+ T-cell immune response to a conserved epitope in the circumsporozoite protein correlates with protection from natural Plasmodium falciparum infection and disease. Nat. Med. 10:406–410 [DOI] [PubMed] [Google Scholar]
- 55.Nganou-Makamdop K, Van Gemert GJ, Arens T, Hermsen CC, Sauerwein RW. 2012. Long term protection after immunization with P. berghei sporozoites correlates with sustained IFNgamma responses of hepatic CD8+ memory T cells. PLoS One 7:e36508. 10.1371/journal.pone.0036508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Olotu A, Moris P, Mwacharo J, Vekemans J, Kimani D, Janssens M, Kai O, Jongert E, Lievens M, Leach A, Villafana T, Savarese B, Marsh K, Cohen J, Bejon P. 2011. Circumsporozoite-specific T cell responses in children vaccinated with RTS,S/AS01E and protection against P. falciparum clinical malaria. PLoS One 6:e25786. 10.1371/journal.pone.0025786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rodrigues MM, Cordey AS, Arreaza G, Corradin G, Romero P, Maryanski JL, Nussenzweig RS, Zavala F. 1991. CD8+ cytolytic T cell clones derived against the Plasmodium yoelii circumsporozoite protein protect against malaria. Int. Immunol. 3:579–585 [DOI] [PubMed] [Google Scholar]
- 58.Romero P, Maryanski JL, Corradin G, Nussenzweig RS, Nussenzweig V, Zavala F. 1989. Cloned cytotoxic T cells recognize an epitope in the circumsporozoite protein and protect against malaria. Nature 341:323–326 [DOI] [PubMed] [Google Scholar]
- 59.Weiss WR, Jiang CG. 2012. Protective CD8+ T lymphocytes in primates immunized with malaria sporozoites. PLoS One 7:e31247. 10.1371/journal.pone.0031247 [DOI] [PMC free article] [PubMed] [Google Scholar]