

Abstract
We report the case of a 36-year-old woman who presented with headache, fever, and amenorrhea. Laboratory analysis revealed hypopituitarism and autoimmune thyroiditis, while a cerebrospinal fluid study suggested concurrent aseptic meningitis. A magnetic resonance image (MRI) scan revealed a 1.0×0.9 cm cystic mass enlarging the sella turcica. Surgical resection via an endoscopic transsphenoidal route was performed. The histological finding of the excised tissue revealed foamy histiocytes with vacuolated cytoplasm, supporting the diagnosis of xanthomatous hypophysitis. Although a residual soft lesion was observed on the MRI image postoperatively, the patient's headache and fever improved. Ten months after surgery, the patient complained of visual impairment and headache, and the residual mass had enlarged into the suprasellar area. High dose (500 mg intravenous) methylprednisolone was administered for 3 days. During the methylprednisolone pulse therapy, the patient's visual acuity and headache improved. A follow-up MRI taken after methylprednisolone therapy showed a marked mass reduction. Our case supports an autoimmune pathophysiology for xanthomatous hypophysitis and suggests that high dose glucocorticoid therapy as a treatment option.
Keywords: Thyroiditis, autoimmune; Glucocorticoids; Xanthomatous hypophysitis
INTRODUCTION
Primary hypophysitis describes chronic inflammation that primarily affects the pituitary gland. It is classified as either a lymphocytic, granulomatous, or xanthomatous hypophysitis based on histologic findings. Among these subtypes, xanthomatous hypophysitis is the rarest form, while lymphocytic hypophysitis is the most common [1]. Although the pathogenesis of primary hypophysitis is not well understood, an autoimmune etiology is considered to be one of the underlying causes [2]. In contrast to lymphocytic hypophysitis, xanthomatous hypophysitis is rarely reported in association with autoimmune diseases or known to improve in response to glucocorticoid therapies.
We report a case of xanthomatous hypophysitis with symptoms of headache, fever, and hypopituitarism in the setting of autoimmune thyroiditis that was initially surgically resected, but then managed using corticosteroid therapy postoperatively to treat recurrence.
CASE REPORT
A 36-year-old woman was referred for the evaluation of a pituitary mass detected by brain magnetic resonance image (MRI). She had a 4-month history of amenorrhea and had suffered from headache for 2 months. One month prior to admission, she visited a different hospital emergency room for headache, fever, and vomiting. She was noted to be febrile with a body temperature of 38.5℃. A cerebrospinal fluid (CSF) examination was performed given concerns for meningitis and the CSF findings displayed an elevated white cell count of 350/mm3 (48% lymphocyte), a glucose level of 2.39 mmol/L (normal range, 2.77 to 4.99), and protein 112.7 g/L (normal range, 200 to 400), suggestive of aseptic meningitis. No bacteria or fungi were grown from culture. There was no leukocytosis (white blood count, 7,200/mm3). Her fever resolved after several days of conservative therapies and supportive care. However, her headache still persisted, prompting further imaging. Brain MRI scans showed a cystic pituitary mass prompting her transfer to our hospital.
On admission, she also had complaints of general weakness, polydipsia, and polyuria. Her urine volume was more than 5 L per day (specific gravity, 1.006). There was no family history of endocrine disease except for her brother who has hyperthyroidism. She was dehydrated and appeared clinically unwell. She had no visual disturbance and no neurologic deficits such as an abnormal extraocular movement. Her body temperature was 36.4℃ with stable vital signs (blood pressure, 102/77 mm Hg; pulse rate, 96 beats/min). Laboratory investigation showed mild hypernatremia (sodium of 147 mmol/L; normal range, 136 to 145). The patient's serum and urinary osmolarity were 304 and 111 mmol/kg, respectively. Morning serum levels of cortisol (38.64 nmol/L; normal range, 49.66 to 717.34) and estradiol (11.01 pmol/L; normal range, 36.7 to 1,618.4) were low. A mildly elevated level of prolactin (2,756.50 pmol/L; normal range, 156.52 to 821.73) was also noted. Her thyroid function test revealed hyperthyroidism with a high free T4 (34.75 pmol/L; normal range, 8.24 to 22.14) and low thyroid stimulating hormone (0.01 mIU/L; normal range, 0.3 to 6.50). While antithyroglobulin antibody (1,457 kIU/L; normal range, <60) and antimicrosomal antibody (1,720 kIU/L; normal range, <60) were high, anti-TSH receptor antibody (0.1 kIU/L; normal range, 0.1 to 1.0) levels were normal (Table 1). Thyroid gland ultrasonography showed a normal volume with diffuse heterogeneous background echotexture of both thyroid glands. Stimulation test with gonadotropin releasing hormone (GnRH) and insulin revealed poor responsiveness to gonadotroph, somatotroph, and corticotrophs. Other routine laboratory findings were all unremarkable. A MRI disclosed a 1.0×0.9 cm sized cystic mass enlarging the sella turcica. The mass had heterogeneous signal intensity and mainly peripheral enhancement was observed after administration of contrast material. There was no extension of the mass into the suprasellar cistern or cavernous sinus (Fig. 1A). After 0.2 mg of desmopressin was given orally twice a day, urine volume, serum sodium, and osmolarity normalized. Glucocorticoids were administrated before neurosurgical exploration was performed. Surgical resection via the endoscopic transsphenoidal route was performed. On the pituitary gland, a friable, encapsulated mass with moderate vascularity was found. Histological examination of the resected lesion showed that the pituitary gland was destroyed by infiltration of lymphoplasma cells and foamy histiocytes, fibrosis, and focal necrosis. Foamy histiocytes were positive for CD68 and negative for CD1a and S-100 protein. There was no evidence of an associated adenoma or granuloma (Fig. 2). A postoperative Gadolinium-enhanced MRI was performed on day one postoperatively and showed a residual soft tissue lesion in the superior aspect of pituitary fossa and pituitary stalk (Fig. 1B). The patient continued to take desmopressin 0.2 mg twice daily and prednisolone 5 mg once daily after surgery. Two months later, the patient underwent an endocrine work-up. Poor responsiveness to gonadotroph, somatotroph, and corticotrophs was still shown on the anterior pituitary stimulation test with GnRH and insulin. Desmopressin was also not able to be withdrawn. In comparison to her previous study, the level of TSH was still under the normal range (0.02 mIU/L), but her free T4 had decreased (5.53 pmol/L) significantly, which is consistent with secondary hypothyroidism, prompting thyroxin replacement (Table 1). At 5 months after operation, the patient still had amenorrhea with low estradiol (11.01 pmol/L) and normal serum follicle stimulating hormone/luteinizing hormone (0.3/1.3 IU/L), and she started estrogen replacement.
Table 1.
Pituitary Function Test
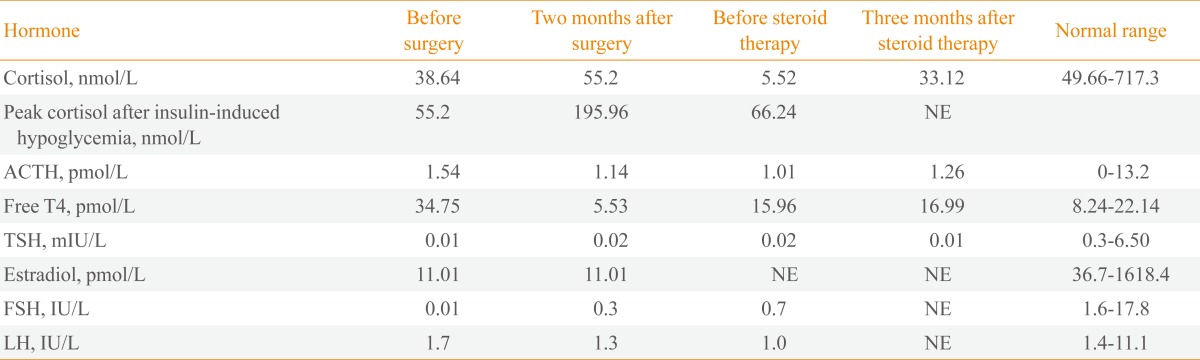
NE, not evaluated; ACTH, adrenocorticotropic hormone; TSH, thyroid-stimulating hormone; FSH, follicle-stimulating hormone; LH, luteinizing hormone.
Fig. 1.
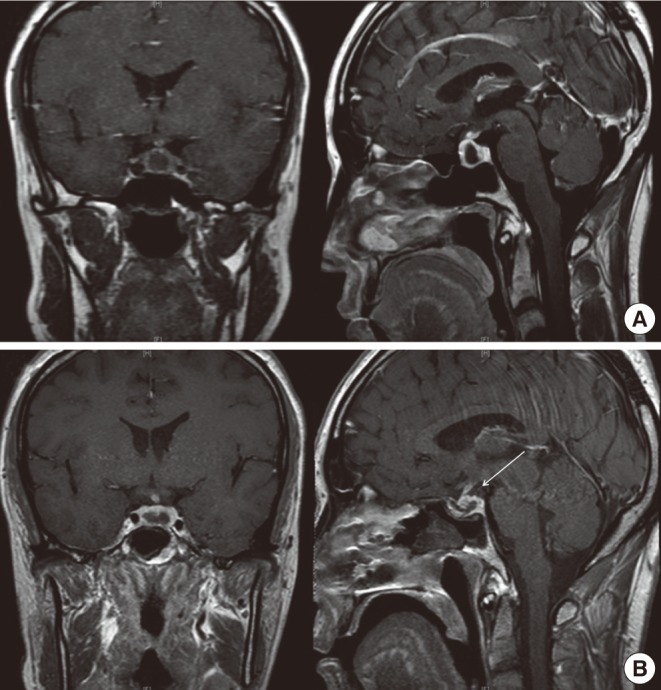
Prepost operative magnetic resonance imaging (MRI) scan, coronal (right), and sagittal (left) T1 weighted images. (A) Preoperative MRI scan following intravenous gadolinium showed a 1×0.9 cm sized peripheral enhancing cystic lesion with high signal intensity in the pituitary fossa. (B) On postoperative day 1, a residual soft tissue lesion (white arrow) was shown.
Fig. 2.
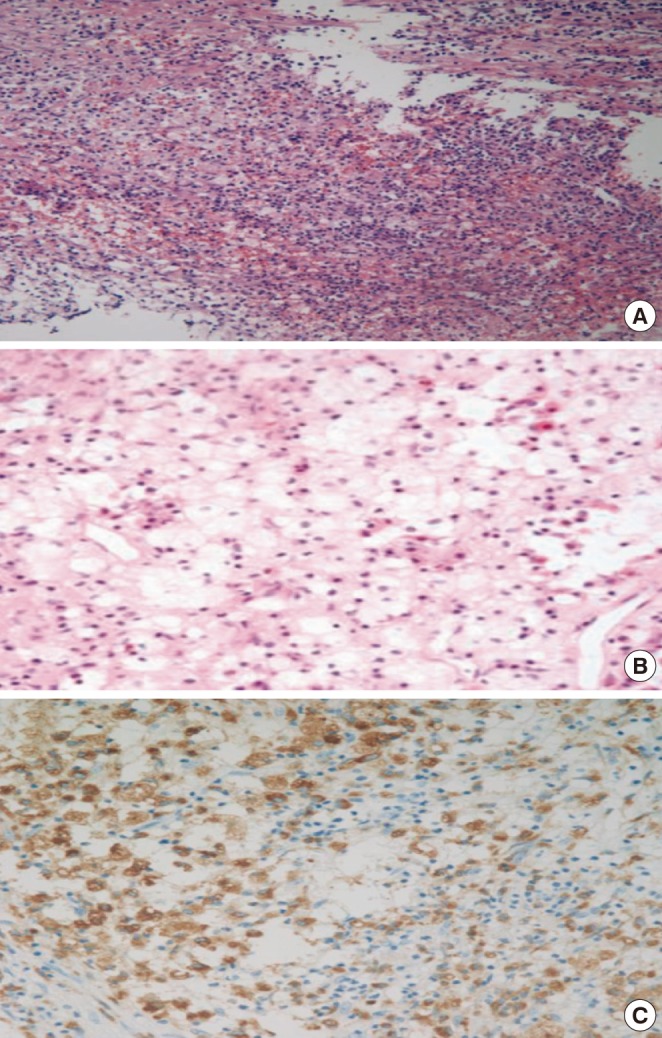
Histology of the resected pituitary gland. (A) Photomicrograph shows that the pituitary gland was destroyed by infiltration of lymphoplasma cells, fibrosis and necrosis (H&E stain, ×200). (B) The pituitary contains a localized aggregates of foamy histiocytes with abundant clear cytoplasm (H&E stain, ×400). (C) Foamy histiocytes were positive for CD68 (CD68, ×400).
At the 10-month postoperative visit, the patient complained of progressive visual impairment and headaches. An MRI performed immediately showed that the residual mass seen on the previous MRI had increased in size and infiltrated the suprasellar lesion (Fig. 3A). The patient refused to undergo surgery again, prompting a trial of high dose steroid therapy. High dose (500 mg intravenous) methylprednisolone was administered for 3 days. During the methyprednisolone pulse therapy, the patient's visual acuity became clear and her headache resolved. An MRI taken 4 days after methyprednisolone initiation revealed a decreased mass size (Fig. 3B). After confirmation of steroid effect on the lesion, high doses of prednisolone (50 mg) were given orally and tapered gradually by 5 to 10 mg per week until her dose reached 10 mg a day. Three months later, a MRI was performed after maintaining prednisolone treatment at 10 mg a day, which demonstrated a marked mass reduction at the sella and suprasellar area (Fig. 3C).
Fig. 3.
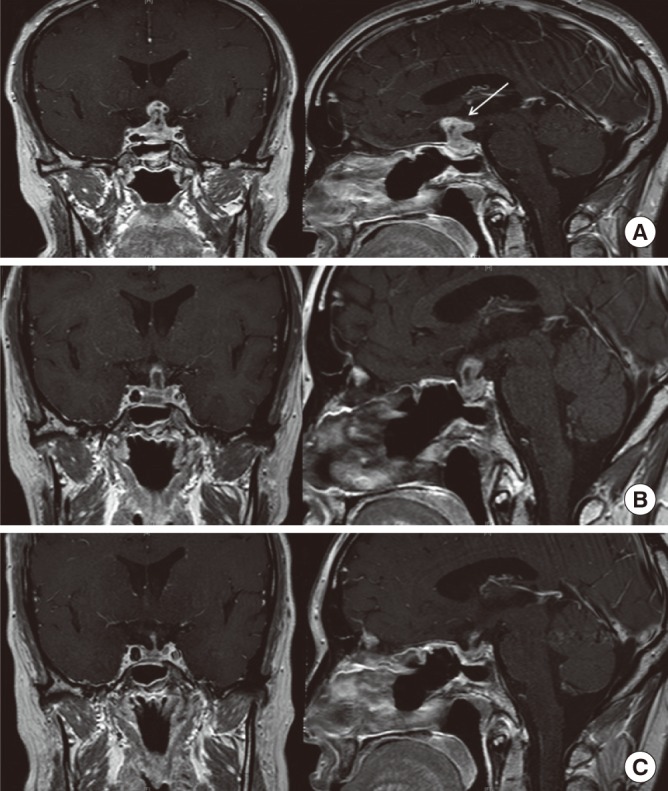
Magnetic resonance imaging (MRI) scan at prepost steroid pulse therapy, coronal (right), and sagittal (left) T1 weighted images. (A) At 10 months after surgery, MRI shows a significantly increasing mass (white arrow) infiltrating into the suprasellar area. (B) Four days after high dose steroid therapy, MRI shows decreased extent of mass at the suprasella area. (C) Three months after steroid therapy, MRI displays marked mass reduction at the sella and suprasella area.
DISCUSSION
Xanthomatous hypophysitis, the least common form of primary hypophysitis, is characterized by the presence of lipid-rich foamy histiocytes with variable numbers of lymphocytes [3]. Unlike the other hypophysitidies, xanthomatous hypophysitis are more likely to be cystic on radiological evaluation and it may be that the inflammation is a response to components of a ruptured cyst [4]. In our case, the lesion was a cystic mass on a MR image and histological examination of the resected lesion showed infiltration of lymphoplasma cells and foamy histiocytes.
The clinical presentation of primary hypophysitis is variable and comprises four categories of symptoms including: sellar compression, hypopituitarism, diabetes insipidus, and hyperprolactinemia [1]. Based on a review of studies where a total of 379 lymphocytic hypophysitis cases were analyzed, symptoms of sellar compression, represented by headache and visual disturbances, were the most common and usually the initial complaint [2]. Endocrine dysfunction was also common due to partial or complete deficit of the anterior pituitary hormones, mainly adrenocorticotropic hormone followed by TSH, gonadotropins, and prolactin. One-third of patients had diabetes insipidus due to deficiency of the posterior pituitary. The rarity of xanthomatous hypophysitis has limited the understanding of their natural history, pathogenesis, and prognosis. The number of reported cases is also not sufficient to characterize clinical presentation. However, xanthomatous hypophysitis commonly presented with headache and amenorrhea upon review of previously reported cases. Gonadal axis impairment and growth hormone deficiency were commonly reported in these cases, while adrenal deficiency and hypothyroidism were less common. Diabetes insipidus was reported in 25% of patients at presentation [1,5,6]. Our patient had all symptoms of sellar compression, hypopituitarism, and diabetes insipidus. Hypopituitarism includes the complete deficiency of the anterior pituitary hormones.
Lymphocytic hypophysitis is often referred to as autoimmune hypophysitis because it displays characteristics suggestive for an autoimmune disease, including improvement of disease in response to immunosuppressive drugs such as glucocorticoids, lymphoplasmacytic nature upon histologic examination, and an association with other autoimmune diseases [7,8]. A review of previously reported lymphocytic hypophysitis cases revealed that 18% of patients with lymphocytic hypophysitis had other better-characterized autoimmune diseases such as Hashimoto's thyroiditis, Graves' disease, and systemic lupus erythematosus [2]. In addition, among the granulomatous subtypes, autoimmune disorders have also been occasionally reported and some cases, particularly those with an underlying systemic granulomatous condition, have been successfully treated with high dose steroids with resolution of clinical and radiological findings [4,9].
However, as far as xanthomatous hypophysitis is concerned, an association with other autoimmune diseases has rarely been reported. Only one case has been reported with ulcerative colitis until recently [5]. Our patient initially presented with thyrotoxicosis, and then progressed to hypothyroidism as time passed. Although a 24-hour thyroid uptake scan was not performed, high titers of antimicrosomal antibodies, ultrasound findings, and the clinical course of thyrotoxicosis followed by hypothyroidism indicate that the thyrotoxicosis in our patients was due to Hashimoto's thyroiditis. Hashimoto's thyroiditis is an auto-immune thyroid disease, which has been reported in patients with lymphocytic hypophysitis as a frequently observed, coexisting autoimmune disorder. There are not enough data reflecting the effects of medical treatment such as glucocorticoids, because it is extremely rare and most cases of xanthomatous hypophysitis have been diagnosed only after surgical resection. Furthermore, no apparent benefit was reported after either pre- or postsurgical therapeutic trials with corticosteroids to date [1, 10]. To our knowledge, this is the first xanthomatous hypophysitis case with steroid responsiveness.
There are several reports of the co-occurrence of aseptic meningitis with lymphocytic hypophysitis. Dissemination of inflammatory cells from the pituitary to the CSF space is one of the possible explanations, although a definitive relationship is unknown. In view of the fact that xanthomatous hypophysitis is due to chronic inflammation with lymphocytes infiltration into the pituitary gland, it is assumed that it is the cause for our patient's aseptic meningitis on presentation.
In most cases of xanthomatous hypophysitis, the pituitary lesion is removed by surgery and the progression of a pituitary lesion after surgery has not been reported. However, in our patient, the small residual lesion seen postoperatively progressed into the suprasellar area over a 10-month interval. Based on our case, xanthomatous hypophysitis could be inferred as a progressive infiltrative disorder. Improvement of pituitary function after surgery has been reported to occur in less than 50% of the cases, which may be attributed to the duration of morbidity, because chronic inflammation can cause irreversible changes to the pituitary gland via tissue destruction and fibrosis [5]. Also, the good response to steroid treatment observed in our case is thought to be due to the relative short duration of lesion progression after surgery.
An autoimmune phenomenon is thought to participate in the pathogenesis of xanthomatous hypophysitis. However, there is limited data supporting an autoimmune etiology at this time, compared to lymphocytic hypophysitis. The patient in our case had Hashimoto's thyroiditis and showed a good response to glucocorticoid pulse therapy. Our case illustrates the possibility of an autoimmune basis in xanthomatous hypophysitis. We suggest that glucocorticoid therapy can be a potential option to manage of xanthomatous hypophysitis not amendable to surgical resection.
Footnotes
No potential conflict of interest relevant to this article was reported.
References
- 1.Gutenberg A, Hans V, Puchner MJ, Kreutzer J, Bruck W, Caturegli P, Buchfelder M. Primary hypophysitis: clinical-pathological correlations. Eur J Endocrinol. 2006;155:101–107. doi: 10.1530/eje.1.02183. [DOI] [PubMed] [Google Scholar]
- 2.Caturegli P, Newschaffer C, Olivi A, Pomper MG, Burger PC, Rose NR. Autoimmune hypophysitis. Endocr Rev. 2005;26:599–614. doi: 10.1210/er.2004-0011. [DOI] [PubMed] [Google Scholar]
- 3.Tashiro T, Sano T, Xu B, Wakatsuki S, Kagawa N, Nishioka H, Yamada S, Kovacs K. Spectrum of different types of hypophysitis: a clinicopathologic study of hypophysitis in 31 cases. Endocr Pathol. 2002;13:183–195. doi: 10.1385/ep:13:3:183. [DOI] [PubMed] [Google Scholar]
- 4.Cheung CC, Ezzat S, Smyth HS, Asa SL. The spectrum and significance of primary hypophysitis. J Clin Endocrinol Metab. 2001;86:1048–1053. doi: 10.1210/jcem.86.3.7265. [DOI] [PubMed] [Google Scholar]
- 5.Aste L, Bellinzona M, Meleddu V, Farci G, Manieli C, Godano U. Xanthomatous hypophysitis mimicking a pituitary adenoma: case report and review of the literature. J Oncol. 2010;2010:195323. doi: 10.1155/2010/195323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burt MG, Morey AL, Turner JJ, Pell M, Sheehy JP, Ho KK. Xanthomatous pituitary lesions: a report of two cases and review of the literature. Pituitary. 2003;6:161–168. doi: 10.1023/b:pitu.0000011177.43408.56. [DOI] [PubMed] [Google Scholar]
- 7.Kristof RA, Van Roost D, Klingmuller D, Springer W, Schramm J. Lymphocytic hypophysitis: non-invasive diagnosis and treatment by high dose methylprednisolone pulse therapy? J Neurol Neurosurg Psychiatry. 1999;67:398–402. doi: 10.1136/jnnp.67.3.398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leung GK, Lopes MB, Thorner MO, Vance ML, Laws ER., Jr Primary hypophysitis: a single-center experience in 16 cases. J Neurosurg. 2004;101:262–271. doi: 10.3171/jns.2004.101.2.0262. [DOI] [PubMed] [Google Scholar]
- 9.de Bruin WI, van 't Verlaat JW, Graamans K, de Bruin TW. Sellar granulomatous mass in a pregnant woman with active Crohn's disease. Neth J Med. 1991;39:136–141. [PubMed] [Google Scholar]
- 10.Niyazoglu M, Celik O, Bakkaloglu DV, Oz B, Tanriover N, Gazioglu N, Kadioglu P. Xanthomatous hypophysitis. J Clin Neurosci. 2012;19:1742–1744. doi: 10.1016/j.jocn.2011.08.041. [DOI] [PubMed] [Google Scholar]