

Abstract
The transcriptional regulator RovA positively regulates transcription of the Yersinia enterocolitica virulence gene inv. Invasin, encoded by inv, is important for establishment of Y. enterocolitica infection. However, a rovA mutant is more attenuated for virulence than an inv mutant, implying that RovA regulates additional virulence genes. When the Y. enterocolitica RovA regulon was defined by microarray analysis, YE1984 and YE1985 were among the genes identified as being upregulated by RovA. Since these genes are homologous to Xenorhabdus nematophila cytotoxin genes xaxA and xaxB, we named them yaxA and yaxB, respectively. In this work, we demonstrate the effects of YaxAB on the course of infection in the murine model. While a yaxAB mutant (ΔyaxAB) is capable of colonizing mice at the same level as the wild type, it slightly delays the course of infection and results in differing pathology in the spleen. Further, we found that yaxAB encode a probable cytotoxin capable of lysing mammalian cells, that both YaxA and YaxB are required for cytotoxic activity, and that the two proteins associate. YaxAB-mediated cell death occurs via osmotic lysis through the formation of distinct membrane pores. In silico tertiary structural analysis identified predicted structural homology between YaxA and proteins in pore-forming toxin complexes from Bacillus cereus (HBL-B) and Escherichia coli (HlyE). Thus, it appears that YaxAB function as virulence factors by inducing cell lysis through the formation of pores in the host cell membrane. This characterization of YaxAB supports the hypothesis that RovA regulates expression of multiple virulence factors in Y. enterocolitica.
INTRODUCTION
Yersinia enterocolitica is an enteric pathogen causing a range of diseases, from mild diarrhea to a more severe systemic bacteremia (1, 2). Y. enterocolitica infection typically results in an acute inflammatory disease of the gastrointestinal tract and/or mesenteric lymph nodes and is characterized by diarrhea, fever, and abdominal pain (1, 2). Normally the infection is cleared in 1 to 2 weeks; however, in some cases, especially when the host is immunocompromised or has high serum iron levels, the infection can become systemic and mortality rates are high (1, 2). Infection with Y. enterocolitica most commonly results from consumption of contaminated food, milk, or water. Livestock populations have been shown to harbor a high carriage rate of Y. enterocolitica (3, 4), and it is thought that this Y. enterocolitica burden in the food supply contributes to human yersiniosis (4).
A number of years ago a murine model of oral infection was established that mimics human systemic Y. enterocolitica infection (5, 6). In this model, Y. enterocolitica survives passage through the stomach, binds to intestinal epithelial cells, and crosses the epithelium, where it colonizes the Peyer's patches. From here, the bacteria can spread to the mesenteric lymph nodes (MLN), spleen, and liver (6). Mice infected with highly pathogenic biovar 1B strains develop a lethal systemic infection (3, 6, 7). During a Y. enterocolitica infection, an inflammatory response from the host results in an influx of macrophages and neutrophils to the sites of infection, aided by the production of proinflammatory cytokines, such as gamma interferon (IFN-γ), tumor necrosis factor alpha (TNF-α), and interleukin-6 (IL-6) (6, 8–11). This influx of immune cells contributes to the pathology of a Y. enterocolitica infection, and lesions of primarily neutrophils surrounding necrotic tissue and bacteria have been reported in the MLN, spleens, and livers of infected mice (6, 10, 12). Similar colonization patterns and pathology are observed in humans with systemic yersiniosis (1, 2).
Y. enterocolitica possesses several virulence factors that allow the bacterium to bind and invade host cells (9, 13, 14). One of these factors is invasin, an outer membrane protein that binds to β1-integrins on the apical surface of intestinal M cells, aiding the bacterial translocation across M cells into the Peyer's patches below (13, 15–17). Our laboratory previously showed that the transcriptional regulator RovA controls expression of inv, the gene encoding invasin (18). RovA is thermoregulated and is responsible for the temperature dependence of inv expression in Y. enterocolitica and Y. pseudotuberculosis (18, 19). Further investigation into inv regulation revealed that two other regulatory proteins, H-NS and YmoA, repress inv transcription (18, 20–22). However, RovA can displace H-NS from the inv promoter (21, 23), suggesting they compete for binding at the inv promoter. YmoA has not been shown to directly bind the inv promoter, but it has been shown to negatively regulate inv expression (20, 22). It is thought that YmoA and H-NS form a repressive complex that binds to the promoter of inv, and that RovA displaces this complex, acting as a derepressor to induce transcription of inv (22).
A strain of Y. enterocolitica with a mutation in rovA is less virulent than an inv mutant, implying that RovA also regulates other Y. enterocolitica genes important for virulence (18, 24). Comparison of gene expression profiles from the wild type and a rovA mutant of Y. enterocolitica using microarray analysis revealed 62 other RovA-regulated genes (25). Two of the genes upregulated in the presence of RovA, YE1984 and YE1985, are located adjacent to each other in the Y. enterocolitica chromosome and are predicted to form an operon (25). These genes are homologous to genes recently described to encode a two-protein cytotoxin from Xenorhabdus nematophila, XaxAB (25, 26). Due to the homology to xaxAB, we have named YE1984 and YE1985 yaxA and yaxB, respectively. Both RovA and H-NS bind to the predicted promoter region of yaxA, and RovA, H-NS, and YmoA regulate transcription of yaxA (25). These data suggest RovA promotes yaxA transcription while H-NS and YmoA inhibit transcription, similar to the mechanism of inv regulation, implying that regulation of RovA and H-NS/YmoA is a global regulatory system used by Y. enterocolitica (25). Since yaxA and yaxB are homologous to genes known to encode a toxin and are regulated by the same system that regulates another defined Y. enterocolitica virulence factor, we investigated whether yaxA and yaxB play a role in Y. enterocolitica pathogenesis and their effect on mammalian cells in culture.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
Bacterial strains and plasmids used in this work are listed in Table 1. All cultures of Y. enterocolitica were grown in LB (1% tryptone, 0.5% yeast extract, 170 mM NaCl; Difco [BD Biosciences], Bedford, MA) at 26°C. All cultures of Escherichia coli were grown in LB at 37°C. Antibiotics were added to growth medium as needed at the following concentrations: nalidixic acid (Nal), 20 μg/ml; kanamycin (Kan), 100 μg/ml (both from Sigma-Aldrich, St. Louis, MO).
Table 1.
Strains and plasmids used in this work
| Strain or plasmid | Relevant genotype/phenotypea | Source or reference |
|---|---|---|
| E. coli | ||
| DH5α | F− ϕ80lacZ ΔM15 Δ(lacZYA-argF)U169 deoP recA1 endA1 hsdR17 (rk− mk−) | Invitrogen |
| S17-1λpir | recA thi pro hsdR hsdM+ RP4::2-Tc::Mu::Kan Tn7 λ pir lysogen, Tpr Strr | 61 |
| Y. enterocolitica | ||
| JB580v | 8081v (r− m+ Nalr) serotype O:8, biovar 1B | 62 |
| YVM1334 | JB580v with deletion of yaxAB | This work |
| MC5 | Clinical isolate, biovar 1A | Gift from M. Cafferkey |
| MC7 | Clinical isolate, biovar 1A | Gift from M. Cafferkey |
| MC8 | Clinical isolate, biovar 1A | Gift from M. Cafferkey |
| WC-A | Biovar 1B | 63 |
| Y295 | Biovar 1B | 64 |
| 657-83 | Reference strain, biovar 1B | 65 |
| 9291-78 | Clinical isolate, biovar 1B | 65, 66 |
| 2400-87 | Biovar 1B | 64 |
| MC22 | Clinical isolate, biovar 3 | 67 |
| MC32 | Clinical isolate, biovar 3 | 64, 67 |
| MC33 | Clinical isolate, biovar 3 | 67 |
| MC34 | Clinical isolate, biovar 3 | 67 |
| MC6 | Clinical isolate, biovar 4 | 67 |
| MC28 | Clinical isolate, biovar 4 | 67 |
| MC51 | Clinical isolate, biovar 4 | 64, 67 |
| Plasmids | ||
| pSR47S | MobRP4 oriR6K cloning vector, Kanr | 68 |
| pGK501 | yaxA/B deletion in pSR47S, Kanr | This work |
| pMWO-005 | Low-copy cloning vector, Kanr | 39 |
| pMWO-034 | Low-copy cloning vector, Kanr | 39 |
| pNW306 | yaxA and yaxB coding sequence in pMWO-034, Kanr | This work |
| pNW307 | yaxA-myc and yaxB-HA coding sequence in pMWO-034, Kanr | This work |
| pNW311 | yaxA-myc coding sequence in pMWO-034, Kanr | This work |
| pNW312 | yaxB-HA coding sequence in pMWO-005, Kanr | This work |
Tpr, trimethoprim resistant; Strr, streptomycin resistant; Kanr, kanamycin resistant; Nalr, nalidixic acid resistant.
Plasmid and strain construction.
All strains and plasmids used in this study are listed in Table 1, and the primers are listed in Table 2. All DNA inserted into plasmid vectors was verified by DNA sequencing (Eton Bioscience, San Diego, CA, or Genewiz, South Plainfield, NJ).
Table 2.
Primers used in this work
| Primer name | Sequencea (5′-3′) |
|---|---|
| YE1984/5del1 | GCGTCGACGTTTTATGGCTGATTTAAATTTC |
| YE1984/5del2 | CGGGATCCTTGTGTCATTGTATTATCCTCATC |
| YE1984/5del3 | CGGGATCCCAAACAGCTGATTTGACTTGTG |
| YE1984/5del4 | ATAAGAATGCGGCCGCCTTCACTTGTGACTCCATCTC |
| Yax001 | GTGCGTCGACGTCTTCTTACCTTTACGGGTCTGC |
| Yax004 | CGGAATTCCGCCTACCTTTACGGGTCTGC |
| Yax005 | CGGGATCCCGAAATCAGCTGTTTGATTAATGA |
| Yax006 | CGGGATCCCGCGAGCGATGTCTGGGACGTCGTATGG GTAAATCAGCTGTTTGATTAATGAC |
| Yax009 | TTACAGATCTTCTTCAGAAATAAGTTTTTGTTCGCCATACACTTTTTTGTATTC |
| Yax010 | GAACAAAAACTTATTTCTGAAGAAGATCTGTAATCAATATTACCGTATTTAATTATTC |
| Yax011 | GGAATTCCTCATAATGAATAATTAAATACG |
| Yax013 | GGGTACCCCGGAAATAAGCACATTTCCA |
| Yax014 | GTGCGTCGCACACAAACACAATTGGCTATTG |
| Yax015 | TGAAGCGGCCGCTCAAATCAGCTGTTTGATTAATGAC |
| MWO-05rev | CAGCCTGAAACAGGCGATGCTGC |
Restriction sites are underlined, and sequences for epitope tags are in boldface.
A deletion of the yaxA and yaxB genes was made in Y. enterocolitica JB580v as previously described (27). Briefly, regions upstream of yaxA and downstream of yaxB were amplified using primers YE1984/5del1 and YE1984/5del2 (upstream) or YE1984/5del3 and YE1984/5del4 (downstream). The products were digested with SalI and BamHI or BamHI and NotI (upstream and downstream, respectively), ligated into pSR47S cut with SalI and NotI, and transformed into S17-1λpir. The resulting plasmid, pGK501, was introduced into Y. enterocolitica by conjugation to create strain YVM1334 (ΔyaxAB). Confirmation that the genes were deleted was determined by PCR using primers outside the region cloned into pGK501.
Constructs to express yaxA and yaxB were generated as follows. Primers Yax004 and Yax005 were used to amplify a fragment that included the ribosomal binding site (RBS) upstream of yaxA as well as yaxA and yaxB from chromosomal Y. enterocolitica DNA. This product was digested with EcoRI and BamHI and ligated into pMWO-034 to generate pNW306. A C-terminal myc tag was added to yaxA by amplifying the gene with a 5′ primer (Yax004) and a 3′ primer that contains the sequence for myc (Yax009). A C-terminal hemagglutinin (HA) tag was added to yaxB using a similar strategy. The downstream primer Yax006, which includes the sequence for an HA tag, was used with primer Yax010 to amplify a product that contained yaxB-HA. Primer Yax010 also contains sequence for the myc tag, so that the ends of these two fragments are homologous. An additional overlap PCR, using these two products as the template DNA, was performed with primers Yax004 and Yax006. The resulting product contained the RBS upstream of yaxA, yaxA-myc, and yaxB-HA. This product was digested with EcoRI and BamHI and ligated into pMWO-034, generating plasmid pNW307. To clone YaxA alone, the sequence of yaxA-myc was amplified using primers Yax001 and Yax011 from plasmid pNW307. The resulting PCR product was digested with SalI and EcoRI and ligated into pMWO-034, producing plasmid pNW311. To clone yaxB-HA without yaxA, primers Yax013 and MWO-05rev were used to generate a product that contained the sequence of yaxB and the in-frame HA tag from plasmid pNW307. The product was digested with SacI and KpnI and ligated into pMWO-005, generating plasmid pNW312.
Animal infection.
All animal experiments were conducted with the approval of the University of North Carolina IACUC (protocol 11-127.0). Six- to 8-week-old female BALB/c mice were purchased from Jackson Laboratory (Bar Harbor, ME) and housed in an approved animal facility with free access to food and water throughout the experiment. Overnight cultures of Y. enterocolitica, grown in LB broth at 26°C with aeration, were normalized to an optical density at 600 nm (OD600) of 3.6 with sterile LB broth. Mice were infected via oral gavage, delivering 5.5 × 108 to 8.0 × 108 Y. enterocolitica cells in 0.1 ml using a 22-gauge ball-tipped feeding needle. Mock-infected mice were given 0.1 ml LB via oral gavage. At the indicated time points, mice were euthanized by CO2 asphyxiation and tissues were harvested. Tissues were weighed and homogenized in sterile PBS. Serial dilutions of the tissue homogenates were plated to determine bacterial load in Peyer's patches, spleens, livers, and mesenteric lymph nodes (MLN). Dilutions from Peyer's patches were plated on Yersinia selective agar (Difco); all other organs were plated on LB agar containing 20 μg/ml nalidixic acid. Plated dilutions and tissue weight was used to determine CFU/gram of tissue. Statistical significance was determined by analyzing the CFU/gram of tissue with a two-tailed Mann-Whitney t test.
Histopathology.
Mice were infected as described above. Uninfected (LB-treated) mice or mice infected for 1, 3, 4, 5, or 6 days were sacrificed by CO2 asphyxiation. The MLN, spleen, and liver were removed and fixed in 10% neutral buffered formalin (Sigma-Aldrich) for 24 h. Tissues were embedded in paraffin, sectioned, and stained with hematoxylin and eosin (H&E) by the UNC Center for Gastrointestinal Biology and Disease Cell Services and Histology Core. Lesions of Y. enterocolitica infection in the spleen were enumerated as follows. A single pathologist (L. B. Borst) evaluated samples of approximately equal size in a blinded fashion for microscopic lesions of Y. enterocolitica. Individual foci of inflammation, necrosis, and intralesional bacteria, regardless of size, were counted as individual lesions, and the total number of lesions was determined using the entire longitudinal section of spleen. Lesions were characterized using cellular morphology on H&E-stained sections. Inflammatory cell infiltrates were characterized using cytologic features as follows. Nondegenerative neutrophils were identified by segmented or ring-shaped nuclei with clumped heterochromatin and clear to slightly eosinophilic cytoplasm; degenerative neutrophils were identified by swollen hypochromatic nuclei, hypersegmented nuclei, and hypereosinophilic or foamy fragmented cytoplasm; and macrophages were identified by a round to oval, often eccentrically placed, nucleus and moderate to abundant foamy pale eosinophilic cytoplasm. Areas of necrosis were identified by homogeneous eosinophilic staining with loss of cellular detail mixed with basophilic cellular and nuclear debris. The data were analyzed with the Student t test.
Bacterial lysates.
Y. enterocolitica or E. coli strains containing plasmids that express yaxA (pNW311), yaxB (pNW312), both yaxA and yaxB (pNW307), or vector (pMWO-034) were grown at 26°C (Y. enterocolitica) or 37°C (E. coli) overnight. Saturated cultures were subcultured into fresh media to 0.2 OD600 units/ml. Anhydrous tetracycline (ATc; Sigma-Aldrich) was added to the growth media at 100 μg/ml. After 5 h of growth at 26 or 37°C, induced bacteria were collected by centrifugation at 1,200 × g for 10 min. Bacterial pellets were resuspended in 200 mM Tris (pH 8.0), 500 mM NaCl, 18% glycerol, 10 mM EDTA at approximately 40 OD600/ml. Bacteria were lysed by sonication with 10 5-s bursts, resting on ice between bursts. Samples were centrifuged (16,000 × g, 20 min) to clear the lysates; any remaining bacteria were removed by filtering the lysate through a 0.2-μm filter. Protein concentration was determined using Bradford reagent (Bio-Rad, Hercules, CA). Lysates were stored at 4°C with no noticeable loss in toxin activity.
Cell culture.
The murine macrophage-like cell line J774.A1 (ATCC TIB-67) was cultured in Dulbecco's modified Eagle medium (DMEM; Gibco [Life Technologies], Carlsbad, CA) supplemented with 10% heat-inactivated fetal bovine serum (FBS; Gibco), 1 mM MEM nonessential amino acids (Gibco), 1 mM sodium pyruvate (Gibco), and 2 mM l-glutamine (Gibco) at 37°C with 5% CO2. The human monocyte cell line THP-1 (ATCC TIB-202) was cultured in RPMI 1640 (Gibco) supplemented with 10% heat-inactivated FBS, 0.05 M β-mercaptoethanol (Gibco), and 2 mM l-glutamine at 37°C under 5% CO2. To differentiate THP-1 cells, 200 nM phorbol 12-myristate 13-acetate (PMA; Sigma-Aldrich) was added to the medium for 72 to 96 h. Medium was replaced with fresh media lacking PMA, and cells were cultured an additional 3 to 5 days before being used in assays (28). The human epithelial cell line HEp-2 (ATCC CCL-23) was cultured in MEM (Gibco) supplemented with 10% heat-inactivated FBS at 37°C under 5% CO2. The human colon epithelial cell line Caco-2 (ATCC HTB-37) was cultured in MEM supplemented with 10% heat-inactivated FBS, 1 mM MEM nonessential amino acids, and 1 mM sodium pyruvate at 37°C under 5% CO2. The hemocyte-like Drosophila melanogaster cell line S2 (Invitrogen [Life Technologies], Carlsbad, CA) was cultured in serum-free Sf-900 II (Gibco) at 26°C in ambient CO2.
To derive macrophages from murine bone marrow (BMM), nonadherent cells from a single-cell suspension of bone marrow harvested from the femurs and tibia of C57BL/6 mice were cultured in DMEM supplemented with 10% heat-inactivated FBS, 1 mM sodium pyruvate, 2 mM l-glutamine, 50 mM β-mercaptoethanol (Gibco), and 40 ng/ml macrophage colony-stimulating factor (M-CSF; Peprotech, Rocky Hill, NJ) at 37°C under 5% CO2. Cells were cultured for 7 days before being used in assays.
Microscopy.
Eukaryotic cells were grown on glass poly-d-lysine-coated coverslips (BD Biosciences), incubated with lysate from bacteria expressing YaxAB or empty vector, fixed to the glass with 4% paraformaldehyde (EMD Millipore, Billerica, MA), and stained with Giemsa stain (IMEB, Inc., San Marcos, CA). Coverslips were mounted with Cytoseal-60 (Richard-Allan Scientific, Kalamazoo, MI). Cells were imaged on an Olympus BX60 microscope. Images were captured digitally using a SPOT-RT slider charge-coupled device (CCD) camera (Diagnostic Instruments, Sterling Heights, MI) and iVision imaging software v.4.0.0 (BioVision Technologies, Exton, PA).
LDH release assay.
Lactate dehydrogenase (LDH) released from mammalian cells was assayed with a CytoTox 96 kit by following the manufacturer's protocol (Promega, Madison, WI). Briefly, cells were seeded into 96-well tissue culture-treated plates approximately 16 h before the assay. Cell density used for each cell type was determined by measuring LDH release from Triton X-100-treated cells; the number of cells that gave at least 2-fold more LDH when treated with Triton X-100 than media were used for assays. Specifically, J774.A1 cells and BMM were seeded at 2 × 105 cells/ml, THP-1 cells at 2 × 104 cells/ml, HEp-2 cells at 1 × 105cells/ml, and Caco-2 cells at 5 × 104cells/ml. Lysates from the various bacterial strains were added to wells at the indicated concentrations, which were based on total protein content in the lysate preparation. Cells were incubated in the presence of lysate for 4 h unless otherwise indicated. Supernatant from the cells was removed and assayed for the presence of LDH. The level of LDH in the media from lysate-treated cells was compared to the level from cells treated with Triton X-100 (assumed to be 100% cell lysis). Where indicated, cells were preincubated with osmoprotectants, including glycine (5 mM), sucrose (30 mM), raffinose (30 mM), polyethylene glycol 600 (PEG 600; 30 mM), PEG 1450 (30 mM), or PEG 3350 (30 mM), for 1 h before the addition of bacterial lysates (29, 30).
Lysis of S2 cells was measured using both the Cytotox 96 kit, as described above, and R-Biopharm's (Darmstadt, Germany) d-lactic acid/l-lactic acid assay, as previously described (31). For the R-Biopharm kit, assays were performed in 240-μl reaction mixtures with 50 μl of culture supernatant added to each reaction mixture. After reactions were initiated by the addition of NAD, absorbance at 340 nm was measured using a BioTek Synergy H1 and Gen5 software (version 1.11; BioTek, Winooski, VT) every 2 min for 1 h. S2 cells were seeded at 1 × 106 cells/ml approximately 16 h before the assay. Cells treated with Triton X-100 were considered 100% lysed, and these readings were used as maximum lysis. Bacterial lysates were added to cells as described for the mammalian cell assays. J774.A1 cells were also tested using the R-Biopharm assay and resulted in similar LDH release, as seen with the Promega kit. Statistical analysis for all LDH release assays was performed with the two-tailed Mann-Whitney t test.
Immunoprecipitation and Western blot analysis.
Cleared bacterial lysates, prepared as described above (Y. enterocolitica expressing YaxA, YaxB, both YaxA and YaxB, or vector), were incubated with mouse anti-HA agarose beads (Sigma-Aldrich) overnight at 4°C. Beads were washed three times with 200 mM Tris (pH 8.0), 500 mM NaCl, 18% glycerol, 10 mM EDTA. Following washes, beads were suspended in 1× SDS buffer and boiled. Proteins in lysates and proteins recovered from anti-HA precipitation were separated by SDS-PAGE and transferred to Immobilon-P polyvinylidene difluoride (PVDF; EMD Millipore) for Western blot analysis. Primary antibodies, monoclonal rabbit anti-HA, and rabbit anti-myc (Cell Signaling Technology, Danvers, MA) were used at a concentration of 1:2,000, and peroxidase-conjugated secondary antibody (Sigma-Aldrich) was used at 1:20,000. A chemiluminescent signal was generated using ECL plus (GE Healthcare, Piscataway, NJ) or Clarity Western ECL substrate (Bio-Rad), and emitted light was captured using a G:Box and GeneSys software (version 1.2.5; Syngene, Frederick, MD).
Y. enterocolitica strain survey for yaxAB.
To determine if the sequences for yaxAB are present in Y. enterocolitica strains from different biovars, chromosomal DNA was obtained from the strains listed in Table 1 by phenol-chloroform extraction. Primers were designed using the sequence from 8081v (32). The upstream primer (Yax014) (Table 2) binds yaxA 3′ of the start codon, and the downstream primer (Yax015) binds the 3′ end of yaxB, including the stop codon. PCR was performed with these primers using MyTaq DNA polymerase (BioLine USA, Boston, MA) according to the supplier's instructions. The JB580v and ΔyaxAB strains were used as positive and negative controls, respectively. PCR products were separated on a 1.5% agarose gel, stained with ethidium bromide, and visualized using the Syngene G:Box and GeneSys software (version 1.2.5; Syngene).
YaxA structural prediction and modeling.
To identify structural motifs in YaxA and YaxB, the following algorithms were used to predict protein motifs and topology: TMpred (ch.embnet.org/software/TMPRED_form.html), using a window of 17 to 33 amino acids; HMMTOP (enzim.hu.hmmtop), which uses a 17- to 25-amino-acid window to predict transmembrane helices; TMHMM (cbs.dtu.dk/services/TMHMM), which models transmembrane helices between 15 and 25 amino acids; and TopPred (http://mobyle.pasteur.fr/cgi-bin/portal.py?#forms::toppred), using a window of 21 amino acids (33–36). To identify tertiary structural homology and predict protein function, the protein sequence was submitted to the Iterative Threading Assembly Refinement (I-TASSER) server (zhanglab.ccmb.med.umich.edu/I-TASSER) (37, 38). I-TASSER generated a three-dimensional model that was viewed and manipulated with PyMOL, version 1.3 (Schrödinger LLC, Portland, OR).
RESULTS
YaxAB affect the course of murine oral Y. enterocolitica infection.
Our laboratory previously showed that yaxA and yaxB, homologs to the genes which encode the cytotoxins XaxA and XaxB (26), are positively regulated by RovA and negatively regulated by H-NS and YmoA (25). To determine if yaxA and yaxB are important in Y. enterocolitica virulence, a strain was constructed with a complete deletion of yaxAB (the ΔyaxAB mutant). We compared the course of an oral infection between this mutant strain and wild-type Y. enterocolitica (JB580v) in BALB/c mice. Both Y. enterocolitica strains were able to colonize Peyer's patches within the first 24 h following infection (data not shown). At the time points examined, 1, 3, and 5 days postinoculation (dpi), equal numbers of CFU were recovered from the Peyer's patches and mesenteric lymph nodes (MLN) of mice infected with either the wild-type or mutant strain (Fig. 1A and data not shown). Likewise, we saw no significant difference in the rate of bacterial dissemination or colonization in the livers of infected mice at 3 and 5 dpi (data not shown). These data imply that YaxAB does not play a role in the establishment of Y. enterocolitica infection. However, in the spleen, there were fewer CFU at 3 dpi in mice infected with the ΔyaxAB strain (median, 553 CFU/g) than in those infected with the wild type (median, 1.7 × 106 CFU/g) (Fig. 1B). At 5 dpi, the difference in bacterial load was reduced (mean, 1.8 × 107 CFU/g in ΔyaxAB mutant-infected mice and 1.4 × 108 CFU/g in wild type-infected mice) (Fig. 1B). While there are differences in the splenic bacterial burden between the wild type- and ΔyaxAB mutant-infected mice, due to the large variation of CFU recovered from the spleens of mice, the data are not statistically significant using the Mann-Whitney t test (P = 0.285). Along with different numbers of recovered bacteria from the spleens, we also noticed a trend of more mice surviving ΔyaxAB mutant infection to day five. At this time point, 38% of mice infected with the wild-type strain had succumbed to infection, while only 17% of mice infected with the ΔyaxAB mutant had died.
Fig 1.
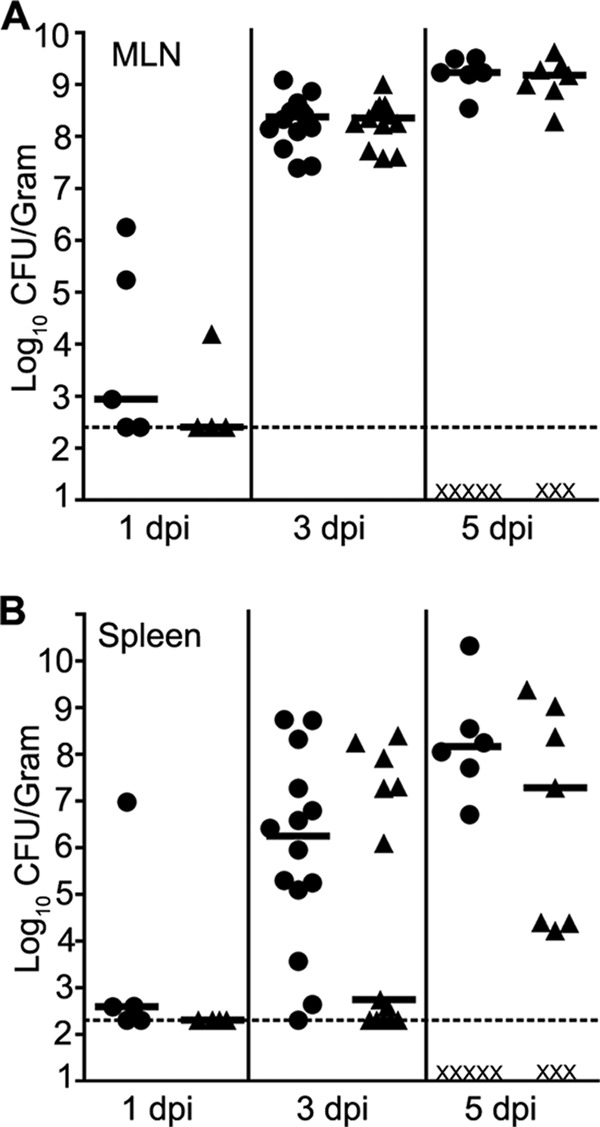
Colonization of BALB/c mice infected intragastrically with Y. enterocolitica. Mice were infected with 5.5 × 108 to 8 × 108 CFU of the wild-type (●) or ΔyaxAB (▲) strain. At the indicated time points, animals were sacrificed and tissues were harvested to determine the colonization of the mesenteric lymph nodes (A) or spleen (B). Each symbol represents one mouse, an X indicates a dead mouse, black bars are the median numbers of CFU recovered per gram of tissue, and dashed lines are set at the limit of detection. Data are from three independent experiments.
YaxAB influence host pathology in response to Y. enterocolitica.
The difference in survival at day 5 between the ΔyaxAB mutant- and wild type-infected mice caused us to investigate whether or not a difference exists in pathology between mice infected with these two strains. We found considerable inflammation in the Peyer's patches and MLN of mice infected with both strains at 1 dpi and in the Peyer's patches, MLN, and spleens at 3 and 5 dpi (data not shown). Lesions consisting of neutrophils surrounding necrotic areas and apoptotic bodies were previously reported for Y. enterocolitica infection (10, 11, 24). At 5 dpi, lesions similar to those previously described and consisting of the same cell types were seen in the spleens of mice infected with either strain; however, more lesions were present in the spleens of mice infected with the mutant strain (mean of 53 lesions per spleen at 5 dpi) than in those infected with wild-type bacteria (mean of 2 lesions per spleen at 5 dpi) or mock infected (no lesions observed) (Fig. 2). Microscopically, these lesions consisted primarily of degenerative and nondegenerative neutrophils and fewer macrophages, which mix with necrotic cellular debris and apoptotic bodies (Fig. 2A to D). The inflammatory infiltrates surround variably large central aggregates of extracellular bacteria (Fig. 2C to E). The spleens of mice infected with the ΔyaxAB mutant strain of Y. enterocolitica had fewer bacteria and more lesions than mice infected with the wild-type strain (Fig. 1B and 2F).
Fig 2.

Infection with the ΔyaxAB mutant results in more splenic lesions than wild-type infection. Shown are H&E-stained splenic sections from a mouse infected with either the wild-type (A) or ΔyaxAB (B to D) strain. (A and B) The image at ×2 magnification. Lesions with similar appearance and cellular makeup are present throughout both spleens (examples are highlighted by arrows); however, more lesions are apparent in the ΔyaxAB mutant-infected spleen (B). The region shown in the black box of panel B is enlarged in panel C. (C) The image at ×10 magnification. The region shown in the box is enlarged in panel D. (D) The image at ×60 magnification. The large colonies of bacteria are identified with an arrowhead and are surrounded by numerous neutrophils (identified by segmented or ring-shaped nuclei) and fewer macrophages (identified by round or oval nuclei and abundant foamy cytoplasm). Apoptotic bodies were occasionally observed (arrow). (E) Splenic lesion boxed in panel B stained with anti-Yersinia antibody (brown) showing bacteria localized within the splenic lesions of ΔyaxAB mutant-infected mice. Magnification, ×10. (F) Splenic lesions were counted per spleen in wild-type-infected (●), ΔyaxAB mutant-infected (▲), or mock-infected (not shown) mice. The number of lesions per spleen is shown. Each symbol represents one mouse, and black bars represent the means. At 5 dpi, there are significantly more lesions per spleen in ΔyaxAB mutant-infected mice than wild-type-infected mice, as determined by one-sample t test.
Further supporting the idea that yaxAB plays a role in virulence, we found conservation of the yaxAB sequence among strains of the pathogenic biovars 1B, 3, and 4 and not in strains from biovar 1A by a PCR screen (data not shown). In every strain tested from biovars 1B, 3, and 4 (Table 1), a PCR product was detected using primers flanking yaxAB in strain 8081v (primers Yax014 and Yax015) (Table 2). As these genes have been maintained in pathogenic strains of Y. enterocolitica, particularly in the highly pathogenic 1B strains, it would seem that the proteins provide some advantage. While the impact on the murine model of systemic infection was modest, YaxAB could play a more significant role in other mammalian infections, including humans. Alternatively, YaxAB may be important for establishing and/or maintaining a Y. enterocolitica reservoir.
YaxAB are cytotoxic in vitro.
Since YaxAB are homologous to a defined cytotoxin (XaxAB), we wanted to determine if YaxAB had similar properties. The expression of yaxA and yaxB under normal laboratory growth conditions is undetectable (25); therefore, in order to produce YaxAB, we induced expression of yaxA and yaxB. The genes encoding YaxA and YaxB were cloned into pMWO-034, a low-copy-number expression vector with an anhydrous tetracycline (ATc)-inducible promoter (39). The yax genes were cloned in tandem, either directly from the chromosomal DNA (pNW306) or with C-terminal epitope tags (YaxA-myc and YaxB-HA; pNW307), and transformed into wild-type Y. enterocolitica. Bacterial lysates were prepared by sonication from mid-log cultures that had been grown with ATc and were used to treat tissue culture cells (YaxAB lysate). Once we established activity in lysates prepared from pNW306 (untagged), we confirmed equal activity in the tagged version of the proteins (pNW307). For the reported experiments, YaxAB lysates were generated from pNW307 so that expression could be verified by visualizing the epitope tags. When J774.A1 murine macrophage-like cells were incubated with YaxAB lysate, destruction of the cells was evident within 4 h by microscopy and LDH release (Fig. 3A and B). Cells incubated with lysate from bacteria harboring pMWO-034 (vector lysate) (Fig. 3B, samples V) showed the same level of LDH release as untreated cells (Fig. 3B, samples U), indicating that these lysates had low or no cytotoxic activity. To ensure native yaxAB expression did not contribute to toxicity, vector lysates were prepared from both wild-type and ΔyaxAB strains and were found to have equally low levels of LDH release (data not shown).
Fig 3.
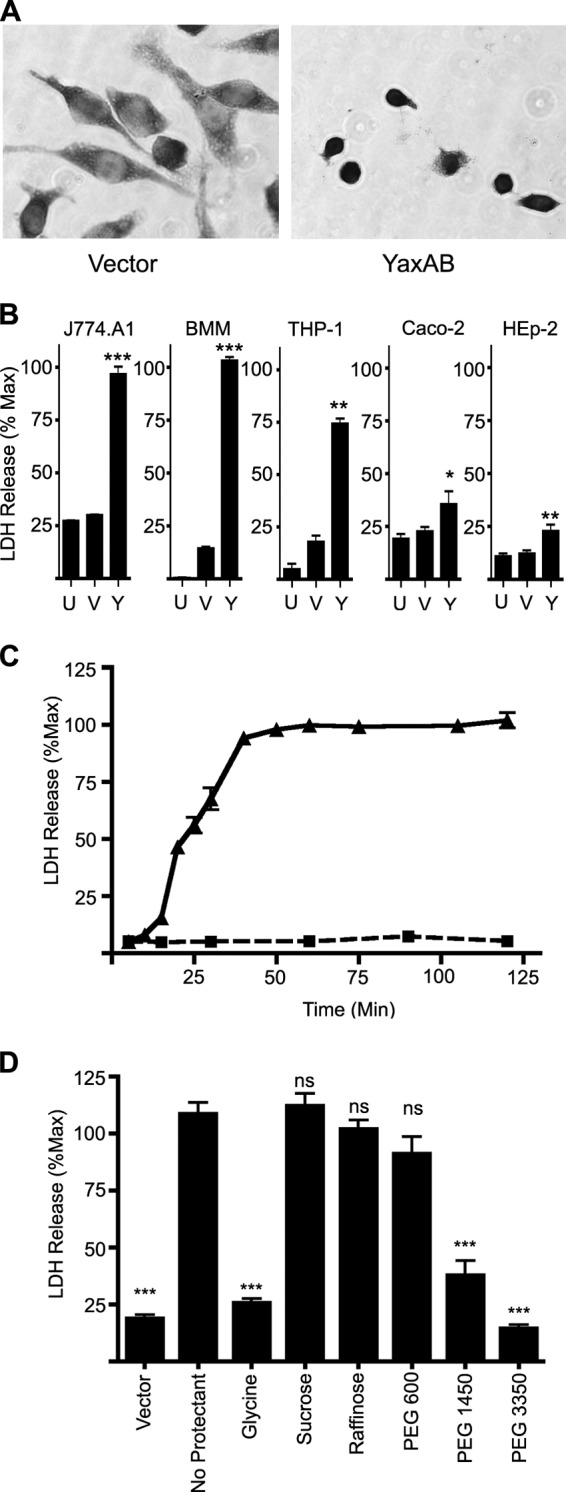
YaxAB kills mammalian cells by osmotic lysis. (A) Light microscopy images (×20) of Giemsa-stained J774.A1 cells exposed to YaxAB lysate for 4 h. (B to D) LDH measured from culture supernatants and represented as a percentage of Triton X-100-treated cells (maximal lysis control). (B) LDH released from the indicated cells after a 4-h incubation with PBS (untreated [U]), vector lysate (V), or YaxAB lysate (Y). (C) LDH released from J774.A1 cells treated with vector lysate (■, dashed line) or YaxAB lysate (▲, solid line). (D) LDH release from J774.A1 cells after 1 h of preincubation with the indicated osmoprotectants, followed by 1 h of incubation with vector or YaxAB lysate. In panels B to D, means ± SEM are shown. *, P < 0.05; **, P < 0.005; ***, P < 0.0001; ns, not significant.
To determine if YaxAB-mediated killing was specific to J774.A1 cells, we incubated other cell types with these same lysates (Fig. 3B, samples Y). YaxAB lysates induced statistically significant LDH release in murine bone marrow-derived macrophages (BMM; 100% maximum), human monocytes derived from THP-1 cells (74.0% maximum), the human epithelial cell line HEp-2 (22.6% maximum), and Caco-2 human colon epithelial cells (35.2% maximum) compared to cells left untreated or treated with vector lysate (Fig. 3B, samples U or V, respectively). Because XaxAB was shown to be an insect toxin, we also tested for YaxAB toxicity on the Drosophila melanogaster phagocytic hemocyte cell line S2. We observed no difference in cell death by LDH release or microscopy between S2 cells incubated with YaxAB lysate or vector lysate (data not shown). Together, these data suggest that Y. enterocolitica YaxAB is cytotoxic to mammalian cells but not insect cells, and that there is a more potent effect on immune cell lines.
YaxAB-mediated toxicity acts through osmotic lysis.
As we had established that YaxAB was toxic to mammalian cells, we wanted to investigate the mechanism by which YaxAB causes cell death. Since YaxAB-mediated killing of J774.A1 cells is robust, this cell line was chosen for further investigation. We tested the ability of YaxAB-containing bacterial lysates to induce LDH release from J774.A1 cells over time and found that YaxAB-induced permeabilization of J774.A1 cells can be detected by LDH release as early as 20 min after addition of YaxAB lysate (Fig. 3C), a time point when cells show no gross sign of lysis via microscopy (data not shown). Levels of LDH release increased with time, and maximal levels were observed within 1 h of YaxAB lysate exposure (Fig. 3C). Low levels of LDH were detected from cells treated with vector lysate and remained at baseline throughout the time course tested (5 to 10% of maximum) (Fig. 3C). Similar results were seen when lysates were incubated with bone marrow-derived macrophages (data not shown).
The time-course LDH release assay indicates that the mechanism of YaxAB-mediated killing is fast acting, with complete lysis of J774.A1 cells within 1 h. One phenomenon that can cause rapid cell death is osmotic lysis. To test if this was the mechanism by which YaxAB acted on J774.A1 cells, cells were preincubated for 1 h prior to the addition of YaxAB lysate with glycine, which nonspecifically blocks osmotic lysis by preventing ion flux across cell membranes (30, 40–42). Glycine protected J774.A1 cells from YaxAB-mediated lysis, as the treated cells displayed LDH release at levels similar to those from cells treated with vector lysates (25.6% compared to 18.7% of the maximum) (Fig. 3D). These data imply that YaxAB is killing target cells through osmotic lysis. While the osmotic protection provided by glycine is not pore size specific, other molecules that do not impact ion flux will block osmotic lysis when the molecules are larger than the pore size, whereas particles smaller than the pore should freely pass through and the cell will still be subject to lysis (30, 43, 44). To test whether or not exposure to YaxAB resulted in the formation of pores and to estimate the pore size, cells were pretreated with molecules of various sizes (sucrose, 0.9 nm; raffinose, 1.1 nm; PEG 600, 2.0 nm; PEG 1450, 2.4 nm; PEG 3350, 3.8 nm) (29, 30) for 1 h, followed by lysate treatment and LDH release assay. Pretreatment with PEG 1450 or PEG 3350 conferred the same protection as that observed with glycine, producing low LDH release levels (37.6 and 14.4% of maximum, respectively) (Fig. 3D). However, sucrose, raffinose, and PEG 600 did not confer any protection to J774.A1 cells from YaxAB-mediated killing. These data indicate that YaxAB form a distinct pore in the target cell membrane, and that the pore size is between 2.0 and 2.4 nm.
Pore-forming toxins can induce K+ flux, leading to capsase-1 activation and cell death (45, 46). Additionally, cells that have activated the NLRP3/caspase-1-mediated cell death pathway also have distinct pores formed in the membrane (30, 41). To determine if the rapid osmotic lysis we see in target cells involves the caspase-1-mediated cell death pathway(s) (pyrotosis), we tested the ability of YaxAB to kill macrophages derived from the bone marrow of caspase-1−/− mice. Immortalized caspase-1−/− bone marrow-derived macrophages (a gift from Edward Miao) had elevated levels of LDH release when exposed to YaxAB lysate (76.5% of maximum) compared to those exposed to vector lysate (47.7% of maximum) or untreated cells (44.4% of maximum) (data not shown). Therefore, YaxAB-mediated cell death occurs in a caspase-1-independent manner. From these data, we conclude that YaxAB-mediated cell death occurs through caspase-1-independent osmotic lysis by the formation of discrete membrane pores.
Toxicity requires both YaxA and YaxB.
To determine whether both YaxA and YaxB are required for toxicity, constructs were generated that expressed epitope-tagged versions of either YaxA or YaxB (plasmid pNW311 expresses YaxA-myc and pNW312 expresses YaxB-HA). These plasmids were transformed into wild-type Y. enterocolitica, and YaxA or YaxB expression was confirmed by Western blotting (Fig. 4A). Lysates from bacteria expressing YaxA-myc (YaxA lysate), YaxB-HA (YaxB lysate), or both YaxA-myc and YaxB-HA (YaxAB lysate) were generated as described above and incubated with J774.A1 cells. When lysates from bacteria expressing either component individually were incubated with cells, the amount of LDH released into the medium was comparable to levels in cells treated with vector lysate (YaxA lysate, 28.1% of maximum; YaxB lysate, 26.5% of maximum; vector lysate, 27.5% of maximum) (Fig. 4B). When YaxA lysate was added to cells, followed 1 h later by YaxB lysate, J774.A1 cells released near-maximal LDH levels (67.2%) (Fig. 4B), demonstrating that both YaxA and YaxB are required for YaxAB-mediated toxicity. Interestingly, when lysate from bacteria expressing YaxB was added to J774.A1 cells prior to the addition of YaxA lysate, there was minimal LDH release (37.1% of maximum) (Fig. 4B). Similar results were reported with XaxA and XaxB from Xenorhabdus (26, 30).
Fig 4.
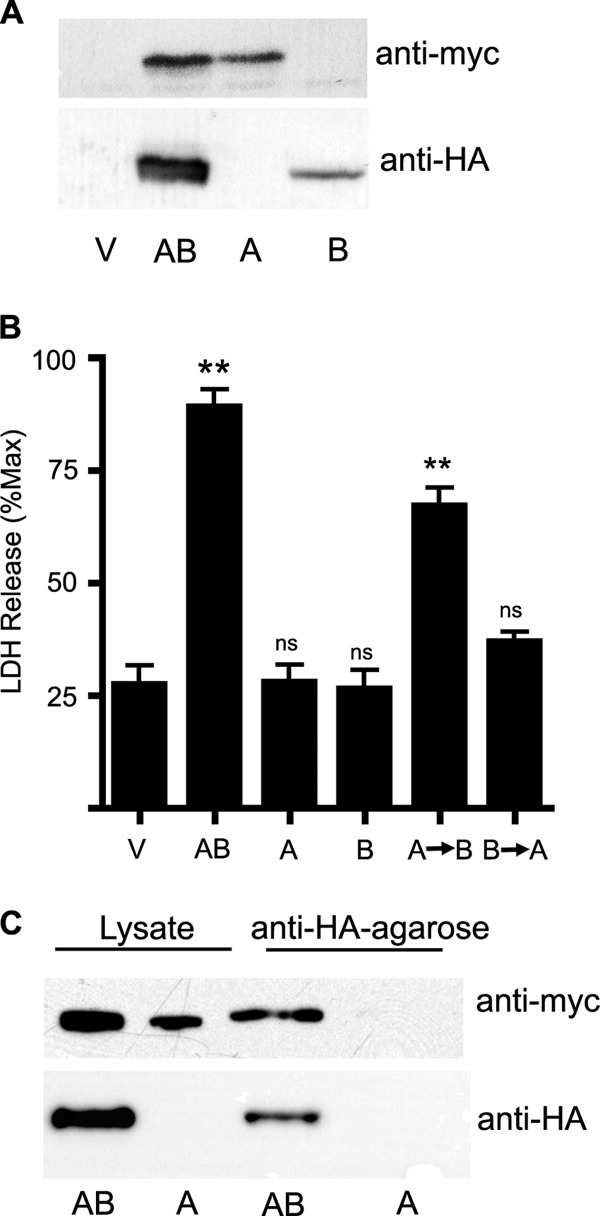
Both YaxA and YaxB are required for lysis and are copurified. (A) Western blot of YaxA-myc and YaxB-HA from bacteria expressing both YaxA and YaxB (AB) or YaxA (A) or YaxB (B) alone. Anti-HA and anti-myc antibodies were used to verify YaxA-myc and YaxB-HA expression. (B) LDH measured from culture supernatants and represented as a percentage of Triton X-100-treated cells. LDH release from J774.A1 cells after 1 h of incubation with vector (V), YaxAB lysate (AB), YaxA lysate (A), or YaxB lysate (B), 1 h of incubation with YaxA lysate followed by 1 h with YaxB lysate (A→B), or 1 h of incubation with YaxB lysate followed by YaxA lysate for 1 h (B→A). Means ± SEM are shown. **, P < 0.005. (C) YaxAB (AB) or YaxA (A) lysates were incubated with agarose beads bound with anti-HA antibody. The supernatant was removed and the beads were washed three times. The washed beads and starting lysate were analyzed by Western blotting as described above.
Since both proteins are required for toxic activity, it was of interest to determine whether YaxA and YaxB directly interact. Lysates from bacteria expressing both YaxA-myc and YaxB-HA were incubated with anti-HA antibody bound to agarose beads. After incubation, the beads were washed, boiled in SDS loading buffer, run on an SDS-PAGE gel, and Western blotted with an anti-HA antibody to confirm that YaxB was pulled down and an anti-myc antibody to assay for coimmunoprecipitation (Fig. 4C). A positive HA signal from samples containing YaxB-HA (Fig. 4C, samples AB) showed that the immunoprecipitation worked as expected. A positive myc signal from the anti-HA beads indicated that the antibody on the beads pulled down YaxA-myc with YaxB-HA. Fractions eluted from anti-HA beads incubated with lysate from bacteria expressing only YaxA-myc did not react with either the anti-HA or the anti-myc antibody by Western blotting (Fig. 4C, samples A), indicating that YaxA detected from YaxAB lysate is the result of interactions between YaxB and YaxA, not nonspecific binding of YaxA to the anti-HA agarose beads. These data confirm that YaxA and YaxB directly interact.
Predicted structure of YaxA homologous to pore-forming toxin.
Our data established that YaxAB induces lysis of target cells by forming pores in the target cell membrane. Further supporting the idea that YaxAB is a cytolysin, the sequence of YaxA has structural homology to known pore-forming toxins. The amino acid sequence of YaxA was analyzed with the iterative threading assembly refinement (I-TASSER) server, which derived a predicted model of YaxA. This model had a confidence score (C-score) of −1.4. The C-score estimates the quality of the model based on the significance of threading template alignments and convergence of the I-TASSER structural assembly refinements (37). At least 90% of the predictions are accurate when models have C-scores of greater than −1.5 (37). As part of the I-TASSER analysis, TM-align was used to compare the predicted structure of YaxA to crystallized proteins with similar structures. TM-align identified one component of a three-protein Bacillus cereus toxin, HblB (PDB code 2NRJ), and the E. coli toxin HlyE (also called ClyA and SheA) (PDB code 1QOY) as highly similar to the predicted structure of YaxA. Using the structures of these toxins, TM-align generated overlays of the predicted structure of YaxA with HblB and YaxA with HlyE (Fig. 5). Examination of these overlays shows that the structure of YaxA predicted by I-TASSER is highly similar to the structure of HlyE, but it has a stronger alignment with HblB (root mean square deviation [RMSD] of 1.24 and 4.31, respectively). YaxA structure prediction based on HblB orients it with five long coils and a head region extending from one side (Fig. 5). In HblB, this head is predicted to contain a transmembrane domain that may insert into the membrane of the target cell (47). The head of HlyE has been shown to undergo a conformational change upon membrane association, causing this region of the protein to swing away from the rest of the protein and into the membrane of a target cell (48). Motif prediction algorithms (TMpred, TopPred, HMMTOP, and TMHMM; see Materials and Methods for details) suggest that the portion of YaxA that aligns with the head region of HblB also contains a transmembrane domain, and the function of this region may be similar to that of this region of HblB and HlyE.
Fig 5.

Structural comparisons of YaxA to HblB and HlyE. (A) I-TASSER structural prediction of YaxA based on tertiary alignment with HblB. The predicted transmembrane domain is shown in pink. (B) Predicted structure of YaxA (blue and pink) overlaid on the crystal structure of HblB (green). (C) Predicted structure of YaxA overlaid on the crystal structure of HlyE (red).
DISCUSSION
Here, we demonstrate that the RovA-regulated genes yaxAB encode proteins which contribute to cytotoxic activity for both human and murine cells in culture. We also demonstrate that YaxAB-mediated toxicity acts through the formation of pores in the membrane of target cells, causing osmotic lysis that can be blocked by osmoprotectants. YaxAB plays a role in the pathogenicity of Y. enterocolitica, as the absence of these genes results in altered pathology and lower levels of bacterial colonization of the spleen. The structural homology of YaxA to components of other pore-forming toxins is especially intriguing, as this mechanism of cytotoxicity appears to be conserved across many bacterial species. Along with the structural similarity, hlyE and yaxAB are both regulated by similar mechanisms involving the homologous proteins SlyA (E. coli) and RovA (Y. enterocolitica) in relieving H-NS-mediated repression (25, 49, 50).
The predicted structural homology of YaxA to the known pore-forming toxins HblB and HlyE supports our data that YaxAB forms pores in the membranes of host target cells. Like YaxAB, HlyE is toxic to macrophage-like cells in culture, and it has been shown to lyse cells via osmotic lysis (51, 52). Additionally, the pores YaxAB and HlyE form in the membrane of target cells are of similar sizes (HlyE pores are 2.5 to 3.0 nm, and YaxAB pores are 2.0 to 2.4 nm) (50–54). While the structural models do not fit between HlyE and YaxA as well as those between HblB and YaxA (Fig. 5), the regulatory and functional similarities between HlyE and YaxA are intriguing and hint at a global mechanism for toxin production and function in these two enteric pathogens.
Curiously, we found no structural or sequence homolog for YaxB other than the previously described XaxB-like proteins in Xenorhabdus and Photorhabdus (26), indicating that its role in the toxicity mechanism is unique to this group of proteins. Our data show that YaxA alone has no cytotoxic activity. HblB also requires other proteins for function (HblL1 and HblL2), but HlyE is toxic without any other proteins (51, 52, 55). This suggests that while there are structural similarities among YaxA, HblB, and HlyE, the YaxAB toxin complex employs a mechanism of toxicity distinct from those of these other two pore-forming toxins. It is interesting that when the proteins were expressed separately, we found full lytic activity only occurred when YaxA was added before YaxB. Similar results were reported for the Xenorhabdus XaxAB and Bacillus HBL toxin complexes (26, 55). Thus, it seems likely that the pore-forming portion of each of these toxins acts similarly upon the host cell, but the mechanism of toxicity and/or how the target cell is exposed to the toxin varies between bacterial species. There is some evidence that XaxB, as well as XaxA, can bind membrane (26), so one explanation for these results is that addition of XaxB (or YaxB) prior to XaxA (or YaxA) leads to membrane binding in a manner that is no longer competent for interaction with the corresponding A component.
The method of release from the bacteria or surface exposure utilized by YaxAB has not been determined, as is the case for the structural homolog, HlyE. There is no evidence that HlyE is secreted through any of the type I to V secretion systems, and it does not contain any described signal sequences or appear to have any signal sequence cleavage or posttranscriptional modifications (50, 56). YaxA and YaxB also contain no defined signal sequence and were not secreted from Y. enterocolitica under any of the conditions tested (N. J. Wagner & V. L. Miller, unpublished data). It is possible that YaxA and YaxB are released spontaneously as a result of bacterial lysis, or that they are released from the bacteria through outer membrane vesicles, as has been suggested for HlyE (56). However, we were unable to detect YaxAB-mediated cell death using live bacteria (N. J. Wagner and V. L. Miller, unpublished).
In this work, the source of YaxAB for in vitro studies was derived from bacterial lysates made from Y. enterocolitica induced to express yaxAB. The plasmid we used to express yaxAB has a very low copy number (approximately 5 plasmids per bacterium [39, 57, 58]), but as we induced gene expression, the levels of protein likely were higher than what is produced from the chromosomal copy of the genes. Therefore, assays performed with induced yaxAB may overstate the impact of these genes in the native bacterium. Additionally, as our experiments were carried out using bacterial lysates as the source of YaxAB, it is conceivable that some other bacterial component acts in conjunction with YaxAB as part of the toxin complex. Attempts to purify YaxA and YaxB to confirm their toxicity were unsuccessful. While our data show that YaxAB have toxic effects, until the purified proteins can be tested, we cannot definitively state that Yax-mediated cytotoxicity is due solely to the presence of YaxAB.
Along with questions about optimal expression of native yaxAB and how YaxAB is released from the bacteria, it remains to be determined how the YaxAB toxin binds to target cells. Since more complete death of macrophage-like cells is observed compared to that of epithelial cells, it may be that the target/receptor of YaxAB is expressed on most or all macrophage (or hematopoietic) cells while it is present in only a subset of cultured epithelial cells. Alternatively, YaxAB could bind nonspecifically to host cells via a universal cell component, such as cholesterol. In support of this idea, the E. coli toxin HlyE binds cholesterol and is thought to target cells for lysis through this interaction (52). Identifying the binding target on host cells will contribute to our understanding of how YaxAB influences virulence; the identity of the receptor may determine if the effect of YaxAB is focused only on immune cells or if it may equally impact other cell types in vivo.
Mice infected with the ΔyaxAB mutant have fewer CFU/gram in the spleen than mice infected with the wild type, yet more lesions are found in the spleens of mice infected with the ΔyaxAB mutant. While these phenotypes are subtle, they suggest that the absence of YaxAB alters the immune response to Y. enterocolitica. Since YaxAB is an apparent cytotoxin, the presence of YaxAB could result in the death of cells recruited to the site of infection, thereby altering the immune response of the host in order to facilitate bacterial survival. This may lead to changes in cytokine expression in the spleens of mice infected with ΔyaxAB strains compared to those infected with wild-type Y. enterocolitica. We attempted to determine if gross changes in cytokine response were occurring in the spleens of mice infected with the wild-type or ΔyaxAB mutant Y. enterocolitica strain by testing for IL-6 and IFN-γ by ELISA. We found that the results from splenic homogenates varied greatly (N. J. Wagner and V. L. Miller, unpublished). With the wide range of cytokine concentrations measured, we did not detect significant differences in these cytokines. It is possible that while there was no change in cytokine levels at the whole-spleen level, the difference in cellular localization seen in the ΔyaxAB mutant-infected spleens results in an altered localized cytokine response. These localized changes could lead to differential cellular recruitment of immune cells to sites of infection, which may explain the increased number of splenic lesions seen in mice infected with the ΔyaxAB mutant. More sensitive techniques to measure local cytokine levels would need to be employed to determine if these differences are occurring in wild-type and ΔyaxAB mutant-infected spleens. In addition to alterations in cell recruitment, the recruited cells could be responding differently to the mutant bacteria. It has been shown that when mice depleted of dendritic cells are infected with Y. enterocolitica, the neutrophil response is altered such that a greater number of neutrophils are recruited to sites of infection (59). Additionally, infected mice depleted of dendritic cells generate more reactive oxygen species (ROS) (both total and per cell) (59). If YaxAB-mediated cell death results in a reduction in the number of dendritic cells and/or macrophages, the immune response could be similarly altered.
While we observed that the course of ΔyaxAB strain infection is altered from that of wild-type Y. enterocolitica, neither the Δinv nor the ΔyaxAB strain shows the level of attenuation of the rovA mutant strain (17, 18, 60). The ΔyaxAB strain shows a defect in colonization of the spleen, and the Δinv strain exhibits delayed colonization of the Peyer's patches (9); however, colonization of the mesenteric lymph nodes was equal between mice infected with wild-type Y. enterocolitica or either of these mutant strains (9) (Fig. 1). This is in contrast to reports of defective colonization of the Peyer's patches, spleen, and mesenteric lymph nodes by the rovA mutant. Additionally, we did not observe the reduced Peyer's patch inflammation in mice infected with the ΔyaxAB strain that has been reported with the rovA mutant strain (18). These data suggest that while the impact of RovA on virulence of Y. enterocolitica depends on yaxAB and inv, neither the inv nor the yaxAB mutant shows the colonization defect in the MLN that is apparent in the rovA mutant. Although it is possible a more severe defect would be apparent in a strain with both mutations, the data from the three single-mutant strains (rovA, inv, and yaxAB) imply that additional RovA-regulated genes contribute to pathogenicity.
ACKNOWLEDGMENTS
We are grateful to the following for contributions that made this work possible: Greer Kaufman for construction of the ΔyaxAB strain, Moriah Beck for advice on YaxA modeling and structural analysis, Ed Maio for immortalized caspase 1−/− macrophages, Carolyn Suitt of the UNC Center for Gastrointestinal Biology and Disease (CGIBD) Imaging and Histology Core for processing and staining histology samples, and Bill Goldman for the use of his microscope. We are indebted to Kim Walker for thoughtful discussion, experimental advice, and critical reading of the manuscript.
This work was supported in part by National Institutes of Health grants AI052167, awarded to V.L.M., and P30-DK-034987, awarded to the CGIBD Imaging and Histology Core.
Footnotes
Published ahead of print 3 September 2013
REFERENCES
- 1.Bottone EJ. 1997. Yersinia enterocolitica: the charisma continues. Clin. Microbiol. Rev. 10:257–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cover TL, Aber RC. 1989. Yersinia enterocolitica. N. Engl. J. Med. 321:16–24 [DOI] [PubMed] [Google Scholar]
- 3.McNally A, Cheasty T, Fearnley C. 2004. Comparison of the biotypes of Yersinia enterocolitica isolated from pigs, cattle and sheep at slaughter and from humans with yersiniosis in Great Britain during 1999–2000. Lett. Appl. Microbiol. 39:103–108 [DOI] [PubMed] [Google Scholar]
- 4.Fredriksson-Ahomaa M, Stolle A, Stephan R. 2007. Prevalence of pathogenic Yersinia enterocolitica in pigs slaughtered at a Swiss abattoir. Int. J. Food Microbiol. 119:207–212 [DOI] [PubMed] [Google Scholar]
- 5.Carter PB. 1975. Animal model of human disease. Yersinia enteritis. Animal model: oral Yersinia enterocolitica infection of mice. Am. J. Pathol. 81:703–706 [PMC free article] [PubMed] [Google Scholar]
- 6.Carter PB. 1975. Pathogenicity of Yersinia enterocolitica for mice. Infect. Immun. 11:164–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carter PB, Varga CF, Keet EE. 1973. New strain of Yersinia enterocolitica pathogenic for rodents. Appl. Microbiol. 26:1016–1018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Autenrieth IB, Vogel U, Preger S, Heymer B, Heesemann J. 1993. Experimental Yersinia enterocolitica infection in euthymic and T-cell-deficient athymic nude C57BL/6 mice: comparison of time course, histomorphology, and immune response. Infect. Immun. 61:2585–2595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pepe JC, Wachtel MR, Wagar E, Miller VL. 1995. Pathogenesis of defined invasion mutants of Yersinia enterocolitica in a BALB/c mouse model of infection. Infect. Immun. 63:4837–4848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Handley SA, Dube PH, Revell PA, Miller VL. 2004. Characterization of oral Yersinia enterocolitica infection in three different strains of inbred mice. Infect. Immun. 72:1645–1656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dube PH, Handley SA, Lewis J, Miller VL. 2004. Protective role of interleukin-6 during Yersinia enterocolitica infection is mediated through the modulation of inflammatory cytokines. Infect. Immun. 72:3561–3570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Autenrieth IB, Hantschmann P, Heymer B, Heesemann J. 1993. Immunohistological characterization of the cellular immune response against Yersinia enterocolitica in mice: evidence for the involvement of T lymphocytes. Immunobiology 187:1–16 [DOI] [PubMed] [Google Scholar]
- 13.Miller VL, Falkow S. 1988. Evidence for two genetic loci in Yersinia enterocolitica that can promote invasion of epithelial cells. Infect. Immun. 56:1242–1248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grützkau A, Hanski C, Hahn H. 1990. Involvement of M cells in the bacterial invasion of Peyer's patches: a common mechanism shared by Yersinia enterocolitica and other enteroinvasive bacteria. Gut 31:1011–1015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Clark MA, Hirst BH, Jepson MA. 1998. M-cell surface beta1 integrin expression and invasin-mediated targeting of Yersinia pseudotuberculosis to mouse Peyer's patch M cells. Infect. Immun. 66:1237–1243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Isberg RR, Leong JM. 1990. Multiple beta 1 chain integrins are receptors for invasin, a protein that promotes bacterial penetration into mammalian cells. Cell 60:861–871 [DOI] [PubMed] [Google Scholar]
- 17.Pepe JC, Miller VL. 1993. The biological role of invasin during a Yersinia enterocolitica infection. Infect. Agents Dis. 2:236–241 [PubMed] [Google Scholar]
- 18.Revell PA, Miller VL. 2000. A chromosomally encoded regulator is required for expression of the Yersinia enterocolitica inv gene and for virulence. Mol. Microbiol. 35:677–685 [DOI] [PubMed] [Google Scholar]
- 19.Nagel G, Lahrz A, Dersch P. 2001. Environmental control of invasin expression in Yersinia pseudotuberculosis is mediated by regulation of RovA, a transcriptional activator of the SlyA/Hor family. Mol. Microbiol. 41:1249–1269 [DOI] [PubMed] [Google Scholar]
- 20.Ellison DW, Young B, Nelson K, Miller VL. 2003. YmoA negatively regulates expression of invasin from Yersinia enterocolitica. J. Bacteriol. 185:7153–7159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Heroven AK, Nagel G, Tran HJ, Parr S, Dersch P. 2004. RovA is autoregulated and antagonizes H-NS-mediated silencing of invasin and rovA expression in Yersinia pseudotuberculosis. Mol. Microbiol. 53:871–888 [DOI] [PubMed] [Google Scholar]
- 22.Ellison DW, Miller VL. 2006. H-NS represses inv transcription in Yersinia enterocolitica through competition with RovA and interaction with YmoA. J. Bacteriol. 188:5101–5112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ellison DW, Miller VL. 2006. Regulation of virulence by members of the MarR/SlyA family. Curr. Opin. Microbiol. 9:153–159 [DOI] [PubMed] [Google Scholar]
- 24.Dube PH, Handley SA, Revell PA, Miller VL. 2003. The rovA mutant of Yersinia enterocolitica displays differential degrees of virulence depending on the route of infection. Infect. Immun. 71:3512–3520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cathelyn JS, Ellison DW, Hinchliffe SJ, Wren BW, Miller VL. 2007. The RovA regulons of Yersinia enterocolitica and Yersinia pestis are distinct: evidence that many RovA-regulated genes were acquired more recently than the core genome. Mol. Microbiol. 66:189–205 [DOI] [PubMed] [Google Scholar]
- 26.Vigneux F, Zumbihl R, Jubelin G, Ribeiro C, Poncet J, Baghdiguian S, Givaudan A, Brehélin M. 2007. The xaxAB genes encoding a new apoptotic toxin from the insect pathogen Xenorhabdus nematophila are present in plant and human pathogens. J. Biol. Chem. 282:9571–9580 [DOI] [PubMed] [Google Scholar]
- 27.Walker KA, Miller VL. 2004. Regulation of the Ysa type III secretion system of Yersinia enterocolitica by YsaE/SycB and YsrS/YsrR. J. Bacteriol. 186:4056–4066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Daigneault M, Preston J, Marriott H, Whyte M. 2010. The identification of markers of macrophage differentiation in PMA-stimulated THP-1 cells and monocyte-derived macrophages. PLoS One 5:e8668. 10.1371/journal.pone.0008668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Scherrer R, Cabrera Beaman T, Gerhardt P. 1971. Macromolecular sieving by the dormant spore of Bacillus cereus. J. Bacteriol. 108:868–873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fink SL, Cookson BT. 2006. Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cell. Microbiol. 8:1812–1825 [DOI] [PubMed] [Google Scholar]
- 31.Walker KA, Maltez V, Hall JD, Vitko NP, Miller VL. 2013. A phenotype at last: essential role for the Yersinia enterocolitica Ysa type III secretion system in a Drosophila S2 cell model. Infect. Immun. 81:2478–2487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thomson NR, Howard S, Wren BW, Holden MTG, Crossman L, Challis GL, Churcher C, Mungall K, Brooks K, Chillingworth T, Feltwell T, Abdellah Z, Hauser H, Jagels K, Maddison M, Moule S, Sanders M, Whitehead S, Quail MA, Dougan G, Parkhill J, Prentice MB. 2006. The complete genome sequence and comparative genome analysis of the high pathogenicity Yersinia enterocolitica strain 8081. PLoS Genet. 2:e206. 10.1371/journal.pgen.0020206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hofmann K, Stoffel W. 1993. TMbase–a database of membrane spanning proteins segments. Biol. Chem. Hoppe Seyler 374:166 [Google Scholar]
- 34.Claros MG, von Heijne G. 1994. TopPred II: an improved software for membrane protein structure predictions. Comput. Appl. Biosci. 10:685–686 [DOI] [PubMed] [Google Scholar]
- 35.Tusnády GE, Simon I. 2001. The HMMTOP transmembrane topology prediction server. Bioinformatics 17:849–850 [DOI] [PubMed] [Google Scholar]
- 36.Krogh A, Larsson B, von Heijne G, Sonnhammer EL. 2001. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 305:567–580 [DOI] [PubMed] [Google Scholar]
- 37.Roy A, Kucukural A, Zhang Y. 2010. I-TASSER: a unified platform for automated protein structure and function prediction. Nat. Protoc. 5:725–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang Y. 2008. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics 9:40. 10.1186/1471-2105-9-40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Obrist MW, Miller VL. 2012. Low copy expression vectors for use in Yersinia sp. and related organisms. Plasmid 68:33–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Frank A, Rauen U, de Groot H. 2000. Protection by glycine against hypoxic injury of rat hepatocytes: inhibition of ion fluxes through nonspecific leaks. J. Hepatol. 32:58–66 [DOI] [PubMed] [Google Scholar]
- 41.Fink SL, Bergsbaken T, Cookson BT. 2008. Anthrax lethal toxin and Salmonella elicit the common cell death pathway of caspase-1-dependent pyroptosis via distinct mechanisms. Proc. Natl. Acad. Sci. U. S. A. 105:4312–4317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brennan MA, Cookson BT. 2000. Salmonella induces macrophage death by caspase-1-dependent necrosis. Mol. Microbiol. 38:31–40 [DOI] [PubMed] [Google Scholar]
- 43.Noronha FS, Cruz JS, Beirão PS, Horta MF. 2000. Macrophage damage by Leishmania amazonensis cytolysin: evidence of pore formation on cell membrane. Infect. Immun. 68:4578–4584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kirby JE, Vogel JP, Andrews HL, Isberg RR. 1998. Evidence for pore-forming ability by Legionella pneumophila. Mol. Microbiol. 27:323–336 [DOI] [PubMed] [Google Scholar]
- 45.Hamon MA, Cossart P. 2011. K+ efflux is required for histone-H3 dephosphorylation by Listeria LLO and other pore forming toxins. Infect. Immun. 79:2839–2846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Thomas J, Epshtein Y, Chopra A, Ordog B, Ghassemi M, Christman JW, Nattel S, Cook JL, Levitan I. 2011. Anthrax lethal factor activates K+ channels to induce IL-1 beta secretion in macrophages. J. Immunol. 186:5236–5243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Madegowda M, Eswaramoorthy S, Burley SK, Swaminathan S. 2008. X-ray crystal structure of the B component of Hemolysin BL from Bacillus cereus. Proteins 71:534–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mueller M, Grauschopf U, Maier T, Glockshuber R, Ban N. 2009. The structure of a cytolytic alpha-helical toxin pore reveals its assembly mechanism. Nature 459:726–730 [DOI] [PubMed] [Google Scholar]
- 49.Oscarsson J, Mizunoe Y, Uhlin BE, Haydon DJ. 1996. Induction of haemolytic activity in Escherichia coli by the slyA gene product. Mol. Microbiol. 20:191–199 [DOI] [PubMed] [Google Scholar]
- 50.del Castillo FJ, Leal SC, Moreno F, del Castillo I. 1997. The Escherichia coli K-12 sheA gene encodes a 34-kDa secreted haemolysin. Mol. Microbiol. 25:107–115 [DOI] [PubMed] [Google Scholar]
- 51.Ludwig A, Bauer S, Benz R, Bergmann B, Goebel W. 1999. Analysis of the SlyA-controlled expression, subcellular localization and pore-forming activity of a 34 kDa haemolysin (ClyA) from Escherichia coli K-12. Mol. Microbiol. 31:557–567 [DOI] [PubMed] [Google Scholar]
- 52.Oscarsson J, Mizunoe Y, Li L, Lai XH, Wieslander A, Uhlin BE. 1999. Molecular analysis of the cytolytic protein ClyA (SheA) from Escherichia coli. Mol. Microbiol. 32:1226–1238 [DOI] [PubMed] [Google Scholar]
- 53.Ludwig A, Tengel C, Bauer S, Bubert A, Benz R, Mollenkopf HJ, Goebel W. 1995. SlyA, a regulatory protein from Salmonella typhimurium, induces a haemolytic and pore-forming protein in Escherichia coli. Mol. Gen. Genet. 249:474–486 [DOI] [PubMed] [Google Scholar]
- 54.Wallace AJ, Stillman TJ, Atkins A, Jamieson SJ, Bullough PA, Green J, Artymiuk PJ. 2000. E. coli hemolysin E (HlyE, ClyA, SheA): X-ray crystal structure of the toxin and observation of membrane pores by electron microscopy. Cell 100:265–276 [DOI] [PubMed] [Google Scholar]
- 55.Beecher DJ, Macmillan JD. 1991. Characterization of the components of hemolysin BL from Bacillus cereus. Infect. Immun. 59:1778–1784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wai SN, Westermark M, Oscarsson J, Jass J, Maier E, Benz R, Uhlin BE. 2003. Characterization of dominantly negative mutant ClyA cytotoxin proteins in Escherichia coli. J. Bacteriol. 185:5491–5499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Stoker NG, Fairweather NF, Spratt BG. 1982. Versatile low-copy-number plasmid vectors for cloning in Escherichia coli. Gene 18:335–341 [DOI] [PubMed] [Google Scholar]
- 58.Cabello F, Timmis K, Cohen SN. 1976. Replication control in a composite plasmid constructed by in vitro linkage of two distinct replicons. Nature 259:285–290 [DOI] [PubMed] [Google Scholar]
- 59.Autenrieth SE, Warnke P, Wabnitz GH, Lucero Estrada C, Pasquevich KA, Drechsler D, Günter M, Hochweller K, Novakovic A, Beer-Hammer S, Samstag Y, Hämmerling GJ, Garbi N, Autenrieth IB. 2012. Depletion of dendritic cells enhances innate anti-bacterial host defense through modulation of phagocyte homeostasis. PLoS Pathog. 8:e1002552. 10.1371/journal.ppat.1002552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dube P, Revell P, Chaplin D, Lorenz R, Miller V. 2001. A role for IL-1 alpha in inducing pathologic inflammation during bacterial infection. Proc. Natl. Acad. Sci. U. S. A. 98:10880–10885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Miller VL, Mekalanos JJ. 1988. A novel suicide vector and its use in construction of insertion mutations: osmoregulation of outer membrane proteins and virulence determinants in Vibrio cholerae requires toxR. J. Bacteriol. 170:2575–2583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kinder SA, Badger JL, Bryant GO, Pepe JC, Miller VL. 1993. Cloning of the YenI restriction endonuclease and methyltransferase from Yersinia enterocolitica serotype O8 and construction of a transformable R−M+ mutant. Gene 136:271–275 [DOI] [PubMed] [Google Scholar]
- 63.Heesemann J. 1987. Chromosomal-encoded siderophores are required for mouse virulence of enteropathogenic Yersinia species. FEMS Microbiol. Lett. 48:229–233 [Google Scholar]
- 64.Beer KB, Miller VL. 1992. Amino acid substitutions in naturally occurring variants of ail result in altered invasion activity. J. Bacteriol. 174:1360–1369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Miller VL, Farmer JJ, Hill WE, Falkow S. 1989. The ail locus is found uniquely in Yersinia enterocolitica serotypes commonly associated with disease. Infect. Immun. 57:121–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Baker PM, Farmer JJ. 1982. New bacteriophage typing system for Yersinia enterocolitica, Yersinia kristensenii, Yersinia frederiksenii, and Yersinia intermedia: correlation with serotyping, biotyping, and antibiotic susceptibility. J. Clin. Microbiol. 15:491–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cafferkey MT, McClean K, Drumm ME. 1989. Production of bacteriocin-like antagonism by clinical isolates of Yersinia enterocolitica. J. Clin. Microbiol. 27:677–680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Merriam JJ, Mathur R, Maxfield-Boumil R, Isberg RR. 1997. Analysis of the Legionella pneumophila fliI gene: intracellular growth of a defined mutant defective for flagellum biosynthesis. Infect. Immun. 65:2497–2501 [DOI] [PMC free article] [PubMed] [Google Scholar]