

Abstract
Interleukin-17A (IL-17A)-producing γδ T cells are known to be activated following Mycobacterium bovis bacillus Calmette-Guérin (BCG) infection. Here, we show that CD30, a member of the tumor necrosis factor (TNF) receptor superfamily, is important for activation of IL-17A-producing γδ T cells after BCG infection. Vγ1− Vγ4− γδ T cells preferentially expressing Vγ6/Vδ1 genes were identified as the major source of IL-17A in the peritoneal cavity during the early stage of BCG infection. The number of IL-17A-producing Vγ1− Vγ4− γδ T cells bearing Vγ6 increased in peritoneal exudate cells (PEC) of wild-type (WT) mice but not in those of CD30 knockout (KO) mice in response to BCG infection. Consistently, CD30 ligand (CD30L) or CD30 expression, predominantly by Vγ1− Vγ4− γδ T cells, was rapidly upregulated after BCG infection. Inhibition of CD30L/CD30 signaling by in vivo administration of a soluble CD30 and immunoglobulin fusion protein (CD30-Ig) severely impaired activation of IL-17A-producing Vγ1− Vγ4− γδ T cells in WT mice, while stimulating CD30L/CD30 signaling by in vivo administration of agonistic anti-CD30 monoclonal antibody (MAb) restored IL-17A production by Vγ1− Vγ4− γδ T cells in CD30L KO mice after BCG infection. These results suggest that CD30 signaling plays an important role in the activation of IL-17A-producing Vγ1− Vγ4− γδ T cells bearing Vγ6 at an early stage of BCG infection.
INTRODUCTION
Unlike conventional T cells, which are exported from the thymus as naive cells and acquire effector functions upon antigen (Ag) encounter in the periphery, some subsets of murine γδ T cells are functionally differentiated into effector cells producing gamma interferon (IFN-γ) or interleukin-17A (IL-17A) within the fetal thymus and are disproportionately distributed in mucosal epithelia such as skin, intestine, uterus, and lung as tissue-associated cells (1–8). IL-17A is a proinflammatory cytokine originally identified from helper CD4+ αβ T cells, Th17 cells, which participate in host defense against various types of pathogens as well as in autoimmune disorders (9–12). Recently, it was found that IL-17A production by γδ T cells rather than CD4+ αβ T cells plays an important role in the immune response to pulmonary Mycobacterium bovis Bacillus Calmette-Guérin (BCG) infection as well as in BCG-induced lung granuloma formation (13–15). We have also reported the importance of IL-17A-producing γδ T cells in other models of infection of mice with Escherichia coli, Candia albicans, or BCG, which are involved in the neutrophil-mediated clearance of the pathogens or tumor cells (16–18). These findings suggest the hypothesis that a novel host defense mechanism mediated by the “innate” IL-17A-producing γδ T cells is acting in innate immunity and initiating an inflammatory response following a microbe invasion.
A CD30 ligand (CD30L; CD153) is a 40-kDa type II membrane-associated glycoprotein belonging to the tumor necrosis factor (TNF) superfamily and is expressed on activated and memory CD4+ T helper cells in addition to macrophages, dendritic cells (DCs), B cells, and unique CD4+ CD3− CD11c− accessory cells (19–22). CD30, the receptor for CD30L, which belongs to the TNF receptor superfamily, was expressed preferentially by activated and memory Th cells but not by resting B or T cells (23–25). CD30L/CD30 signaling was initially thought to be involved in Th2 cell responses and Th2-associated diseases (26–29). However, a number of recent studies indicated that CD30L/CD30 signaling is also linked to Th1 or regulatory T cell responses and their associated diseases (30–35). We have recently found that CD30L/CD30 signaling executed by T cell-T cell interaction plays a novel role in Th1 and Th17 cell differentiation in vitro and in vivo (36–38). In this regard, CD30L/CD30 signaling may be dispensable for differentiation of a specific Th cell subset in a physiological pathway, while it may be required for the activation and/or amplification of T cells irrespective of the T cell subset in the periphery.
Mycobacterial infection by such bacilli as BCG and Mycobacterium tuberculosis has been widely discussed with respect to their adaptive immune response, which mainly depends on IFN-γ production by CD4+ Th1 cells (39, 40). Recently, we and others reported that CD30L and CD30 play an important role in acquired immunity against mycobacterial infection by amplifying the Th1 response (32, 36). However, the potential role of CD30L/CD30 signaling in controlling host defense through activation of IL-17A-producing T cells against mycobacterial infection is unknown. In this study, we found that the numbers of IL-17A-producing Vγ1− Vγ4− γδ T cells bearing Vγ6 significantly decreased in peritoneal exudate cells (PEC) of CD30-deficient mice at an early stage of intraperitoneal (i.p.) infection with BCG. Consistently, the expression level of CD30L or CD30 was selectively upregulated after BCG infection on Vγ1− Vγ4− γδ T cells, which are the “innate” source of IL-17A in PEC during the early stage of BCG infection. These findings demonstrate the important role of CD30 signaling in the activation of Vγ1− Vγ4− γδ T cells bearing Vγ6 producing IL-17A in the innate immune response to BCG infection.
MATERIALS AND METHODS
Mice.
C57BL/6 (B6) male mice were purchased from Japan KBT Inc. (Shizuoka, Japan). CD30 knockout (KO) (B6.129P2-Tnfrsf8<tm1Mak>/J) mice were purchased from the Jackson Laboratory. The generation and preliminary characterization of CD30L KO (BALB/c background) mice were described previously (41), and those mice were backcrossed for 10 or more generations to B6 mice. All mice were maintained under specific pathogen-free conditions and were offered food and water ad libitum. Age- and gender-matched mice were used for all experiments. This study was approved by the Committee of Ethics on Animal Experiments in the Faculty of Medicine, Kyushu University. Experiments were carried out under the control of the Guidelines for Animal Experiments.
Microorganisms.
Lyophilized M. bovis BCG (Tokyo strain) was purchased from Kyowa Pharmaceuticals and dissolved in 7H9 medium (Difco) supplemented with albumin-dextrose-catalase enrichment (Difco). The viable bacterial numbers were determined using a 7H10 (Difco) plate supplemented with oleic acid-albumin-dextrose-catalase enrichment (Difco). Small aliquots of BCG suspended in 7H9 medium containing 20% glycerol were stored at −80°C until use. Before use, the bacteria were washed twice with phosphate-buffered saline (PBS) containing 0.05% Tween 80 and resuspended in PBS. Mice were infected i.p. with 1 × 106 CFU of BCG in 200 μl of PBS.
Bacterial growth.
At the indicated time after infection, PEC were subjected to lavage with 1 ml of ice-cold Hanks balanced salt solution (HBSS) and harvested after gentle massage. The spleen and liver were removed and separately placed in homogenizers containing 1 ml of HBSS. For BCG infection, those samples were spread on Middlebrook 7H10 medium enriched with oleic acid-albumin-dextrose-catalase, and colonies were counted after incubation for 3 weeks at 37°C.
Flow cytometry and antibodies.
Single cells were isolated from PEC as previously described (7). Briefly, PEC from infected mice were obtained by lavage with 5 ml of HBSS. After washing, PEC were preincubated with an Fcγ receptor-blocking monoclonal antibody (MAb) (CD16/32 [clone 2.4G2]; BD Biosciences, San Diego, CA) for 20 min at 4°C to prevent antibody binding to the Fc receptor. The total cells were then stained with various combinations of MAbs. We purchased the following MAbs: anti-CD11b-fluorescein isothiocyanate (FITC) (clone M1/70; BD Biosciences), anti-NK1.1-phycoerythrin (PE) (clone PK136; BD Biosciences), anti-F4/80-allophycocyanin (APC) (clone BM8; eBioscience, San Diego, CA), anti-Gr1-PE (clone RB6-8C5; BD Biosciences), anti-CD45R-APC (clone RA3-6B2; eBioscience), anti-major histocompatibility complex (MHC) class II-peridinin chlorophyll protein (PerCP)-eFluor 710 (clone M5/114.15.2; eBioscience), anti-CD3e-PE (clone 145-2C11; Biolegend, San Diego, CA), anti-T cell receptor beta (TCRβ)-PE (clone H57-597; eBioscience), anti-CD4-PerCP or anti-CD4-APC (clone RM4-5; BD Biosciences), anti-CD8α-PerCP or anti-CD8α-FITC (clone RM53-6.7; BD Biosciences), anti-TCRγδ-APC (clone GL3; Biolegend), anti-TCR Vγ1.1-PE or anti-TCR Vγ1.1-FITC (clone 2.11; Biolegend), anti-TCR Vγ2-PE or anti-TCR Vγ2-FITC (clone UC3-10A6; Biolegend), anti-CD30-PE (clone Mcd30.1; BD Biosciences), and anti-CD153-PE (clone RM153; BD Biosciences). The stained cells were acquired and analyzed in a FACSCalibur flow cytometer (BD Biosciences). The data were analyzed using CellQuest software (BD Biosciences). For flow cytometry, neutrophils were defined as CD11b+ Gr-1high F4/80low and macrophages as CD11b+ F4/80high Gr-1low (see Fig. S1 in the supplemental material).
Cell purification.
PEC from BCG-infected wild-type (WT) mice were isolated as previously described (7) and prepared in RPMI 1640 containing 10% fetal bovine serum (FBS), 100 U/ml penicillin, 100 μl/ml streptomycin, and 50 μm 2-mercaptoethanol (2-ME). After staining with anti-MHC class II-PerCP-eFluor 710 (clone M5/114.15.2; eBioscience), anti-TCRγδ-PE (clone eBioGL3; eBioscience), and a mixture of anti-TCR Vγ1-FITC (clone 2.11; Biolegend) and anti-TCR Vγ4-FITC (clone UC3-10A6; Biolegend) MAbs, γδ T cell subsets in PEC were confirmed by FACSCalibur flow cytometry (BD Biosciences, San Diego, CA). Next, Vγ1− Vγ4− and Vγ1+ Vγ4+ γδ T cells were sorted respectively by analysis using a fluorescence-activated cell sorter (FACS) Aria II system (BD Biosciences, San Diego, CA) at a purity of more than 97%. In some experiments, γδ T cells in PEC from WT or CD30 KO mice on day 7 after BCG infection were sorted using a FACS Aria II system at a purity of more than 95%.
Intracellular cytokine staining.
PEC were suspended in RPMI 1640 containing 10% FBS, 100 U/ml penicillin, 100 μl/ml streptomycin, and 50 μm 2-ME. PEC were stimulated with 25 ng/ml phorbol myristate acetate (PMA) (P-8139; Sigma-Aldrich) and 1 μg/ml ionomycin (I-0634; Sigma-Aldrich) for 5 h at 37°C in 5% CO2 or left unstimulated. Brefeldin A (BFA) (Sigma-Aldrich) (10 μg/ml) was added for the last 4 h. After incubation, the cells were washed with FACS buffer and surface stained with various combinations of MAbs for 30 min at 4°C. Intracellular staining was then performed according to the manufacturer's instructions (BD Biosciences). Briefly, 100 μl of BD Cytofix/Cytoperm solution (BD Biosciences) was added to the cell suspension with mild mixing and then placed for 20 min at 4°C. The fixed cells were washed twice with 500 μl of BD Perm/Wash buffer (BD Biosciences) and stained intracellularly with anti-IL-17A-PE (clone TC11-18H10; BD Biosciences) and anti-IFN-γ-FITC (clone XMG1.2; BD Biosciences) for 30 min at 4°C. After washing, the cells were analyzed on a FACSCalibur flow cytometer (BD Biosciences).
RNA purification and reverse transcription-PCR (RT-PCR).
Total RNA of the sorted γδ T cells was purified using an RNeasy minikit (Qiagen), and cDNA was synthesized using Superscript II (Invitrogen) according to the manufacturer's instructions. For analysis of the Vγ repertoire, combinations of the following primers were used. The forward primers were as follows: for Vγ1/2, 5′-ACACAGCTATACATTGGTAC-3′; for Vγ2, 5′-CGGCAAAAAACAAATCAACAG-3′; for Vγ4, 5′-TGTCCTTGCAACCCCTACCC-3′; for Vγ5, 5′-TGTCCTTGCAACCCCTACCC-3′; for Vγ6, 5′-GGAATTCAAAAGAAAACATTGTCT-3′; for Vγ7, 5′-AAGCTAGAGGGGTCCTCTGC-3′; for Vδ1, 5′-ATTCAGAAGGCAACAATGAAAG-3′; for Vδ2, 5′-AGTTCCCTGCAGATCCAAGC-3′; for Vδ3, 5′-TTCCTGGCTATTGCCTCTGAC-3′; for Vδ4, 5′-CCGCTTCTCTGTGAACTTCC-3′; for Vδ5, 5′-CAGATCCTTCCAGTTCATCC-3′; for Vδ6, 5′-TCAAGTCCATCAGCCTTGTC-3′; for Vδ7, 5′-CGCAGAGCTGCAGTGTAACT-3′; and for Vδ8, 5′-AAGGAAGATGGACGATTCAC-3′. The reverse primers were as follows: for Cγ, 5′-CTTATGGAGATTTGTTTCAGC-3′; and for Cδ, 5′-CGAATTCCACAATCTTCT-3′. For β-actin, the primers were 5′-TGGAATCCTGTGGCATCCATGAAAC-3′ and 5′-TAAAACGCAGCTCAGTAACAGTCCG-3′. PCR was performed on a PCR thermal cycler (TaKaRa Corp., Tokyo, Japan).
ELISAs.
Supernatants of PEC from the mice at the indicated times after BCG inoculation were obtained by centrifugation at 440 × g for 3 min at 4°C. IL-17A, IFN-γ, IL-6, IL-12 p70, TNF alpha (TNF-α), and IL-23 secretion in the supernatants was measured by using a DuoSet enzyme-linked immunosorbent assay (ELISA) development system (R&D Systems) according to the manufacturer's instructions.
In vivo treatment of mice with antibodies.
Agonistic anti-CD30 MAbs (clone 30.1) were obtained by growing hybridoma cells in Cell Line CL-1000 (BD Biosciences, San Diego, CA) with serum-free medium (medium 101; Nissui Pharmaceutical, Tokyo, Japan). Abs were collected using a HiTrap Protein G HP column (Amersham Biosciences, Piscataway, NJ). The purity of the preparation was confirmed by SDS-PAGE; the concentration of Abs was determined by the Lowry method. The MAbs, diluted to 1 mg/ml in PBS, were stored at −80°C until use. For in vivo activation, 100 μg of agonistic anti-CD30 MAb or isotype control Armenian hamster IgG1 (eBio299Arm; BD Biosciences) was injected i.p. into WT and CD30L KO mice simultaneously with bacterial infection.
Soluble CD30 and immunoglobulin fusion protein (CD30-Ig) was obtained using a previously described method (22). Secreted CD30-Ig protein obtained by growing NIH 3T3 cells transfected with CD30-Ig fusion cDNA expression vector pBMGNeo in serum-free medium (medium 101; Nissui Pharmaceutical) was purified using a HiTrap Protein G HP column (Amersham Biosciences, Piscataway, NJ) and analyzed by SDS-PAGE for purity. CD30-Ig, diluted to 1 mg/ml in PBS, was stored at −80°C until use. For in vivo neutralization, 200 μg of CD30-Ig or isotype control mouse IgG1 κ (MG1-45; Biolegend) was injected i.p. into WT mice simultaneously with bacterial infection.
Statistics.
Statistical significance was evaluated by Student's t test using Prism software (GraphPad, San Diego, CA). P values of <0.05 were considered to represent a significant difference.
RESULTS
Bacterial growth, cytokine production, and cell accumulation in PEC of CD30 KO mice after BCG infection.
We examined the bacterial growth in PEC, liver, and spleen at an early stage (on days 3, 7, and 14) after i.p. inoculation with 1 × 106 CFU of BCG in CD30 KO mice with a C57BL/6 background. As shown in Fig. 1A, the number of bacteria in PEC in CD30 KO mice was significantly higher than that in WT mice on day 7 after BCG infection, but there was no difference between WT and CD30 KO mice in liver and spleen.
Fig 1.

Kinetics of bacterial growth, cytokine production, and cell accumulation in PEC after i.p. infection with BCG. WT (B6) and CD30 KO mice were inoculated i.p. with 1 × 106 CFU of BCG. (A) On days 3, 7, and 14 postinfection, bacterial counts in PEC, liver, and spleen were determined. Data are shown as means and standard deviations (SD) of the results determined for five mice at each time point. Significant differences in P values were calculated using Student's t test (*, P < 0.05). (B) Secretion of IL-17A, IFN-γ, IL-23, IL-12 p70, IL-6, and TNF-α from the supernatants of PEC at the indicated time points after BCG inoculation was measured by ELISA. (C) Kinetics of absolute numbers of neutrophils and macrophages in PEC of infected mice as analyzed by flow cytometry. PEC were harvested on the indicated days after inoculation with BCG and surface stained with various MAbs. The absolute numbers of neutrophils (CD11b+ Gr-1high F4/80low) and macrophages (CD11b+ F4/80high Gr-1low) were calculated by multiplying the total number of PEC by the percentages of each subset in the PEC. Data in panels B and C are shown as means ± SD of the results determined for three mice at each time point. Significant differences in P values were calculated using Student's t test (*, P < 0.05; **, P < 0.01). Data are representative of three independent experiments.
We next monitored cytokine levels in PEC supernatants after infection with BCG. In WT mice, IL-17A secretion increased rapidly and reached a peak on day 7 after infection. The levels of IL-17A secretion decreased markedly in CD30 KO mice on days 3 and 7 after BCG infection compared to those in WT mice (Fig. 1B). IFN-γ secretion progressively increased after infection and appeared to be attenuated until 14 days postinfection (Fig. 1B). We also examined the kinetics of IL-23, which has been reported to promote IL-17A production by γδ T cells against E. coli or C. albicans infection (17, 42). However, IL-23 secretion was detected at low levels during the course of BCG infection, and there were no significant differences between CD30 KO and WT mice at those levels (Fig. 1B). In addition, there were also no significant differences between CD30 KO and WT mice in the levels of IL-12 p70, IL-6, and TNF-α in PEC supernatants during the early stage of BCG infection (Fig. 1B).
We also examined the kinetics of infiltration of leukocytes by FACS analysis in PEC on days 3, 7, and 14 after BCG infection. Consistent with increased IL-17A secretion, the number of CD11b+ Gr-1high F4/80low cells corresponding to neutrophils increased after BCG infection, and the frequency and absolute number of neutrophils decreased significantly in CD30 KO mice compared to WT mice on days 7 and 14 after infection (Fig. 1C; see also Fig. S1 in the supplemental material). The levels of CD11b+ F4/80high Gr-1low cells corresponding to macrophages also decreased in CD30 KO mice on days 7 and 14 after infection (Fig. 1C; see also Fig. S1).
Kinetics of γδ T cells in PEC of CD30 KO mice after BCG infection.
γδ T cells are reported to play a key role in the early immune response to BCG infection (43). In the next experiment, we investigated the kinetics of γδ T cells in CD30 KO mice at an early stage of BCG infection. As shown in Fig. 2A, the frequency and absolute number of γδ T cells reached a peak on day 7 after BCG infection, and cell frequency as well as cell number was significantly lower in CD30 KO mice than in WT mice on days 3, 7, and 14 after infection (Fig. 2A). In contrast, there was no significant difference between the two PEC preparations in the absolute numbers of αβ T cells, NK cells, NKT cells, or Β cells at an early stage of BCG infection (Fig. 2B).
Fig 2.

Kinetics of γδ T cell subsets in PEC of CD30 KO mice during the early stage of BCG infection. WT or CD30 KO mice were inoculated i.p. with 1 × 106 CFU of BCG, and PEC were harvested on days 0, 3, 7, and 14 post-BCG infection. (A) The frequencies and absolute numbers of γδ T cells in PEC. (B) Absolute numbers of αβ TCR+ cells, NK1.1+ CD3− cells, NK1.1+ CD3+ T cells, and B220+ CD3− cells in PEC. (C and D) Frequencies (C) and absolute numbers (D) of Vγ1− Vγ4−, Vγ1+, and Vγ4+ γδ T cells in PEC. The analysis gate was set on γδ TCR+ cells. The absolute number of each subset of γδ T cells was calculated by multiplying the total γδ T cell number by the percentage in each dot plot figure. The numbers on the dot plots are expressed as means and SD of the results determined for at least three mice per group and per day, with data from one representative mouse shown. Student's t tests were performed to determine statistical differences between the results from any two groups (*, P < 0.05; **, P < 0.01). Data are representative of three independent experiments.
To further determine which Vγ repertoire of γδ T cells is affected in CD30 KO mice, each subset of Vγ from PEC was assessed by staining with available anti-Vγ1 and Vγ4 MAbs. It was found that the Vγ1− Vγ4− subset was the major subset of γδ T cells distributed in PEC during the early stage of BCG infection and that their frequency and absolute number were significantly lower in CD30 KO mice than in WT mice on days 3, 7, and 14 after BCG infection, while the absolute numbers in the Vγ1+ or Vγ4+ γδ T cell subset in CD30 KO mice did not differ from those in WT mice during this early phase of infection (Fig. 2C and D). Therefore, it is suggested that Vγ1− Vγ4− γδ T cells selectively showed a poor response in PEC of CD30 KO mice after BCG infection.
Because of the lack of availability of a Vγ6-specific MAb, we identified the repertoire of Vγ1− Vγ4− γδ T cells expanding in the peritoneal exudate of mice infected with BCG by RT-PCR. After staining with anti-TCRγδ MAb and a mixture of anti-Vγ1 and anti-Vγ4 MAbs, Vγ1− Vγ4− and Vγ1+ Vγ4+ γδ T cell populations were clearly confirmed by flow cytometry and then these two subsets were purified at a purity of more than 97% (Fig. 3A). As shown in Fig. 3B, the Vγ1− Vγ4− subset of γδ T cells exclusively expressed Vγ2 and Vγ6 of the Vγ gene transcripts and Vδ1 of the Vδ gene transcripts. In contrast, the Vγ1+ Vγ4+ subset expressed Vγ1, Vγ2, and Vγ4 of the Vγ gene transcripts and Vδ4, Vδ5, and Vδ7 of the Vδ gene transcripts. Since Vγ2 transcript is generally expressed in a single γδ T cell clone expressing other Vγ and Vγ6 chains exclusively paired with Vδ1 (3, 44), these observations proved that Vγ1− Vγ4− γδ T cells in peritoneal exudate preferentially expressed the Vγ6 chain paired with Vδ1. Next, total RNA was extracted from γδ T cells sorted from PEC of mice infected with BCG, and TCR V gene expression was analyzed by RT-PCR. As shown in Fig. 3C, γδ T cells in the peritoneal exudate of WT mice expressed high levels of TCR Vγ1, Vγ2, Vγ4, and Vγ6 mRNA but lacked expression of TCR Vγ5 or Vγ7 mRNA. Notably, only TCR Vγ6 and Vδ1 expression levels were downregulated in CD30 KO mice after BCG infection. Therefore, these findings further confirmed that CD30 signaling was required for regulating functions of Vγ1− Vγ4− γδ T cells bearing Vγ6 in peritoneal exudate after BCG infection.
Fig 3.

Identification of the TCR Vγ and Vδ gene repertoire of γδ T cells by RT-PCR. (A) γδ T cell subsets in PEC from WT mice (n = 5) that had been i.p. infected with BCG 7 days previously were confirmed by FACSCalibur flow cytometry. The numbers on the dot plot (left) indicate the percentages of Vγ1+ Vγ4+ or Vγ1− Vγ4− γδ T cells in PEC. These two subsets of γδ T cells were then sorted respectively using a FACS Aria II system (purity > 97%). (B) The TCR Vγ and Vδ gene repertoires of Vγ1− Vγ4− or Vγ1+ Vγ4+ γδ T cells were analyzed by RT-PCR and agarose gel electrophoresis. (C) Total RNA was extracted from γδ T cells sorted from PEC of WT or CD30 KO mice (n = 5) that had been infected with BCG 7 days previously at a purity of more than 95%, and TCR Vγ and Vδ gene expression levels were analyzed by RT-PCR and agarose gel electrophoresis. mRNA levels were normalized using β-actin mRNA as the reference. Data are representative of three independent experiments.
Kinetics of IL-17A- or IFN-γ-producing T cells in PEC of CD30 KO mice after BCG infection.
We next examined the ability of γδ T cells or αβ T cells to produce cytokines after BCG infection by intracellular cytokine FACS analysis. Consistent with our previous report (7), IL-17A-producing γδ T cells could be detected in PEC of naive WT mice even in the absence of PMA and ionomycin stimulation; the numbers of such “innate” IL-17A-producing γδ T cells increased immediately after BCG infection and reached a peak on day 7 and then returned to the baseline by day 14 after BCG infection. The frequency and absolute number of the IL-17A+ γδ T cells markedly decreased in CD30 KO mice compared to WT mice on days 3, 7, and 14 after BCG infection (Fig. 4A; see also Fig. S2A in the supplemental material). Such a difference between WT and CD30 KO mice was more obvious in IL-17A+ γδ T cells in response to PMA and ionomycin stimulation (Fig. 4A; see also Fig. S2A). In contrast, very few CD4+ αβ T cells producing IL-17A were detected during the early stage of BCG infection without further stimulation (Fig. 4A; see also Fig. S2B). These results indicated that IL-17A production in PEC was mainly derived from γδ T cells during the early stage of BCG infection and that CD30 signaling plays an important role in the activation of IL-17A-producing γδ T cells in the early response to BCG infection.
Fig 4.

Kinetics of IL-17A- or IFN-γ-producing T cells in PEC of CD30 KO mice during the early stage of BCG infection. WT or CD30 KO mice infected i.p. with 1 × 106 CFU of BCG and PEC were harvested on days 0, 3, 7, and 14 postinfection. Cells were cultured for 5 h without (−) or with PMA/ionomycin (P/I) stimulation, and BFA was added for the last 4 h. After incubation, intracellular flow cytometric analysis was performed on PEC to measure the IL-17A-producing (IL-17A+) or IFN-γ-producing (IFN-γ+) T cells. (A) Absolute numbers of IL-17A+ γδ T or IL-17A+ CD4 T cells without P/I stimulation (top panels) or with P/I stimulation (botton panels). (B) Absolute numbers of IFN-γ+ γδ T or IFN-γ+ CD4 T cells without P/I stimulation (top panel) or with P/I stimulation (bottom panel). (C and D) Frequencies (C) and absolute numbers (D) of IL-17A+ Vγ1− Vγ4− γδ T cells without or with P/I stimulation. (E and F) Frequencies (E) and absolute numbers (F) of IFN-γ+ Vγ1− Vγ4− or IFN-γ+ Vγ1+ Vγ4+ γδ T cells with P/I stimulation. Data are shown after gating on γδ TCR+ cells. The numbers on the dot plots are expressed as means and SD of the results determined for at least three mice per group and per day, with data from one representative mouse shown. Student's t tests were performed to determine statistical differences between two groups (*, P < 0.05; **, P < 0.01). Data are representative of three independent experiments.
Next, we examined the kinetics of IFN-γ-producing T cells in PEC after BCG infection. IFN-γ production was hardly detected by analysis of either γδ T cells or CD4+ αβ T cells in the absence of PMA/ionomycin stimulation during the first 7 days of BCG infection (Fig. 4B). As BCG infection progressed, the numbers of both γδ T cells and CD4+ αβ T cells producing IFN-γ increased; the numbers of IFN-γ+ CD4+ αβ T cells were apparently over 10 times greater than those of IFN-γ+ γδ T cells on day 14 after BCG infection in the absence or presence of PMA/ionomycin stimulation, suggesting that these sharply increased CD4+ αβ T cells serve as the main producer of IFN-γ in PEC to participate in the following immune response to BCG infection (Fig. 4B). The frequency and absolute number of IFN-γ+ CD4+ αβ T cells significantly decreased in CD30 KO mice compared to WT mice on day 14 after BCG infection in the absence or presence of PMA/ionomycin stimulation (Fig. 4B; see also Fig. S2B in the supplemental material). Thus, consistent with our previous report (36), CD30 signaling was also involved in the activation of IFN-γ-producing CD4+ αβ T cells at a relatively late stage of BCG infection.
To further analyze the Vγ repertoire of IL-17A- or IFN-γ-producing γδ T cells, PEC isolated from BCG-infected mice were surface stained with anti-TCRγδ MAb and a mixture of anti-TCR Vγ1 and Vγ4 MAbs and then intracellularly stained with anti-IL-17A or anti-IFN-γ. As shown in Fig. 4C, most of the IL-17A-producing γδ T cells were Vγ1− Vγ4− γδ T cells bearing Vγ6. Compared to the results seen with WT mice, the frequency and cell number of IL-17A+ Vγ1− Vγ4− γδ T cells in PEC of CD30 KO mice significantly decreased on days 3, 7, and 14 after infection (Fig. 4C and D). Given that Vγ1+ γδ T cells do not produce IL-17A in response to BCG infection (reference 15 and data not shown), the IL-17A+ Vγ1+ Vγ4+ γδ T cells (i.e., the Vγ4+ γδ T cells producing IL-17A) showed a relatively higher frequency in CD30 KO mice in the presence of PMA/ionomycin. However, there was no significant difference between CD30 KO mice and WT mice in the absolute numbers of IL-17A+ Vγ4+ γδ T cells in PEC after BCG infection (data not shown). These results indicated that CD30 signaling selectively promotes the activation of IL-17A-producing Vγ1− Vγ4− γδ T cells bearing Vγ6 in PEC of mice after BCG infection. Next, by analyzing the Vγ repertoire of IFN-γ-producing γδ T cells, we found that not only Vγ1− Vγ4− but also Vγ1+ Vγ4+ γδ T cells in PEC produced IFN-γ upon PMA/ionomycin stimulation and that these two subsets of γδ T cells producing IFN-γ were significantly reduced in number in CD30 KO mice on day 14 after BCG infection (Fig. 4E and F).
CD8+ αβ T cells are also reported to participate in immune responses to M. tuberculosis (45). Recently, IL-17-producing CD8+ αβ T cells (Tc17 cells) have been identified in mice and their enrichment in the antitumor immune response has been reported (46, 47). Therefore, we examined CD8+ αβ T cells in the early phase of BCG infection. As shown in Fig. S3 in the supplemental material, IL-17A+ CD8+ αβ T cells were not detected in mice infected with BCG even in the presence of PMA and ionomycin stimulation, and there was no significant difference between CD30 KO and WT mice in the percentages and cell numbers of IFN-γ-producing CD8+ αβ T cells after BCG infection. Thus, it appears that CD30 signaling may not contribute to the early activation of CD8+ αβ T cells after BCG infection.
Taken together, these results indicate that CD30 signaling was primarily involved in the activation of IL-17A-producing γδ T cells, mainly, Vγ1− Vγ4− γδ T cells bearing Vγ6, in the early immune response to BCG infection. In addition, it also contributes to the activation of IFN-γ-producing CD4+ αβ T cells in the peripheral tissues at a relatively late stage of BCG infection.
Expression of CD30L and CD30 on peritoneal γδ T cells after BCG infection.
To further demonstrate that CD30L/CD30 signaling was selectively involved in the activation of Vγ1− Vγ4− γδ T cells, we examined the expression of CD30L and CD30 on the γδ T cells of PEC from WT mice after BCG infection. As shown in Fig. 5, CD30L and CD30 were predominantly expressed on Vγ1− Vγ4− γδ T cells presumably bearing Vγ6 of PEC after BCG infection. The expression levels of both CD30L and CD30 were rapidly upregulated in mice previously infected with BCG compared to those in naive mice and were further enhanced on γδ T cells after 24 h culture in vitro without any stimulation (Fig. 5A and B). It should be noted that the numbers of CD30L-expressing Vγ1− Vγ4− γδ T cells and CD30-expressing Vγ1− Vγ4− γδ T cells were much greater than those of CD30L+ Vγ1+ or Vγ4+ T cells and CD30+ Vγ1+ and Vγ4+ γδ T cells, respectively, on specific days after BCG infection (Fig. 5C).
Fig 5.

Expression of CD30L and CD30 on γδ T cells of PEC. Cells were isolated from PEC of naive mice or mice inoculated i.p. with 1 × 106 CFU of BCG. CD30L and CD30 expression on subsets of γδ T cells before and/or after 24 h of culture in RPMI medium without any stimulation was analyzed by flow cytometry. (A and B) Kinetics of CD30L expression (A) and CD30 expression (B) on the repertoire of γδ T cells in PEC after BCG infection. Data are expressed as the increase in expression level over that before infection. Each number in a quadrant indicates the percentage of each quadrant. The numbers on the dot plots are expressed as means of the results determined for three mice per group and per day, with data from one representative mouse shown. (C) Absolute numbers of subsets of CD30L+ or CD30+ γδ T cells of PEC on indicated day after BCG infection. Data are representative of two independent experiments.
Since CD30L and CD30 are reported to be expressed on cells other than γδ T cells, we additionally examined their expression on F4/80high CD11b+ CD3− macrophages, CD11c+ CD3− DCs, B220+ CD3− B cells, NK1.1+ CD3− NK cells, NK1.1+ CD3+ NKT cells, and CD4+/CD8+ CD3+ T cells in PEC from BCG-infected WT mice, and none of these cells exhibited detectable CD30 expression on cell surfaces (data not shown). Similarly, only a few NK cells, NKT cells, and CD4+/CD8+ T cells expressed CD30L. Neither macrophages nor DCs nor B cells from PEC expressed CD30L after BCG infection (data not shown). Taken together, these results strongly suggested that CD30L/CD30 signaling plays an important role in controlling the innate immune response to BCG by the specific Vγ1− Vγ4− subset-bearing Vγ6 γδ T cells.
Effects of soluble CD30-Ig on IL-17A-producing γδ T cells in PEC of WT mice infected with BCG.
To further verify that CD30 signaling plays an important role in the activation of IL-17A-producing γδ T cells after infection, we examined the effect of the inhibition of CD30 signaling caused by administering soluble CD30-Ig in vivo on the activation of IL-17A-producing γδ T cells after BCG infection in WT mice. Mice administered CD30-Ig showed markedly decreased numbers of IL-17A-producing γδ T cells (Fig. 6A and B), mainly, IL-17A+ Vγ1− Vγ4− γδ T cells (Fig. 6C and D), in PEC after BCG infection, while there were no significant changes in the frequency and absolute number of IFN-γ-producing γδ T cells in CD30-Ig-treated mice (Fig. 6A and B). These results suggested that de novo synthesis of CD30 is important for the activation of the early-appearing γδ T cells producing IL-17A after BCG infection.
Fig 6.
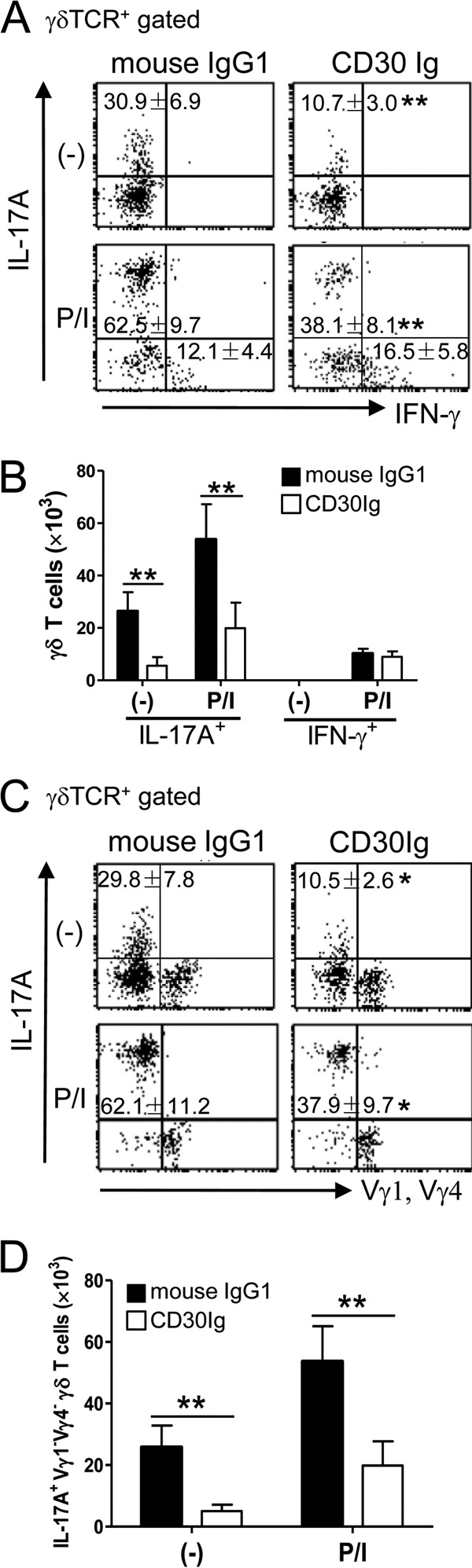
Effects of in vivo treatment with CD30-Ig on early activation of IL-17A+ γδ T cells after BCG infection. WT mice were inoculated i.p. with 1 × 106 CFU of BCG and simultaneously treated with CD30-Ig or mouse IgG1 (200 mg/head) as a control. On day 3 postinfection, PEC were harvested and cultured with or without PMA/ionomycin (P/I) stimulation for 5 h, and BFA was added for the last 4 h. After incubation, IL-17A and IFN-γ production of γδ T cells was measured by flow cytometry. (A and B) Frequencies (A) and absolute numbers (B) of IL-17A+ or IFN-γ+ γδ T cells in PEC. (C and D) Frequencies (C) and absolute numbers (D) of IL-17A+ Vγ1− Vγ4− γδ T cells in PEC. Data are shown after gating on γδ TCR+ cells. The numbers on the dot plots are expressed as means and SD of the results determined for five mice per group, with data from one representative mouse shown. Significant differences were calculated by Student's t test (*, P < 0.05; **, P < 0.01). Data are representative of three independent experiments.
Effects of agonistic anti-CD30 MAb on IL-17A-producing γδ T cells in PEC of mice infected with BCG.
Since CD30L is the exclusive ligand for CD30, CD30L KO mice show similar phenotypes in terms of susceptibility to bacterial infection or inflammatory diseases (38, 41, 48). We found that the appearance of IL-17A-producing γδ T cells was impaired in PEC of CD30L KO mice on day 7 after BCG infection. Administration of agonistic anti-CD30 MAb not only restored the early appearance of IL-17A-producing γδ T cells in CD30L KO mice but also stimulated these cells in WT mice (Fig. 7A and B). Furthermore, the frequency and absolute number of IL-17A+ Vγ1− Vγ4− γδ T cells significantly increased in both CD30L KO and WT mice administered agonistic anti-CD30 MAb (Fig. 7C and D). There were no differences in the IFN-γ-producing γδ T cells between agonistic anti-CD30 MAb-treated mice and the control group in WT/CD30L KO mice (Fig. 7A and B). These results strongly prove that CD30L/CD30 signaling selectively contributes to the activation of the Vγ1− Vγ4− subset of γδ T cells bearing Vγ6, which is the main source of IL-17A during the early stage of BCG infection.
Fig 7.

Effects of in vivo treatment with agonistic anti-CD30 MAb on early activation of IL-17A+ γδ T cells after BCG infection. WT or CD30L KO mice were infected i.p. with 1 × 106 CFU of BCG and simultaneously treated with agonistic anti-CD30 MAb (clone CD30.1) or hamster IgG1 (100 mg/head) as a control. IL-17A or IFN-γ production of γδ T cells was measured in PEC on day 3 postinfection by flow cytometry. (A and B) Frequencies (A) and absolute numbers (B) of IL-17A+ or IFN-γ+ γδ T cells in PEC. (C and D) Frequencies (C) and absolute numbers (D) of IL-17A+ Vγ1− Vγ4− γδ T cells in PEC. Data are shown after gating on γδ TCR+ cells. The numbers on the dot plots are expressed as means and SD of the results determined for four mice per group, with data from one representative mouse shown. Significant differences were calculated by Student's t test (*, P < 0.05; **, P < 0.01). Data are representative of three independent experiments.
DISCUSSION
IL-17A-producing γδ T cells are known to be activated at an early stage of BCG infection (13). Here we show that the appearance of IL-17A-producing γδ T cells preferentially expressing TCR Vγ6/Vδ1 genes was selectively impaired in PEC of CD30 KO mice at an early stage of BCG infection. CD30L or CD30 expression was upregulated predominantly on a Vγ1− Vγ4− subset of γδ T cells in PEC after BCG infection. In vivo blocking of CD30 signaling by CD30-Ig inhibited the appearance of IL-17A-producing Vγ1− Vγ4− γδ T cells in WT mice, whereas in vivo stimulation of CD30 signaling with agonistic anti-CD30 MAb increased the number of IL-17A-producing Vγ1− Vγ4− γδ T cells after infection in CD30L KO mice as well as WT mice. Taken together, IL-17A-producing Vγ1− Vγ4− γδ T cells bearing Vγ6 may be activated in a CD30L/CD30-dependent manner as an innate immunity against BCG infection.
A noteworthy finding in the present study is that CD30 signaling plays an important role in the activation of the subset of IL-17A-producing Vγ1− Vγ4−-bearing Vγ6 γδ T cells in innate immunity against BCG infection. There are several potential mechanisms that may explain the novel role of CD30 signaling in regulating IL-17A response by the unique subset of γδ T cells in the early response to BCG infection. CD30 is a member of the TNF receptor family that has the potential to induce TNF receptor-associated factor 2 (TRAF-2)-mediated NF-κB activation and can recruit TRAF-1 (49–51). It probably promotes T cell survival (50, 51). Thus, we speculate that CD30 signaling may enhance γδ T cell survival after BCG infection. It is widely accepted that Toll-like receptors (TLRs) are responsible for the innate immune response and act by recognizing bacterial products that induce proinflammatory cytokines and chemokine production. Martin et al. (52) recently reported that IL-17A-producing γδ T cells, which express TLR2 as well as TLR1 and dectin-1, selectively expanded in the early response to M. tuberculosis invasion. A previous study has shown that Vγ6+ Vδ1+ γδ T cells in PEC express TLR2 mRNA (53). In the present study, we found that expression of CD30L or CD30 was upregulated selectively on a subset of Vγ1− Vγ4−-bearing Vγ6 γδ T cells after BCG infection. Therefore, it is alternatively possible that CD30 signaling in synergy with TLR2 plays an important role in the activation of IL-17A-producing Vγ1− Vγ4−-bearing Vγ6 γδ T cells in the innate immune response to BCG infection. In addition, we previously reported that early IL-23 production and IL-23-triggered Tyk2 signaling were essential for IL-17A production by γδ T cells after E. coli or C. albicans infection (17, 42). However, in this study, there was no significant difference between CD30 KO and WT mice in the level of IL-23 secretion in PEC after BCG infection. Moreover, expression of CD30L or CD30 was not detected on either macrophages or DCs. Therefore, impaired IL-17A production in CD30 KO mice may not be caused by impaired accessory functions of macrophages/DCs.
Here we found that the expression level of CD30L or CD30 on a subset of Vγ1− Vγ4−-bearing Vγ6 γδ T cells was rapidly upregulated after BCG infection. It remains unknown why the Vγ1− Vγ4− subset selectively expressed CD30L/CD30. We have recently reported that Hes 1 induced by Notch signaling, which is expressed in IL-17A-producing γδ T cells, can regulate the IL-17A-producing function of γδ T cells in the peripheral tissues (54). Notch signaling is capable of inducing IL-7Rα expression in T cells (55), and mediating IL-7 signaling can promote CD30L expression (56). More recently, IL-17A-producing γδ T cells were reported to express high levels of IL-7Rα on their surface (57). Here we found that the Vγ1− Vγ4− γδ T cells, which preferentially express Vγ6/Vδ1 genes, act as the major IL-17A-producing γδ T cells predominantly expanded in the early response to BCG infection. Therefore, it is possible that the Notch-Hes/IL-7Rα axis may contribute to induce CD30L/CD30 expression preferentially on IL-17A-producing Vγ1− Vγ4− γδ T cells bearing Vγ6 in response to BCG infection. On the other hand, Vγ6+ Vδ1+ γδ T cells induced by infection bear truly invariant TCRs in a nucleotide canonical sequence with no apparent N-region contribution (44, 58, 59). A recent report showed that γδ T cells can recognize a microbially encoded B cell antigen and induce IL-17A production (60), raising the possibility that Vγ6+ Vδ1+ γδ T cells have the capability to recognize some specific self-Ags induced by mycobacterial infection to initiate a swift IL-17 response. At present, whether IL-17A-producing γδ T cells recognize such a unique Ag with a restricted Vγ gene repertoire is still unknown.
In the present study, we observed that γδ T cells capable of producing IL-17A precede those producing IFN-γ in appearance following BCG infection. Furthermore, γδ T cells have the capacity to produce IL-17A without further stimulation, while they required in vitro stimulation with PMA/ionomycin for IFN-γ production. It has been recently reported that Ag-naive γδ T cells produce IL-17A, while Ag-experienced γδ T cells produce IFN-γ (6). More recently, IL-17A+ γδ T cells were reported to develop earlier than IFN-γ+ γδ T cells from double-negative thymocytes in ontogeny (61). In this regard, it is possible that γδ T cells producing IL-17A may precede γδ T cells producing IFN-γ in appearance following BCG infection, which may recapitulate their ontogeny.
We previously reported that “innate” CD4+ αβ T cells producing IFN-γ are activated and play a role in early protection against Listeria monocytogenes (48). However, in the present study, the “innate” CD4+ αβ T cells producing IFN-γ were hardly detected during the first 7 days of BCG infection without further stimulation. Instead, IFN-γ+ CD4+ αβ T cells, here speculated to correspond to M. tuberculosis-specific Th1 cells, activated/proliferated in PEC as infection progressed and served as the overwhelming source of IFN-γ to join in the following response to BCG infection. We found here that CD30 signaling was involved in the activation of IFN-γ-producing CD4+ αβ T cells, which is consistent with our previous report on the Ag-specific Th1 response to BCG infection (36). Additionally, we also found that numbers of not only the Vγ1− Vγ4− subset but also the Vγ1+ Vγ4+ subset of γδ T cells producing IFN-γ were reduced, though to a very low degree, in CD30-deficient mice on day 14 after BCG infection. Since Vγ1+ or Vγ4+ γδ T cells marginally expressed CD30L or CD30 after BCG infection, given previous reports that innate production of IL-17A plays an important role in priming of the response of IFN-γ-producing T cells following BCG infection (14, 62), it is possible that the reduction of the numbers of IFN-γ-producing γδ T cells in CD30 KO mice may have been due to the attenuated production of IL-17A but may not directly link to CD30 ablation. Taking the results together, our report suggests that CD30 signaling-dependent activation of γδ T cells producing IL-17A may promote a subsequent expansion of IFN-γ-producing T cells that predominantly drives a Th1 response to BCG infection in the peripheral tissues.
It may alternatively be possible that CD30 signaling is necessary for intrathymic differentiation of innate γδ T cells since basal levels of innate IL-17A-producing Vγ1− Vγ4− γδ T cells were lower in the peritoneal cavity of naive CD30 KO mice. However, in the present study, inhibition of CD30L/CD30 signaling by administering soluble CD30-Ig in vivo markedly decreased the numbers of IL-17A-producing Vγ1− Vγ4− γδ T cells among PEC of WT mice after BCG infection. Furthermore, stimulating CD30L/CD30 signaling by administering agonistic anti-CD30 MAb restored IL-17A production by Vγ1− Vγ4− γδ T cells in CD30L KO mice after BCG infection. These results suggested that de novo synthesis of CD30 is important for the activation of the early-appearing γδ T cells producing IL-17A after BCG infection.
In conclusion, our report suggests that activation of IL-17A-producing Vγ1− Vγ4− γδ T cells bearing TCR Vγ6 leading the innate immune response to BCG infection is controlled by a CD30L/CD30-dependent mechanism.
Supplementary Material
ACKNOWLEDGMENTS
We thank Mihoko Ohkubo, Miki Kijima, and Akiko Yano for their excellent technical assistance.
This work was supported by a Grant-in-Aid for Scientific Research from the Japan Society for Promotion of Science; by the program of Founding Research Centers for Emerging and Reemerging Infectious Diseases launched as a project commissioned by the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan; and by the Takeda Science Foundation (to Y.Y.).
We have no financial conflict of interest.
This work was performed in Division of Host Defense, Medical Institute of Bioregulation, Kyushu University, Fukuoka, Japan.
Footnotes
Published ahead of print 5 August 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.00887-13.
REFERENCES
- 1.Heilig JS, Tonegawa S. 1986. Diversity of murine gamma genes and expression in fetal and adult T lymphocytes. Nature 322:836–840 [DOI] [PubMed] [Google Scholar]
- 2.Havran WL, Allison JP. 1988. Developmentally ordered appearance of thymocytes expressing different T-cell antigen receptors. Nature 335:443–445 [DOI] [PubMed] [Google Scholar]
- 3.Hayday AC. 2000. [gamma][delta] cells: a right time and a right place for a conserved third way of protection. Annu. Rev. Immunol. 18:975–1026 [DOI] [PubMed] [Google Scholar]
- 4.Bonneville M, O'Brien RL, Born WK. 2010. Gammadelta T cell effector functions: a blend of innate programming and acquired plasticity. Nat. Rev. Immunol. 10:467–478 [DOI] [PubMed] [Google Scholar]
- 5.Ribot JC, deBarros A, Pang DJ, Neves JF, Peperzak V, Roberts SJ, Girardi M, Borst J, Hayday AC, Pennington DJ, Silva-Santos B. 2009. CD27 is a thymic determinant of the balance between interferon-gamma- and interleukin 17-producing gammadelta T cell subsets. Nat. Immunol. 10:427–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jensen KD, Su X, Shin S, Li L, Youssef S, Yamasaki S, Steinman L, Saito T, Locksley RM, Davis MM, Baumgarth N, Chien YH. 2008. Thymic selection determines gammadelta T cell effector fate: antigen-naive cells make interleukin-17 and antigen-experienced cells make interferon gamma. Immunity 29:90–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shibata K, Yamada H, Nakamura R, Sun X, Itsumi M, Yoshikai Y. 2008. Identification of CD25+ gamma deltaT cells as fetal thymus-derived innate IL-17 producers. J. Immunol. 181:5940–5947 [DOI] [PubMed] [Google Scholar]
- 8.Turchinovich G, Hayday AC. 2011. Skint-1 identifies a common molecular mechanism for the development of interferon-gamma-secreting versus interleukin-17-secreting gammadelta T cells. Immunity 35:59–68 [DOI] [PubMed] [Google Scholar]
- 9.Ye P, Rodriguez FH, Kanaly S, Stocking KL, Schurr J, Schwarzenberger P, Oliver P, Huang W, Zhang P, Zhang J, Shellito JE, Bagby GJ, Nelson S, Charrier K, Peschon JJ, Kolls JK. 2001. Requirement of interleukin 17 receptor signaling for lung CXC chemokine and granulocyte colony-stimulating factor expression, neutrophil recruitment, and host defense. J. Exp. Med. 194:519–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang W, Na L, Fidel PL, Schwarzenberger P. 2004. Requirement of interleukin-17A for systemic anti-Candida albicans host defense in mice. J. Infect. Dis. 190:624–631 [DOI] [PubMed] [Google Scholar]
- 11.Kolls JK, Lindén A. 2004. Interleukin-17 family members and inflammation. Immunity 21:467–476 [DOI] [PubMed] [Google Scholar]
- 12.Kikly K, Liu L, Na S, Sedgwick JD. 2006. The IL-23/Th(17) axis: therapeutic targets for autoimmune inflammation. Curr. Opin. Immunol. 18:670–675 [DOI] [PubMed] [Google Scholar]
- 13.Lockhart E, Green AM, Flynn JL. 2006. IL-17 production is dominated by gammadelta T cells rather than CD4 T cells during Mycobacterium tuberculosis infection. J. Immunol. 177:4662–4669 [DOI] [PubMed] [Google Scholar]
- 14.Umemura M, Yahagi A, Hamada S, Begum MD, Watanabe H, Kawakami K, Suda T, Sudo K, Nakae S, Iwakura Y, Matsuzaki G. 2007. IL-17-mediated regulation of innate and acquired immune response against pulmonary Mycobacterium bovis bacilli Calmette-Guerin infection. J. Immunol. 178:3786–3796 [DOI] [PubMed] [Google Scholar]
- 15.Okamoto Yoshida Y, Umemura M, Yahagi A, O'Brien RL, Ikuta K, Kishihara K, Hara H, Nakae S, Iwakura Y, Matsuzaki G. 2010. Essential role of IL-17A in the formation of a mycobacterial infection-induced granuloma in the lung. J. Immunol. 184:4414–4422 [DOI] [PubMed] [Google Scholar]
- 16.Shibata K, Yamada H, Hara H, Kishihara K, Yoshikai Y. 2007. Resident Vdelta1+ gammadelta T cells control early infiltration of neutrophils after Escherichia coli infection via IL-17 production. J. Immunol. 178:4466–4472 [DOI] [PubMed] [Google Scholar]
- 17.Dejima T, Shibata K, Yamada H, Hara H, Iwakura Y, Naito S, Yoshikai Y. 2011. Protective role of innate interleukin-17A-producing γδ T cells in the lung at the early stage of systemic Candidiasis in mice. Infect. Immun. 79:4503–4510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Takeuchi A, Dejima T, Yamada H, Shibata K, Nakamura R, Eto M, Nakatani T, Naito S, Yoshikai Y. 2011. IL-17 production by γδ T cells is important for the antitumor effect of Mycobacterium bovis bacillus Calmette-Guérin treatment against bladder cancer. Eur. J. Immunol. 41:246–251 [DOI] [PubMed] [Google Scholar]
- 19.Smith CA, Gruss HJ, Davis T, Anderson D, Farrah T, Baker E, Sutherland GR, Brannan CI, Copeland NG, Jenkins NA, Grabstein KH, Gliniak B, McAlister IB, Fanslow W, Alderson M, Falk B, Gimpel S, Gillis S, Din WS, Goodwin RG, Armitage RJ. 1993. CD30 antigen, a marker for Hodgkin's lymphoma, is a receptor whose ligand defines an emerging family of cytokines with homology to TNF. Cell 73:1349–1360 [DOI] [PubMed] [Google Scholar]
- 20.Shimozato O, Takeda K, Yagita H, Okumura K. 1999. Expression of CD30 ligand (CD153) on murine activated T cells. Biochem. Biophys. Res. Commun. 256:519–526 [DOI] [PubMed] [Google Scholar]
- 21.Kennedy MK, Willis CR, Armitage RJ. 2006. Deciphering CD30 ligand biology and its role in humoral immunity. Immunology 118:143–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lane PJ, Gaspal FM, Kim MY. 2005. Two sides of a cellular coin: CD4 (+) CD3- cells regulate memory responses and lymph-node organization. Nat. Rev. Immunol. 5:655–660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dürkop H, Latza U, Hummel M, Eitelbach F, Seed B, Stein H. 1992. Molecular cloning and expression of a new member of the nerve growth factor receptor family that is characteristic for Hodgkin's disease. Cell 68:421–427 [DOI] [PubMed] [Google Scholar]
- 24.Ellis TM, Simms PE, Slivnick DJ, Jäck HM, Fisher RI. 1993. CD30 is a signal-transducing molecule that defines a subset of human activated CD45RO+ T cells. J. Immunol. 151:2380–2389 [PubMed] [Google Scholar]
- 25.Bowen MA, Lee RK, Miragliotta G, Nam SY, Podack ER. 1996. Structure and expression of murine CD30 and its role in cytokine production. J. Immunol. 156:442–449 [PubMed] [Google Scholar]
- 26.Romagnani S, Del Prete G, Maggi E, Chilosi M, Caligaris-Cappio F, Pizzolo G. 1995. CD30 and type 2 T helper (Th2) responses. J. Leukoc. Biol. 57:726–730 [DOI] [PubMed] [Google Scholar]
- 27.Bengtsson A. 2001. The role of CD30 in atopic disease. Allergy 56:593–603 [DOI] [PubMed] [Google Scholar]
- 28.Tarkowski M. 2003. Expression and a role of CD30 in regulation of T-cell activity. Curr. Opin. Hematol. 10:267–271 [DOI] [PubMed] [Google Scholar]
- 29.Polte T, Behrendt AK, Hansen G. 2006. Direct evidence for a critical role of CD30 in the development of allergic asthma. J. Allergy Clin. Immunol. 118:942–948 [DOI] [PubMed] [Google Scholar]
- 30.Bengtsson A, Johansson C, Linder MT, Halldén G, van der Ploeg I, Scheynius A. 1995. Not only Th2 cells but also Th1 and Th0 cells express CD30 after activation. J. Leukoc. Biol. 58:683–689 [DOI] [PubMed] [Google Scholar]
- 31.Munk ME, Kern P, Kaufmann SH. 1997. Human CD30+ cells are induced by Mycobacterium tuberculosis and present in tuberculosis lesions. Int. Immunol. 9:713–720 [DOI] [PubMed] [Google Scholar]
- 32.Flórido M, Borges M, Yagita H, Appelberg R. 2004. Contribution of CD30/CD153 but not of CD27/CD70, CD134/OX40L, or CD137/4-1BBL to the optimal induction of protective immunity to Mycobacterium avium. J. Leukoc. Biol. 76:1039–1046 [DOI] [PubMed] [Google Scholar]
- 33.Gerli R, Lunardi C, Vinante F, Bistoni O, Pizzolo G, Pitzalis C. 2001. Role of CD30+ T cells in rheumatoid arthritis: a counter-regulatory paradigm for Th1-driven diseases. Trends Immunol. 22:72–77 [DOI] [PubMed] [Google Scholar]
- 34.Saraiva M, Smith P, Fallon PG, Alcami A. 2002. Inhibition of type 1 cytokine-mediated inflammation by a soluble CD30 homologue encoded by ectromelia (mousepox) virus. J. Exp. Med. 196:829–839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dai Z, Li Q, Wang Y, Gao G, Diggs LS, Tellides G, Lakkis FG. 2004. CD4+CD25+ regulatory T cells suppress allograft rejection mediated by memory CD8+ T cells via a CD30-dependent mechanism. J. Clin. Invest. 113:310–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tang C, Yamada H, Shibata K, Muta H, Wajjwalku W, Podack ER, Yoshikai Y. 2008. A novel role of CD30L/CD30 signaling by T-T cell interaction in Th1 response against mycobacterial infection. J. Immunol. 181:6316–6327 [DOI] [PubMed] [Google Scholar]
- 37.Sun X, Yamada H, Shibata K, Muta H, Tani K, Podack ER, Yoshikai Y. 2010. CD30 ligand/CD30 plays a critical role in Th17 differentiation in mice. J. Immunol. 185:2222–2230 [DOI] [PubMed] [Google Scholar]
- 38.Sun X, Yamada H, Shibata K, Muta H, Tani K, Podack ER, Iwakura Y, Yoshikai Y. 2010. CD30 ligand is a target for a novel biological therapy against colitis associated with Th17 responses. J. Immunol. 185:7671–7680 [DOI] [PubMed] [Google Scholar]
- 39.Flynn JL, Chan J. 2001. Immunology of tuberculosis. Annu. Rev. Immunol. 19:93–129 [DOI] [PubMed] [Google Scholar]
- 40.Kaufmann SH. 2001. How can immunology contribute to the control of tuberculosis? Nat. Rev. Immunol. 1:20–30 [DOI] [PubMed] [Google Scholar]
- 41.Blazar BR, Levy RB, Mak TW, Panoskaltsis-Mortari A, Muta H, Jones M, Roskos M, Serody JS, Yagita H, Podack ER, Taylor PA. 2004. CD30/CD30 ligand (CD153) interaction regulates CD4+ T cell-mediated graft-versus-host disease. J. Immunol. 173:2933–2941 [DOI] [PubMed] [Google Scholar]
- 42.Nakamura R, Shibata K, Yamada H, Shimoda K, Nakayama K, Yoshikai Y. 2008. Tyk2-signaling plays an important role in host defense against Escherichia coli through IL-23-induced IL-17 production by γδ T cells. J. Immunol. 181:2071–2075 [DOI] [PubMed] [Google Scholar]
- 43.Inoue T, Yoshikai Y, Matsuzaki G, Nomoto K. 1991. Early appearing gamma/delta-bearing T cells during infection with Calmette Guérin bacillus. J. Immunol. 146:2754–2762 [PubMed] [Google Scholar]
- 44.Lafaille JJ, DeCloux A, Bonneville M, Takagaki Y, Tonegawa S. 1989. Junctional sequences of T cell receptor gamma delta genes: implications for gamma delta T cell lineages and for a novel intermediate of V-(D)-J. joining. Cell 59:859–870 [DOI] [PubMed] [Google Scholar]
- 45.Flynn JL, Goldstein MM, Triebold KJ, Koller B, Bloom BR. 1992. Major histocompatibility complex class I-restricted T cells are required for resistance to Mycobacterium tuberculosis infection. Proc. Natl. Acad. Sci. U. S. A. 89:12013–12017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hinrichs CS, Kaiser A, Paulos CM, Cassard L, Sanchez-Perez L, Heemskerk B, Wrzesinski C, Borman ZA, Muranski P, Restifo NP. 2009. Type 17 CD8+ T cells display enhanced antitumor immunity. Blood 114:596–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li J, Huang ZF, Xiong G, Mo HY, Qiu F, Mai HQ, Chen QY, He J, Chen SP, Zheng LM, Qian CN, Zeng YX. 2011. Distribution, characterization, and induction of CD8+ regulatory T cells and IL-17-producing CD8+ T cells in nasopharyngeal carcinoma. J. Transl. Med. 9:189. 10.1186/1479-5876-9-189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Umeda K, Sun X, Guo Y, Yamada H, Shibata K, Yoshikai Y. 2011. Innate memory phenotype CD4+ T cells play a role in early protection against infection by Listeria monocytogenes in a CD30L-dependent manner. Microbiol. Immunol. 55:645–656 [DOI] [PubMed] [Google Scholar]
- 49.Ansieau S, Scheffrahn I, Mosialos G, Brand H, Duyster J, Kaye K, Harada J, Dougall B, Hübinger G, Kieff E, Herrmann F, Leutz A, Gruss HJ. 1996. Tumor necrosis factor receptor-associated factor (TRAF)-1, TRAF-2, and TRAF-3 interact in vivo with the CD30 cytoplasmic domain; TRAF-2 mediates CD30-induced nuclear factor kappa B activation. Proc. Natl. Acad. Sci. U. S. A. 93:14053–14058 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 50.Lee SY, Lee SY, Kandala G, Liou ML, Liou HC, Choi Y. 1996. CD30/TNF receptor-associated factor interaction: NF-kappa B activation and binding specificity. Proc. Natl. Acad. Sci. U. S. A. 93:9699–9703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Duckett CS, Gedrich RW, Gilfillan MC, Thompson CB. 1997. Induction of nuclear factor kappa B by the CD30 receptor is mediated by TRAF1 and TRAF2. Mol. Cell. Biol. 17:1535–1542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Martin B, Hirota K, Cua DJ, Stockinger B, Veldhoen M. 2009. Interleukin-17-producing γδ T cells selectively expand in response to pathogen products and environmental signals. Immunity 31:321–330 [DOI] [PubMed] [Google Scholar]
- 53.Mokuno Y, Matsuguchi T, Takano M, Nishimura H, Washizu J, Ogawa T, Takeuchi O, Akira S, Nimura Y, Yoshikai Y. 2000. Expression of toll-like receptor 2 on gamma delta T cells bearing invariant Vγ6/Vδ1 induced by Escherichia coli infection in mice. J. Immunol. 165:931–940 [DOI] [PubMed] [Google Scholar]
- 54.Shibata K, Yamada H, Sato T, Dejima T, Nakamura M, Ikawa T, Hara H, Yamasaki S, Kageyama R, Iwakura Y, Kawamoto H, Toh H, Yoshikai Y. 2011. Notch-Hes1 pathway is required for the development of IL-17-producing γδ T cells. Blood 118:586–593 [DOI] [PubMed] [Google Scholar]
- 55.González-García S, García-Peydró M, Martín-Gayo E, Ballestar E, Esteller M, Bornstein R, de la Pompa JL, Ferrando AA, Toribio ML. 2009. CSL-MAML-dependent Notch1 signaling controls T lineage-specific IL-7R{alpha} gene expression in early human thymopoiesis and leukemia. J. Exp. Med. 206:779–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kim MY, Anderson G, White A, Jenkinson E, Arlt W, Martensson IL, Erlandsson L, Lane PJ. 2005. OX40 ligand and CD30 ligand are expressed on adult but not neonatal CD4+CD3− inducer cells: evidence that IL-7 signals regulate CD30 ligand but not OX40 ligand expression. J. Immunol. 174:6686–6691 [DOI] [PubMed] [Google Scholar]
- 57.Hou L, Jie Z, Desai M, Liang Y, Soong L, Wang T, Sun J. 2013. Early IL-17 production by intrahepatic T cells is important for adaptive immune responses in viral hepatitis. J. Immunol. 190:621–629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nandi D, Allison JP. 1991. Phenotypic analysis and gamma delta-T cell receptor repertoire of murine T cells associated with the vaginal epithelium. J. Immunol. 147:1773–1778 [PubMed] [Google Scholar]
- 59.Heyborne K, Fu YX, Kalataradi H, Reardon C, Roark C, Eyster C, Vollmer M, Born W, O'Brien R. 1993. Evidence that urine V gamma 5 and V gamma 6 gamma delta-TCR+ lymphocytes are derived from a common distinct lineage. J. Immunol. 151:4523–4527 [PubMed] [Google Scholar]
- 60.Zeng X, Wei YL, Huang J, Newell EW, Yu H, Kidd BA, Kuhns MS, Waters RW, Davis MM, Weaver CT, Chien YH. 2012. γδ T cells recognize a microbial encoded B cell antigen to initiate a rapid antigen-specific interleukin-17 response. Immunity 37:524–534. 10.1016/j.immuni.2012.06.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Malhotra N, Narayan K, Cho OH, Sylvia KE, Yin C, Melichar H, Rashighi M, Lefebvre V, Harris JE, Berg LJ, Kang J, Immunological Genome Project Consortium 2013. A network of high-mobility group box transcription factors programs innate interleukin-17 production. Immunity 38:681–693. 10.1016/j.immuni.2013.01.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gopal R, Lin Y, Obermajer N, Slight S, Nuthalapati N, Ahmed M, Kalinski P, Khader SA. 2012. IL-23-dependent IL-17 drives Th1-cell responses following Mycobacterium bovis BCG vaccination. Eur. J. Immunol. 42:364–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.