

Abstract
Neutrophils are the first cells to infiltrate to the site of Leishmania promastigote infection, and these cells help to reduce parasite burden shortly after infection is initiated. Several clinical reports indicate that neutrophil recruitment is sustained over the course of leishmaniasis, and amastigote-laden neutrophils have been isolated from chronically infected patients and experimentally infected animals. The goal of this study was to compare how thioglycolate-elicited murine neutrophils respond to L. amazonensis metacyclic promastigotes and amastigotes derived from axenic cultures or from the lesions of infected mice. Neutrophils efficiently internalized both amastigote and promastigote forms of the parasite, and phagocytosis was enhanced in lipopolysaccharide (LPS)-activated neutrophils or when parasites were opsonized in serum from infected mice. Parasite uptake resulted in neutrophil activation, oxidative burst, and accelerated neutrophil death. While promastigotes triggered the release of tumor necrosis factor alpha (TNF-α), uptake of amastigotes preferentially resulted in the secretion of interleukin-10 (IL-10) from neutrophils. Finally, the majority of promastigotes were killed by neutrophils, while axenic culture- and lesion-derived amastigotes were highly resistant to neutrophil microbicidal mechanisms. This study indicates that neutrophils exhibit distinct responses to promastigote and amastigote infection. Our findings have important implications for determining the impact of sustained neutrophil recruitment and amastigote-neutrophil interactions during the late phase of cutaneous leishmaniasis.
INTRODUCTION
Leishmania parasites are obligate intracellular protozoa that cause leishmaniasis, a neglected tropical disease responsible for extensive morbidity and mortality in the developing world. Infection is initiated when metacyclic promastigotes are deposited into the skin by the bite of a female sandfly, and parasitism of host neutrophils, dendritic cells (DCs), and macrophages rapidly ensues. In macrophages, promastigotes convert into amastigotes, the parasite stage that replicates in mammalian hosts. Leishmania amastigotes are able to modify macrophage functions and resist macrophage microbicidal activity, resulting in the establishment of an environment that is permissive for parasite growth (1–3). Parasite-mediated manipulation of multiple signaling pathways in other cell types, such as DCs, is also well established, and disruption of innate immune cell function ultimately hinders the formation of a potent, effective T helper cell response. Consequently, amastigote replication continues unabated in the context of low-grade inflammation and tissue damage (4, 5).
Neutrophils rapidly recruit to the site of infection after metacyclic promastigotes are delivered into the skin, either via their natural sandfly vector or by needle injection (6, 7). After contacting each other, Leishmania promastigotes and neutrophils may each undergo one of several fates. For example, L. major promastigotes can survive inside neutrophils and ultimately use these cells as Trojan horses to facilitate silent infection of macrophages (8). In contrast, neutrophils respond to L. amazonensis promastigotes by undergoing several forms of cell death; many cells encountering parasites rapidly die by NETosis, a specialized form of death that results in parasite entrapment and degradation (9), while the remaining neutrophils die largely by apoptosis (10).
In numerous mouse models, antibody-mediated neutrophil depletion has been extensively used to determine how these cells contribute to the pathogenesis of various infectious diseases (11–13). However, there is currently a lack of consensus regarding the function of neutrophils during Leishmania promastigote infection, as these cells have been implicated in both promoting and inhibiting disease progression in different studies (14, 15). Despite reporting contradictory roles for neutrophils in controlling infection, depletion studies nevertheless emphasize the importance of these cells in the early disease process of cutaneous leishmaniasis.
According to several clinical reports, neutrophil recruitment to the site of infection is not limited to the promastigote-mediated phase of disease but continues throughout the course of chronic leishmaniasis as well. In L. tropica-infected patients, neutrophils were recovered from lesions ranging from 1 to 36 months in duration (16). Neutrophils were also observed in the ulcerated lesions of patients chronically infected with L. major, and in some patients, these cells were the predominant immune cell type at the site of infection (17). Interestingly, BALB/c mice infected with L. major display a progressive increase in the number of intralesional neutrophils throughout the first 6 weeks of infection (18), suggesting that persistent neutrophil recruitment may be a characteristic feature of chronic cutaneous leishmaniasis.
Amastigote-laden neutrophils have been isolated from numerous infected hosts, including experimentally infected macaques and naturally infected humans, dogs, and foxes (17, 19–21). However, the immunological ramifications of amastigote-neutrophil interactions remain largely uncharacterized. We have recently demonstrated that L. amazonensis amastigotes are highly resistant to the antimicrobial effects of purified human histone proteins (22), which are known to be released together with other microbicidal agents when neutrophils undergo NETosis (23). Currently, it is unclear whether neutrophils recognize amastigotes and influence amastigote clearance or persistence (24).
In this study, we aimed to examine the interaction between L. amazonensis amastigotes and peritoneal neutrophils obtained from C57BL/6 mice. We demonstrate that neutrophils efficiently internalized both the amastigote and promastigote forms of the parasite, particularly when parasites were opsonized with Leishmania-specific antibodies. Parasite uptake resulted in neutrophil activation and oxidative burst, but neutrophils differed in their responses to amastigotes and promastigotes in several ways, including cytokine secretion and pathogen clearance. Specifically, neutrophils responded to promastigotes by releasing tumor necrosis factor alpha (TNF-α) and by killing the majority of parasites. In contrast, neutrophils failed to efficiently kill amastigotes and preferentially released interleukin-10 (IL-10) in response to this stage of parasite. Therefore, the role of neutrophils during leishmaniasis may differ depending on the stage of parasite encountered. These findings have important implications for understanding the pathogenic mechanisms of immune system dysfunction and chronic parasite persistence during experimental cutaneous leishmaniasis.
MATERIALS AND METHODS
Mice.
Female C57BL/6 and BALB/c mice were purchased from Taconic Farms (Germantown, NY). C57BL/6 mice were the source of the majority of neutrophils in this study, while BALB/c mice were predominately used for the maintenance of parasite infectivity and for isolating lesion-derived amastigotes. Neutrophils from BALB/c mice were used as a control for the clearance of lesion-derived amastigotes shown in Fig. S4 in the supplemental material. B6(Cg)-Ncf1m1J/J mice deficient in the gp47 subunit of NADPH oxidase were obtained from The Jackson Laboratory (Bar Harbor, ME) and bred on campus. Mice were maintained under specific-pathogen-free conditions and used at 6 to 12 weeks of age, according to protocols approved by the Animal Care and Use Committee of the University of Texas Medical Branch (Galveston, TX).
Parasite cultivation.
The infectivity of L. amazonensis (strains RAT/BA/74/LV78 and MHOM/BR/77/LTB0016) was maintained by regular passage through BALB/c mice. Strain RAT/BA/74/LV78 was used for all experiments using promastigotes and amastigotes. Strain MHOM/BR/77/LTB0016 was used in the infection of mice to generate immune serum. Promastigotes were cultured at 26°C in M199 containing 40 mM HEPES, 10% heat-inactivated fetal bovine serum (FBS), 0.1% hemin in 50:50 H2O and triethanolamine (Frontier Scientific, Logan, UT), 0.1 mM adenine (pH 7.5), 5 mM l-glutamine, and 50 μg/ml gentamicin. Metacyclic promastigotes were purified as described previously (25) by using the monoclonal antibody 3A1, which was generously provided by Norma Andrews (University of Maryland). All experiments using promastigote groups utilized metacyclic promastigotes purified in this way. Axenic amastigotes were cultured at 32°C in Grace's insect cell culture medium (Invitrogen, Carlsbad, CA), pH 5.2, supplemented with 20% FBS and 25 μg/ml gentamicin. Lesion-derived amastigotes were collected from the footpads of infected BALB/c mice through mechanical tissue disruption, followed by 3 washes and incubation in amastigote medium. Lesion-derived amastigotes were used within 48 h of isolation from infected footpads. Prior to use, lesion-derived amastigotes were washed an additional 3 times to remove any residual tissue components. Fresh parasite lysates were prepared through 2 freeze-thaw cycles followed by sonication for 15 min.
Production of luciferase-expressing parasites.
Circular pSP72-YNEO-αIR-LUC1.2 was generously provided by Barbara Papadopoulou (Laval University, Quebec, Canada). Logarithmic-phase RAT/BA/74/LV78 promastigotes were transfected with 35 μg plasmid, as reported previously (26), resulting in episomal expression of firefly luciferase. Following a 24-h rest period, selection for luciferase-expressing promastigotes was performed via titration of G418 (Invitrogen). Luciferase-expressing amastigotes were derived from logarithmic-phase promastigote cultures. To maintain selective pressure, luciferase-expressing promastigotes and amastigotes were grown in normal parasite medium containing G418 (50 μg/ml).
Generation of immune serum and parasite opsonization.
C57BL/6 mice were infected with L. amazonensis MHOM/BR/77/LTB0016 promastigotes in the rear footpads for 12 weeks. Infected mice and age- and sex-matched naive mice were subsequently sacrificed, and serum was collected, heat inactivated, and stored at −20°C. To ensure suitable anti-Leishmania antibody concentrations, antibody titers were determined via direct enzyme-linked immunosorbent assay (ELISA). In experiments utilizing opsonized amastigotes, parasites were incubated in naive or immune serum (10%) for 20 min at room temperature prior to infection.
Neutrophil collection.
Peritoneal exudate cells were obtained from mice 5 h after injection with 3% thioglycolate (Sigma-Aldrich, St. Louis, MO). Thioglycolate was removed, and neutrophils were purified via density gradient centrifugation with Percoll (Sigma-Aldrich). Neutrophil purity (>95%) was validated by fluorescence-activated cell sorting (FACS) and examination of morphology after staining; cell viability was routinely >95%, as monitored by trypan blue exclusion. Prior to treatment or coculture with parasites, neutrophils were plated in tissue culture-treated polystyrene. Because L. amazonensis poorly tolerates high temperatures, all neutrophil-parasite cocultures were maintained at 32°C.
Neutrophil phagocytosis of parasites.
Parasites were labeled with carboxyfluorescein succinimidyl ester (CFSE) (Sigma-Aldrich), as described previously (27). Neutrophils were cocultured with CFSE-labeled amastigotes or promastigotes at a multiplicity of infection (MOI) of 5 for 4 h at 32°C with 5% CO2. Cells were collected, stained with allophycocyanin (APC)-conjugated anti-Ly6G (BD Biosciences, San Jose, CA), and analyzed by FACS. Neutrophils were identified based on forward/side scatter characteristics and Ly6G positivity. Parasite-carrying neutrophils were identified based on CFSE positivity. In some experiments, neutrophils were treated with lipopolysaccharide (LPS), cytochalasin D (Sigma-Aldrich), or granulocyte-macrophage colony-stimulating factor (GM-CSF) (PeproTech, Oak Park, CA), and parasites were opsonized in heat-inactivated naive or immune serum prior to coculture. Data were collected using an Accuri C6 flow cytometer (Accuri Cytometers Inc., Ann Arbor, MI). Flow cytometry data were subsequently analyzed using CFlow version 1.0.227.4 (Accuri Cytometers Inc.) or FlowJo version 7.6.1 (Tree Star, Ashland, OR).
Electron microscopy (EM).
Following 4 h of coculture with amastigotes, neutrophils were fixed in Ito's fixative (2.5% formaldehyde prepared from paraformaldehyde, 0.1% glutaraldehyde, 0.03% CaCl2, and 0.03% trinitrophenol in 0.05 M cacodylate buffer, pH 7.3) at room temperature for 15 min and then overnight at 4°C. After washing in 0.1 M cacodylate buffer, samples were postfixed in 1% osmium tetroxide in the same buffer for 1 h and en bloc stained with 1% aqueous uranyl acetate for 20 min at 60°C. After dehydration in a graded series of ethanol solutions, samples were embedded in Poly/Bed 812 (Polysciences, Warrington, PA). Ultrathin sections were cut on a Leica EM UC7 ultramicrotome (Leica Microsystems, Buffalo Grove, IL), stained with lead citrate, and examined using a Philips 201 transmission electron microscope (Philips Electron Optics, Eindhoven, The Netherlands) at 60 kV.
Measurement of neutrophil activation and oxidative burst.
Neutrophil-parasite cocultures were incubated for 4 h at 32°C and 5% CO2. After 4 h, some neutrophils were blocked with anti-CD16/CD32 and stained with peridinin chlorophyll protein (PerCP)-Cy5.5-conjugated anti-CD11b (eBioscience, San Diego, CA) and APC-conjugated anti-Ly6G, and samples were analyzed by FACS. Separate cell groups were stained with APC-conjugated anti-Ly6G and dihydrorhodamine 123 (1 μM; Sigma-Aldrich), which converts to fluorescent rhodamine 123 (Rho 123) when oxidized. In some experiments, 1 μM N-formyl-methionyl-leucyl-phenylalanine (fMLP) (Sigma-Aldrich) was added for the last 5 min of incubation prior to measurement of oxidative burst. The oxidation reaction was stopped on ice, and neutrophil reactive oxygen species (ROS) production was analyzed by gating on Ly6G+ cells and measuring the mean fluorescence intensity (MFI) of Rho 123 by FACS. To determine whether the parasite-mediated oxidative burst was restricted to infected cells, amastigotes were labeled with PKH26 (Sigma-Aldrich) according to the manufacturer's instructions. The MFIs of Rho123 in PKH26+ (infected) and PKH26− (bystander) neutrophils were then compared.
Neutrophil cytokine detection.
To minimize protease activity, neutrophils were treated with 50 μg/ml aprotinin (Sigma-Aldrich) prior to treatment with parasites at an MOI of 5. Supernatants were collected after 24 h, and the cytokine concentration was measured via ELISA (eBioscience). After treatment with tetramethylbenzidine substrate and stop solution, optical density (OD) values at 450 nm were measured with a Multiskan Ascent ELISA reader (Labsystems, Helsinki, Finland).
Measurement of neutrophil apoptosis.
Neutrophils were cocultured with amastigotes in the presence or absence of GM-CSF (20 ng/ml). After 18 h, neutrophils were collected and stained with APC-conjugated anti-Ly6G and the annexin V:FITC apoptosis detection kit I (BD Biosciences). Early apoptosis in Ly6G+ neutrophils was quantified by FACS based on positive staining for annexin V and negative staining for propidium iodide (PI). To determine whether changes in apoptosis were restricted to infected cells, CFSE-labeled amastigotes were cocultured with neutrophils, followed by phycoerythrin (PE)-conjugated annexin V staining.
Parasite killing by neutrophils.
Luciferase-expressing amastigotes or promastigotes were cocultured with neutrophils at an MOI of 0.1. In some experiments, to better simulate lesion-derived amastigotes, axenic parasites were precoated with heat-inactivated serum from infected mice prior to coculture with neutrophils. At 0, 6, and 18 h postinfection, cocultures were lysed and frozen at −80°C prior to analysis. Parasite burdens were estimated by mixing lysates with luciferase assay substrate (Promega Corporation, Madison, WI) and measuring photon emission on a Veritas microplate luminometer (Turner BioSystems Inc., Sunnyvale, CA). Parasite survival was estimated by comparing the baseline photon emission at 0 h to the signal intensities at subsequent time points.
Statistical analysis.
Differences between two groups were determined by using the two-tailed Student's t test. Graphs were prepared by using GraphPad Prism 4.0 (GraphPad Software, San Diego, CA). The difference between two groups was considered significant when the P value was ≤0.05.
RESULTS
Neutrophils internalize L. amazonensis promastigotes and amastigotes.
To investigate the interaction between neutrophils and Leishmania amastigotes, we opted to use thioglycolate-elicited neutrophils from C57BL/6 mice and L. amazonensis amastigotes. We selected this particular system to dissect amastigote-neutrophil interactions for several reasons. First, C57BL/6 mice are traditionally viewed as a resistant strain in regard to Leishmania infection (28), and C57BL/6 neutrophils have been shown to respond to L. major by secreting biologically active IL-12p70 (29). Thioglycolate-elicited neutrophils can be isolated with high yield and purity, which is advantageous for conducting a detailed analysis of neutrophil function (30). Finally, we opted to examine neutrophil responses to L. amazonensis because these parasites are easily propagated as amastigotes in vitro, induce a nonhealing disease phenotype in both C57BL/6 and BALB/c mice, and have been shown to have potent immunosuppressive effects on numerous cell types (31, 32).
Neutrophil uptake of metacyclic promastigotes is a critical feature of the initial phase of Leishmania infection. As infection progresses, neutrophils may also encounter amastigotes liberated from ruptured macrophages. However, despite the popularity of murine models of cutaneous and visceral leishmaniasis, reports of amastigote uptake by mouse neutrophils are largely absent from the literature. We compared neutrophil phagocytosis of CFSE-labeled axenic amastigotes and metacyclic promastigotes. After 4 h of coculture, we observed that approximately 8.6% of neutrophils engulfed amastigotes, while 7.9% of neutrophils internalized promastigotes. Phagocytosis of parasites was inhibited in neutrophils that were pretreated with cytochalasin D (20 μM), confirming that parasite uptake was mediated via an actin polymerization-dependent mechanism. Of note, parasite opsonization in heat-inactivated serum collected from L. amazonensis-infected mice markedly enhanced neutrophil phagocytosis of both promastigotes and amastigotes (Fig. 1A). Opsonization with naive mouse serum also enhanced parasite uptake, but to a lesser extent that that with serum from infected animals (see Fig. S1 in the supplemental material). Phagocytosis of amastigotes was also significantly enhanced in neutrophils treated with LPS (100 ng/ml) or GM-CSF (200 ng/ml) (Fig. 1B). Electron microscopy was used to confirm amastigote internalization. Ultrastructural analysis indicated that internalized amastigotes were housed within tight, membrane-bound vacuoles (Fig. 2). The presence of intact flagellar remnants in some internalized amastigotes (Fig. 2, arrow) suggested that parasites were not damaged by neutrophils during phagocytosis. In some instances, neutrophils carrying 4 or more parasites were also observed (image not shown).
Fig 1.

Neutrophil phagocytosis of L. amazonensis parasites. (A) Thioglycolate-elicited peritoneal neutrophils were cocultured with CFSE-labeled axenic amastigotes (AxAm) or metacyclic promastigotes (Pm) for 4 h. In some groups, neutrophils were pretreated with cytochalasin D (Cyto D) (20 μM) or parasites were opsonized in serum from infected mice. Neutrophils were identified by forward/side scatter characteristics and by Ly6G positivity. CFSE positivity in the boxes shown represents Ly6G+ neutrophils carrying parasites. Values are mean percentages of CFSE+ cells ± 1 standard deviation (SD). (B) Percentages of CFSE+ neutrophils (PMN) carrying amastigotes after 4 h in medium alone (Med) or in the presence of LPS (100 ng/ml) or GM-CSF (200 ng/ml). Data are pooled from 3 independent repeats and are shown as means ± standard errors. *, statistically significant differences (P < 0.05) between the groups.
Fig 2.

Ultrastructural analysis of amastigote uptake by neutrophils. Neutrophils were cocultured with serum-coated amastigotes for 4 h, fixed, and prepared for analysis via electron microscopy. A characteristic neutrophil is depicted, exhibiting a multilobular nucleus (N), electron-dense granules (arrowheads), and 2 intracellular amastigotes (asterisks). A flagellar remnant is also clearly visible in the amastigote on the left (arrow). Bar, 2 μm.
Amastigote infection triggers neutrophil activation and oxidative burst.
Activated neutrophils characteristically upregulate CD11b on their surface, which reflects their ability to execute a number of important functions, including phagocytosis, degranulation, apoptosis, and oxidative burst (33–36). As shown in Fig. 3A, neutrophil coculture with axenic amastigotes or promastigotes resulted in an appreciable upregulation of surface CD11b on infected neutrophils. However, the extent of CD11b upregulation did not differ for neutrophils cocultured with promastigotes or amastigotes. In contrast, neutrophil coculture with lesion-derived amastigotes resulted in a significant increase in CD11b upregulation over that of axenic amastigotes (Fig. 3A).
Fig 3.

Neutrophil activation and oxidative burst after contact with parasites. (A) Mean fluorescence intensity (MFI) of CD11b on neutrophils resting in medium (Med) or cocultured with metacyclic promastigotes (Pm), axenic amastigotes (AxAm), or lesion-derived amastigotes (Am). (B) MFI of rhodamine 123 (Rho 123) in dihydrorhodamine 123-labeled Ly6G+ cells after 4 h of coculture with parasites. (C) Oxidative burst in Ly6G+ Rho123+ neutrophils, indicating that the extent of burst on a per-cell basis does not differ for neutrophils cocultured with promastigotes or amastigotes. (D) ROS production in neutrophils infected with PKH26-labeled axenic amastigotes for 4 h, showing that the majority of ROS was generated in PKH26hi (amastigote-laden) cells. All data are pooled from at least 2 independent experiments and shown as means ± standard errors. * (P < 0.05), ** (P < 0.01), and *** (P < 0.001) indicate statistically significant differences between groups. NS, not significant.
Because reactive oxygen species (ROS) are a critical component of the microbicidal armament of neutrophils and because promastigote-induced ROS production in neutrophils has been reported (37), we investigated whether amastigotes also trigger a neutrophil oxidative burst. To do this, we labeled resting and parasite-laden neutrophils with dihydrorhodamine 123, a cell-permeative dye that converts into fluorescent rhodamine 123 (Rho 123) when oxidized (38). As shown in Fig. 3B, neutrophil coculture with amastigotes or promastigotes for 4 h resulted in a significant increase in rhodamine 123 fluorescence compared to that in cells resting in medium. While promastigotes tended to elicit more ROS, the difference between the amastigote- and promastigote-mediated oxidative bursts was not significant, regardless of whether the entire neutrophil population (Fig. 3B) or the Rho123+ population (Fig. 3C) was examined. In contrast, lesion-derived amastigotes did elicit significantly more neutrophil oxidative burst than their axenically cultured counterparts (Fig. 3B). Dihydrorhodamine 123-labeled parasites had no detectable dye oxidation, confirming that the ROS measured in our assay was neutrophil-derived (data not shown).
We examined whether an oxidative burst was occurring in infected or bystander neutrophils by coculturing cells with PKH26-labeled amastigotes. After 4 h of infection, neutrophils could be clearly gated based on PKH26 positivity, and we observed that amastigote-laden (PKH26hi) cells were the major producers of ROS (Fig. 3D). The neutrophil oxidative burst in response to parasites was dependent upon the presence of intact parasites, as parasite lysates failed to increase ROS production above control levels (see Fig. S2A in the supplemental material). These findings collectively indicate that parasite internalization is required for a neutrophil oxidative burst in response to L. amazonensis.
To determine whether parasites were able to alter the oxidative burst elicited by an external signal such as N-formyl-methionyl-leucyl-phenylalanine (fMLP), we compared neutrophil production of ROS in response to amastigotes, fMLP, or both stimuli combined. Coculture with parasites plus fMLP treatment resulted in substantially greater ROS production than amastigote or fMLP treatment alone (see Fig. S2B in the supplemental material). To ensure that the observed increase in ROS was due to phagocyte NADPH oxidase rather than mitochondrial damage or other ROS sources, we compared ROS production in neutrophils from wild-type (WT) mice and mice deficient in the gp47 subunit of the NADPH oxidase complex. gp47−/− neutrophils produced appreciably less ROS in response to fMLP or amastigotes than WT neutrophils, confirming that the majority of the ROS detected in our assay was derived from the NADPH oxidase (see Fig. S2C in the supplemental material).
Promastigotes and amastigotes trigger differential cytokine release from neutrophils.
Although neutrophils release only small quantities of cytokines, neutrophil-derived mediators have been shown to play an important role in the pathogenesis of multiple conditions, including arthritis (39), cancer (40), and HIV infection (41). We measured cytokine release by neutrophils after 24 h of coculture with axenic amastigotes or metacyclic promastigotes. Compared to baseline cytokine secretion by resting neutrophils, we observed that promastigotes promoted TNF-α release but failed to induce IL-10 secretion. In contrast, amastigotes induced considerably less TNF-α and preferentially induced the release of IL-10 (Fig. 4A and B). While a previous study reported some IL-12p40 and IL-12p70 release from L. major-infected murine neutrophils (29), the concentration of IL-12 in the supernatants of L. amazonensis-infected neutrophils was below the level of detection, regardless of whether axenic amastigotes or metacyclic promastigotes were utilized (data not shown).
Fig 4.

Neutrophil cytokine production in response to parasites. Neutrophils were cocultured with axenic amastigotes (AxAm) or metacyclic promastigotes (Pm) for 24 h. Production of TNF-α (A) and IL-10 (B) was analyzed by ELISA. Results are pooled from 2 independent repeats and shown as means ± standard errors. * (P < 0.05), ** (P < 0.01), and *** (P < 0.001) indicate statistically significant differences between groups. NS, not significant.
Amastigote infection accelerates neutrophil apoptosis.
The life span of circulating neutrophils is typically short (8 to 20 h), but neutrophil survival can be altered by recruitment to a site of inflammation, contact with prosurvival signals, or phagocytosis of infectious cargo (42, 43). Infection with promastigotes from several Leishmania species has been shown to alter neutrophil life span, but the outcome (prolonged life span versus accelerated cell death) may be largely context dependent. For example, L. major promastigotes have been shown to inhibit apoptosis of human peripheral blood neutrophils in vitro (30). In contrast, the majority of L. major-laden neutrophils isolated from recently infected mice were apoptotic (6). To determine how amastigotes influence neutrophil longevity, we cocultured neutrophils with axenic or lesion-derived amastigotes at an MOI of 5 for 18 h. Cells were then stained with FITC-conjugated annexin V to measure phosphatidylserine (PS) surface exposure, an indicator of apoptosis. A significantly greater percentage of cells cocultured with amastigotes exposed PS compared to uninfected neutrophils, suggesting that amastigote infection accelerated neutrophil apoptosis. The percentage of apoptotic neutrophils did not differ for cocultures containing axenic or lesion-derived amastigotes (Fig. 5A). Infecting neutrophils with amastigotes at a lower dose (an MOI of 2) resulted in similar increases in neutrophil apoptosis (data not shown). We also examined neutrophil death in response to promastigotes and observed that promastigote-infected neutrophils remaining after 18 h exhibited a similar acceleration in apoptosis (see Fig. S3A in the supplemental material). However, we noted that the majority of promastigote-infected neutrophils were not recoverable after 18 h, suggesting that apoptosis was not the primary form of neutrophil death (see Fig. S3B in the supplemental material). These findings support a previous report that L. amazonensis promastigotes potently trigger early neutrophil NETosis prior to the onset of apoptosis (9). We also noted that GM-CSF, which prolongs the neutrophil life span by activating the phosphatidylinositol 3-kinase (PI3K) and extracellular signal-regulated kinase pathways (44), partially reversed axenic amastigote-mediated apoptosis (Fig. 5B).
Fig 5.

Accelerated neutrophil apoptosis after L. amazonensis amastigote uptake. (A) Percentages of PS+ PI− neutrophils after 18 h of culture in medium (Med) or after coculture with axenic amastigotes (AxAm) or lesion-derived amastigotes (Am). (B) Neutrophil apoptosis in medium alone or in response to axenic amastigotes in the presence or absence of GM-CSF (20 ng/ml). All data in panels A and B are pooled from at least 2 independent repeats and shown as means ± standard errors. NS, not significant. * (P < 0.05) and *** (P < 0.001) indicate statistically significant differences between the groups. (C) Comparison of apoptosis in resting neutrophils, CFSE+ (parasite-carrying) neutrophils, and CFSE− (bystander) neutrophils. PS surface exposure in neutrophils was measured via binding of annexin V (AnxV). The percentages of apoptotic cells were calculated by dividing the number of PS+ cells by the total number of cells for each group. Values are mean percentages of apoptotic cells ± 1 SD. Shown are representative results of one of three independent repeats.
To determine whether amastigote-mediated apoptosis was restricted to infected cells, amastigotes were labeled with CFSE prior to coculture with neutrophils. As shown in Fig. 5C, resting cells in cultures lacking amastigotes and CFSE− cells from neutrophil-amastigote cocultures had similar percentages of annexin V+ cells at 18 h (16.9% versus 18.9%). In contrast, nearly 40% of CFSE+ neutrophils carrying parasites were apoptotic. In all of our experiments, fewer than 3% of neutrophils stained positively for propidium iodide (a marker of necrosis) at 18 h, implying that neutrophil necrosis was unaffected by parasite infection during our observation period (data not shown). Therefore, amastigote infection decreased the life span of parasite-carrying neutrophils, rather than that of bystander neutrophils, via accelerated neutrophil apoptosis.
Amastigotes are more resistant to neutrophil microbicidal mechanisms than promastigotes.
Neutrophil uptake and elimination of L. amazonensis promastigotes have been previously documented (9), but it is unclear whether neutrophils can destroy amastigotes in a similar manner. To address this issue, we cocultured luciferase-expressing promastigotes or amastigotes with neutrophils and tracked the loss of luciferase activity in serial samples over time (Fig. 6A). To ensure efficient parasite internalization by neutrophils, we used a very low infection dose (MOI of 0.1). Consistent with previous reports (9), we found that neutrophils killed approximately 55% of metacyclic promastigotes within 6 h of coculture and that more than 65% of promastigotes were killed by 18 h. Importantly, the luciferase activity in cocultures containing axenic amastigotes failed to decline over the 18-h period assayed (Fig. 6B). Axenic amastigote-dependent luciferase activity remained nearly constant even when parasites and neutrophils were cocultured for as long as 40 h (data not shown), indicating a remarkable resistance of axenic amastigotes against neutrophil microbicidal defenses. To validate and expand upon these findings, we also examined the survival of lesion-derived amastigotes following coculture with neutrophils. While we noted some killing of lesion-derived amastigotes after 18 h, the extent of amastigote survival still surpassed that of metacyclic promastigotes (Fig. 6C). Clearance of lesion-derived amastigotes was comparable for neutrophils obtained from C57BL/6 and BALB/c mice (see Fig. S4 in the supplemental material). We also noted that coating axenic amastigotes in immune serum prior to coculture with neutrophils had a small but appreciable effect on parasite clearance at 18 h, suggesting that host tissue components may aid in the partial elimination of amastigotes (Fig. 6C). Taken together, these results indicate that amastigotes, regardless of their source, demonstrated a clear survival advantage during interaction with neutrophils compared to metacyclic promastigotes.
Fig 6.
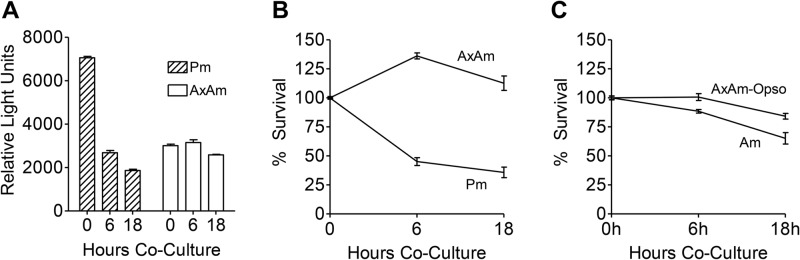
Neutrophil killing of promastigotes and amastigotes. Luciferase-expressing metacyclic promastigotes (Pm) and axenic amastigotes (AxAm) were cocultured with neutrophils (at an MOI of 0.1) for 0, 6, or 18 h. Cells were lysed and treated with luciferin substrate to elicit photon emission. Photon emission of 0-h samples was used as a reference for 100% parasite survival, and the subsequent decay in signal at 6 h and 18 h was used to estimate the extent of parasite killing. (A) Representative graph showing photon intensity in relative light units. (B) Survival of axenic amastigotes and metacyclic promastigotes cocultured with neutrophils for 6 h and 18 h. (C) Survival of lesion-derived amastigotes (Am) and axenic amastigotes opsonized in fresh serum from infected mice (AxAm-Opso) after 6 h and 18 h of coculture with neutrophils. The data in panels B and C are pooled from at least 2 independent experiments and shown as means ± the errors.
DISCUSSION
During the early stages of infection, neutrophils may aid in the elimination of many promastigotes, but surviving parasites may acquire a distinct advantage in the subsequent infection of macrophages (7, 8). The complicated role of neutrophils during promastigote infection is exemplified by the presence of several contradicting studies that followed disease progression in neutrophil-depleted mice. In response to neutrophil depletion with the monoclonal antibody RB6-8C5, Chen et al. noted no difference in the progression of L. major infection in resistant C3H/HeJ mice, while depleted BALB/c mice were less able to control infection than nondepleted animals (14). In contrast, Tacchini-Cottier et al. observed that depletion of neutrophils with the monoclonal antibody NIMP-R14 reduced the severity of L. major infection in BALB/c mice, while depletion in resistant C57BL/6 mice failed to alter the time required for lesion resolution (15). The discrepancies between these two studies may be due in part to the use of antibodies with different neutrophil specificities (RB6-8C5 also depletes eosinophils, inflammatory monocytes, and several other immune cell populations, while NIMP-R14 also depletes inflammatory monocytes) and different L. major strains (45).
Given that promastigotes encounter neutrophils predominantly prior to contacting monocytes or macrophages, it is somewhat surprising that this stage of parasite is relatively susceptible to neutrophil microbicidal mechanisms in our in vitro studies. It is possible that the neutrophil-promastigote interaction in vivo is complicated by additional vector, host, or parasite components that improve parasite resistance against killing by neutrophils (7, 46). It is also possible that the purification of metacyclic promastigotes from parasite cultures may weaken the natural defenses of this parasite against neutrophils in vitro. Alternatively, some promastigote killing by neutrophils may favorably alter the inflammatory milieu at the site of infection, resulting in improved survival conditions for the remaining promastigotes. For example, NETosing neutrophils have been shown to be a potent stimulus for type I interferon release from plasmacytoid DCs (47), and we have previously demonstrated that type I interferon signaling can promote parasite survival and disease pathology (10). Regardless of the underlying mechanisms, it is evident that promastigotes are relatively susceptible to neutrophil microbicidal defense in our study.
Despite a few observations documenting contact between neutrophils and Leishmania amastigotes in several mammalian hosts, the outcome of this interaction is unclear. Here we report that amastigotes of L. amazonensis successfully survive within murine neutrophils despite triggering neutrophil activation and apoptosis. L. amazonensis is particularly adept at modifying the mammalian immune response to establish chronic persistence. This is best exemplified by the uncommon clinical manifestation of infection known as diffuse cutaneous leishmaniasis (DCL). DCL patients exhibit selective anergy against Leishmania antigens and cannot control parasite replication and dissemination throughout the skin, resulting in the appearance of multiple, disfiguring lesions that are often refractory to treatment (31). L. amazonensis infection in mice is similarly characterized by progressively growing lesions, poor T helper cell responses, and unchecked parasite growth, even in mouse strains (e.g., C57BL/6 and C3H) that are genetically resistant to Leishmania major (48). Unlike many other species, L. amazonensis amastigotes thrive under axenic conditions, permitting extensive opportunities to study this stage of parasite in vitro. These characteristics make L. amazonensis an excellent tool to study pathogenesis and immunoevasion in the context of chronic infection (31).
We have previously demonstrated a pathogenic role for antibodies and B cells during L. amazonensis infection (49). It has also been reported that macrophage uptake of antibody-coated L. mexicana amastigotes can result in the robust secretion of IL-10, contributing to parasite immunoevasion (50). Similarly, mice lacking IgG (due to a deletion of the Ig heavy chain) are more resistant to L. major infection, and passive transfer of parasite-specific antibodies to IgG-deficient animals resulted in increased IL-10 production, larger lesions, and increased parasite burden (51). Here, we demonstrate that coating of L. amazonensis amastigotes in serum from infected mice greatly enhanced parasite uptake by neutrophils (Fig. 1A), but serum coating had only marginal effects on neutrophil-mediated killing of axenic amastigotes in vitro (Fig. 6C). These results provide further evidence to support the notion that parasite-specific antibodies are not a major protective component of the immune response during Leishmania amastigote infection.
Phagocytosis of certain pathogens and subsequent respiratory burst can result in a Mac-1-dependent acceleration in neutrophil apoptosis through a process known as phagocytosis-induced cell death (PICD) (52). In contrast, many pathogens such as Francisella tularensis, Mycobacterium tuberculosis, and Chlamydia pneumoniae can prolong the neutrophil life span as a part of their immunoevasion strategy (53–55). In this study, we observed that L. amazonensis amastigotes do not utilize an antiapoptotic strategy when infecting neutrophils (Fig. 5A). The mechanism that L. amazonensis parasites employ to accelerate neutrophil apoptosis remains undetermined. We are currently investigating whether parasites trigger neutrophil apoptosis through a PICD-like mechanism by utilizing anti-CD11b and anti-CD18 antibodies and mice deficient in the phagocyte respiratory burst (such as gp47−/− mice).
The ability of neutrophils to modify the functions of other immune cell types is an essential area for future investigation, particularly in the context of Leishmania infection. Importantly, neutrophils can display antigen-presenting functions and prime T cells (56, 57). L. amazonensis-infected DCs are particularly poor at priming and activating T cells and fail to trigger a strong adaptive immune response (5). However, parasite infection does ultimately result in the generation of antigen-specific T cells and B cells, possibly indicating that antigen presentation may proceed through alternative or atypical mechanisms. The ability of neutrophils to present parasite antigens and prime T cells directly is largely unexplored at this time.
There is ample evidence that neutrophils can modulate DC function during leishmaniasis. For example, early DC recruitment during L. major infection was dependent upon CCL3 secretion by neutrophils, and CCL3 blockade delayed the development of a protective adaptive response (58). Additionally, L. major-loaded neutrophils isolated from infected mice were efficiently internalized by dermal DCs, and parasites delivered through this mechanism were less efficient in activating DCs and priming T cells than free parasites (6).
Neutrophils can also aid or hinder macrophages in clearing Leishmania infection. Murine macrophages infected with L. amazonensis displayed enhanced microbicidal activity when cocultured with neutrophils (59). In contrast, delivery of L. major to macrophages via apoptotic human neutrophils can result in anti-inflammatory cytokine production, favoring parasite growth (8). These findings suggest that neutrophils can positively or negatively affect the function of other immune cell types and that the outcome of this interaction may vary greatly depending upon neutrophil activation and life status.
It is interesting to note some discrepancies between our findings and those from several previous reports. Specifically, the newly described method for culturing axenic L. major amastigotes yields parasites that are not readily internalized by human neutrophils (60). Additionally, it has been shown that L. donovani amastigotes derived from infected hamsters are degraded by human neutrophils (24). These disparities suggest that differences in the host and parasite species, as well the methods used to isolate parasites, may contribute to dramatic differences in the interaction between Leishmania amastigotes and host neutrophils.
While this study is an important first step in improving our understanding of neutrophil-amastigote interactions, it must be emphasized that there are some noteworthy limitations to the work presented here. Specifically, the in vitro nature of our experiments makes it difficult to fully understand the role of neutrophils at the site of parasite infection. It is well established that neutrophils are highly sensitive and responsive to the inflammatory environment around them (42), and this environment is obviously difficult to simulate in vitro. Therefore, we are currently investigating alterations in amastigote infection and disease pathogenesis in neutropenic mice.
It is important to consider that the skin is a unique organ with specialized cells and architecture, and it stands to reason that cells responding to an infection in the skin may behave differently than those responding to infection elsewhere. For this study, we used neutrophils derived from the peritoneal cavities of thioglycolate-stimulated mice because this technique allows for the isolation of sufficient neutrophils to conduct detailed functional analyses. At this time, it is difficult to determine whether the antiparasitic response of peritoneal neutrophils closely resembles the behavior of neutrophils responding to parasites in the skin in vivo.
It was surprising to observe that lesion-derived amastigotes triggered more neutrophil activation and were more susceptible to neutrophil-mediated killing than their axenically cultured counterparts (Fig. 6C). We suspect that these differences are largely due to host components that remain associated with lesion-derived amastigotes (anti-Leishmania antibodies, complement components, etc.). However, it is currently unclear which host components are responsible for the improvement in lesion-derived amastigote clearance that we observed. Additionally, the mechanical process of isolating amastigotes from the footpads of infected mice is relatively vigorous, and lesion-derived parasites may require additional time to fully recover and prime their antineutrophil defenses.
This study, for the first time, examines in detail how murine neutrophils respond to Leishmania amastigotes and promastigotes. Here, we provide evidence that both promastigotes and amastigotes are efficiently internalized by mouse neutrophils and similarly trigger neutrophil activation and an oxidative burst. However, we observed that amastigotes are highly resistant to neutrophil microbicidal mechanisms and induce anti-inflammatory IL-10 release, while promastigotes trigger more TNF-α secretion and are more susceptible to killing by neutrophils. Collectively, this study supports and expands upon our previous understanding of the role of neutrophils during leishmaniasis (10, 22) and highlights the possible cross talk between neutrophils and other immune cells involved in parasite recognition and clearance.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by NIH grant R56AI043003 to L. Soong. E. Carlsen was supported by NIH predoctoral training grant T32AI007526 (principal investigator, A. Barrett) and the UTMB McLaughlin Endowment Pre-Doctoral Fellowship. C. Henard was supported by NIH postdoctoral training grant T32AI00753613 (principal investigator, C. White, Jr.).
We greatly appreciate the gift of monoclonal antibody 3A1 from Norma Andrews (University of Maryland). We also thank E. Carlsen's Dissertation Supervisory Committee members (Peter Melby, Janice Endsley, Gustavo Valbuena, Judy Aronson, and Stephanie Watowich) for helpful discussions and Mardelle Susman for assisting in manuscript preparation.
Footnotes
Published ahead of print 5 August 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.00770-13.
REFERENCES
- 1.Wanasen N, Soong L. 2008. l-Arginine metabolism and its impact on host immunity against Leishmania infection. Immunol. Res. 41:15–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sacks D, Noben-Trauth N. 2002. The immunology of susceptibility and resistance to Leishmania major in mice. Nat. Rev. Immunol. 2:845–858 [DOI] [PubMed] [Google Scholar]
- 3.Osorio y Fortea J, Prina E, de La Llave E, Lecoeur H, Lang T, Milon G. 2007. Unveiling pathways used by Leishmania amazonensis amastigotes to subvert macrophage function. Immunol. Rev. 219:66–74 [DOI] [PubMed] [Google Scholar]
- 4.Mougneau E, Bihl F, Glaichenhaus N. 2011. Cell biology and immunology of Leishmania. Immunol. Rev. 240:286–296 [DOI] [PubMed] [Google Scholar]
- 5.Soong L. 2008. Modulation of dendritic cell function by Leishmania parasites. J. Immunol. 180:4355–4360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ribeiro-Gomes FL, Peters NC, Debrabant A, Sacks DL. 2012. Efficient capture of infected neutrophils by dendritic cells in the skin inhibits the early anti-Leishmania response. PLoS Pathog. 8:e1002536. 10.1371/journal.ppat.1002536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Peters NC, Egen JG, Secundino N, Debrabant A, Kimblin N, Kamhawi S, Lawyer P, Fay MP, Germain RN, Sacks D. 2008. In vivo imaging reveals an essential role for neutrophils in leishmaniasis transmitted by sand flies. Science 321:970–974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van Zandbergen G, Klinger M, Mueller A, Dannenberg S, Gebert A, Solbach W, Laskay T. 2004. Neutrophil granulocyte serves as a vector for Leishmania entry into macrophages. J. Immunol. 173:6521–6525 [DOI] [PubMed] [Google Scholar]
- 9.Guimaraes-Costa AB, Nascimento MT, Froment GS, Soares RP, Morgado FN, Conceicao-Silva F, Saraiva EM. 2009. Leishmania amazonensis promastigotes induce and are killed by neutrophil extracellular traps. Proc. Natl. Acad. Sci. U. S. A. 106:6748–6753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xin L, Vargas-Inchaustegui DA, Raimer SS, Kelly BC, Hu J, Zhu L, Sun J, Soong L. 2010. Type I IFN receptor regulates neutrophil functions and innate immunity to Leishmania parasites. J. Immunol. 184:7047–7056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shi C, Hohl TM, Leiner I, Equinda MJ, Fan X, Pamer EG. 2011. Ly6G+ neutrophils are dispensable for defense against systemic Listeria monocytogenes infection. J. Immunol. 187:5293–5298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Archer NK, Harro JM, Shirtliff ME. 2013. Clearance of Staphylococcus aureus nasal carriage is T cell dependent and mediated through interleukin-17A expression and neutrophil influx. Infect. Immun. 81:2070–2075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ribes S, Regen T, Meister T, Tauber SC, Schutze S, Mildner A, Mack M, Hanisch UK, Nau R. 2013. Resistance of the brain to Escherichia coli K1 infection depends on MyD88 signaling and the contribution of neutrophils and monocytes. Infect. Immun. 81:1810–1819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen L, Zhang ZH, Watanabe T, Yamashita T, Kobayakawa T, Kaneko A, Fujiwara H, Sendo F. 2005. The involvement of neutrophils in the resistance to Leishmania major infection in susceptible but not in resistant mice. Parasitol. Int. 54:109–118 [DOI] [PubMed] [Google Scholar]
- 15.Tacchini-Cottier F, Zweifel C, Belkaid Y, Mukankundiye C, Vasei M, Launois P, Milon G, Louis JA. 2000. An immunomodulatory function for neutrophils during the induction of a CD4+ Th2 response in BALB/c mice infected with Leishmania major. J. Immunol. 165:2628–2636 [DOI] [PubMed] [Google Scholar]
- 16.Dabiri S, Hayes MM, Meymandi SS, Basiri M, Soleimani F, Mousavi MR. 1998. Cytologic features of “dry-type” cutaneous leishmaniasis. Diagn. Cytopathol. 19:182–185 [DOI] [PubMed] [Google Scholar]
- 17.Daboul MW. 2010. Role of neutrophils in cutaneous leishmaniasis. East Mediterr. Health J. 16:1055–1058 [PubMed] [Google Scholar]
- 18.Lopez Kostka S, Dinges S, Griewank K, Iwakura Y, Udey MC, von Stebut E. 2009. IL-17 promotes progression of cutaneous leishmaniasis in susceptible mice. J. Immunol. 182:3039–3046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Souza-Lemos C, de-Campos SN, Teva A, Corte-Real S, Fonseca EC, Porrozzi R, Grimaldi G., Jr 2008. Dynamics of immune granuloma formation in a Leishmania braziliensis-induced self-limiting cutaneous infection in the primate Macaca mulatta. J. Pathol. 216:375–386 [DOI] [PubMed] [Google Scholar]
- 20.Tenorio MD, Oliveira e Sousa L, Paixao MD, Alves MF, Paulan SD, Lima FL, Jusi MM, Tasca KI, Machado RZ, Starke-Buzetti WA. 2011. Visceral leishmaniasis in a captive crab-eating fox Cerdocyon thous. J. Zoo Wildl. Med. 42:608–616 [DOI] [PubMed] [Google Scholar]
- 21.Vercosa BL, Melo MN, Puerto HL, Mendonca IL, Vasconcelos AC. 2012. Apoptosis, inflammatory response and parasite load in skin of Leishmania (Leishmania) chagasi naturally infected dogs: a histomorphometric analysis. Vet. Parasitol. 189:162–170 [DOI] [PubMed] [Google Scholar]
- 22.Wang Y, Chen Y, Xin L, Beverley SM, Carlsen ED, Popov V, Chang KP, Wang M, Soong L. 2011. Differential microbicidal effects of human histone proteins H2A and H2B on Leishmania promastigotes and amastigotes. Infect. Immun. 79:1124–1133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lu T, Kobayashi SD, Quinn MT, Deleo FR. 2012. A NET outcome. Front. Immunol. 3:365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chang KP. 1981. Leishmanicidal mechanisms of human polymorphonuclear phagocytes. Am. J. Trop. Med. Hyg. 30:322–333 [DOI] [PubMed] [Google Scholar]
- 25.Courret N, Prina E, Mougneau E, Saraiva EM, Sacks DL, Glaichenhaus N, Antoine JC. 1999. Presentation of the Leishmania antigen LACK by infected macrophages is dependent upon the virulence of the phagocytosed parasites. Eur. J. Immunol. 29:762–773 [DOI] [PubMed] [Google Scholar]
- 26.Roy G, Dumas C, Sereno D, Wu Y, Singh AK, Tremblay MJ, Ouellette M, Olivier M, Papadopoulou B. 2000. Episomal and stable expression of the luciferase reporter gene for quantifying Leishmania spp. infections in macrophages and in animal models. Mol. Biochem. Parasitol. 110:195–206 [DOI] [PubMed] [Google Scholar]
- 27.Vargas-Inchaustegui DA, Xin L, Soong L. 2008. Leishmania braziliensis infection induces dendritic cell activation, ISG15 transcription, and the generation of protective immune responses. J. Immunol. 180:7537–7545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Perez H, Arredondo B, Gonzalez M. 1978. Comparative study of American cutaneous leishmaniasis and diffuse cutaneous leishmaniasis in two strains of inbred mice. Infect. Immun. 22:301–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Charmoy M, Megnekou R, Allenbach C, Zweifel C, Perez C, Monnat K, Breton M, Ronet C, Launois P, Tacchini-Cottier F. 2007. Leishmania major induces distinct neutrophil phenotypes in mice that are resistant or susceptible to infection. J. Leukoc. Biol. 82:288–299 [DOI] [PubMed] [Google Scholar]
- 30.Baron EJ, Proctor RA. 1982. Elicitation of peritoneal polymorphonuclear neutrophils from mice. J. Immunol. Methods 49:305–313 [DOI] [PubMed] [Google Scholar]
- 31.Soong L. 2012. Subversion and utilization of host innate defense by Leishmania amazonensis. Front. Immunol. 3:58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xin L, Li K, Soong L. 2008. Down-regulation of dendritic cell signaling pathways by Leishmania amazonensis amastigotes. Mol. Immunol. 45:3371–3382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lau D, Mollnau H, Eiserich JP, Freeman BA, Daiber A, Gehling UM, Brummer J, Rudolph V, Munzel T, Heitzer T, Meinertz T, Baldus S. 2005. Myeloperoxidase mediates neutrophil activation by association with CD11b/CD18 integrins. Proc. Natl. Acad. Sci. U. S. A. 102:431–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li Z. 1999. The alphaMbeta2 integrin and its role in neutrophil function. Cell Res. 9:171–178 [DOI] [PubMed] [Google Scholar]
- 35.Coxon A, Rieu P, Barkalow FJ, Askari S, Sharpe AH, von Andrian UH, Arnaout MA, Mayadas TN. 1996. A novel role for the beta 2 integrin CD11b/CD18 in neutrophil apoptosis: a homeostatic mechanism in inflammation. Immunity 5:653–666 [DOI] [PubMed] [Google Scholar]
- 36.Schymeinsky J, Mocsai A, Walzog B. 2007. Neutrophil activation via beta2 integrins (CD11/CD18): molecular mechanisms and clinical implications. Thromb Haemost. 98:262–273 [PubMed] [Google Scholar]
- 37.Horta MF, Mendes BP, Roma EH, Noronha FS, Macedo JP, Oliveira LS, Duarte MM, Vieira LQ. 2012. Reactive oxygen species and nitric oxide in cutaneous leishmaniasis. J. Parasitol. Res. 2012:203818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rothe G, Emmendörffer A, Oser A, Roesler J, Valet G. 1991. Flow cytometric measurement of the respiratory burst activity of phagocytes using dihydrorhodamine 123. J. Immunol. Methods 138:133–135 [DOI] [PubMed] [Google Scholar]
- 39.Katayama M, Ohmura K, Yukawa N, Terao C, Hashimoto M, Yoshifuji H, Kawabata D, Fujii T, Iwakura Y, Mimori T. 2013. Neutrophils are essential as a source of IL-17 in the effector phase of arthritis. PLoS One 8:e62231. 10.1371/journal.pone.0062231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tecchio C, Scapini P, Pizzolo G, Cassatella MA. 2013. On the cytokines produced by human neutrophils in tumors. Semin. Cancer Biol. 23:159–170 [DOI] [PubMed] [Google Scholar]
- 41.Scapini P, Lapinet-Vera JA, Gasperini S, Calzetti F, Bazzoni F, Cassatella MA. 2000. The neutrophil as a cellular source of chemokines. Immunol. Rev. 177:195–203 [DOI] [PubMed] [Google Scholar]
- 42.Luo HR, Loison F. 2008. Constitutive neutrophil apoptosis: mechanisms and regulation. Am. J. Hematol. 83:288–295 [DOI] [PubMed] [Google Scholar]
- 43.Colotta F, Re F, Polentarutti N, Sozzani S, Mantovani A. 1992. Modulation of granulocyte survival and programmed cell death by cytokines and bacterial products. Blood 80:2012–2020 [PubMed] [Google Scholar]
- 44.Klein JB, Rane MJ, Scherzer JA, Coxon PY, Kettritz R, Mathiesen JM, Buridi A, McLeish KR. 2000. Granulocyte-macrophage colony-stimulating factor delays neutrophil constitutive apoptosis through phosphoinositide 3-kinase and extracellular signal-regulated kinase pathways. J. Immunol. 164:4286–4291 [DOI] [PubMed] [Google Scholar]
- 45.Ribeiro-Gomes FL, Sacks D. 2012. The influence of early neutrophil-Leishmania interactions on the host immune response to infection. Front. Cell Infect. Microbiol. 2:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wanderley JL, Pinto da Silva LH, Deolindo P, Soong L, Borges VM, Prates DB, de Souza AP, Barral A, Balanco JM, do Nascimento MT, Saraiva EM, Barcinski MA. 2009. Cooperation between apoptotic and viable metacyclics enhances the pathogenesis of Leishmaniasis. PLoS One 4:e5733. 10.1371/journal.pone.0005733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Garcia-Romo GS, Caielli S, Vega B, Connolly J, Allantaz F, Xu Z, Punaro M, Baisch J, Guiducci C, Coffman RL, Barrat FJ, Banchereau J, Pascual V. 2011. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci. Transl. Med. 3:73ra20. 10.1126/scitranslmed.3001201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ji J, Masterson J, Sun J, Soong L. 2005. CD4+CD25+ regulatory T cells restrain pathogenic responses during Leishmania amazonensis infection. J. Immunol. 174:7147–7153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wanasen N, Xin L, Soong L. 2008. Pathogenic role of B cells and antibodies in murine Leishmania amazonensis infection. Int. J. Parasitol. 38:417–429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Thomas BN, Buxbaum LU. 2008. FcγRIII mediates immunoglobulin G-induced interleukin-10 and is required for chronic Leishmania mexicana lesions. Infect. Immun. 76:623–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Miles SA, Conrad SM, Alves RG, Jeronimo SM, Mosser DM. 2005. A role for IgG immune complexes during infection with the intracellular pathogen Leishmania. J. Exp. Med. 201:747–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang B, Hirahashi J, Cullere X, Mayadas TN. 2003. Elucidation of molecular events leading to neutrophil apoptosis following phagocytosis: cross-talk between caspase 8, reactive oxygen species, and MAPK/ERK activation. J. Biol. Chem. 278:28443–28454 [DOI] [PubMed] [Google Scholar]
- 53.Schwartz JT, Barker JH, Kaufman J, Fayram DC, McCracken JM, Allen LA. 2012. Francisella tularensis inhibits the intrinsic and extrinsic pathways to delay constitutive apoptosis and prolong human neutrophil lifespan. J. Immunol. 188:3351–3363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Blomgran R, Desvignes L, Briken V, Ernst JD. 2012. Mycobacterium tuberculosis inhibits neutrophil apoptosis, leading to delayed activation of naive CD4 T cells. Cell Host Microbe 11:81–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.van Zandbergen G, Gieffers J, Kothe H, Rupp J, Bollinger A, Aga E, Klinger M, Brade H, Dalhoff K, Maass M, Solbach W, Laskay T. 2004. Chlamydia pneumoniae multiply in neutrophil granulocytes and delay their spontaneous apoptosis. J. Immunol. 172:1768–1776 [DOI] [PubMed] [Google Scholar]
- 56.Ashtekar AR, Saha B. 2003. Poly's plea: membership to the club of APCs. Trends Immunol. 24:485–490 [DOI] [PubMed] [Google Scholar]
- 57.Tillack K, Breiden P, Martin R, Sospedra M. 2012. T lymphocyte priming by neutrophil extracellular traps links innate and adaptive immune responses. J. Immunol. 188:3150–3159 [DOI] [PubMed] [Google Scholar]
- 58.Charmoy M, Brunner-Agten S, Aebischer D, Auderset F, Launois P, Milon G, Proudfoot AE, Tacchini-Cottier F. 2010. Neutrophil-derived CCL3 is essential for the rapid recruitment of dendritic cells to the site of Leishmania major inoculation in resistant mice. PLoS Pathog. 6:e1000755. 10.1371/journal.ppat.1000755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.de Souza Carmo EV, Katz S, Barbieri CL. 2010. Neutrophils reduce the parasite burden in Leishmania (Leishmania) amazonensis-infected macrophages. PLoS One 5:e13815. 10.1371/journal.pone.0013815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wenzel UA, Bank E, Florian C, Forster S, Zimara N, Steinacker J, Klinger M, Reiling N, Ritter U, van Zandbergen G. 2012. Leishmania major parasite stage-dependent host cell invasion and immune evasion. FASEB J. 26:29–39 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.