

Abstract
Staphylococcus aureus infections are an important public health concern due to their increasing incidence and high rates of mortality. The success of S. aureus as a pathogen is highly related to its enormous capacity to evade the host immune response. The critical role of tumor necrosis factor alpha (TNF-α) in the initial host defense against systemic staphylococcal infection has been demonstrated in experimental models and may partially explain the lack of significant benefits observed in clinical trials attempting to neutralize this cytokine in septic patients. S. aureus protein A plays a key role in regulating inflammation through its ability to bind and signal through the TNF-α receptor 1 (TNFR1). In this study, we demonstrate that S. aureus, via protein A-mediated signaling, induces early shedding of TNFR1, which precedes the secretion of TNF-α in vitro and in vivo. The results obtained using a protein A-deficient mutant and tnfr1−/− mice strongly suggest that the increased levels of soluble TNFR1 present during experimental S. aureus infection may neutralize circulating TNF-α and impair the host inflammatory response. Early shedding of TNFR1 induced by protein A may constitute a novel mechanism by which S. aureus subverts the host immune response.
INTRODUCTION
Staphylococcus aureus is a major hospital- and community-acquired pathogen (1–3). The incidence of severe sepsis and septic shock is steadily increasing, and worldwide mortality ranges from 30% to 50% (4, 5). The increasing worldwide prevalence of antibiotic-resistant strains, which renders staphylococcal infections very difficult to treat and eradicate, is a serious public health concern (6–8).
S. aureus infections are characterized by a profound inflammatory response, which contributes significantly to pathogenesis but is also required for bacterial clearance (9). Among the proinflammatory cytokines induced, tumor necrosis factor alpha (TNF-α) has been shown to be crucial for the eradication of bacteria in several experimental models (10, 11). TNF-α-induced signaling is modulated by the availability of its cognate receptor, TNF receptor 1 (TNFR1), at the cell surface. The abundance of TNFR1 is positively controlled in response to several stimuli by mobilization from intracellular compartments and can be negatively regulated by shedding of the extracellular domain from the surface (12–14). TNFR1 is cleaved from the cell surface by ADAM17 (15–17), a mechanism that decreases the amount of receptor available to respond to the ligand and terminates TNF-α signaling, thus modulating inflammation (18, 19).
Among the many factors that determine the pathogenicity of S. aureus, protein A, a surface protein, acts as an exceptionally complex virulence factor, contributing to the success of S. aureus as a pathogen by interfering with immune clearance and also by inducing inflammation. In addition to its roles in immunoglobulin G binding (20, 21), platelet activation (22), and B cell activation (23, 24), protein A plays a key role in stimulating inflammation through its ability to bind and signal through TNFR1 (25). TNFR1 signaling triggered by protein A initiates the production of interleukin 8 (IL-8) and the recruitment of neutrophils to the site of infection (26). Protein A induces other proinflammatory cytokines and chemokines, such as type I interferon (IFN) and CXCL10, in airway epithelial cells and macrophages, which also contribute to neutrophil recruitment and bacterial clearance (27–29).
We have shown previously that S. aureus protein A activates ADAM17 in airway epithelial cells (30). Considering that this protease is a key molecule in the regulation of TNF-α–TNFR1 signaling, and given the central role of this cascade in the immune responses orchestrated by macrophages, this study was aimed at investigating the role of S. aureus protein A in the activation of ADAM17 in macrophages, the induction of soluble TNFR1 (sTNFR1) in vivo, and the regulation of TNF-α availability during systemic staphylococcal infection.
MATERIALS AND METHODS
Recombinant proteins and bacterial strains.
Full-length protein A cloned from S. aureus strain Newman, the IgG binding domains (EC region), the carboxyl-terminal polymorphic region (X), and IgG binding domain D and its corresponding L17A mutant were expressed as glutathione S-transferase (GST) fusion proteins in Escherichia coli BL21 and were purified as described previously (29). The recombinant proteins were dialyzed against phosphate-buffered saline (PBS) and were used at a concentration of 2.5 μM for in vitro stimulation. Potentially remaining traces of lipopolysaccharide (LPS) were removed using Detoxi-Gel endotoxin-removing gel and columns (Pierce, Holmdel, NJ). The proteins were proved to be free of LPS by testing of their stimulatory capacities in the presence or absence of polymyxin B.
S. aureus strain Newman and the spa-deficient isogenic mutant (deficient in the spa gene, which encodes protein A) were grown in Trypticase soy broth (Britania, Buenos Aires, Argentina) and were suspended in RPMI 1640 medium (Life Technologies, Grand Island, NY) at a concentration of 1 × 109 CFU/ml. A clinical isolate of Escherichia coli (isolate 515; generously provided by D. Centron, IMPaM, UBA-CONICET, Buenos Aires, Argentina) and Pseudomonas aeruginosa PAO-1 (31) were grown in Luria-Bertani agar (Britania, Buenos Aires, Argentina) and were suspended in RPMI 1640 medium (Life Technologies, Grand Island, NY) at a concentration of 1 × 109 CFU/ml. Lactococcus lactis MG1363 carrying the pKS80 vector containing full-length spa or an empty-vector control (provided by Tim Foster, Trinity College, Dublin, Ireland) was grown in M17 medium supplemented with 0.5% glucose and 5 μg/ml erythromycin at 30°C without agitation (29, 32, 33). Cells were harvested by centrifugation at 10,000 rpm for 10 min, and the pellet was suspended in RPMI 1640 medium (Life Technologies, Grand Island, NY) at a concentration of 5 × 108 CFU/ml.
Cell culture.
RAW 264.7 cells (a mouse macrophage cell line) and THP-1 cells (a human monocytic/macrophage cell line) were cultured in RPMI 1640 medium (Life Technologies, Grand Island, NY) supplemented with 10% fetal bovine serum, 0.11 mg/ml pyruvate (Sigma-Aldrich, St. Louis, MO), 0.29 mg/ml GlutaMAX (Life Technologies, Grand Island, NY), and 1× nonessential amino acids (Life Technologies, Grand Island, NY). RAW 264.7 cells cultured to confluence were weaned from serum 24 h before exposure to stimuli. THP-1 cells were used at 106/ml. Where indicated, biochemical inhibitors were added 30 min prior to stimulation and during stimulation.
Detection of TNF-α and soluble TNFR1.
TNF-α and soluble TNFR1 were quantified in culture supernatants by enzyme-linked immunosorbent assays (ELISA) using DuoSet antibody pairs (R&D Systems, Minneapolis, MN).
Flow cytometry.
After stimulation, cells were washed three times with PBS and were stained with an antibody against ADAM17 (Santa Cruz Biotechnology, Dallas, TX) followed by an Alexa Fluor 488-conjugated secondary antibody. The cells were analyzed in a Partec PAS III flow cytometer, and the data were analyzed with WinMDI software.
In vivo studies.
Six-week-old BALB/c mice obtained from the Department of Microbiology, School of Medicine, University of Buenos Aires animal facility were used unless otherwise indicated. C57BL/6 tnfrp55−/− (tnfr1−/−) mice were obtained from the Max von Pettenkofer Institute, Munich, Germany. Breeding colonies were established at the animal facility of the National University of San Luis (San Luis, Argentina). Mice were inoculated intraperitoneally with 200 μl of either recombinant protein A (at a dose of 0.125 nmol/g, based on a mouse model of LPS-induced endotoxemia [34] and our previous studies [29]), wild-type S. aureus, a protein A-deficient mutant (2 × 108 CFU/ml), E. coli 515 (2 × 108 CFU/ml), or PBS as control. Blood samples were obtained by puncture of the retro-orbital plexus at different times after inoculation. Serum was stored at −80°C for subsequent TNF-α and sTNFR1 quantification. Peritoneal cells were harvested using 8 ml of cold RPMI 1640 medium supplemented with 2% fetal bovine serum. The cells were then washed by centrifugation and were used for RNA extraction or surface antigen staining and flow cytometry analysis.
For neutrophil detection, peritoneal-cell suspensions were stained with phycoerythrin (PE)-labeled anti-CD45 and fluorescein isothiocyanate (FITC)-labeled anti-Ly6G (BD Pharmingen, San Jose, CA) as described previously (26). Negative-control samples were incubated with irrelevant isotype-matched antibodies. Cells were gated based on their forward-scatter (FSC)/side-scatter (SSC) profiles and were analyzed for the double expression of CD45 and Ly6G with WinMDI software.
Animal studies were approved by the Institutional Committee for the Care and Use of Laboratory Animals (CICUAL) of the University of Buenos Aires.
Western blotting.
Cells were lysed using 60 mM n-octyl-β-d-glucopyranoside in TBS (0.1 M Tris-HCl and 0.15 M NaCl [pH 7.8]) containing a protease inhibitor cocktail (Sigma-Aldrich, St. Louis, MO), 1 mM sodium orthovanadate, and 100 mM sodium fluoride. Proteins were separated on 8% bis-acrylamide gels, transferred to a nitrocellulose membrane, and blocked with 5% milk in TBST (50 mM Tris [pH 7.5], 150 mM NaCl, and 0.05% Tween) for 1 h at room temperature. Immunodetection was performed using antibodies against phosphorylated extracellular signal-regulated kinases 1 and 2 (phospho-ERK1/2) (Santa Cruz Biotechnology, Dallas, TX) or actin (Sigma-Aldrich, St. Louis, MO), followed by secondary antibodies conjugated to horseradish peroxidase (Santa Cruz Biotechnology, Dallas, TX). Images were analyzed with ImageJ software.
Real-time PCR.
RNA was isolated using TRIzol reagent (Life Technologies, Grand Island, NY). cDNA was made from 1 μg of RNA using ImProm-II reverse transcriptase (Promega, Fitchburg, WI). For quantitative real-time PCR, amplification was performed in an Applied Biosystems thermal cycler. The following primers were used for amplification: for mouse TNF-α, 5′-ATG AGC ACA GAA AGC ATG ATC-3′ and 5′-TAC AGG CTT GTC ACT CGA ATT-3′; for mouse ADAM17, 5′-ATG TGA GCA GTT TCC CGA ACG-3′ and 5′-ATC AAG CTT CTC AAG TCG CGG-3′; and for mouse glyceraldehyde-3-phosphate dehydrogenase (GAPDH), 5′-GAA GGT GGT GAA GCA GGC AT-3′ and 5′-TCG AAG GTG GAA GAG TGG GA-3′. GAPDH was used as a control for standardization.
Statistics.
Data from samples with normal distribution were analyzed using the Student t test. Data from samples that did not follow normal distribution were analyzed with the nonparametric Mann-Whitney test or the Wilcoxon test from GraphPad Prism software, version 4.0. A P value lower than 0.05 was considered statistically significant.
RESULTS
S. aureus protein A induces TNFR1 shedding in macrophages.
We investigated the ability of protein A to activate ADAM17 in macrophages and to induce TNFR1 shedding. Significant increases in the concentrations of soluble TNFR1 (sTNFR1) were observed in culture supernatants from macrophages stimulated with protein A (Fig. 1A and B). We have demonstrated previously that the interaction of protein A with the epidermal growth factor receptor (EGFR) mediates ADAM17 activation and TNFR1 shedding in airway epithelial cells (30). Thus, we next investigated whether the amino-terminal portion of protein A, known to interact with the EGFR, was involved in sTNFR1 production in macrophages. TNFR1 shedding was observed when macrophages were stimulated with recombinant GST-tagged proteins corresponding to the amino-terminal region containing the IgG binding domains (EC fragment) of protein A, as opposed to stimulation with the carboxyl-terminal region (X fragment) (Fig. 1A). The interaction of leucine 17, located in the conserved amino-terminal domain of protein A, is necessary for the recognition of protein A by the EGFR and the subsequent phosphorylation of ADAM17 on threonine by ERK1/2 in airway epithelial cells (30). Therefore, we determined the role of protein A-EGFR-mediated signaling in the shedding of TNFR1 from macrophages by using a mutated (L17A) form of protein A. Cells stimulated with the L17A mutant showed reduced levels of sTNFR1 (Fig. 1C). In agreement with these findings, ERK1/2 phosphorylation was observed starting at 15 min after stimulation (Fig. 1D), and chemical inhibition of ERK1/2 significantly reduced the levels of sTNFR1 induced in response to protein A (Fig. 1E). In agreement with our previous findings in airway epithelial cells (28), ADAM17 was abundantly expressed at the surfaces of macrophages (Fig. 1F); no further amounts of the enzyme were mobilized in response to protein A (Fig. 1G); and no transcriptional regulation was observed at the times studied (Fig. 1H).
Fig 1.
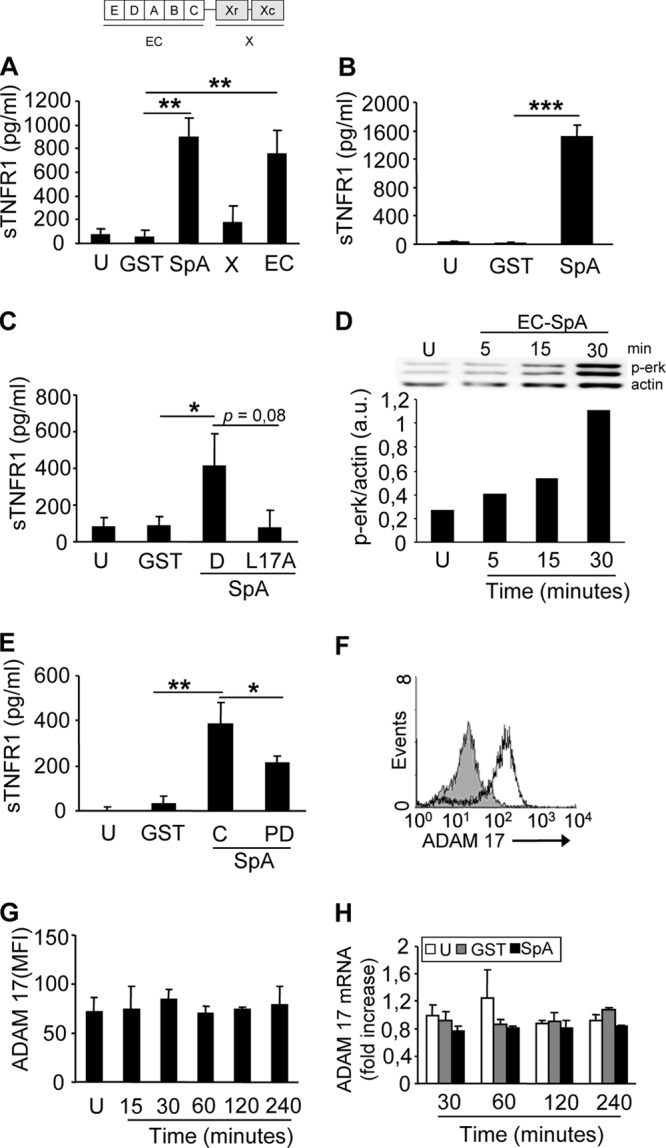
Activation of ADAM17 and TNFR1 shedding in response to protein A. (A through C) RAW 264.7 cells (A and C) and THP-1 cells (B) were stimulated with the amino-terminal portion of protein A (EC), the polymorphic region of protein A (X), full-length protein A (SpA), IgG binding domain D (D), its corresponding L17A mutant (L17A), GST (control), or medium alone (unstimulated [U]), and the induction of sTNFR1 was determined by ELISA. (D) RAW 264.7 cells were stimulated with the amino-terminal portion of protein A (EC) or with medium alone (U), and phospho-ERK1/2 was detected at different time points by immunoblotting. Bars show the densitometric quantification of phospho-ERK relative to actin, which was used to normalize the protein charge. a.u., arbitrary units. (E) RAW 264.7 cells were stimulated with protein A in the absence (control [C]) or presence (PD) of the MEK1/2 inhibitor PD98058 (10 μM), and the induction of sTNFR1 was determined 30 min later by ELISA. (F) The expression of ADAM17 on the surfaces of unstimulated RAW 264.7 cells was determined by flow cytometry. Open histogram, cells stained with an antibody to ADAM17; shaded histogram, cells stained with normal goat serum as a control. (G) RAW 264.7 cells were stimulated with recombinant protein A for the times indicated or with medium alone (U) and were stained for surface expression of ADAM17. MFI, mean fluorescence intensity. (H) RAW 264.7 cells were stimulated for the times indicated with protein A, GST (control), or medium alone (U), and the transcription of ADAM17 was quantified by real-time PCR and was standardized to GAPDH expression. Data in panels A through C, E, G, and H are means and standard deviations of cumulative data from three independent experiments (n, 3 for each experiment). Asterisks indicate significant differences (*, P < 0.05; **, P < 0.01; ***, P < 0.001) by Student's t test.
Release of sTNFR1 precedes TNF-α production in S. aureus-stimulated macrophages.
The kinetics of TNF-α production and TNFR1 shedding in response to whole live S. aureus was investigated in macrophages. S. aureus induced TNF-α production starting at 1 h after stimulation, and significantly larger amounts of this cytokine were detected 1 h later (Fig. 2A). Although the response was significantly diminished, cells stimulated with a protein A-deficient isogenic mutant still produced TNF-α (Fig. 2A). Interestingly, shedding of TNFR1 occurred as soon as 30 min after the exposure of macrophages to S. aureus and was highly dependent on protein A expression (Fig. 2B). The role of protein A in the induction of both TNF-α and sTNFR1 was confirmed by stimulating macrophages with Lactococcus lactis expressing protein A (L. lactis SpA). As was found with S. aureus, sTNFR1 released in response to L. lactis SpA was detected before TNF-α production (Fig. 2C and D). In contrast, early shedding of TNFR1 was not observed when macrophages were stimulated with an equal dose of the Gram-negative bacterium Escherichia coli or Pseudomonas aeruginosa (Fig. 2E and F).
Fig 2.
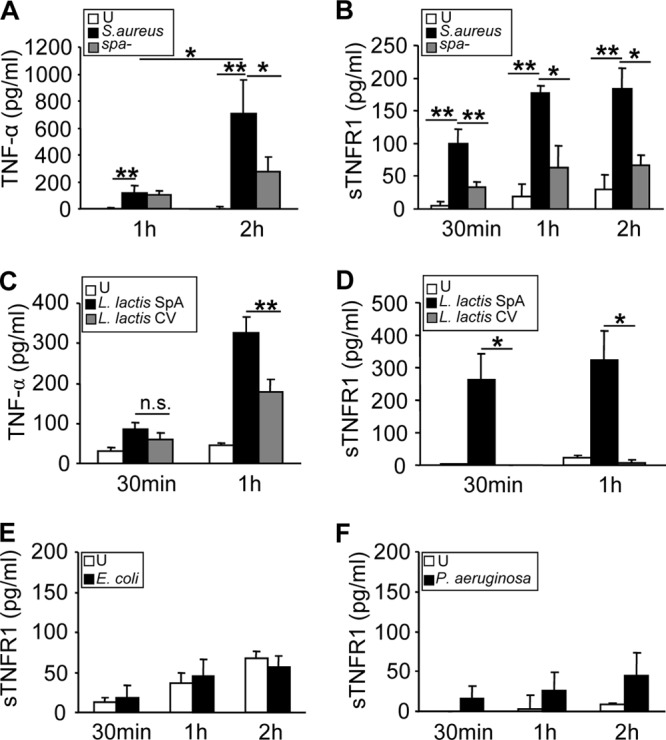
TNF-α production and shedding of TNFR1 in response to protein A. RAW 264.7 cells were stimulated for the times indicated with wild-type S. aureus or a protein A-deficient mutant (spa mutant) (A and B), L. lactis expressing protein A (L. lactis SpA) or L. lactis containing a control vector (L. lactis CV) (C and D), E. coli (E), Pseudomonas aeruginosa (PAO-1) (F), or medium alone (unstimulated [U]), and the induction of TNF-α (A and C) and sTNFR1 (B, D, E, and F) was determined by ELISA. Data are means and standard deviations of cumulative data from three independent experiments (n, 3 for each experiment). Asterisks indicate significant differences (*, P < 0.05; **, P < 0.01) by Student's t test.
S. aureus protein A induces shedding of TNFR1 in vivo.
To demonstrate the biological relevance of the in vitro findings, we determined whether protein A could induce TNFR1 shedding in vivo. Mice were inoculated intraperitoneally with protein A, and the amounts of sTNFR1 and TNF-α in serum were quantified over time. A significant increase in the level of circulating sTNFR1 was observed as soon as 30 min after inoculation (Fig. 3A), and this response was sustained at least up to 4 h (Fig. 3C, E, and G). TNF-α levels in serum increased 1 h after inoculation (median, 716 pg/ml) (Fig. 3D) and peaked 1 h later (median, 1,800 pg/ml) (Fig. 3F). These results show that in agreement with the kinetics seen in vitro, after in vivo inoculation of protein A, significant levels of sTNFR1 were found in the circulation before TNF-α was detected.
Fig 3.
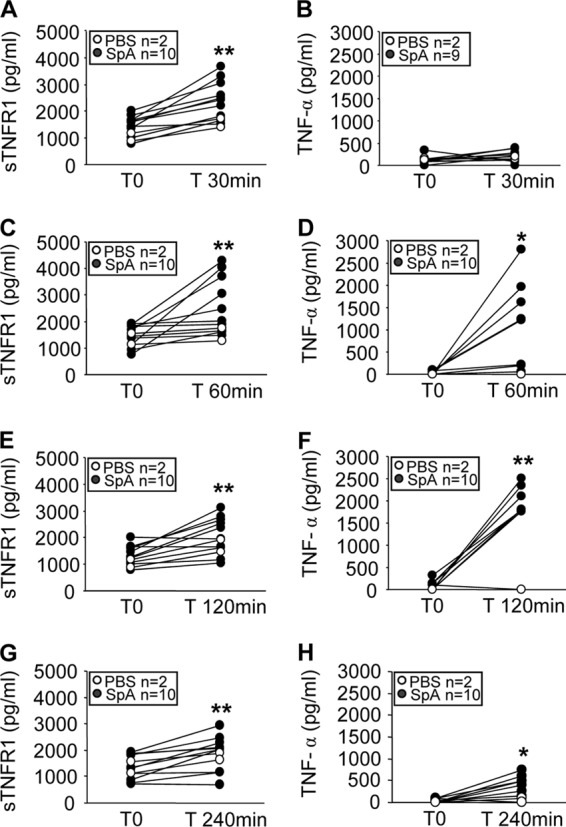
Induction of sTNFR1 in response to protein A. Groups of mice were inoculated intraperitoneally with recombinant protein A (filled circles) or PBS (control) (open circles), and at different time points, sTNFR1 (A, C, E, and G) and TNF-α (B, D, F, and H) levels in serum were quantified by ELISA. Each pair of dots represents an individual mouse at the time points indicated. Asterisks indicate significant differences (*, P < 0.05; **, P < 0.01) by the Wilcoxon matched-pairs test.
Soluble TNFR1 released in response to protein A neutralizes TNF-α during systemic S. aureus infection.
In order to evaluate the kinetics of TNFR1 shedding and TNF-α induction during staphylococcal infection, groups of mice were inoculated intraperitoneally with S. aureus or the spa-deficient strain, and the levels of sTNFR1 and TNF-α in serum were analyzed over time. A significant increase in the amount of circulating sTNFR1 was observed starting at 1 h after challenge with wild-type S. aureus (Fig. 4A). By 2 h after inoculation with S. aureus, the amount of sTNFR1 in the circulation had increased ∼2,000 pg over basal levels, whereas no differences were observed for mice inoculated with the spa-deficient mutant (Fig. 4A). Conversely, a significant increase in the levels of circulating TNF-α was observed for mice inoculated with the spa-deficient mutant 4 h after inoculation, whereas this cytokine could not be detected in sera from mice inoculated with wild-type S. aureus (Fig. 4B).
Fig 4.
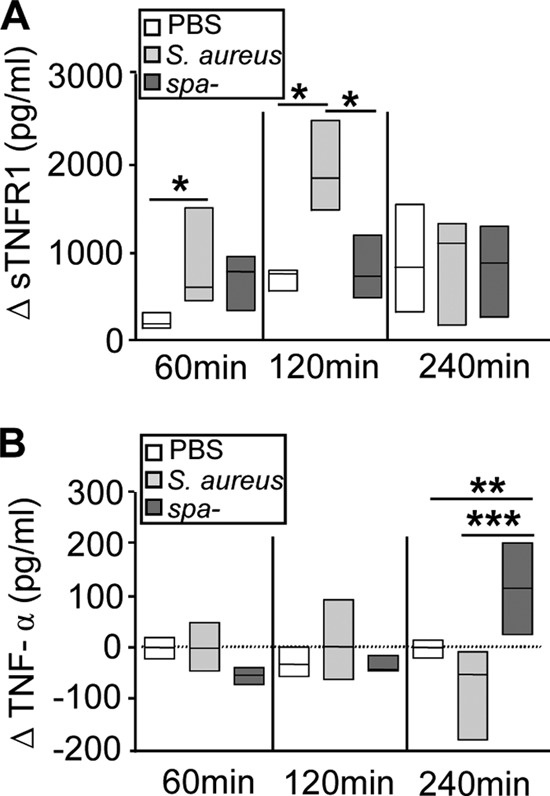
Shedding of TNFR1 during S. aureus infection. Groups of mice were inoculated intraperitoneally with wild-type S. aureus (light shaded boxes), a protein A-deficient mutant (spa-deficient mutant) (dark shaded boxes), or PBS (control) (open boxes). At different time points, sTNFR1 (A) and TNF-α (B) levels in serum were quantified by ELISA. Each box represents an individual group of mice, and horizontal lines show the median value for each group. Asterisks indicate significant differences (*, P < 0.05; **, P < 0.01; ***, P < 0.001) by the Mann-Whitney test for nonparametric data. (A) The change in sTNFR1 levels (ΔsTNFR1) was calculated for each individual mouse at the indicated time points as the sTNFR1 level after inoculation minus the sTNFR1 level prior to inoculation. (B) The change in TNF-α levels (ΔTNF-α) was calculated for each individual mouse at the indicated time points as the TNF-α level after inoculation minus the TNF-α level prior to inoculation. Basal levels of sTNFR1 and TNF-α prior to inoculation (median values) were 1,500 pg/ml and 45 pg/ml, respectively.
We confirmed that S. aureus infection was indeed able to induce TNF-α production in vivo by quantifying TNF-α transcription in peritoneal infiltrates. Significant increases in TNF-α mRNA levels were observed in mice infected with either S. aureus or the spa-deficient mutant by 2 and 4 h after inoculation (Fig. 5).
Fig 5.
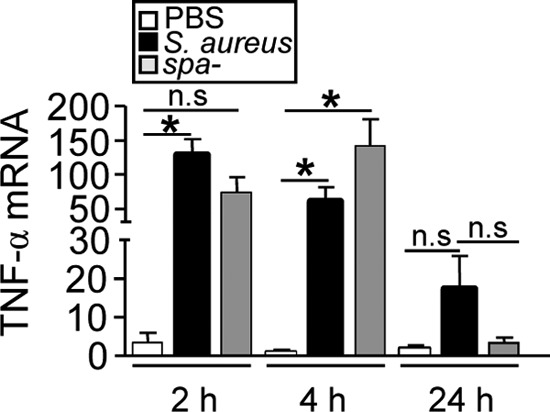
TNF-α expression during S. aureus infection. Mice were inoculated intraperitoneally with wild-type S. aureus, a protein A-deficient mutant (spa-deficient mutant), or PBS (control), and TNF-α expression by peritoneal cells at the times indicated was quantified by real-time PCR and was normalized to GAPDH expression. Each bar represents the mean and standard deviation for 3 to 5 mice. Asterisks indicate significant differences (*, P < 0.05) by Student's t test. n.s., not significant.
Based on the data obtained using the wild-type strain and the spa-deficient mutant, we hypothesized that the absence of detectable TNF-α during infection with S. aureus may be due to its neutralization by the sTNFR1 induced in response to protein A. In agreement with this hypothesis, we observed that mice inoculated with E. coli did not present an increase in circulating sTNFR1 levels and showed increased levels of TNF-α in serum at 2 and 4 h after challenge (data not shown). These results agree with previous observations for mice challenged with LPS, where TNF-α was found in the circulation at 2 h postinoculation, while sTNFR1 was detected only at 24 h (35). To further demonstrate the neutralization of TNF-α by sTNFR1 in vivo, we compared the levels of TNF-α in serum from TNFR1-deficient (tnfr1−/−) and wild-type (C57BL/6) mice under basal conditions and after S. aureus inoculation. In agreement with the results obtained for BALB/c mice, C57BL/6 mice challenged with the spa-deficient mutant had levels of TNF-α in the circulation significantly higher than those in mice challenged with wild-type S. aureus (Fig. 6A). Basal levels of circulating TNF-α were similar in wild-type and tnfr1−/− mice (Fig. 6B). Although the levels of circulating TNF-α increased in both groups after S. aureus inoculation, the amounts of TNF-α detected in the sera of tnfr1−/− mice were double those found in wild-type mice (Fig. 6B). However, statistical significance was not achieved, probably due to the lack of positive regulation of TNF-α via TNFR1 signaling in tnfr1−/− mice. This hypothesis is supported by the fact that the concentrations of TNF-α in sera from tnfr1−/− mice challenged with S. aureus were indeed lower than the amounts found in wild-type mice challenged with the spa-deficient mutant (Fig. 6A and B).
Fig 6.
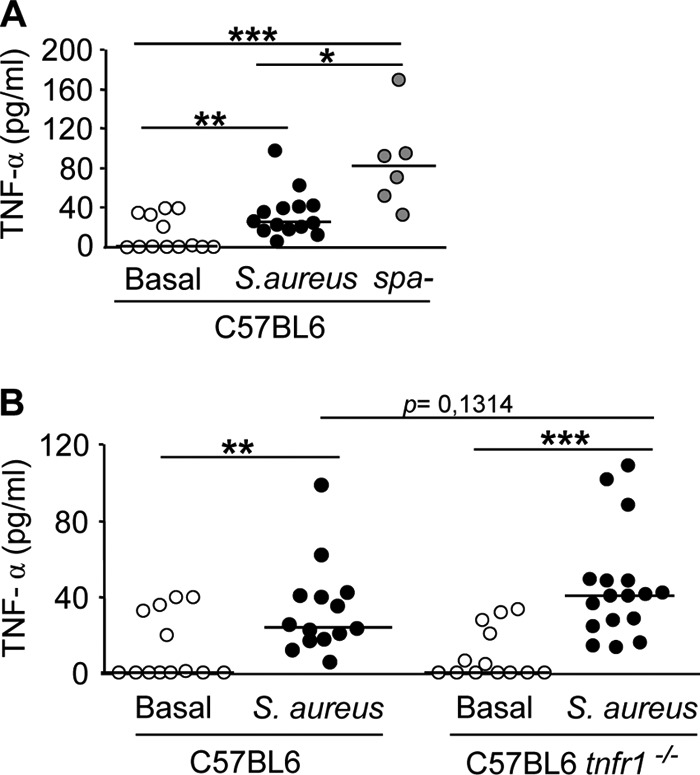
TNF-α induction during systemic S. aureus infection in TNFR1-deficient mice. Groups of C57BL6 (wild-type) and tnfr1−/− mice were inoculated intraperitoneally with wild-type S. aureus (A and B) or the spa-deficient mutant (A), and 4 h later, TNF-α levels in serum were quantified by ELISA. Each symbol represents an individual mouse, and horizontal lines show the median value for each group. Asterisks indicate significant differences (*, P < 0.05; **, P < 0.01; ***, P < 0.001) by the Mann-Whitney test for nonparametric data.
Early shedding of TNFR1 decreases inflammation during S. aureus infection.
We then determined the consequences of the early shedding of TNFR1 for the inflammatory status of mice systemically infected with S. aureus. We observed that 24 h after inoculation, the levels of circulating TNF-α were significantly lower in mice inoculated with wild-type S. aureus than in those inoculated with the spa-deficient mutant (Fig. 7A). The levels of IL-1β, a proinflammatory cytokine regulated by TNF-α, were also significantly lower in mice inoculated with wild-type S. aureus than in mice inoculated with the spa-deficient mutant (Fig. 7B). The levels of IL-6, a cytokine that has dual pro- and anti-inflammatory functions, were not affected (Fig. 7C). Moreover, the percentage of neutrophils recruited to the infection site 24 h after inoculation was significantly lower in mice inoculated with wild-type S. aureus than in mice inoculated with the spa-deficient mutant (Fig. 7D).
Fig 7.
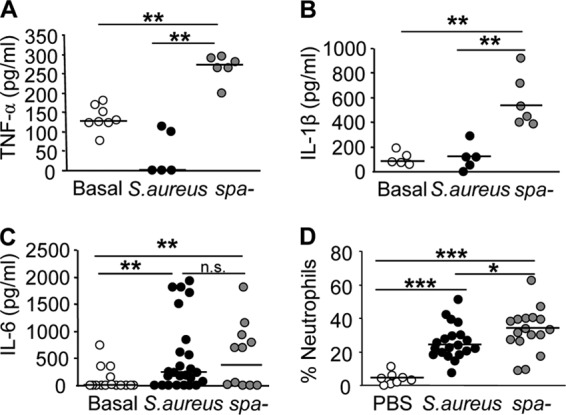
Inflammatory response induced during systemic S. aureus infection. Groups of mice were inoculated intraperitoneally with wild-type S. aureus (filled circles) or a protein A-deficient mutant (spa-deficient mutant) (shaded circles). Open circles indicate values for mice inoculated with PBS as a control. Twenty-four hours later, the levels of TNF-α (A), IL-1β (B), and IL-6 (C) in serum were quantified by ELISA, and the percentage of neutrophils in the peritoneum was determined by flow cytometry (D). Each symbol represents an individual mouse, and horizontal lines show the median value for each group. Asterisks indicate significant differences (*, P < 0.05; **, P < 0.01; ***, P < 0.001) by the Mann-Whitney test for nonparametric data.
DISCUSSION
Despite the critical role of TNF-α in the eradication of bacteria, excessively high levels of this cytokine may lead to tissue damage, organ failure, and death, a frequent scenario for septic patients (36). For this reason, TNF-α has been neutralized in many clinical trials in an attempt to prevent the undesirable effects of this cytokine (37). The different approaches considered have included the use of monoclonal antibodies to TNF-α (38) and the TNF receptor fused to the Fc portion of IgG (39). The outcomes of these trials, however, failed to show any significant beneficial effect on the evolution of septic patients, indicating not only that other mediators of inflammation are involved in sepsis but also that, clearly, certain levels of TNF-α are required for bacterial eradication (10, 40). TNF-α supports host antibacterial defenses through several mechanisms, including potentiation of the killing of bacteria by neutrophils and upregulation of vascular and adhesion molecules, which are necessary for neutrophil recruitment (40). In this regard, treatment of inflammatory diseases, such as lupus or rheumatoid arthritis, with neutralizing antibodies to TNF-α has led to increased risks of infectious diseases (41, 42), including infections caused by S. aureus (43).
One of the major strategies through which S. aureus causes infection in humans is evasion of the host immune response, for which it has an enormous capacity (21, 44). In the present study, we demonstrated that protein A induces the early shedding of TNFR1 in vivo during systemic S. aureus infection. Receptor cleavage precedes the release of TNF-α both in vitro and in vivo. The results obtained in experiments using a protein A-deficient mutant and tnfr1−/− mice strongly suggest that the increased levels of circulating sTNFR1 induced by S. aureus may neutralize circulating TNF-α and interfere with the host inflammatory response in the initial phase of infection.
TNF-α signaling is finely regulated by the availability of TNFR1 at the cell surface. The cleavage of the receptor ectodomain by ADAM17 culminates TNFR1 signaling and produces a soluble receptor that potentially neutralizes circulating TNF-α. The importance of downregulating TNF-α signaling has been demonstrated in patients with TNFR-associated periodic syndrome (TRAPS). These patients exhibit a mutation in TNFR1 that impairs its cleavage from the cell surface, reducing the levels of sTNFR1 in serum to half of those found in normal individuals, which leads to a chronic inflammatory condition characterized by fever and cutaneous and articular inflammation (45). Whereas downregulation of TNF-α signaling is important, excessive liberation of sTNFR1 may lead to an imbalance of the immune response. In this regard, the presence of high levels of circulating sTNFR1 and other soluble molecules, such as sTNFR2 and IL-1 receptor antagonist (IL-1Ra), is a marker of poor prognosis for septic patients. It has been proposed that these molecules may contribute to an anti-inflammatory state by blocking the action of TNF-α and IL-1β, thus impairing bacterial clearance (46). To date, no experimental studies aimed at investigating the release of these decoy molecules during S. aureus infection in vivo have been performed.
Activation of ADAM17 and shedding of TNFR1 by immune cells have been reported in response to septic stimuli in vitro (13, 47) and in animal models of LPS-induced endotoxemia (19, 35). Shedding of TNFR1, which occurs between 6 and 24 h after LPS inoculation, was critical for the neutralization of TNF-α signaling and the arrest of inflammation (19). The results presented in this study suggest that S. aureus may exploit this physiological downregulatory mechanism to its own benefit and that the cleavage of TNFR1 at an early time point during infection may serve to neutralize the action of TNF-α. Whereas TNF-α induction depends on the recognition of several S. aureus antigens, such as teichoic acids, peptidoglycan, lipoproteins, and protein A (9), we demonstrated that early shedding of TNFR1 is highly dependent on protein A expression, underscoring the role of this virulence factor in the evasion of the immune response. The ability of protein A to activate ADAM17 rapidly seems crucial in this process, since TNFR1 is already available at the cell surface and thus can be cleaved before TNF-α is induced. Whereas blockade of TNF-α using a neutralizing antibody given in combination with antibiotics 3 days after infection reduces staphylococcal arthritis and sepsis (48), early studies with experimental models using recombinant TNF-α have demonstrated that this cytokine contributes to the initial host defense against S. aureus (49). Moreover, it has also been reported that inhibition of endogenous TNF-α increases mortality during S. aureus infection (11, 48), demonstrating the important role of this cytokine during the initial host response.
Interestingly, P. aeruginosa and E. coli did not induce early shedding of TNFR1, indicating that receptor cleavage is not a general immune evasion mechanism used by bacteria and highlighting the importance of distinguishing between Gram-positive and Gram-negative bacteria during the study of ADAM17 and TNF-α/TNFR1 signaling modulation in sepsis. In this regard, it is noteworthy that the majority of clinical trials attempting to modulate TNF-α-driven immune responses have not taken into account during data analysis the potential differences in results according to the etiology of sepsis. Moreover, the most commonly used experimental model of sepsis is the murine cecal ligation and puncture model, in which peritonitis is induced by a mixture of anaerobic and facultatively anaerobic Gram-negative and Gram-positive bacteria (50–52). Whereas this model might be very useful for understanding the pathophysiology of sepsis with a peritoneal focus, studies investigating the immune responses induced by single pathogens are also needed. Indeed, it has been demonstrated that ADAM17 deficiency leads to protection against lethal shock by LPS (53, 54), whereas iRhom2-deficient mice, in which ADAM17 cannot be activated, failed to control the replication of the Gram-positive bacterium Listeria monocytogenes (55). Moreover, several experimental studies reveal that TNF-α-neutralizing therapy may adversely affect survival in Gram-positive models of peritonitis, cerebritis, and bacteremia (40).
The results presented here also seem to indicate that a given virulence factor may have differential roles in pathogenesis according to the type of infection. Protein A can act either as a proinflammatory or as an anti-inflammatory molecule. In the context of pulmonary infection, the inflammatory response induced by protein A via TNFR1 initiates IL-8 signaling and neutrophil recruitment to the lungs (Fig. 8), with deleterious consequences for pulmonary function (26). The early action of TNF-α in the lung is very limited, because airway epithelial cells do not produce significant levels of this cytokine, and prior recruitment of immune cells is required for TNF-α levels to peak. In contrast, in a systemic infection, immune cells are readily available to produce TNF-α, a potent contributor to bacterial clearance. In this scenario, the early shedding of TNFR1 induced by protein A may constitute a novel strategy by which S. aureus subverts the host immune response by neutralizing the action of TNF-α (Fig. 8). The study of signaling cascades that are critical for the initiation of the host immune response and that are altered by bacterial components may allow the identification of novel alternative targets for S. aureus infection therapy.
Fig 8.
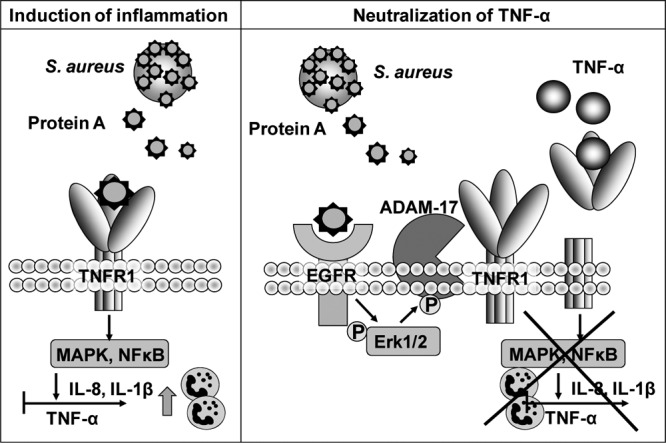
Protein A induces pro- and anti-inflammatory signaling cascades. (Left) Protein A interacts with TNF-α receptor 1 (TNFR1) in airway epithelial and immune cells. Activation of TNFR1 causes the induction of IL-8 by airway epithelial cells and of TNF-α and IL-1β by immune cells, with consequent recruitment of neutrophils to the infection site. (Right) The interaction of protein A with the epidermal growth factor receptor (EGFR) mediates ADAM17 activation and TNFR1 shedding in airway epithelial and immune cells. The release of soluble TNFR1 in response to protein A neutralizes the action of TNF-α and decreases the amount of receptor that is available to respond to this cytokine, thus impairing the downstream inflammatory cascade.
ACKNOWLEDGMENTS
This work was supported by grants from the Agencia Nacional de Promoción de la Ciencia y la Tecnología, Argentina (PICT 07-01012, PICT 10-2152, and PICT11-2263), and the Secretaría de Ciencia y Técnica, Universidad de Buenos Aires, Buenos Aires, Argentina (UBACyT 20020090200180), to M.G.
We thank Daniela Ureta for technical assistance with flow cytometry.
Footnotes
Published ahead of print 3 September 2013
REFERENCES
- 1.Rasmussen RV, Fowler VG, Jr, Skov R, Bruun NE. 2011. Future challenges and treatment of Staphylococcus aureus bacteremia with emphasis on MRSA. Future Microbiol. 6:43–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van Hal SJ, Jensen SO, Vaska VL, Espedido BA, Paterson DL, Gosbell IB. 2012. Predictors of mortality in Staphylococcus aureus bacteremia. Clin. Microbiol. Rev. 25:362–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lam SW, Bauer SR, Neuner EA. 2012. Predictors of septic shock in patients with methicillin-resistant Staphylococcus aureus bacteremia. Int. J. Infect. Dis. 16:e453–e456 [DOI] [PubMed] [Google Scholar]
- 4.Kotsaki A, Giamarellos-Bourboulis EJ. 2012. Emerging drugs for the treatment of sepsis. Expert Opin. Emerg. Drugs 17:379–391 [DOI] [PubMed] [Google Scholar]
- 5.Klevens RM, Morrison MA, Nadle J, Petit S, Gershman K, Ray S, Harrison LH, Lynfield R, Dumyati G, Townes JM, Craig AS, Zell ER, Fosheim GE, McDougal LK, Carey RB, Fridkin SK. 2007. Invasive methicillin-resistant Staphylococcus aureus infections in the United States. JAMA 298:1763–1771 [DOI] [PubMed] [Google Scholar]
- 6.Lowy FD. 2007. Secrets of a superbug. Nat. Med. 13:1418–1420 [DOI] [PubMed] [Google Scholar]
- 7.Boucher H, Miller LG, Razonable RR. 2010. Serious infections caused by methicillin-resistant Staphylococcus aureus. Clin. Infect. Dis. 51(Suppl 2):S183–S197 [DOI] [PubMed] [Google Scholar]
- 8.Ippolito G, Leone S, Lauria FN, Nicastri E, Wenzel RP. 2010. Methicillin-resistant Staphylococcus aureus: the superbug. Int. J. Infect. Dis. 14(Suppl 4):S7–S11 [DOI] [PubMed] [Google Scholar]
- 9.Fournier B, Philpott DJ. 2005. Recognition of Staphylococcus aureus by the innate immune system. Clin. Microbiol. Rev. 18:521–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abraham E. 1999. Why immunomodulatory therapies have not worked in sepsis. Intensive Care Med. 25:556–566 [DOI] [PubMed] [Google Scholar]
- 11.Nakane A, Okamoto M, Asano M, Kohanawa M, Minagawa T. 1995. Endogenous gamma interferon, tumor necrosis factor, and interleukin-6 in Staphylococcus aureus infection in mice. Infect. Immun. 63:1165–1172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xanthoulea S, Pasparakis M, Kousteni S, Brakebusch C, Wallach D, Bauer J, Lassmann H, Kollias G. 2004. Tumor necrosis factor (TNF) receptor shedding controls thresholds of innate immune activation that balance opposing TNF functions in infectious and inflammatory diseases. J. Exp. Med. 200:367–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bell JH, Herrera AH, Li Y, Walcheck B. 2007. Role of ADAM17 in the ectodomain shedding of TNF-α and its receptors by neutrophils and macrophages. J. Leukoc. Biol. 82:173–176 [DOI] [PubMed] [Google Scholar]
- 14.Scheller J, Chalaris A, Garbers C, Rose-John S. 2011. ADAM17: a molecular switch to control inflammation and tissue regeneration. Trends Immunol. 32:380–387 [DOI] [PubMed] [Google Scholar]
- 15.Reddy P, Slack JL, Davis R, Cerretti DP, Kozlosky CJ, Blanton RA, Shows D, Peschon JJ, Black RA. 2000. Functional analysis of the domain structure of tumor necrosis factor-alpha converting enzyme. J. Biol. Chem. 275:14608–14614 [DOI] [PubMed] [Google Scholar]
- 16.Gooz M. 2010. ADAM-17: the enzyme that does it all. Crit. Rev. Biochem. Mol. Biol. 45:146–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Levine SJ. 2004. Mechanisms of soluble cytokine receptor generation. J. Immunol. 173:5343–5348 [DOI] [PubMed] [Google Scholar]
- 18.Westlund KN, Zhang L, Ma F, Oz HS. 2012. Chronic inflammation and pain in a tumor necrosis factor receptor (TNFR) (p55/p75−/−) dual deficient murine model. Transl. Res. 160:84–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yagi H, Soto-Gutierrez A, Navarro-Alvarez N, Nahmias Y, Goldwasser Y, Kitagawa Y, Tilles AW, Tompkins RG, Parekkadan B, Yarmush ML. 2010. Reactive bone marrow stromal cells attenuate systemic inflammation via sTNFR1. Mol. Ther. 18:1857–1864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moks T, Abrahmsen L, Nilsson B, Hellman U, Sjoquist J, Uhlen M. 1986. Staphylococcal protein A consists of five IgG-binding domains. Eur. J. Biochem. 156:637–643 [DOI] [PubMed] [Google Scholar]
- 21.Foster TJ. 2005. Immune evasion by staphylococci. Nat. Rev. Microbiol. 3:948–958 [DOI] [PubMed] [Google Scholar]
- 22.Hartleib J, Kohler N, Dickinson RB, Chhatwal GS, Sixma JJ, Hartford OM, Foster TJ, Peters G, Kehrel BE, Herrmann M. 2000. Protein A is the von Willebrand factor binding protein on Staphylococcus aureus. Blood 96:2149–2156 [PubMed] [Google Scholar]
- 23.Sasso EH, Silverman GJ, Mannik M. 1989. Human IgM molecules that bind staphylococcal protein A contain VHIII H chains. J. Immunol. 142:2778–2783 [PubMed] [Google Scholar]
- 24.Silverman GJ, Goodyear CS. 2006. Confounding B-cell defences: lessons from a staphylococcal superantigen. Nat. Rev. Immunol. 6:465–475 [DOI] [PubMed] [Google Scholar]
- 25.Gómez MI, O'Seaghdha M, Magargee M, Foster TJ, Prince AS. 2006. Staphylococcus aureus protein A activates TNFR1 signaling through conserved IgG binding domains. J. Biol. Chem. 281:20190–20196 [DOI] [PubMed] [Google Scholar]
- 26.Gómez MI, Lee A, Reddy B, Muir A, Soong G, Pitt A, Cheung A, Prince A. 2004. Staphylococcus aureus protein A induces airway epithelial inflammatory responses by activating TNFR1. Nat. Med. 10:842–848 [DOI] [PubMed] [Google Scholar]
- 27.Martin FJ, Gómez MI, Wetzel DM, Memmi G, O'Seaghdha M, Soong G, Schindler C, Prince A. 2009. Staphylococcus aureus activates type I IFN signaling in mice and humans through the Xr repeated sequences of protein A. J. Clin. Invest. 119:1931–1939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gómez MI, Sokol SH, Muir AB, Soong G, Bastien J, Prince AS. 2005. Bacterial induction of TNF-α converting enzyme expression and IL-6 receptor alpha shedding regulates airway inflammatory signaling. J. Immunol. 175:1930–1936 [DOI] [PubMed] [Google Scholar]
- 29.Garofalo A, Giai C, Lattar S, Gardella N, Mollerach M, Kahl BC, Becker K, Prince AS, Sordelli DO, Gómez MI. 2012. The length of the Staphylococcus aureus protein A polymorphic region regulates inflammation: impact on acute and chronic infection. J. Infect. Dis. 206:81–90 [DOI] [PubMed] [Google Scholar]
- 30.Gómez MI, Seaghdha MO, Prince AS. 2007. Staphylococcus aureus protein A activates TACE through EGFR-dependent signaling. EMBO J. 26:701–709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stover CK, Pham XQ, Erwin AL, Mizoguchi SD, Warrener P, Hickey MJ, Brinkman FS, Hufnagle WO, Kowalik DJ, Lagrou M, Garber RL, Goltry L, Tolentino E, Westbrock-Wadman S, Yuan Y, Brody LL, Coulter SN, Folger KR, Kas A, Larbig K, Lim R, Smith K, Spencer D, Wong GK, Wu Z, Paulsen IT, Reizer J, Saier MH, Hancock RE, Lory S, Olson MV. 2000. Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature 406:959–964 [DOI] [PubMed] [Google Scholar]
- 32.O'Brien L, Kerrigan SW, Kaw G, Hogan M, Penadés J, Litt D, Fitzgerald DJ, Foster TJ, Cox D. 2002. Multiple mechanisms for the activation of human platelet aggregation by Staphylococcus aureus: roles for the clumping factors ClfA and ClfB, the serine-aspartate repeat protein SdrE and protein A. Mol. Microbiol. 44:1033–1044 [DOI] [PubMed] [Google Scholar]
- 33.O'Brien LM, Walsh EJ, Massey RC, Peacock SJ, Foster TJ. 2002. Staphylococcus aureus clumping factor B (ClfB) promotes adherence to human type I cytokeratin 10: implications for nasal colonization. Cell. Microbiol. 4:759–770 [DOI] [PubMed] [Google Scholar]
- 34.Sha T, Sunamoto M, Kitazaki T, Sato J, Ii M, Iizawa Y. 2007. Therapeutic effects of TAK-242, a novel selective Toll-like receptor 4 signal transduction inhibitor, in mouse endotoxin shock model. Eur. J. Pharmacol. 571:231–239 [DOI] [PubMed] [Google Scholar]
- 35.Bobrowski WF, McDuffie JE, Sobocinski G, Chupka J, Olle E, Bowman A, Albassam M. 2005. Comparative methods for multiplex analysis of cytokine protein expression in plasma of lipopolysaccharide-treated mice. Cytokine 32:194–198 [DOI] [PubMed] [Google Scholar]
- 36.Hotchkiss RS, Karl IE. 2003. The pathophysiology and treatment of sepsis. N. Engl. J. Med. 348:138–150 [DOI] [PubMed] [Google Scholar]
- 37.Marshall JC. 2003. Such stuff as dreams are made on: mediator-directed therapy in sepsis. Nat. Rev. Drug Discov. 2:391–405 [DOI] [PubMed] [Google Scholar]
- 38.Abraham E, Anzueto A, Gutierrez G, Tessler S, San Pedro G, Wunderink R, Dal Nogare A, Nasraway S, Berman S, Cooney R, Levy H, Baughman R, Rumbak M, Light RB, Poole L, Allred R, Constant J, Pennington J, Porter S. 1998. Double-blind randomised controlled trial of monoclonal antibody to human tumour necrosis factor in treatment of septic shock. NORASEPT II Study Group. Lancet 351:929–933 [PubMed] [Google Scholar]
- 39.Fisher CJ, Jr, Agosti JM, Opal SM, Lowry SF, Balk RA, Sadoff JC, Abraham E, Schein RM, Benjamin E. 1996. Treatment of septic shock with the tumor necrosis factor receptor:Fc fusion protein. The Soluble TNF Receptor Sepsis Study Group. N. Engl. J. Med. 334:1697–1702 [DOI] [PubMed] [Google Scholar]
- 40.Lorente JA, Marshall JC. 2005. Neutralization of tumor necrosis factor in preclinical models of sepsis. Shock 24(Suppl 1):107–119 [DOI] [PubMed] [Google Scholar]
- 41.Chen YF, Jobanputra P, Barton P, Jowett S, Bryan S, Clark W, Fry-Smith A, Burls A. 2006. A systematic review of the effectiveness of adalimumab, etanercept and infliximab for the treatment of rheumatoid arthritis in adults and an economic evaluation of their cost-effectiveness. Health Technol. Assess. 10(42):iii-iv, xi-xiii, 1–229 [DOI] [PubMed] [Google Scholar]
- 42.Woolacott NF, Khadjesari ZC, Bruce IN, Riemsma RP. 2006. Etanercept and infliximab for the treatment of psoriatic arthritis: a systematic review. Clin. Exp. Rheumatol. 24:587–593 [PubMed] [Google Scholar]
- 43.Marques M, Rodrigues S, Mariz E, Pinto J, Videira T, Brito J, Reis C, Simoes-Ventura F, Magro F. 2010. Severe gram positive bacterial infection in an ulcerative colitis patient treated with Infliximab. J. Crohns Colitis 4:110–113 [DOI] [PubMed] [Google Scholar]
- 44.Kim HK, Thammavongsa V, Schneewind O, Missiakas D. 2012. Recurrent infections and immune evasion strategies of Staphylococcus aureus. Curr. Opin. Microbiol. 15:92–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McDermott MF, Aksentijevich I, Galon J, McDermott EM, Ogunkolade BW, Centola M, Mansfield E, Gadina M, Karenko L, Pettersson T, McCarthy J, Frucht DM, Aringer M, Torosyan Y, Teppo AM, Wilson M, Karaarslan HM, Wan Y, Todd I, Wood G, Schlimgen R, Kumarajeewa TR, Cooper SM, Vella JP, Amos CI, Mulley J, Quane KA, Molloy MG, Ranki A, Powell RJ, Hitman GA, O'Shea JJ, Kastner DL. 1999. Germline mutations in the extracellular domains of the 55 kDa TNF receptor, TNFR1, define a family of dominantly inherited autoinflammatory syndromes. Cell 97:133–144 [DOI] [PubMed] [Google Scholar]
- 46.de Pablo R, Monserrat J, Reyes E, Diaz-Martin D, Rodriguez Zapata M, Carballo F, de la Hera A, Prieto A, Alvarez-Mon M. 2011. Mortality in patients with septic shock correlates with anti-inflammatory but not proinflammatory immunomodulatory molecules. J. Intensive Care Med. 26:125–132 [DOI] [PubMed] [Google Scholar]
- 47.Robertshaw HJ, Brennan FM. 2005. Release of tumour necrosis factor alpha (TNFα) by TNFα cleaving enzyme (TACE) in response to septic stimuli in vitro. Br. J. Anaesth. 94:222–228 [DOI] [PubMed] [Google Scholar]
- 48.Fei Y, Wang W, Kwiecinski J, Josefsson E, Pullerits R, Jonsson IM, Magnusson M, Jin T. 2011. The combination of a tumor necrosis factor inhibitor and antibiotic alleviates staphylococcal arthritis and sepsis in mice. J. Infect. Dis. 204:348–357 [DOI] [PubMed] [Google Scholar]
- 49.Vaudaux P, Grau GE, Huggler E, Schumacher-Perdreau F, Fiedler F, Waldvogel FA, Lew DP. 1992. Contribution of tumor necrosis factor to host defense against staphylococci in a guinea pig model of foreign body infections. J. Infect. Dis. 166:58–64 [DOI] [PubMed] [Google Scholar]
- 50.Hubbard WJ, Choudhry M, Schwacha MG, Kerby JD, Rue LW, III, Bland KI, Chaudry IH. 2005. Cecal ligation and puncture. Shock 24(Suppl 1):52–57 [DOI] [PubMed] [Google Scholar]
- 51.Dejager L, Pinheiro I, Dejonckheere E, Libert C. 2011. Cecal ligation and puncture: the gold standard model for polymicrobial sepsis? Trends Microbiol. 19:198–208 [DOI] [PubMed] [Google Scholar]
- 52.Secher T, Vasseur V, Poisson DM, Mitchell JA, Cunha FQ, Alves-Filho JC, Ryffel B. 2009. Crucial role of TNF receptors 1 and 2 in the control of polymicrobial sepsis. J. Immunol. 182:7855–7864 [DOI] [PubMed] [Google Scholar]
- 53.Horiuchi K, Kimura T, Miyamoto T, Takaishi H, Okada Y, Toyama Y, Blobel CP. 2007. TNF-α-converting enzyme (TACE/ADAM17) inactivation in mouse myeloid cells prevents lethality from endotoxin shock. J. Immunol. 179:2686–2689 [DOI] [PubMed] [Google Scholar]
- 54.Long C, Wang Y, Herrera AH, Horiuchi K, Walcheck B. 2010. In vivo role of leukocyte ADAM17 in the inflammatory and host responses during E. coli-mediated peritonitis. J. Leukoc. Biol. 87:1097–1101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McIlwain DR, Lang PA, Maretzky T, Hamada K, Ohishi K, Maney SK, Berger T, Murthy A, Duncan G, Xu HC, Lang KS, Haussinger D, Wakeham A, Itie-Youten A, Khokha R, Ohashi PS, Blobel CP, Mak TW. 2012. iRhom2 regulation of TACE controls TNF-mediated protection against Listeria and responses to LPS. Science 335:229–232 [DOI] [PMC free article] [PubMed] [Google Scholar]