

Abstract
Autophagy plays a crucial role in the maintenance of cellular nutrient balance and the function of organelles such as mitochondria or the endoplasmic reticulum, which are important in intracellular metabolism, insulin release, and insulin sensitivity. In the insulin-producing pancreatic β-cells, autophagy is important in the maintenance of β-cell mass, structure, and function. Mice with deficiencies in β-cell-specific autophagy show reduced β-cell mass and defects in insulin secretion that lead to hypoinsulinemia and hyperglycemia but not diabetes. However, these mice developed diabetes when bred with ob/ob mice, suggesting that autophagy-deficient β-cells have defects in dealing with the increased metabolic stress imposed by obesity. These results also imply that autophagy deficiency in β-cells could be a factor in the progression from obesity to diabetes. Another important function of autophagy is in hypothalamic neurons for the central control of energy expenditure, appetite, and body weight. In addition, mice with autophagy deficiencies in the target tissues of insulin have yielded diverse phenotypes. Taken together, these results suggest that autophagy is important in the control of whole body energy and nutrient homeostasis, and its dysregulation could play a role in the development of metabolic disorders and diabetes.
Keywords: Autophagy, Metabolism, Diabetes
INTRODUCTION
Autophagy, literally 'self-eating,' is a catabolic process characterized by the lysosomal degradation of a cell's own material or organelles for the maintenance of cellular energy balance and organelle function. Among the diverse types of autophagy, macroautophagy (hereafter referred to as autophagy) involves the rearrangement of subcellular membranes to form autophagosomes, which are then delivered to lysosomes and form autophagolysosomes where the sequestered material is degraded and recycled [1].
While autophagy plays a critical role in the clearance of degenerated proteins and senescent organelles as well as in the maintenance of cellular homeostasis during energy starvation or stress, dysregulated autophagy has been implicated in the pathogenesis of neurodegenerative diseases, cancer, aging, and infection [2].
Here, we summarize current understandings of the molecular mechanism of autophagy, focusing on recent results that have examined the role of autophagy in diverse tissues involved in the control of body metabolism and energy homeostasis.
MOLECULAR AND CELLULAR MECHANISM OF AUTOPHAGY
The UNC51-like kinase 1 (ULK1) complex is crucial in autophagy. In nutrient-rich conditions, mammalian target of rapamycin complex 1 (mTORC1) kinase is incorporated into the ULK1-autophagy-related gene (Atg) 13-FIP200 complex and phosphorylates ULK1. Inhibition of mTORC1 by nutrient deprivation or rapamycin induces its dissociation from the ULK complex. Dephosphorylated ULK1 is enzymatically active and phosphorylates mAtg13 and FIP200 to initiate the autophagic process (Fig. 1) [3].
Fig. 1.
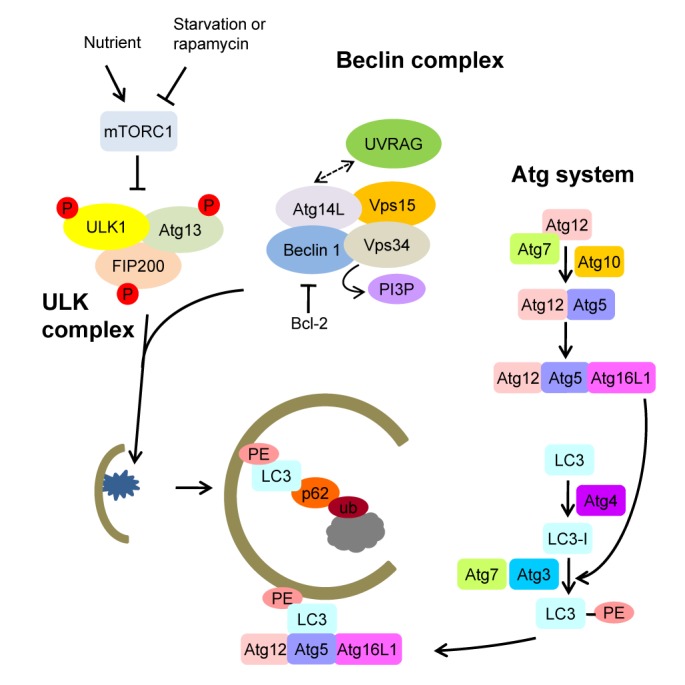
Steps of autophagy induction and autophagosome formation. Mammalian target of rapamycin complex 1 (mTORC1) inhibition by nutrient deprivation or rapamycin treatment induces the dephosphorylation of UNC51-like kinase 1 (ULK1), which phosphorylates autophagy-related gene 13 (Atg13) and FIP200. When autophagy is activated, Beclin 1 is liberated from Bcl-2 and induces autophagosome formation with Vps34, Vps15, and Atg14L or autophagosome maturation with Vps34, Vps15, and ultraviolet radiation resistance-associated gene (UVRAG). Phosphatidylinositol-3-phosphate (PI3P) produced by Vps34 is important in the recruitment of Atg proteins to initiate autophagosome formation. The Atg system is similar to the ubiquitination system. Atg12 and light chain 3 (LC3) are ubiquitin-like proteins. Atg7 is similar to E1. Atg10 and Atg 3 are E2-like enzymes. The Atg12-Atg5-Atg16L1 complex behaves like E3 ligase. LC3-II, formed by LC3 conjugation to its lipid target (phosphatidylethanolamine, PE), is a receptor for p62 which binds to ubiquitinated proteins for proteolytic degradation.
Bcl-2-interacting myosin-like coiled-coil protein (Beclin 1), which was initially identified as a Bcl-2 binding protein, is also critically involved in the initiation of autophagy. Beclin 1 forms complexes with Vps34, Vps15, and Atg14L to induce autophagosome formation or with Vps34, Vps15, and ultraviolet radiation resistance-associated gene (UVRAG) to induce autophagosome maturation [4]. After the dissociation of the Beclin-1 complex from Bcl-2 in autophagy-inducing conditions such as nutrient deprivation, Vps34, a class III phosphatidylinositol 3-kinase within the complex, is activated and produces phosphatidylinositol-3-phosphate (PI3P). PI3P then recruits double FYVE-containing protein 1 (DFCP1) and Atg proteins, which play crucial roles in the formation of the autophagosome cradle (Fig. 1) [5].
The Atg system is critical for autophagosome completion and is similar to the ubiquitination system. Atg12 as a ubiquitin-like protein that is conjugated to Atg5 and then to Atg16L1 through the concerted action of Atg7, an E1-like enzyme, and Atg10, an E2-like enzyme. Atg8, also called microtubule-associated protein 1 light chain 3 (LC3), is another ubiquitin-like protein that is converted to LC3-I immediately after synthesis by Atg4. LC3-I is conjugated to its lipid target, phosphatidylethanolamine (PE), through Atg7, Atg3 acting as another E2-like enzyme and Atg12-Atg5-Atg16L1 complex [6,7]. After processing, lipidated LC3 (LC3-II) is localized to the membranes of autophagosomes. Autophagolysosomes are then conjugated to lysosomes to form autophagolysosomes, where the proteolysis of enclosed cytoplasmic contents or organelles occurs. LC3 is a receptor for p62. Hence, unwanted ubiquitinated proteins can be selectively eliminated by binding to p62 and becoming conjugated to LC3-II of autophagosomes [8]. p62, a substrate of specific autophagy, accumulates in autophagy-deficient conditions. Thus, p62 can be employed as an index of autophagic flux (Fig. 1).
ROLE OF AUTOPHAGY IN PANCREATIC β-CELL PHYSIOLOGY
In an attempt to study the role of autophagy in endocrine tissues, mice with β-cell-specific deletion of autophagy-related 7 (Atg7) have been produced (Atg7Δβ-cell). Atg7Δβ-cell mice showed hyperglycemia and glucose intolerance but not diabetes [9,10]. The insulinogenic index was significantly lower in Atg7Δβ-cell mice compared to control mice, suggesting impaired β-cell function in vivo. Morphologically, β-cell mass was decreased, which was attributed to increased β-cell death and reduced β-cell proliferation. Insulin release and transient glucose-induced cytosolic Ca2+ were significantly attenuated in autophagy-deficient β-cells compared to wild-type β-cells, suggesting functional defects in addition to compromised viability [10].
Confocal microscopy showed inclusion bodies in autophagy-deficient β-cells which contained ubiquitin material and p62 [10], a polyubiquitin-binding adaptor protein [11], attesting to the importance of autophagy in the removal of insoluble or large long-lived ubiquitinated proteins [12]. Electron microscopy showed vacuolar degeneration along with mitochondrial swelling and endoplasmic reticulum (EPrefaceR) distension [10]. These results suggest that autophagy is crucial in the maintenance of β-cell structure, mass and function, although the role of autophagy in the development of diabetes is not clear from this study.
ROLE OF β-CELL AUTOPHAGY IN OBESITY-INDUCED DIABETES
To study the role of β-cell autophagy in diabetes, we studied the ER because ER distention was observed in autophagy-deficient β-cells [10] and ER stress is important in the development of diabetes [13,14]. When we examined the expression of genes involved in the ER stress response or the unfolded protein response (UPR), we found that their expression was significantly reduced in autophagy-deficient β-cells despite ER distention suggesting the presence of ER stress (Fig. 2A) [15]. While these results were contrary to expectations, a deficient UPR in the face of ER stress could be a sign of cellular decompensation or maladaptation [16]. Indeed, autophagy-deficient β-cells were more susceptible to treatment with ER stressors such as thapsigargin or lipids in vitro (Fig. 2B) [15]. Autophagy-deficient β-cells were also more susceptible to the in vivo ER stress imposed by obesity compared to autophagy-competent β-cells. Thus, when Atg7Δβ-cell mice were bred to ob/w mice, β-cell apoptosis was more pronounced in Atg7Δβ-cell-ob/ob mice compared to control Atg7Δβ-cell-ob/w or Atg7F/F-ob/ob mice. In addition, there was evidence of the increased accumulation of reactive oxygen species and decreased β-cell mass in the pancreatic islets of Atg7Δβ-cell-ob/ob mice (Fig. 3A, B) [15]. Accordingly, Atg7Δβ-cell-ob/ob mice developed severe diabetes and markedly deteriorated glucose intolerance, while littermate Atg7Δβ-cell-ob/w mice or Atg7F/F-ob/ob mice showed only mild hyperglycemia (Fig. 3C, D) [15]. These results suggest that compromised β-cell autophagy due to genetic causes, environmental insults, or aging could be a factor in the transition from obesity to diabetes. While closely related, obesity and diabetes are not the same disease process and β-cell autophagy status may help determine the progression from obesity to diabetes.
Fig. 2.

Expression of unfolded protein response (UPR)-related genes in autophagy-deficient islets. Pancreatic islets were isolated from Atg7Δβ-cell and Atg7F/F mice. (A) Real-time reverse transcription polymerase chain reaction was performed using primer sets specific for diverse genes of UPR. (B) Susceptibility of autophagy-deficient islet cells to endoplasmic reticulum (ER) stressors. Primary pancreatic islets were treated with thapsigargin (Tg; left) or palmitic acid (PA; right), and cell death was determined by measuring released oligonucleosomes in the culture supernatant. Autophagy-deficient β-cells were more susceptible to ER stress-induced cell death, probably due to insufficient UPR gene expression. Atg, autophagy-related gene. aP<0.05; bP<0.01; cP<0.001. Adapted from Quan W, et al. Diabetologia 2012;55:392-403, with permission from Springer Science+Business Media [15].
Fig. 3.

Blood glucose level and β-cells of Atg7Δβ-cell-ob/ob mice. (A, B) Apoptotic β-cell number (A) and β-cell mass (B) are shown. (C) Fasting blood glucose levels were determined in Atg7F/F-ob/w (n=7), Atg7Δβ-cell-ob/w (n=6), Atg7F/F-ob/ob (n=5), and Atg7Δβ-cell-ob/ob mice (n=4). (D) Intraperitoneal glucose tolerance test was performed, and the results from Atg7Δβ-cell-ob/ob mice were compared with those of other types of mice (n=4, each group). Atg, autophagy-related gene. aP<0.05; bP<0.01; cP<0.001. Adapted from Quan W, et al. Diabetologia 2012;55:392-403, with permission from Springer Science+Business Media [15].
ROLE OF HYPOTHALAMIC AUTOPHAGY IN THE CONTROL OF WHOLE BODY ENERGY BALANCE AND APPETITE
The role of hypothalamic autophagy in the control of whole body energy balance has also been studied. Mice with deleted Atg7 specifically in their orexigenic agouti-related peptide (AgRP) neurons (Atg7ΔAgRP mice) were lean, which could be attributed to an increase of proopiomelanocortin (POMC) expression and anorexigenic α-melanocyte stimulating hormone production, or to impaired AgRP augmentation in response to fatty acids during starvation in autophagy-deficient AgRP neurons [17]. In contrast, mice with anorexigenic POMC neuron-specific Atg7 deletions (Atg7ΔPOMC mice) were obese [18-20], which was attributed to increased food intake and reduced energy expenditure. Atg7ΔPOMC mice were also resistant to intracerebroventricular administration of leptin [18,20]. Interestingly, the number of POMC neurons was not diminished in the hypothalamus of Atg7ΔPOMC mice compared to control mice [18,20], which is in contrast to the diminished pancreatic β-cell mass in Atg7Δβ-cell mice [9,10]. Instead, signal transducer and activator of transcription 3 (STAT3) activation in the POMC neurons by leptin was diminished in Atg7ΔPOMC mice, although the mechanism of deficient STAT3 activation is not clear [20]. Thus, the role of hypothalamic autophagy on whole body metabolism, appetite, and energy expenditure depends on the types of neurons affected.
METABOLIC IMPACT OF AUTOPHAGY IN THE INSULIN TARGET TISSUES
Mice with autophagy deficiencies in insulin target tissues such as skeletal muscle, liver, or adipose tissues have been generated and studied. Animals with targeted disruption of Atg7 in the liver have been reported to show accumulation of lipids due to defects in the lipid-disposal process known as 'lipophagy' [21]. In addition, the conditional knockout of autophagy genes in adipose tissues results in defects in adipocyte differentiation [22,23]. We have demonstrated that autophagy deficiencies in the skeletal muscle or liver lead to leanness and resistance to diet-induced obesity and diabetes. Such phenotypes were attributed to mitochondrial dysfunction due to autophagy deficiencies in insulin target tissues and the subsequent mitochondrial stress response inducing the release of FGF21 as a 'mitokine' [24]. These results are in contrast to a previous paper which suggested that autophagy deficiency is an element in the pathogenesis of insulin resistance and diabetes [25]. Such discrepancies could be due to differences in the methods, severity, or duration of gene targeting, the age of the experimental animals, and the mode of autophagy affected.
The concept of 'mitokines' has been suggested by a Caenorhabditis elegans model that shows increased longevity and has a disruption in the mitochondrial electron transport chain [26]. In addition, a recent paper reported a significant prolongation of lifespan in transgenic mice overexpressing FGF21 [27]. The relationship between autophagy, mitochondria, and 'mitokines' could be a hot topic not only in the field of metabolism and diabetes, but also in aging and longevity studies.
CONCLUSIONS
The roles of autophagy in whole body metabolism and the development of metabolic syndrome or diabetes have been explored by many investigators. The majority of previous studies investigating the role of autophagy in body metabolism and diabetes employed site-specific knockout mouse models. While such models have provided important data regarding the impact of dysregulated autophagy in metabolic disorders, they may not reflect natural physiological or disease conditions. Further studies employing more physiological models are likely to provide valuable information on the role of autophagy in the control of body metabolism and the pathogenesis of metabolic disorders.
Because autophagy plays a crucial role in other body systems besides the control of metabolism, the information obtained by studying the relationship between autophagy and body metabolism could lead to the development of a novel class of drugs that can be employed not only against diabetes or metabolic syndrome, but also cancer, neurodegeneration, and aging.
ACKNOWLEDGMENTS
This study was supported by the Samsung Biomedical Research Institute Grant (SP1-B2-051-2) and the Korea Healthcare Technology R&D Project, Ministry for Health, Welfare & Family Affairs, Korea (A080967). M.S.L. is a recipient of a Global Research Laboratory Grant (K21004000003-10A0500-00310) and Bio&Medical Technology Development Program (20110019335) of the National Research Foundation of Korea.
Footnotes
No potential conflict of interest relevant to this article was reported.
References
- 1.Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science. 2000;290:1717–1721. doi: 10.1126/science.290.5497.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mizushima N. The role of the Atg1/ULK1 complex in autophagy regulation. Curr Opin Cell Biol. 2010;22:132–139. doi: 10.1016/j.ceb.2009.12.004. [DOI] [PubMed] [Google Scholar]
- 4.Matsunaga K, Saitoh T, Tabata K, Omori H, Satoh T, Kurotori N, Maejima I, Shirahama-Noda K, Ichimura T, Isobe T, Akira S, Noda T, Yoshimori T. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat Cell Biol. 2009;11:385–396. doi: 10.1038/ncb1846. [DOI] [PubMed] [Google Scholar]
- 5.Hamasaki M, Yoshimori T. Where do they come from? Insights into autophagosome formation. FEBS Lett. 2010;584:1296–1301. doi: 10.1016/j.febslet.2010.02.061. [DOI] [PubMed] [Google Scholar]
- 6.Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728–741. doi: 10.1016/j.cell.2011.10.026. [DOI] [PubMed] [Google Scholar]
- 7.Nakatogawa H, Oh-oka K, Ohsumi Y. Lipidation of Atg8: how is substrate specificity determined without a canonical E3 enzyme? Autophagy. 2008;4:911–913. doi: 10.4161/auto.6646. [DOI] [PubMed] [Google Scholar]
- 8.Ichimura Y, Kumanomidou T, Sou YS, Mizushima T, Ezaki J, Ueno T, Kominami E, Yamane T, Tanaka K, Komatsu M. Structural basis for sorting mechanism of p62 in selective autophagy. J Biol Chem. 2008;283:22847–22857. doi: 10.1074/jbc.M802182200. [DOI] [PubMed] [Google Scholar]
- 9.Ebato C, Uchida T, Arakawa M, Komatsu M, Ueno T, Komiya K, Azuma K, Hirose T, Tanaka K, Kominami E, Kawamori R, Fujitani Y, Watada H. Autophagy is important in islet homeostasis and compensatory increase of beta cell mass in response to high-fat diet. Cell Metab. 2008;8:325–332. doi: 10.1016/j.cmet.2008.08.009. [DOI] [PubMed] [Google Scholar]
- 10.Jung HS, Chung KW, Won Kim J, Kim J, Komatsu M, Tanaka K, Nguyen YH, Kang TM, Yoon KH, Kim JW, Jeong YT, Han MS, Lee MK, Kim KW, Shin J, Lee MS. Loss of autophagy diminishes pancreatic beta cell mass and function with resultant hyperglycemia. Cell Metab. 2008;8:318–324. doi: 10.1016/j.cmet.2008.08.013. [DOI] [PubMed] [Google Scholar]
- 11.Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, Mizushima N, Iwata J, Ezaki J, Murata S, Hamazaki J, Nishito Y, Iemura S, Natsume T, Yanagawa T, Uwayama J, Warabi E, Yoshida H, Ishii T, Kobayashi A, Yamamoto M, Yue Z, Uchiyama Y, Kominami E, Tanaka K. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. 2007;131:1149–1163. doi: 10.1016/j.cell.2007.10.035. [DOI] [PubMed] [Google Scholar]
- 12.Kirkin V, McEwan DG, Novak I, Dikic I. A role for ubiquitin in selective autophagy. Mol Cell. 2009;34:259–269. doi: 10.1016/j.molcel.2009.04.026. [DOI] [PubMed] [Google Scholar]
- 13.Back SH, Scheuner D, Han J, Song B, Ribick M, Wang J, Gildersleeve RD, Pennathur S, Kaufman RJ. Translation attenuation through eIF2alpha phosphorylation prevents oxidative stress and maintains the differentiated state in beta cells. Cell Metab. 2009;10:13–26. doi: 10.1016/j.cmet.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, Tuncman G, Gorgun C, Glimcher LH, Hotamisligil GS. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004;306:457–461. doi: 10.1126/science.1103160. [DOI] [PubMed] [Google Scholar]
- 15.Quan W, Hur KY, Lim Y, Oh SH, Lee JC, Kim KH, Kim GH, Kim SW, Kim HL, Lee MK, Kim KW, Kim J, Komatsu M, Lee MS. Autophagy deficiency in beta cells leads to compromised unfolded protein response and progression from obesity to diabetes in mice. Diabetologia. 2012;55:392–403. doi: 10.1007/s00125-011-2350-y. [DOI] [PubMed] [Google Scholar]
- 16.Merksamer PI, Trusina A, Papa FR. Real-time redox measurements during endoplasmic reticulum stress reveal interlinked protein folding functions. Cell. 2008;135:933–947. doi: 10.1016/j.cell.2008.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kaushik S, Rodriguez-Navarro JA, Arias E, Kiffin R, Sahu S, Schwartz GJ, Cuervo AM, Singh R. Autophagy in hypothalamic AgRP neurons regulates food intake and energy balance. Cell Metab. 2011;14:173–183. doi: 10.1016/j.cmet.2011.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Coupé B, Ishii Y, Dietrich MO, Komatsu M, Horvath TL, Bouret SG. Loss of autophagy in pro-opiomelanocortin neurons perturbs axon growth and causes metabolic dysregulation. Cell Metab. 2012;15:247–255. doi: 10.1016/j.cmet.2011.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kaushik S, Arias E, Kwon H, Lopez NM, Athonvarangkul D, Sahu S, Schwartz GJ, Pessin JE, Singh R. Loss of autophagy in hypothalamic POMC neurons impairs lipolysis. EMBO Rep. 2012;13:258–265. doi: 10.1038/embor.2011.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Quan W, Kim HK, Moon EY, Kim SS, Choi CS, Komatsu M, Jeong YT, Lee MK, Kim KW, Kim MS, Lee MS. Role of hypothalamic proopiomelanocortin neuron autophagy in the control of appetite and leptin response. Endocrinology. 2012;153:1817–1826. doi: 10.1210/en.2011-1882. [DOI] [PubMed] [Google Scholar]
- 21.Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, Tanaka K, Cuervo AM, Czaja MJ. Autophagy regulates lipid metabolism. Nature. 2009;458:1131–1135. doi: 10.1038/nature07976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Singh R, Xiang Y, Wang Y, Baikati K, Cuervo AM, Luu YK, Tang Y, Pessin JE, Schwartz GJ, Czaja MJ. Autophagy regulates adipose mass and differentiation in mice. J Clin Invest. 2009;119:3329–3339. doi: 10.1172/JCI39228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang Y, Goldman S, Baerga R, Zhao Y, Komatsu M, Jin S. Adipose-specific deletion of autophagy-related gene 7 (atg7) in mice reveals a role in adipogenesis. Proc Natl Acad Sci U S A. 2009;106:19860–19865. doi: 10.1073/pnas.0906048106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim KH, Jeong YT, Oh H, Kim SH, Cho JM, Kim YN, Kim SS, Kim do H, Hur KY, Kim HK, Ko T, Han J, Kim HL, Kim J, Back SH, Komatsu M, Chen H, Chan DC, Konishi M, Itoh N, Choi CS, Lee MS. Autophagy deficiency leads to protection from obesity and insulin resistance by inducing Fgf21 as a mitokine. Nat Med. 2013;19:83–92. doi: 10.1038/nm.3014. [DOI] [PubMed] [Google Scholar]
- 25.Yang L, Li P, Fu S, Calay ES, Hotamisligil GS. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab. 2010;11:467–478. doi: 10.1016/j.cmet.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Durieux J, Wolff S, Dillin A. The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell. 2011;144:79–91. doi: 10.1016/j.cell.2010.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang Y, Xie Y, Berglund ED, Coate KC, He TT, Katafuchi T, Xiao G, Potthoff MJ, Wei W, Wan Y, Yu RT, Evans RM, Kliewer SA, Mangelsdorf DJ. The starvation hormone, fibroblast growth factor-21, extends lifespan in mice. Elife. 2012;1:e00065. doi: 10.7554/eLife.00065. [DOI] [PMC free article] [PubMed] [Google Scholar]