

Abstract
Salmonella enterica serovar Typhimurium is a model organism used to explore the virulence strategies underlying Salmonella pathogenesis. Although intestinal mucus is the first line of defense in the intestine, its role in protection against Salmonella is still unclear. The intestinal mucus layer is composed primarily of the Muc2 mucin, a heavily O-glycosylated glycoprotein. The core 3-derived O-glycans of Muc2 are synthesized by core 3 β1,3-N-acetylglucosaminyltransferase (C3GnT). Mice lacking these glycans still produce Muc2 but display a thinner intestinal mucus barrier. We began our investigations by comparing Salmonella-induced colitis and mucus dynamics in Muc2-deficient (Muc2−/−) mice, C3GnT−/− mice, and wild-type C57BL/6 (WT) mice. Salmonella infection led to increases in luminal Muc2 secretion in WT and C3GnT−/− mice. When Muc2−/− mice were infected with Salmonella, they showed dramatic susceptibility to infection, carrying significantly higher cecal and liver pathogen burdens, and developing significantly higher barrier disruption and higher mortality rates, than WT mice. We found that the exaggerated barrier disruption in infected Muc2−/− mice was invA dependent. We also tested the susceptibility of C3GnT−/− mice and found that they carried pathogen burdens similar to those of WT mice but developed exaggerated barrier disruption. Moreover, we found that Muc2−/− mice were impaired in intestinal alkaline phosphatase (IAP) expression and lipopolysaccharide (LPS) detoxification activity in their ceca, potentially explaining their high mortality rates during infection. Our data suggest that the intestinal mucus layer (Muc2) and core 3 O-glycosylation play critical roles in controlling Salmonella intestinal burdens and intestinal epithelial barrier function, respectively.
INTRODUCTION
Salmonella enterica subspecies 1 serovar Typhimurium is a Gram-negative enteric bacterial pathogen that is a leading clinical cause of food-borne and waterborne diarrheal disease (1). An intracellular pathogen, S. Typhimurium is known to use virulence factors encoded on Salmonella pathogenicity island 1 (SPI-1), such as invA, to infect and/or translocate across the epithelial cells that line the luminal surface of the mammalian intestine. This virulence strategy has been studied extensively in vitro and is also known to be involved in the ability of S. Typhimurium to cause both mucosal inflammation and diarrhea in infected hosts (2, 3). Despite our detailed understanding of this aspect of S. Typhimurium pathogenesis, much less is known about how orally delivered S. Typhimurium circumvents the various luminal defenses and intestinal barriers that protect the targeted epithelium, such as the overlying mucus layer.
In large part, the dearth of knowledge in this area reflects the inability of oral S. Typhimurium infection of mice to provide a relevant model for the enterocolitis caused by Salmonella species (4). Despite the rapid invasion of intestinal epithelial cells in tissue culture by S. Typhimurium, very few orally gavaged S. Typhimurium bacteria are found to directly infect the intestinal epithelium in vivo, resulting in minimal intestinal inflammation. Recently, recognition that the resistance of mice to oral S. Typhimurium infection might reflect commensal-microbe-based colonization resistance led to testing of the impact of antibiotic pretreatment. Prior exposure to the antibiotic streptomycin was found to remove competing commensal microbes within mice, facilitating heavy S. Typhimurium colonization of the murine large bowel, leading to increased contact with the intestinal epithelium and dramatic cecal and colonic inflammation (5). However, the two mouse strains most commonly used for the Salmonella enterocolitis model (C57/BL6 and BALB/c) are known to possess a mutation in their nramp1 genes, leaving these mice highly susceptible to S. Typhimurium and succumbing rapidly to infection (6). To circumvent this limitation, we have recently described a model using the attenuated S. Typhimurium ΔaroA mutant strain, which still causes severe colitis but typically causes no mortality, even in highly susceptible mouse strains (7). Most studies employing the enterocolitis model have focused on dissecting the virulence strategies of S. Typhimurium or exploring the specific host factors that drive the resulting inflammation. In contrast, studies have yet to address how S. Typhimurium interacts with, and ultimately crosses, the intestinal mucus layer to reach the underlying epithelium. The mucus barrier is formed predominantly by Muc2, a prominent secretory mucin that overlies the intestinal epithelium. Produced within specialized goblet cells, Muc2 possesses a protein core that is heavily O-glycosylated, with its numerous carbohydrate chains making up 80% of its mass (8). In large part, the function of Muc2 depends on its glycosylation patterns (9, 10, 11). Among the most abundant of these oligosaccharides are the core 3-derived O-glycans, which are synthesized by β1,3-N-acetylglucosaminyltransferase (C3GnT) (12). While loss of C3GnT does not prevent Muc2 from forming the mucus layer, C3GnT-deficient (C3GnT−/−) mice produce a thinner mucus layer than normal, leaving them more susceptible to chemically induced forms of colitis (13).
Once secreted by goblet cells, Muc2 undergoes rapid and dramatic expansion, forming a gel-like layer on the intestinal epithelial surface. This insoluble layer provides a physical barrier that appears to protect the underlying epithelium from direct contact with commensal microbes as well as from many pathogenic insults (14). In addition to secreted mucins, the mucus barrier also contains carbohydrates, antimicrobial peptides, immunoglobulins, electrolytes, lipids, and other intestinal proteins, making it a complex biochemical matrix acting as an important host defense barrier. Recently, intestinal alkaline phosphatase (IAP), a brush border enzyme expressed on the apical sides of enterocytes (and thus at the base of the mucus layer), has emerged as an important gut mucosal defense factor due to its ability to detoxify bacterial lipopolysaccharide (LPS) by removing the phosphate group from LPS and limiting LPS-mediated activation of the innate immune receptor Toll-like receptor 4 (TLR4). Furthermore, IAP-mediated LPS detoxification plays a role in preventing systemic translocation of LPS across the intestinal barrier (15–21). In the absence of this detoxification, systemic translocation of LPS triggers exaggerated inflammatory responses that can ultimately prove fatal to the host through proinflammatory cytokine-induced septic shock (22, 23, 24).
The mucus barrier provides partial protection against several enteric bacterial pathogens, including Yersinia enterocolitica, Shigella flexneri, and Citrobacter rodentium (25, 26, 27). Despite this protection, these and other microbes do ultimately cross the mucus barrier and infect the underlying epithelial cells, raising the questions of how this subversion occurs and whether bacterial pathogens ultimately use the mucus layer as part of their pathogenic strategies. To better define S. Typhimurium interactions with the intestinal mucus layer, we infected Muc2−/− and C3GnT−/− mice with ΔaroA S. Typhimurium. We found that Muc2 plays an important role in limiting the extent of Salmonella colonization of the intestinal lumen, the subsequent mortality of infected hosts, the interactions of S. Typhimurium with the intestinal epithelium, and its translocation across the intestinal epithelium. Furthermore, Muc2−/− mice had less IAP expression and significantly less LPS detoxification activity in their cecal tissues than WT mice. We suggest that LPS-triggered inflammatory responses at systemic sites, such as the liver, could be a potential basis for the increased mortality seen in Muc2−/− mice. We noted that lack of core 3 O-glycosylation (C3GnT−/− mice) did not impact the pathogen burdens but resulted in epithelial barrier dysfunction, whereas lack of the entire mucus layer (Muc2−/− mice) caused increased epithelial barrier dysfunction as well as heavier colonization. We also found that in the absence of the mucus layer, as seen in Muc2−/− mice, the barrier dysfunction was dramatically more invA dependent than in WT mice. Our study thus demonstrates not only the protective nature of intestinal mucus but also surprising interactions with S. Typhimurium that have an impact on its virulence characteristics.
MATERIALS AND METHODS
Mice.
C57BL/6 mice were purchased from the Centre for Disease Modeling (CDM) Facility (University of British Columbia), while Muc2−/− and C3GnT−/− mice (on the C57BL/6 background) were bred in our animal facility. Mice were kept in sterilized, filter-topped cages, handled in tissue culture hoods, and fed autoclaved food and water under specific-pathogen-free conditions. The protocols used in the study were approved by the University of British Columbia's Animal Care Committee and were in direct accordance with guidelines provided by the Canadian Council on the Use of Laboratory Animals.
Bacterial strains and infection of mice.
The S. Typhimurium wild-type (WT) strain SL1344, the SL1344 ΔinvA strain, and the SL1344 ΔaroA strain (6, 28, 29) were grown, with shaking (200 rpm), at 37°C in Luria-Bertani (LB) broth supplemented with 100 μg/ml streptomycin. Approximately 24 h prior to infection, 6- to 8-week-old mice were treated with 20 mg of streptomycin by oral gavage. After streptomycin treatment, mice were infected with the strains mentioned above at a dose of 3 × 107 CFU in 100 μl of phosphate-buffered saline (PBS; pH 7.2) by oral gavage.
Tissue collection and histology.
Infected mice were anesthetized using isoflurane and were euthanized by cervical dislocation. For bacterial enumeration, tissues (cecum, liver, spleen, mesenteric lymph nodes [MLN]) and luminal contents were collected in 1 ml of sterile PBS, stored on ice, and homogenized with a Mixer Mill 301 homogenizer (Retsch, Newtown, PA). CFU counts were determined by serial 6-fold dilution of homogenized tissues that had been plated onto LB agar plates supplemented with 100 μg/ml streptomycin and incubated overnight at 37°C. Colony counts were normalized to the weight of the tissue to obtain CFU/g. For histology, cecal tissues were fixed in 10% neutral buffered formalin (Fisher Scientific) overnight and were then transferred to 70% ethanol. Fixed tissues were embedded in paraffin and were cut into 5-μm sections. Tissues were stained with hematoxylin-eosin (H&E) according to standard techniques by the University of British Columbia Histology Laboratory (Vancouver, BC, Canada). To preserve the mucus layer, sections of cecal tissue were fixed in water-free ethanol-Carnoy's fixative (60% ethanol, 30% chloroform, and 10% acetic acid) (all reagents were purchased from Fisher Scientific), and after 3 h of storage at 4°C, samples were transferred to 100% ethanol for subsequent processing. Fixed tissues were embedded in paraffin and were cut into 5-μm sections.
Tissue pathology scoring.
The extent of tissue pathology in cecal tissues from uninfected and infected mice was scored using hematoxylin-eosin-stained sections, as described previously (5). Briefly, two observers blinded to the experimental conditions scored cecal tissues for mucosal pathology, including polymorphonuclear leukocyte (PMN) infiltration (scores 0 to 4), goblet cell numbers/depletion (scores 0 to 3), epithelial integrity (scores 0 to 3), and submucosal edema (scores 0 to 3), as outlined previously (2). PMN infiltration was scored at a magnification of ×400 (10 high-power fields), and the average number of cells per high-power field was calculated. Scores were defined, as described previously (5, 7), as follows: 0, <5 cells/high-power field; 1, 5 to 20 cells/high-power field; 2, 21 to 60 cells/high-power field; 3, 61 to 100 cells/high-power field; 4, >100 cells/high-power field.
Immunohistochemistry.
For immunohistochemical detection, sections fixed with Carnoy's fixative (for Muc2 and IAP staining) and formalin-fixed sections (for LPS staining) were deparaffinized and boiled for 20 min in citrate buffer (10 mM sodium citrate, 0.05% Tween 20 [pH 6.0]) for antigen retrieval. The sections were then blocked for 20 min using blocking buffer (2% goat serum, 1% bovine serum albumin [BSA], 0.1% Triton X-100, 0.05% Tween 20 in 0.1 M PBS [pH 7.2]) to prevent nonspecific antibody binding. Sections were incubated overnight at 4°C with rabbit anti-Muc2 (dilution, 1:200; Santa Cruz Biotechnology), an anti-LPS antibody (1:50; Salmonella O antisera; BD Biosciences), or a rabbit anti-IAP antibody (1:200; Abcam). Alexa Fluor 568-conjugated goat anti-rabbit IgG and Alexa Fluor 488-conjugated goat anti-rabbit IgG (both from Molecular Probes) were used as secondary antibodies at a dilution of 1:2,000. Tissues were mounted using ProLong Gold Antifade reagent (Molecular Probes) containing 4′,6-diamidino-2-phenylindole (DAPI) for DNA staining. Images were captured using a Zeiss Axio Imager microscope equipped with an AxioCam HRm camera operating through AxioVision software.
In vivo intestinal permeability.
As described previously (27), mice used in the study were administered 150 μl of 80-mg/ml FITC (fluorescein isothiocyanate)-dextran (Sigma-Aldrich) by oral gavage. Mice were anesthetized using isoflurane, and blood samples were collected using cardiac puncture. The collected samples were immediately transferred to 3% acid-citrate-dextrose (ACD) containing 20 mM citric acid, 110 nM sodium citrate, 5 mM dextrose (protocol provided by Harald Schulze, Shivdasani laboratory, Dana-Farber Cancer Institute [DFCI]). The concentration of FITC-dextran in serum was measured using a fluorometer (PerkinElmer Life Sciences) (excitation wavelength, 490 nm; emission wavelength, 530 nm).
RNA extraction and quantitative RT-PCR.
Cecal tissues stored in RNAlater (Qiagen) at −80°C were thawed on ice, and total RNA was extracted using a Qiagen RNeasy kit as per the manufacturer's instructions. Total RNA was quantified using a NanoDrop spectrophotometer (ND1000). One microgram of RNA was reverse transcribed using an Omniscript reverse transcription (RT) kit (Qiagen). For quantitative PCR, cDNA was diluted 1:5 in RNase- and DNase-free water, and 5 μl of diluted cDNA was added to a PCR mixture (10 μl of Bio-Rad SYBR green supermix, primers at a final concentration of 300 nM; final reaction volume, 20 μl). 18S rRNA was used as a housekeeping gene. Quantitative PCR was carried out using a Bio-Rad MiniOpticon or Opticon 2 system. The sequences for the primers used in this study are as follows: Muc2 forward, 5′ CTGACCAAGAGCGAACACAA 3′; Muc2 reverse, 5′ CATGACTGGAAGCAACTGGA 3′; IL-6 forward, 5′ GAGGATACCACTCCCAACAGACC 3′; IL-6 reverse, 5′ AAGTGCATCATCGTTGTTCAT 3′; TNF-α forward, 5′ CATCTTCTCAAAATTCGAGTGACAA 3′; TNF-α reverse, 5′ TGGGAGTAGACAAGGTACAACCC 3′; IL-1β forward, 5′ CAGGATGAGGACATGAGCACC 3′; IL-1β reverse, 5′ CTCTGCAGACTCAAACTCCAC 3′.
The PCR cycling conditions used were as follows: denaturation at 94°C for 30 s, annealing for 30 s at 55°C for the Muc2 and tumor necrosis factor alpha (TNF-α) genes and at 60°C for the interleukin-6 (IL-6) and IL-1β genes, and extension at 72°C for 45 s, for a total of 30 cycles. There was an initial denaturation step at 95°C for 5 min before PCR cycling, and there was an extension step of 72°C for 10 min after the final cycle. Data were analyzed using Gene Expression Macro OM 3.0 software (Bio-Rad).
LPS dephosphorylation activity analysis.
As described previously (18, 24), to assess crude LPS detoxification activity, cecal tissues were homogenized in 500 μl of homogenization buffer, followed by the addition of 5 μl of phenylmethylsulfonyl fluoride (PMSF), a protease inhibitor. Homogenates were centrifuged at 13,200 rpm (4°C for 15 min) to remove the insoluble contents. Bradford analysis was conducted according to the manufacturer's instructions to determine the protein concentrations in the lysates. Eighty microliters of the lysate was incubated with 30 μl of 2.5-mg/ml Escherichia coli 055:B5 LPS (L2880; Sigma) or Salmonella enterica serotype Typhimurium LPS (L6511; Sigma) at room temperature for 2 h. A malachite green solution was prepared as described previously (30). Forty microliters of the malachite green solution was added to the reaction mixture, which was then incubated for 10 min. The plates were read at an absorbance of 595 nm, and data were analyzed using Microsoft Excel.
Statistical analysis.
All the results shown in this study are plotted as mean values with standard errors of the means (SEM). Statistical analysis was performed with GraphPad Prism, version 4.00 for Windows (GraphPad Software, San Diego, CA, USA), using nonparametric Mann-Whitney t tests. A P value of ≤0.05 was indicative of statistical significance.
RESULTS
S. Typhimurium infection of WT mice alters expression of intestinal glycans and the major secretory mucin Muc2.
To investigate intestinal mucin dynamics and glycosylation patterns over the course of S. Typhimurium infection, we infected wild-type (WT) mice with ΔaroA S. Typhimurium, since we have shown previously that this strain of S. Typhimurium causes significant colitis but does not kill even susceptible murine hosts (7). We collected cecal tissues over a 7-day time course and stained them with periodic acid–Schiff's reagent (PAS) as well as with alcian blue. PAS stains neutral carbohydrates (pink/magenta), whereas alcian blue stains acidic carbohydrates (deep blue), while tissues containing both acidic and neutral mucins stain dark blue/purple (31). Assessment of uninfected tissues identified distinct pink staining (neutral carbohydrates) on the epithelial surface (presumably secreted mucins) and blue staining (acidic carbohydrates) within the goblet cells. In contrast, cecal tissues collected at day 3 postinfection (3 dpi) and 7 dpi revealed significant changes in mucin staining, with dark blue/purple coloration throughout the tissues, indicative of changes in the distribution and expression pattern of neutral and acidic mucins during Salmonella infection. Specifically on 3 dpi, we noted an increase in PAS/alcian blue staining in goblet cells as well as within the cecal lumen, suggesting that mucin levels were both increased in tissues and secreted into the cecal lumen. Interestingly, by 7 dpi, the PAS/alcian blue staining of goblet cells was dramatically reduced, suggesting that the mucin content within goblet cells was reduced. In contrast, staining of mucus was seen primarily in the cecal lumen, suggesting a relative increase in the proportion of secreted mucins versus mucins contained in goblet cells by this stage of the infection (Fig. 1a).
Fig 1.
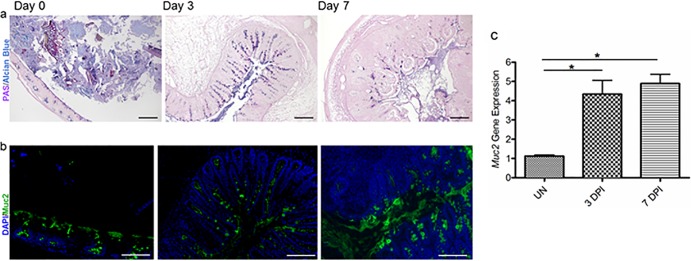
S. Typhimurium ΔaroA infection results in increased mucin secretion in WT mice. (a) Representative PAS/alcian blue staining of Carnoy's solution-fixed cecal tissues at day 0 (uninfected), 3 dpi, and 7 dpi. Original magnification, ×100; bar, 100 μm. (b) Representative Muc2 immunostaining in the cecum using an anti-Muc2 antibody (green) and a DAPI counterstain (for cellular DNA) (blue) at day 0 (uninfected), 3 dpi, and 7 dpi. Original magnification, ×200; bars, 50 μm. (c) S. Typhimurium infection induced significantly higher levels of transcription of the mucin gene Muc2 in the cecal tissues of WT mice than in uninfected samples. Error bars represent SEM from three independent experiments (9 mice per group). Asterisks indicate significant differences (*, P < 0.05) by the Mann-Whitney test.
Since Muc2 is the major secreted mucin within the colon and has been shown previously to protect against enteric bacterial infections (27), we wondered if Salmonella infection induced any changes in Muc2 expression. We noted that by 3 dpi, the amount of Muc2 secreted was relatively larger than that for uninfected mice, as assessed by immunostaining. Muc2 was seen on the mucosal surface and within the lumen, and an increase in Muc2 staining intensity inside goblet cells was also noted. On 7 dpi, we found more Muc2 within the cecal lumen (secreted), and more goblet cells within the cecal crypts staining positive for Muc2, than on 3 dpi (Fig. 1b). We also noted a significant increase in the level of Muc2 gene expression over the course of infection (Fig. 1c). These results suggest that Salmonella infection leads to increases in the expression and secretion of Muc2 glycoprotein in WT mice, potentially as a host defense mechanism.
Muc2−/− mice display increased susceptibility to S. Typhimurium infection.
It has been reported previously that Muc2 plays a protective role against Citrobacter rodentium and dextran sulfate sodium (DSS)-induced colitis (27, 32). To assess whether Muc2 protects against S. Typhimurium infection, we infected WT and Muc2−/− mice with ΔaroA S. Typhimurium and compared body weights and survival over the following 9 days. While infected WT mice showed only modest weight loss and ultimately none of the WT mice succumbed to infection, exposure to S. Typhimurium was more damaging to Muc2−/− mice. Between 6 and 7 dpi, 50% of the Muc2−/− mice succumbed to infection, and by 7 dpi, the remaining mice had lost 15 to 20% of their starting body weight and displayed other signs of morbidity, such as hunched posture, inactivity, and piloerection of their fur (Fig. 2). As a result, all the remaining infected Muc2−/− mice were euthanized on 7 dpi.
Fig 2.
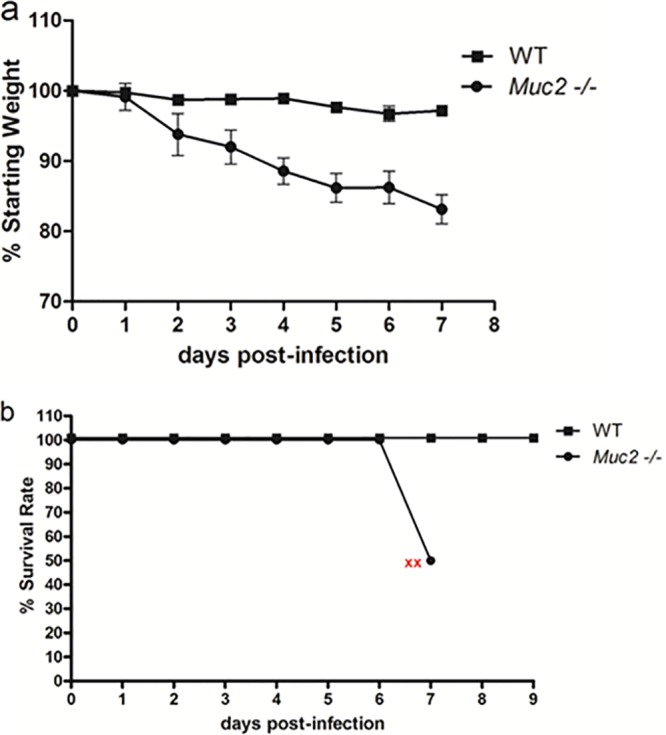
Muc2−/− mice exhibit dramatically higher susceptibility to ΔaroA S. Typhimurium infection than WT mice. (a) Body weights of WT and Muc2−/− mice from 0 to 7 dpi, plotted as percentages of the starting weight. (b) Survival curves of WT and Muc2−/− mice following Salmonella infection. Red x's indicate the humane endpoint for the remaining Muc2−/− mice. Error bars represent SEM from three independent experiments with 9 mice per group.
Muc2−/− mice carry higher S. Typhimurium burdens than WT mice.
Based on the increased Muc2 immunostaining (Fig. 1a) noted for infected WT mice, we speculated that increased Muc2 secretion during Salmonella infection might play a role in controlling pathogenic bacterial burdens. We therefore enumerated the S. Typhimurium bacteria within cecal tissues and within the cecal lumen, while also assessing pathogen translocation and/or replication outside the gut, by collecting liver, spleen, and MLN tissues. We observed significantly higher pathogen loads in the ceca of Muc2−/− mice than in those of WT mice. Similarly, we recovered higher pathogen burdens from the cecal contents (lumens) of Muc2−/− mice than from those of WT mice. We also noted higher pathogen burdens in the livers of Muc2−/− mice than in those of WT mice, suggesting that the higher intestinal burden also affected the numbers of microbes reaching (or proliferating within) the liver. In contrast, S. Typhimurium was recovered from the spleens and MLN of Muc2−/− mice at levels comparable to those for WT mice (Fig. 3), suggesting that Muc2−/− mice do not suffer any overt or widespread defects in controlling S. Typhimurium burdens at other systemic sites.
Fig 3.

Quantification of ΔaroA S. Typhimurium bacteria recovered from the ceca, intestinal lumens, livers, MLN, and spleens of WT and Muc2−/− mice. Bacterial burdens carried at 7 dpi in WT and Muc2−/− mice are shown. Each data point represents one animal, and the results from 3 independent experiments (9 mice per group) are pooled. Horizontal lines represent means, and error bars represent SEM. Asterisks indicate significant differences (**, P < 0.01) by the Mann-Whitney test.
Muc2−/− mice exhibit a level of colitis similar to that for WT mice but suffer exaggerated epithelial barrier disruption.
Next, we sought the cause of the heightened mortality seen in Salmonella-infected Muc2−/− mice. We undertook histological analysis and pathology scoring to determine whether, in addition to higher pathogen burdens, these mice suffered more-severe tissue damage, potentially explaining their dramatically higher mortality rates. Intestinal pathology was evaluated using previously described histopathological scoring methods (5, 7), and surprisingly, no significant differences in cecal histopathology were observed between infected Muc2−/− and WT mice. Histological analysis revealed that Salmonella infection elicited pronounced inflammation within the cecal tissues of both WT and Muc2−/− mice (Fig. 4a). We observed profound edema and PMN infiltration into the cecal submucosa and mucosa, along with significant damage to epithelial integrity, marked by erosions, crypt loss, and damage to crypt structure, by 3 dpi. This pathology was found to be slightly worse in both mouse strains by 7 dpi, resulting in pathology scores slightly higher than those seen at 3 dpi (Fig. 4b). Since tissue damage was comparable for WT and Muc2−/− mice, we next tested whether the lack of mucus led to increased damage to the intestinal epithelial barrier in Muc2−/− mice. Using oral FITC-dextran gavage, we found that the level of FITC-dextran translocation in uninfected Muc2−/− mice was modestly, but not significantly, greater than that seen in WT mice. In contrast, while S. Typhimurium infection caused no significant impairment of epithelial barrier function in WT mice, infected Muc2−/− mice demonstrated significantly greater (3-fold) intestinal permeability (P < 0.01) (Fig. 4c).
Fig 4.

Histology, tissue pathology, and epithelial barrier integrity assessment for ΔaroA S. Typhimurium-infected WT and Muc2−/− mice. (a) H&E-stained cecal sections (original magnification, ×50; bars, 100 μm) of WT and Muc2−/− mice at day 0 (uninfected), 3 dpi, and 7 dpi. (b) Tissue pathology scores. Mucosal pathology scoring includes epithelial barrier integrity, PMN infiltration, and submucosal edema. Each bar represents the average score for 6 to 7 tissues, scored under blinded conditions. (c) FITC-dextran intestinal permeability assay for uninfected (UN) WT and Muc2−/− mice and for the same mice at 7 dpi. Bars represent the average values for 9 mice per group, pooled from 3 independent experiments. The asterisk indicates a significant difference (*, P < 0.05) by the Mann-Whitney test.
S. Typhimurium-infected C3GnT−/− mice show impaired epithelial barrier integrity but bacterial burdens comparable to those of WT mice.
Since Muc2 is a heavily O-glycosylated glycoprotein, we next sought to test whether its critical role in host defense against oral S. Typhimurium reflected the Muc2 protein itself or, instead, some aspect of its glycosylation. We therefore infected mice lacking core 3-derived O-glycans (C3GnT−/− mice) to determine whether they had a phenotype similar to that of infected Muc2−/− mice (i.e., increased pathogen burdens and barrier disruption). Since C3GnT−/− mice still produce intestinal mucus, we noted that, like WT mice, they displayed increased accumulation of luminal mucus following infection by S. Typhimurium (see Fig. S1 in the supplemental material). Despite this response, we also noted that Salmonella-infected C3GnT−/− mice showed 8 to 10% greater weight loss than WT mice over the course of infection (Fig. 5a). To address the basis for their increased weight loss, we looked at intestinal and systemic S. Typhimurium burdens. Surprisingly, the pathogen burdens for C3GnT−/− mice and WT mice were comparable (no statistically significant difference) (Fig. 5c), suggesting that the loss of core 3 O-glycosylation did not significantly affect Salmonella colonization. Correspondingly, histological analysis and pathology scoring also failed to reveal any significant differences between C3GnT−/− and WT mice (see Fig. S2 in the supplemental material). Salmonella-infected C3GnT−/− mice displayed levels of infiltrating inflammatory cells, damage to epithelial structure, and submucosal edema similar to those seen in WT mice. Interestingly, C3GnT−/− mice did display significantly impaired epithelial integrity relative to that of infected WT mice, as assessed by the FITC-dextran assay (Fig. 5b), suggesting that core 3-derived O-glycans do play a significant role in protecting intestinal barrier function.
Fig 5.

Analysis of the susceptibility of C3GnT−/− mice to ΔaroA S. Typhimurium infection. (a) Body weights of WT and C3GnT−/− mice followed until 7 dpi. (b) FITC-dextran intestinal permeability assay of uninfected (UN) WT and C3GnT−/− mice and of the same mice at 7 dpi. Damage to the intestinal barrier was assessed by measuring FITC-dextran in serum, collected by cardiac puncture, 4 h following oral administration. Asterisks indicate significant differences (**, P < 0.01) by the Mann-Whitney test. (c) Colonization of Salmonella-infected WT and C3GnT−/− mice. Each data point represents one animal, and the results from 3 independent experiments (8 to 10 mice per group) are pooled. Horizontal bars represent means; error bars represent SEM.
The Muc2 layer acts as a physical barrier to limit Salmonella contact with the intestinal epithelium.
Based on the impact of Muc2 on both S. Typhimurium pathogen burdens and the protection of intestinal barrier function, we hypothesized that Muc2 provides a physical barrier to limit Salmonella interactions with the underlying epithelium. To investigate the localization of S. Typhimurium relative to the mucus layer and epithelium, we fixed tissues with Carnoy's fixative and stained serial sections for Muc2 and Salmonella LPS. Immunostaining revealed that in WT mice, the mucus layer provided a distinct barrier, keeping the vast majority of the S. Typhimurium bacteria within the cecal lumen and distant from the epithelial surface. In contrast, in Muc2−/− mice, Salmonella bacteria were seen in close proximity, and even adherent, to the epithelial surface (Fig. 6).
Fig 6.

Muc2 provides a physical barrier between the host epithelial surface and S. Typhimurium. (a) Salmonella LPS staining (red) and DAPI counterstain (blue), showing Salmonella bacteria localized to the cecal lumen. (b) Muc2 immunostaining (green) and DAPI counterstain (blue). The thick mucus layer can be seen between the epithelial surface and the lumen. Immunostaining for both markers was done on serial sections of cecal tissue collected from WT mice infected with Salmonella. Original magnification, ×200; bars, 50 μm. (c) In contrast to the results for WT mice (panel a), Salmonella bacteria can be seen in close proximity to the epithelial surface in the cecal tissues of Muc2−/− mice. Original magnification, ×200; bar, 50 μm. (d) Higher magnification of the inset in panel c. Original magnification, ×630; bar, 5 μm.
The increased intestinal barrier dysfunction in S. Typhimurium-infected Muc2−/− mice is invA dependent.
Cell culture studies have shown previously that S. Typhimurium uses Salmonella pathogenicity island 1 (SPI-1), including InvA, an inner membrane protein component of the SPI-1 type 3 secretion system (T3SS), to infect intestinal epithelial cells and cause barrier disruption (2, 3). Since infected Muc2−/− mice suffered from exaggerated barrier disruption, we wondered if this was dependent on the actions of InvA, one of the important proteins involved in Salmonella virulence. To test this hypothesis, we first infected WT and Muc2−/− mice with wild-type and ΔinvA (on the wild-type background) strains of Salmonella Typhimurium and then euthanized the mice at 3 dpi. Interestingly, wild-type S. Typhimurium caused severe cecal pathology and inflammation in both mouse strains, and, as we found with ΔaroA S. Typhimurium, Muc2−/− mice carried heavier pathogen burdens than WT mice. Moreover, infection caused significant barrier disruption in Muc2−/− mice, whereas no disruption was seen in WT mice. When WT mice were infected with ΔinvA Salmonella, the resulting pathogen burdens in the cecal lumen were similar to those seen with wild-type Salmonella, whereas ΔinvA Salmonella numbers in the cecal tissues, as well as within systemic tissues, were modestly but significantly lower than those of wild-type Salmonella. Interestingly, the resulting cecal pathology was only modestly reduced, and no intestinal barrier disruption was noted.
Infected Muc2−/− mice were found to carry ΔinvA Salmonella burdens higher than those of wild-type mice, though significantly lower than wild-type Salmonella burdens, in their cecal lumens. The level of pathogen translocation into cecal tissues was dramatically reduced, and Salmonella numbers at systemic sites, such as the liver and spleen, were also reduced, to roughly the same degrees as those seen in ΔinvA Salmonella-infected WT mice. Notably, in contrast to the cecitis suffered by WT mice, no significant cecal pathology was seen in infected Muc2−/− mice, and despite their high luminal pathogen burdens, ΔinvA S. Typhimurium did not cause any barrier disruption (Fig. 7 and 8). Taken together, these findings suggest that although the ability of S. Typhimurium to cause cecal pathology in WT mice is partially dependent on invA, the dependence on invA for inducing cecal pathology is dramatically greater in the absence of a mucus layer (Muc2−/− mice), suggesting that the interactions of Salmonella with the mucus layer may play a modulatory role in its pathogenesis. Furthermore, while Muc2−/− mice infected with wild-type S. Typhimurium showed Salmonella bacteria in close proximity or adherent to the cecal epithelial surface, mice infected with ΔinvA S. Typhimurium showed few, if any, of these bacteria adherent to the epithelial surface (see Fig. S3 in the supplemental material). Moreover, Muc2−/− mice showed no mortality over the time course of infection with ΔinvA S. Typhimurium (data not shown), suggesting that pathogen translocation out of the cecum likely plays an important role in the high mortality rates suffered by these mice.
Fig 7.

Analysis of invA-dependent susceptibility of Muc2−/− mice. (a) FITC-dextran intestinal permeability assay for WT mice and Muc2−/− mice infected with wild-type Salmonella Typhimurium SL1344 and a ΔinvA Salmonella strain (SL1344 background). (b and c) Colonization of WT and Muc2−/− mice after infection with the wild-type or ΔinvA Salmonella strain is shown for the cecum and lumen (b) and for the liver, spleen, and MLN (c). Horizontal bars represent means; error bars represent SEM. Asterisks indicate significant differences (*, P < 0.05; **, P < 0.01) by the Mann-Whitney test for 9 mice per group in 3 independent experiments.
Fig 8.
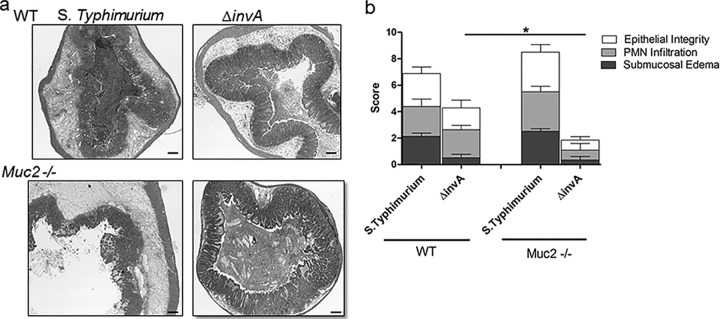
Histological and pathological analyses of WT and Muc2−/− mice upon infection with wild-type and ΔinvA Salmonella strains. (a) Representative H&E-stained images for WT and Muc2−/− mice infected with wild-type and ΔinvA Salmonella strains. Original magnification, ×50; bars, 100 μm. (b) Tissue pathology scores. Pathology scoring included damage to epithelial integrity, PMN infiltration, and submucosal edema. Each bar represents the average for 9 to 10 tissues. The asterisk indicates a significant difference (*, P < 0.05) by the Mann-Whitney test.
IAP expression and LPS detoxification are impaired in Muc2−/− mice.
It was surprising that Muc2−/− mice succumbed to ΔaroA S. Typhimurium, considering that infection by this pathogen is not normally lethal, even to severely immunodeficient mice (7, 33). Considering that their symptoms (hunched appearance, piloerection, and reduced activity) observed during Salmonella infection are typically signs of a systemic disease (34), we decided to compare inflammatory responses within the livers of WT and Muc2−/− mice. Interestingly, levels of production of the cytokines IL-6, TNF-α, and IL-1β were significantly higher in Muc2−/− mice than in WT mice (see Fig. S4b in the supplemental material), while histologically, the livers of Muc2−/− mice displayed much greater signs of inflammation and tissue damage (see Fig. S4a in the supplemental material).
We noted previously that liver injury during S. Typhimurium infection is mediated largely by LPS-based activation of the innate receptor TLR4 (35). Interestingly, LPS produced by Gram-negative bacteria within the intestine is typically detoxified by intestinal alkaline phosphatase (IAP), an enzyme expressed by enterocytes (15–20). We therefore wondered if there were any differences in IAP expression or function between WT and Muc2−/− mice that could lead to exaggerated inflammatory signaling in response to S. Typhimurium bacteria that translocate out of the gut and reach the liver. We found IAP-positive staining on much of the intestinal epithelium of infected WT mice, whereas the staining was comparatively reduced on the epithelial surfaces of Muc2−/− mice (Fig. 9). To address whether this reduced staining had any functional impact, we assessed the capacities of cecal tissues from the two mouse strains to detoxify LPS. Strikingly, we identified significantly lower LPS dephosphorylation activity in the cecal tissues of Muc2−/− mice than in those of WT mice, suggesting that impaired LPS detoxification could underlie the high mortality rates suffered by infected Muc2−/− mice.
Fig 9.

Muc2−/− mice are impaired in IAP staining and activity. (a) Representative IAP immunostaining (red) in the ceca of WT and Muc2−/− mice at day 7 after infection with ΔaroA S. Typhimurium. The DAPI counterstain is shown in blue. Original magnification, ×400; bars, 20 μm. (b) Analysis of ex vivo LPS dephosphorylation activity in the cecal tissues of WT and Muc2−/− mice infected with ΔaroA S. Typhimurium (assessed at 7 dpi). Homogenized cecal tissues were incubated with Salmonella LPS or Escherichia coli LPS for 2 h, and the malachite green assay was used to measure activity (absorbance at 595 nm). Activity was calculated as relative LPS dephosphorylation activity/mg of protein (normalized to the value for uninfected controls in each group). There were 9 mice per group. Asterisks indicate significant differences (**, P < 0.01) by the Mann-Whitney test.
DISCUSSION
The intestinal mucus layer is composed predominantly of the secreted mucin Muc2. Synthesized and secreted by goblet cells, Muc2 is a heavily O-glycosylated glycoprotein that forms a gel-like and viscous mesh-like layer overlying the intestinal epithelium. Intestinal mucus is the initial structural barrier encountered by enteric bacterial pathogens and, as such, provides the first line of defense for the host against these and other noxious agents. The presence of the mucus layer has thus necessitated the development of specific virulence strategies allowing enteric pathogens to cross the mucus layer and reach the epithelium, such as flagellum-based motility and even mucus degradation (36). Aside from functioning in host defense, over the course of the evolutionary dialogue between pathogens and the mucus layer, the mucus layer has also been subverted by some microbes to aid in their pathogenesis. For example, mucus can provide attachment sites for pathogenic bacteria (37, 38), as well as providing an energy/food source for adherent bacteria (39, 40). However, despite our growing understanding of the strategies used by enteric pathogens to cross the mucus barrier, it is not entirely clear how they physically interact with the mucus layer.
S. Typhimurium is a leading cause of enterocolitis in humans and is used as a model organism for studying bacterial pathogenesis and host responses to intracellular bacterial infections (1). Pretreatment of mice with streptomycin, followed by infection with S. Typhimurium, provides a relevant model for studying Salmonella-induced intestinal disease in humans (5). The two most commonly used mouse strains are C57/BL6 and BALB/c, but since these strains suffer from a loss-of-function mutation in their nramp1 genes, they are extremely susceptible to Salmonella infection (6). Recently we have shown the applicability of the attenuated ΔaroA strain of S. Typhimurium for studying colitis in these mouse strains (7). Our present studies found that Muc2 plays a critical role in host defense against S. Typhimurium. Muc2−/− mice showed dramatically heightened susceptibility to Salmonella infection compared to that of WT mice, carrying much heavier pathogen burdens both in their intestinal lumens and in mucosal tissues. Our results recall earlier studies with the bacterial pathogen C. rodentium, where the loss of Muc2 also led to dramatically heavier intestinal pathogen burdens. In both infection models, the level of Muc2 expression/secretion in WT mice was increased during infection, potentially promoting host defense by removing bacteria from the mucosal surface (27). In Muc2−/− mice, numerous S. Typhimurium bacteria were seen in close proximity (or adherent) to the intestinal epithelium, whereas in WT mice, the majority of the Salmonella bacteria were segregated from the epithelial surface by the overlying mucus layer, suggesting that aside from the function of mucus as a physical barrier, mucus-mediated flushing can play an important role in controlling pathogen burdens in the gut.
The exaggerated pathogen burdens carried by Muc2−/− mice were accompanied by significant weight loss and other signs of morbidity, requiring the euthanization of all Muc2−/− mice by 7 dpi. This severe response was unexpected, since ΔaroA S. Typhimurium typically does not cause serious morbidity or any mortality in other mouse strains, including severely immunodeficient RAG1−/− mice, which can carry very high systemic burdens of this mutant strain (33). We hypothesize, however, that the susceptibility of the Muc2−/− mice to this mutant reflects not only their heavy pathogen burdens but also the exaggerated barrier dysfunction they suffer during infection. A number of studies have shown that S. Typhimurium can cause tight-junction disruption in infected epithelial cells, resulting in increased epithelial permeability (41–44). Despite this in vitro phenotype, we and others have not been able to detect overt intestinal barrier dysfunction in other mouse strains orally infected by S. Typhimurium. The current findings thus indicate that loss of Muc2 leaves the intestinal epithelium unusually susceptible to S. Typhimurium-driven barrier disruption.
To better define the mechanisms behind the barrier disruption seen in infected Muc2−/− mice, we infected WT and Muc2−/− mice with a Salmonella strain lacking Salmonella pathogenicity island 1 (SPI-1)-dependent type III secretion. invA is the first gene in the invABC operon, is located in SPI-1, and is required, at least in vitro, for the invasion of epithelial cells by S. Typhimurium (3, 45). When Muc2−/− mice were infected with the ΔinvA mutant, barrier function was not disrupted, and very minor histological damage was observed. Taken together with the observation that systemic ΔinvA pathogen burdens were dramatically reduced in Muc2−/− mice, these data suggest that the susceptibility of the Muc2−/− mice was dependent on invA. In contrast, the ΔinvA mutant was still able to cross the intestinal epithelia of WT mice and cause significant cecal pathology, though less than that seen with wild-type Salmonella. These results suggest that potential interactions with intestinal mucus provide Salmonella (in this case, an invA mutant) with increased opportunities for uptake and translocation out of the gut lumen, potentially by dendritic cells or macrophages, through pathogen-driven but non-SPI-1-dependent mechanisms (46, 47). In contrast, in the absence of mucus, SPI-1-dependent mechanisms appear to play a more important role in the crossing of the intestinal epithelial barrier by Salmonella. These results suggest that S. Typhimurium bacteria adherent to the mucosal surface may be expressing the genes involved in the SPI-1 virulence system and that there may be differential expression of this system in Muc2−/− mice versus WT mice, dictated by the direct interactions between Salmonella bacteria and the epithelial surface. Since in Muc2−/− mice infected with the ΔinvA strain, there is no close adherence to the epithelial surface, the expression of this virulence system may be impaired, thereby reducing the pathology and pathophysiology seen in these mice.
Aside from the mucus layer, there are other factors that determine host susceptibility to an enteric bacterial pathogen, including the enzyme intestinal alkaline phosphatase (IAP). Recently there has been renewed interest in IAP activity and its role in promoting intestinal mucosal defense (15). Several studies have shown that LPS dephosphorylation mediated by IAP protects the host against LPS-induced inflammation as well as reducing the systemic translocation of enteric bacteria (15–24). To investigate whether IAP activity was playing a role in the increased susceptibility/morbidity of Muc2−/− mice in our model, we stained for IAP and found that Muc2−/− mice had reduced IAP expression as well as reduced LPS dephosphorylation activity (a measure of the activity of IAP) within their cecal tissues. While the basis for this impairment is unclear, a recent study has reported altered expression of other enzymes in the intestinal epithelia of Muc2−/− mice (48). However, it is not clear at this point whether these changes in epithelial cell function reflect a direct role for Muc2 mucin or, alternatively, result from increased microbial interactions with the epithelium.
Interestingly, we also noted a dramatic increase in proinflammatory cytokine gene levels within the livers of Muc2−/− mice. We speculate that impaired intestinal barrier function in concert with reduced LPS detoxification within the ceca of Muc2−/− mice leads to the translocation of Salmonella bacteria carrying highly proinflammatory LPS to the liver, resulting in increased inflammation (through TLR4-LPS signaling) and exaggerated damage to the liver, ultimately contributing to the higher mortality observed in these mice. These results are in line with our previous studies showing the importance of TLR4 signaling in mediating inflammatory responses in the liver (35). Our study also sheds light on the complex interactions between host factors (mucus layer, IAP) and pathogen factors (virulence genes) that ultimately determine the outcome of an infection.
To better define how Muc2 plays such a critical role in controlling S. Typhimurium pathogenesis, we infected mice lacking different components of this glycoprotein. Muc2 is a heavily O-glycosylated mucin, and the impact of its glycosylation was noted by a recent study examining different glycosylation patterns of Muc2 in mice and humans (49, 50). In agreement with previous findings (13), we found that core 3 O-glycosylation plays a major role in protecting intestinal barrier function. Mice lacking core 3-derived O-glycans possess a thinner intestinal mucus layer, and although they do not develop any spontaneous gut diseases, they do show increased susceptibility to DSS-induced colitis (13). Infecting these C3GnT−/− mice, we found no significant differences from WT mice in infection-induced histology, pathology, or S. Typhimurium burdens. However, these mice still suffered from greater epithelial barrier disruption during infection than WT mice. This suggests that the core 3 glycosylation component of the mucus layer may play a role in controlling Salmonella-driven disruption of epithelial barrier integrity, whereas the Muc2 protein and its remaining glycosylation are the key factors in controlling S. Typhimurium burdens and overt intestinal pathology.
Increased release of mucus into the intestinal lumen, as seen during Salmonella infection in WT and C3GnT−/− mice, may help protect the epithelium by limiting pathogen contact and barrier disruption. This may reflect a unique action of secreted mucins, since it has been shown previously that Muc1−/− mice were not more susceptible than WT mice to S. Typhimurium infection (51), whereas in this study, we show increased susceptibility of Muc2−/− mice to S. Typhimurium infection. We believe that such a protective role may be a generalized defense against many enteric bacterial pathogens. Indeed, there have been reports of Muc2 interactions with other enteric pathogens, including Campylobacter jejuni (52) and the attaching and effacing (A/E) pathogen Citrobacter rodentium (53).
There has been growing recognition of the important role played by the mucus barrier in regulating the severity of infectious diseases, but the specifics of how enteric bacterial pathogens interact with mucus and mucus components remain unclear. This study unravels the importance of Muc2, a major secreted mucin, during infection with yet another enteric pathogen, S. Typhimurium, and it is the first study to provide insight into the importance of core 3 O-glycosylation during Salmonella infection. This study also provides insights into the potential role of mucus in modulating Salmonella pathogenesis. Considering that the mucus layer acts as a frontline defense barrier, further investigation of interactions between enteric pathogens and the mucus layer may aid in the development of therapeutic strategies.
Supplementary Material
ACKNOWLEDGMENTS
We thank Kirk Bergstrom for helpful discussions. We thank Martin Stahl for careful proofreading of the manuscript.
This study was funded by operating grants from the Crohn's and Colitis Foundation of Canada (CCFC) and the Canadian Institutes of Health Research (CIHR) awarded to B.A.V. M.Z. was funded by a Child and Family Research Institute (CFRI) postdoctoral fellowship. K.B. was funded by a CFRI graduate studentship. B.A.V. is the Children with Intestinal and Liver Disorders (CHILD) Foundation Chair in Pediatric IBD Research and the Canada Research Chair in Pediatric Gastroenterology (tier 2).
Footnotes
Published ahead of print 22 July 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.00854-13.
REFERENCES
- 1.Ohl ME, Miller SI. 2001. Salmonella: a model for bacterial pathogenesis. Annu. Rev. Med. 52:259–274 [DOI] [PubMed] [Google Scholar]
- 2.Galán JE, Ginocchio C, Costeas P. 1992. Molecular and functional characterization of the Salmonella invasion gene invA: homology of InvA to members of a new protein family. J. Bacteriol. 174:4338–4349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Galán JE, Curtiss R., III 1989. Cloning and molecular characterization of genes whose products allow Salmonella typhimurium to penetrate tissue culture cells. Proc. Natl. Acad. Sci. U. S. A. 86:6383–6387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Santos RL, Zhang S, Tsolis RM, Kingsley RA, Adams LG, Bäumler AJ. 2001. Animal models of Salmonella infections: enteritis versus typhoid fever. Microbes Infect. 3:1335–1344 [DOI] [PubMed] [Google Scholar]
- 5.Barthel M, Hapfelmeier S, Quintanilla-Martínez L, Kremer M, Rohde M, Hogardt M, Pfeffer K, Rüssmann H, Hardt WD. 2003. Pretreatment of mice with streptomycin provides a Salmonella enterica serovar Typhimurium colitis model that allows analysis of both pathogen and host. Infect. Immun. 71:2839–2858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vidal S, Tremblay ML, Govoni G, Gauthier S, Sebastiani G, Malo D, Skamene E, Olivier M, Jothy S, Gros P. 1995. The Ity/Lsh/Bcg locus: natural resistance to infection with intracellular parasites is abrogated by disruption of the Nramp1 gene. J. Exp. Med. 182:655–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Månsson LE, Montero M, Zarepour M, Bergstrom KS, Ma C, Huang T, Man C, Grassl GA, Vallance BA. 2012. MyD88 signaling promotes both mucosal homeostatic and fibrotic responses during Salmonella-induced colitis. Am. J. Physiol. Gastrointest. Liver Physiol. 303:G311–G323 [DOI] [PubMed] [Google Scholar]
- 8.Johansson ME, Larsson JM, Hansson GC. 2011. The two mucus layers of colon are organized by the MUC2 mucin, whereas the outer layer is a legislator of host-microbial interactions. Proc. Natl. Acad. Sci. U. S. A. 108(Suppl 1):4659–4665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pusztai A, Ewen SW, Grant G, Peumans WJ, Van Damme EJ, Coates ME, Bardocz S. 1995. Lectins and also bacteria modify the glycosylation of gut surface receptors in the rat. Glycoconj. J. 12:22–35 [DOI] [PubMed] [Google Scholar]
- 10.Robbe-Masselot C, Herrmann A, Carlstedt I, Michalski JC, Capon C. 2008. Glycosylation of the two O-glycosylated domains of human MUC2 mucin in patients transposed with artificial urinary bladders constructed from proximal colonic tissue. Glycoconj. J. 25:213–224 [DOI] [PubMed] [Google Scholar]
- 11.Holmén JM, Olson FJ, Karlsson H, Hansson GC. 2002. Two glycosylation alterations of mouse intestinal mucins due to infection caused by the parasite Nippostrongylus brasiliensis. Glycoconj. J. 19:67–75 [DOI] [PubMed] [Google Scholar]
- 12.Iwai T, Inaba N, Naundorf A, Zhang Y, Gotoh M, Iwasaki H, Kudo T, Togayachi A, Ishizuka Y, Nakanishi H, Narimatsu H. 2002. Molecular cloning and characterization of a novel UDP-GlcNAc:GalNAc-peptide β1,3-N-acetylglucosaminyltransferase (β3Gn-T6), an enzyme synthesizing the core 3 structure of O-glycans. J. Biol. Chem. 277:12802–12809 [DOI] [PubMed] [Google Scholar]
- 13.An G, Wei B, Xia B, McDaniel JM, Ju T, Cummings RD, Braun J, Xia L. 2007. Increased susceptibility to colitis and colorectal tumors in mice lacking core 3-derived O-glycans. J. Exp. Med. 204:1417–1429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim YS, Ho SB. 2010. Intestinal goblet cells and mucins in health and disease: recent insights and progress. Curr. Gastroenterol. Rep. 12:319–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lallès JP. 2010. Intestinal alkaline phosphatase: multiple biological roles in maintenance of intestinal homeostasis and modulation by diet. Nutr. Rev. 68:323–332 [DOI] [PubMed] [Google Scholar]
- 16.Poelstra K, Bakker WW, Klok PA, Hardonk MJ, Meijer DK. 1997. A physiologic function for alkaline phosphatase: endotoxin detoxification. Lab. Invest. 76:319–327 [PubMed] [Google Scholar]
- 17.Poelstra K, Bakker WW, Klok PA, Kamps JA, Hardonk MJ, Meijer DK. 1997. Dephosphorylation of endotoxin by alkaline phosphatase in vivo. Am. J. Pathol. 151:1163–1169 [PMC free article] [PubMed] [Google Scholar]
- 18.Goldberg RF, Austen WG, Jr, Zhang X, Munene G, Mostafa G, Biswas S, McCormack M, Eberlin KR, Nguyen JT, Tatlidede HS, Warren HS, Narisawa S, Millán JL, Hodin RA. 2008. Intestinal alkaline phosphatase is a gut mucosal defense factor maintained by enteral nutrition. Proc. Natl. Acad. Sci. U. S. A. 105:3551–3556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bates JM, Akerlund J, Mittge E, Guillemin K. 2007. Intestinal alkaline phosphatase detoxifies lipopolysaccharide and prevents inflammation in zebrafish in response to the gut microbiota. Cell Host Microbe 2:371–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Beumer C, Wulferink M, Raaben W, Fiechter D, Brands R, Seinen W. 2003. Calf intestinal alkaline phosphatase, a novel therapeutic drug for lipopolysaccharide (LPS)-mediated diseases, attenuates LPS toxicity in mice and piglets. J. Pharmacol. Exp. Ther. 307:737–744 [DOI] [PubMed] [Google Scholar]
- 21.Fukata M, Abreu MT. 2007. TLR4 signalling in the intestine in health and disease. Biochem. Soc. Trans. 35:1473–1478 [DOI] [PubMed] [Google Scholar]
- 22.van Veen SQ, van Vliet AK, Wulferink M, Brands R, Boermeester MA, van Gulik TM. 2005. Bovine intestinal alkaline phosphatase attenuates the inflammatory response in secondary peritonitis in mice. Infect. Immun. 73:4309–4314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bentala H, Verweij WR, Huizinga-Van der Vlag A, van Loenen-Weemaes AM, Meijer DK, Poelstra K. 2002. Removal of phosphate from lipid A as a strategy to detoxify lipopolysaccharide. Shock 18:561–566 [DOI] [PubMed] [Google Scholar]
- 24.Ghosh S, DeCoffe D, Brown K, Rajendiran E, Estaki M, Dai C, Yip A, Gibson DL. 2013. Fish oil attenuates omega-6 polyunsaturated fatty acid-induced dysbiosis and infectious colitis but impairs LPS dephosphorylation activity causing sepsis. PLoS One 8:e55468. 10.1371/journal.pone.0055468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mantle M, Basaraba L, Peacock SC, Gall DG. 1989. Binding of Yersinia enterocolitica to rabbit intestinal brush border membranes, mucus, and mucin. Infect. Immun. 57:3292–3299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nutten S, Sansonetti P, Huet G, Bourdon-Bisiaux C, Meresse B, Colombel JF, Desreumaux P. 2002. Epithelial inflammation response induced by Shigella flexneri depends on mucin gene expression. Microbes Infect. 4:1121–1124 [DOI] [PubMed] [Google Scholar]
- 27.Bergstrom KS, Kissoon-Singh V, Gibson DL, Ma C, Montero M, Sham HP, Ryz N, Huang T, Velcich A, Finlay BB, Chadee K, Vallance BA. 2010. Muc2 protects against lethal infectious colitis by disassociating pathogenic and commensal bacteria from the colonic mucosa. PLoS Pathog. 6:e1000902. 10.1371/journal.ppat.1000902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Menendez A, Arena ET, Guttman JA, Thorson L, Vallance BA, Vogl W, Finlay BB. 2009. Salmonella infection of gallbladder epithelial cells drives local inflammation and injury in a model of acute typhoid fever. J. Infect. Dis. 200:1703–1713 [DOI] [PubMed] [Google Scholar]
- 29.Chatfield SN, Strahan K, Pickard D, Charles IG, Hormaeche CE, Dougan G. 1992. Evaluation of Salmonella typhimurium strains harbouring defined mutations in htrA and aroA in the murine salmonellosis model. Microb. Pathog. 12:145–151 [DOI] [PubMed] [Google Scholar]
- 30.Baykov AA, Evtushenko OA, Avaeva SM. 1988. A malachite green procedure for orthophosphate determination and its use in alkaline phosphatase-based enzyme immunoassay. Anal. Biochem. 171:266–270 [DOI] [PubMed] [Google Scholar]
- 31.Yamabayashi S. 1987. Periodic acid-Schiff-alcian blue: a method for the differential staining of glycoproteins. Histochem. J. 19:565–571 [DOI] [PubMed] [Google Scholar]
- 32.Van der Sluis M, De Koning BA, De Bruijn AC, Velcich A, Meijerink JP, Van Goudoever JB, Büller HA, Dekker J, Van Seuningen I, Renes IB, Einerhand AW. 2006. Muc2-deficient mice spontaneously develop colitis, indicating that MUC2 is critical for colonic protection. Gastroenterology 131:117–129 [DOI] [PubMed] [Google Scholar]
- 33.Hess J, Ladel C, Miko D, Kaufmann SH. 1996. Salmonella typhimurium aroA− infection in gene-targeted immunodeficient mice: major role of CD4+ TCR-αβ cells and IFN-γ in bacterial clearance independent of intracellular location. J. Immunol. 156:3321–3326 [PubMed] [Google Scholar]
- 34.Özkaya H, Akcan AB, Aydemir G, Aydinöz S, Razia Y, Gammon ST, McKinney J. 2012. Salmonella typhimurium infections in BALB/c mice: a comparison of tissue bioluminescence, tissue cultures and mice clinical scores. New Microbiol. 35:53–59 [PubMed] [Google Scholar]
- 35.Vazquez-Torres A, Vallance BA, Bergman MA, Finlay BB, Cookson BT, Jones-Carson J, Fang FC. 2004. Toll-like receptor 4 dependence of innate and adaptive immunity to Salmonella: importance of the Kupffer cell network. J. Immunol. 172:6202–6208 [DOI] [PubMed] [Google Scholar]
- 36.McGuckin MA, Lindén SK, Sutton P, Florin TH. 2011. Mucin dynamics and enteric pathogens. Nat. Rev. Microbiol. 9:265–278 [DOI] [PubMed] [Google Scholar]
- 37.Lillehoj EP, Hyun SW, Kim BT, Zhang XG, Lee DI, Rowland S, Kim KC. 2001. Muc1 mucins on the cell surface are adhesion sites for Pseudomonas aeruginosa. Am. J. Physiol. Lung Cell. Mol. Physiol. 280:L181–L187 [DOI] [PubMed] [Google Scholar]
- 38.Rajkumar R, Devaraj H, Niranjali S. 1998. Binding of Shigella to rat and human intestinal mucin. Mol. Cell. Biochem. 178:261–268 [DOI] [PubMed] [Google Scholar]
- 39.Aristoteli LP, Willcox MD. 2003. Mucin degradation mechanisms by distinct Pseudomonas aeruginosa isolates in vitro. Infect. Immun. 71:5565–5575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Berg RD. 1996. The indigenous gastrointestinal microflora. Trends Microbiol. 4:430–435 [DOI] [PubMed] [Google Scholar]
- 41.Boyle EC, Brown NF, Finlay BB. 2006. Salmonella enterica serovar Typhimurium effectors SopB, SopE, SopE2 and SipA disrupt tight junction structure and function. Cell. Microbiol. 8:1946–1957 [DOI] [PubMed] [Google Scholar]
- 42.Finlay BB, Falkow S. 1990. Salmonella interactions with polarized human intestinal Caco-2 epithelial cells. J. Infect. Dis. 162:1096–1106 [DOI] [PubMed] [Google Scholar]
- 43.Jepson MA, Collares-Buzato CB, Clark MA, Hirst BH, Simmons NL. 1995. Rapid disruption of epithelial barrier function by Salmonella typhimurium is associated with structural modification of intercellular junctions. Infect. Immun. 63:356–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jepson MA, Schlecht HB, Collares-Buzato CB. 2000. Localization of dysfunctional tight junctions in Salmonella enterica serovar Typhimurium-infected epithelial layers. Infect. Immun. 68:7202–7208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Coombes BK, Coburn BA, Potter AA, Gomis S, Mirakhur K, Li Y, Finlay BB. 2005. Analysis of the contribution of Salmonella pathogenicity islands 1 and 2 to enteric disease progression using a novel bovine ileal loop model and a murine model of infectious enterocolitis. Infect. Immun. 73:7161–7169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yrlid U, Svensson M, Håkansson A, Chambers BJ, Ljunggren HG, Wick MJ. 2001. In vivo activation of dendritic cells and T cells during Salmonella enterica serovar Typhimurium infection. Infect. Immun. 69:5726–5735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kalupahana RS, Mastroeni P, Maskell D, Blacklaws BA. 2005. Activation of murine dendritic cells and macrophages induced by Salmonella enterica serovar Typhimurium. Immunology 115:462–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang K, Popova NV, Yang WC, Lozonschi I, Tadesse S, Kent S, Bancroft L, Matise I, Cormier RT, Scherer SJ, Edelmann W, Lipkin M, Augenlicht L, Velcich A. 2008. Interaction of Muc2 and Apc on Wnt signaling and in intestinal tumorigenesis: potential role of chronic inflammation. Cancer Res. 68:7313–7322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Larsson JM, Karlsson H, Crespo JG, Johansson ME, Eklund L, Sjövall H, Hansson GC. 2011. Altered O-glycosylation profile of MUC2 mucin occurs in active ulcerative colitis and is associated with increased inflammation. Inflamm. Bowel Dis. 17:2299–2307 [DOI] [PubMed] [Google Scholar]
- 50.Larsson JM, Karlsson H, Sjövall H, Hansson GC. 2009. A complex, but uniform O-glycosylation of the human MUC2 mucin from colonic biopsies analyzed by nanoLC/MSn. Glycobiology 19:756–766 [DOI] [PubMed] [Google Scholar]
- 51.McAuley JL, Linden SK, Png CW, King RM, Pennington HL, Gendler SJ, Florin TH, Hill GR, Korolik V, McGuckin MA. 2007. MUC1 cell surface mucin is a critical element of the mucosal barrier to infection. J. Clin. Invest. 117:2313–2324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tu QV, McGuckin MA, Mendz GL. 2008. Campylobacter jejuni response to human mucin MUC2: modulation of colonization and pathogenicity determinants. J. Med. Microbiol. 57:795–802 [DOI] [PubMed] [Google Scholar]
- 53.Lindén SK, Florin TH, McGuckin MA. 2008. Mucin dynamics in intestinal bacterial infection. PLoS One 3:e3952. 10.1371/journal.pone.0003952 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.