

Abstract
To protect against invading bacteria, oral epithelial cells appear to use two effector antimicrobial peptides (AMPs): calprotectin (S100A8-S100A9 heterodimer [S100A8/A9]) in the cytosol and cathelicidin antimicrobial protein (CAMP) in endosomes. We sought to learn whether innate immunity might be augmented benignly to increase resistance against invasive bacteria. Epithelial cells were transiently transfected with mRNA constructs containing either the CAMP, S100A8, and S100A9 open reading frames, A8-IRES-A9 (fusion sequence), or A8-nIRES-A9 (fusion with native internal ribosome entry site [IRES] sequence). CAMP, S100A8, and S100A9 protein levels generally peaked between 16 and 44 h after mRNA transfection, depending on the construct; CAMP was processed to LL-37 over time. Following transfection with the respective mRNAs, CAMP and S100A8/A9 each independently increased resistance of epithelial cells to invasion by Listeria and Salmonella for up to 48 h; tandem S100A8/A9 constructs were also effective. Cotransfection to express S100A8/A9 and CAMP together augmented resistance, but synergy was not seen. Independent of the new proteins produced, transfection reduced cell viability after 48 h by 20%, with only 2% attributable to apoptosis. Taken together, these results suggest that epithelial cell resistance to invasive pathogens can be augmented by transient transfection of antimicrobial mRNAs into epithelial cells.
INTRODUCTION
Mucosal epithelia provide the first line of defense against the invasion of microbes. Indeed, epithelial cells offer both a physical barrier and molecular-based antimicrobial resistance in the absence of any assistance from the mucosal immune system. Using a cell-autonomous mechanism to confer resistance to invading bacteria, epithelial innate immunity is provided by endogenous expression of effector antimicrobial peptides (AMPs), including cathelicidin antimicrobial protein (CAMP) and its active proteolytic cleavage product (LL-37), calprotectin (S100A8 complexed to S100A9 [S100A8/A9]), β-defensins, S100A7, secretory leukocyte peptidase inhibitor (SLPI), lipocalin 2 (LCN2), and lysozyme (1, 2). Most antimicrobial peptides/proteins function primarily outside eukaryotic cells, often with proinflammatory and other off-target effects. Upon secretion or release from eukaryotic cells, the antimicrobial peptides can interact and antagonize microorganisms, usually by membrane intercalation and internalization to localize with subcellular microbial targets. Of these antimicrobial peptides/proteins, only CAMP (LL-37) (3) and, as we have shown, S100A8/A9 (4–6) are known to function within epithelial cells to inhibit bacterial invasion.
Human CAMP is a member of a large family of cationic antimicrobial peptides, expressed in many species, that have broad-spectrum activity against bacteria, fungi, and enveloped viruses and also show immunomodulatory effects (7). Following excision of the signal peptide, human cathelicidin precursor protein (hCAP18), encoded by CAMP, is commonly stored in neutrophil granules. Activation of hCAP18 is controlled by the serine proteases stratum corneum tryptic enzyme (SCTE; kallikrein 5) and stratum corneum chymotryptic protease (SCCE; kallikrein 7) (8). After processing, the 37-residue active peptide (LL-37) mediates a wide range of biological responses, such as direct killing of microorganisms, chemotaxis and chemokine induction, regulation of inflammatory responses, and adjuvant, angiogenic, and wound-healing effects (9). LL-37 appears to be directly antimicrobial in the phagolysosomes of neutrophils and macrophages and at sites of acute inflammation (9), showing broad-spectrum antimicrobial activity against Listeria monocytogenes (10), Salmonella enterica serovar Typhimurium (11), and Streptococcus groups A, B, and C (12).
CAMP expression can be induced by 1,25-dihydroxyvitamin D3 (13), pathogens (9), or lipopolysaccharide (LPS) (14). Decreased CAMP/LL-37 production accompanies increased invasion and colonization by pathogens in epithelial cells, which characterizes diseases such as morbus Kostmann (15) and atopic dermatitis (16).
Calprotectin is a heterodimeric complex of calcium-binding proteins S100A8 (MRP8 or calgranulin A; 10.8 kDa) and S100A9 (MRP14 or calgranulin B; 13.2 kDa) (6). S100A8 and S100A9 are members of the S100 family of proteins (17). S100 family members are characterized by their two EF-hand calcium-binding motifs; these proteins are involved in cell growth, cell differentiation, cell cycle progression, cell survival, protein phosphorylation, transcription, cancer development, and inflammatory diseases (18). Calprotectin shows broad-spectrum antimicrobial activity against Candida albicans and bacteria, including Capnocytophaga sputigena, Escherichia coli, Staphylococcus epidermidis, Listeria monocytogenes, S. Typhimurium, and Porphyromonas gingivalis (4, 6, 19–21). After stable transfection to express the calprotectin complex, an epithelial cell line (KB) showed increased resistance to invasion by Listeria monocytogenes and Salmonella enterica serovar Typhimurium (6).
The structural basis for the contribution of S100A8/A9 to resistance to invasion resides, at least in part, in the integrity of the S100A9 calcium-binding EF hands (6). When expressed in the cell in complex with S100A8, S100A9 E36Q and E78Q mutants showed increased bacterial invasion and were predicted to cause loss of the calcium-induced positive face in the S100A8/A9 complex (6). Some intracellular pathogens appear to have strategies to avoid antibacterial calprotectin. To facilitate intraepithelial survival in the cytoplasm, Listeria mobilizes calprotectin to colocalize with cytoplasmic microtubules, appearing to subvert anti-Listeria activity and autonomous cellular immunity (5).
Mucosal epithelial cells can therefore protect against and suppress invasive pathogens mainly by using two intracellular antimicrobial effector systems: CAMP/LL-37, largely in endosomes (3, 22), and calprotectin (S100A8/S100A9), in the cytosol (6). We hypothesized that S100A8/A9 and CAMP can function to augment innate intraepithelial resistance to invading bacterial pathogens. To test this hypothesis, we developed a novel approach to cellular production of neoproteins, using transient delivery of specific antimicrobial effector mRNAs (e.g., CAMP and S100A8/S100A9). mRNA transfections for therapeutic purposes are of considerable current interest to replace or augment expression of proteins through systemic or ex vivo administration (23). When specifically targeted to the native source of the protein of interest, mRNA transfections have the potential to replace, restore, or augment innate immune effector proteins and the mucosal immune barrier to infection. We now report the delivery of CAMP and S100A8/S100A9 mRNAs into human keratinocytes and the functional changes in resistance to bacterial invasion.
MATERIALS AND METHODS
Cell culture.
Human KB cells (ATCC CCL-17; American Type Culture Collection) were cultured in modified Eagle's medium (MEM; Mediatech Inc., Herndon, VA) supplemented with 10% fetal bovine serum (FBS) at 37°C in a 5% CO2 incubator.
Bacteria.
L. monocytogenes ATCC 10403S (provided by Daniel Portnoy, University of California, Berkeley, CA) and S. Typhimurium ATCC 14028 (provided by Carol Wells, University of Minnesota) were grown in brain heart infusion medium (Difco) and on tryptic soy agar (Difco) at 37°C. Listeria and Salmonella cells were harvested from log phase and stationary phase, respectively, at an absorbance at 620 nm of 0.4 to 0.6, and used to infect KB cells.
Plasmid construction.
The open reading frame (ORF) for EGFP (see Fig. S2 in the supplemental material) and ORFs containing Kozak sequences for human CAMP, S100A8, and S100A9 (all primer pairs are listed in Table S1) were amplified by PCR. The PCR products were cloned into pGEM4Z.2bgUTR.150A (24) (provided by C. H. June and Y. Zhao, University of Pennsylvania) via NotI and HindIII sites. To create a single mRNA for transcription, the S100A8 ORF containing a Kozak sequence was cloned into the first multiple-cloning site (MCS) of the pIRES vector (Clontech, Mountain View, CA) via NheI and XhoI sites; the S100A9 ORF was cloned into the second MCS of the pIRES vector via XbaI and NotI sites. Amplified A8-IRES-A9 fragments were also cloned into pGEM4Z.2bgUTR.150A to generate the plasmid pGEM4Z.A8-IRES-A9.2bgUTR.150A.
The internal ribosome entry site (IRES) in the pIRES vector is a partially disabled sequence (25, 26). To construct the native IRES (nIRES), pGEM4Z.A8-IRES-A9.2bgUTR.150A was used as a template and amplified with primers 5′ AAA CGT CTA GGC CCC CCG AAC C 3′ and 5′ TTT AAC CTC GAC TAA ACA CAT GTA AAG CAT GTG C 3′. The product was purified and self-ligated to generate plasmid pGEM4Z.A8-IRES/mut1-A9.2bgUTR.150A. Primers 5′ ACC ATG ACT TGC AAA ATG TCG CAG CTG 3′ and 5′ ATT ATC ATC GTG TTT TTC AAA GGA AAA CCA C 3′ were used to amplify a fragment from pGEM4Z.S100A8-IRES/mut1-S100A9.2bgUTR as the template. The fragments were purified and self-ligated to generate plasmid pGEM4Z.A8-nIRES-A9.2bgUTR.150A.
In vitro transcription.
Templates were amplified from corresponding plasmid constructs and extracted from gels. Purified fragments were treated with 0.5% SDS and 100 μg/ml protease K at 50°C for 30 min and then extracted with phenol-chloroform-isoamyl alcohol (25:24:1) and then with chloroform. After ethanol precipitation, templates were dissolved in 10 mM Tris-HCl and stored at −80°C. Using an mMESSAGE mMACHINE T7 Ultra kit (Life Technologies, Grand Island, NY) or a ScriptCap m7G capping system/ScriptCap 2′-O-methyltransferase kit (Cell Script, Madison, WI), mRNAs were synthesized and then purified using a MEGAclear kit (Life Technologies) followed by ethanol precipitation.
mRNA transfections.
At approximately 60 to 90% confluence, cells were transfected with mRNAs by using the TransIT-mRNA kit reagent (Mirus Biol LLC, Madison, WI) according to the manufacturer's instructions.
qRT-PCR.
See the supplemental material for details on quantitative real-time PCR (qRT-PCR) analysis.
Western blot analysis.
Cells were extracted using mammalian cell-PE LBTM buffer (GBiosciences, Maryland Heights, MO). The extracted proteins were separated by SDS-PAGE, transferred onto nitrocellulose membranes, and incubated with one of the following: rabbit anti-β-actin (DB070; Delta Biolabs, Gilroy, CA), mouse anti-LL37 (sc-166770; Santa Cruz Biotechnology), mouse anti-S100A8 (sc-48352; Santa Cruz Biotechnology), rabbit anti-S100A9 (sc-20173; Santa Cruz Biotechnology), or rabbit anti-poly(ADP-ribose) polymerase (anti-PARP) (9542S; Cell Signaling). Rabbit primary antibodies were then incubated with horseradish peroxidase (HRP)-conjugated goat anti-rabbit antibodies, whereas mouse primary antibodies were incubated with HRP-conjugated goat anti-mouse antibodies (secondary antibodies) (Santa Cruz Biotechnology). Immunoreactions were visualized using SuperSignal West Pico chemiluminescence substrate (Thermo Scientific, Rockford, IL) and exposed to Amersham Hyperfilm ECL film (GE Healthcare Biosciences, Piscataway, NJ). Protein bands were quantified by Quantity One analysis (Bio-Rad, Hercules, CA).
Immunofluorescence.
See the supplemental material for details on immunofluorescence.
Bacterial invasion assay.
Bacterial invasion was determined by the antibiotic protection assay (6). Briefly, KB cells (1 × 105 to 1.2 × 105 cells) were seeded overnight in 24-well plates. Cells were then incubated with L. monocytogenes ATCC 10403S or S. Typhimurium ATCC 14028 at a multiplicity of infection (MOI) of 100:1 or 1:1, respectively. After 2 h of incubation, the monolayers were washed with Dulbecco's phosphate-buffered saline (DPBS) (Sigma) and incubated for 1.5 h in MEM supplemented with 10% FBS containing 100 μg/ml gentamicin (Sigma), and cells were lysed by incubation with sterile distilled water for 15 min. Released bacteria were diluted, plated with a spiral plater (Spiral Biotech, Bethesda, MD), and incubated overnight at 37°C, and then the number of CFU of intracellular bacteria was enumerated on a New Brunswick C-110 colony counter (New Brunswick, NJ).
In vitro toxicity assay.
The toxic effects of mRNA delivery in vitro were analyzed by quantitatively determining cell viability using a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay (in vitro toxicity assay kit; Sigma, St. Louis, MO). Briefly, MTT solution (5 mg/ml) was added to each well in an amount equal to 10% of the culture medium volume. Cells were then incubated for 2 h at 37°C, absorbance was measured at a wavelength of 570 nm, and the percentage of viable cells was calculated as the ratio of absorbances of transfected and untransfected cells.
Analysis of apoptosis.
To evaluate cell apoptotic status, transfected cells were evaluated for PARP cleavage by use of rabbit anti-PARP in Western blots as described above. Cells were also analyzed by flow cytometry using an annexin V-fluorescein isothiocyanate (FITC) apoptosis detection kit (MBL International Corp., Watertown, MA) according to the manufacturer's recommendations. For both analyses of apoptosis, cells were harvested by trypsinization at times up to 72 h following transfection and then washed with 2.5% FBS in PBS. Cells harvested for detection of annexin by flow cytometry were incubated with annexin V-FITC plus propidium iodide in binding buffer at room temperature for 5 min in the dark. A total of 50,000 events were counted using a Becton, Dickinson FACSCalibur flow cytometer (BD Biosciences, San Jose, CA), and the percentage of annexin V-FITC-positive cells (considered apoptotic [27]) was analyzed with CellQuest software, placing the FITC signal in FL1 and the propidium iodide signal in FL2.
Statistical analysis.
Statistical comparisons were done with Student's t test for comparisons between two groups or with one-way analysis of variance (ANOVA) followed by Bonferroni's post hoc test for multiple comparisons, using Excel software (Microsoft, Redmond, WA).
RESULTS
Optimizing mRNA stability.
Capping at the 5′ end of mRNA enhances the stability of the expressed message. An anti-reverse cap analogue (ARCA) can cap at an efficacy of up to 80%, whereas the capping enzyme (CE) can increase the capping efficiency to 100% (25). To optimize the capping strategy, we compared the mMESSAGE mMACHINE T7 Ultra system (Life Technologies), which generates ARCA-capped mRNA, with the mScript RNA system (Cell Script), which uses the 2′-O-methyltransferase CE to generate a cap 0 or cap 1, depending on the addition of a methyl group at the 2′-O position of the penultimate (5′ end) nucleotide of the transcript. CAMP mRNAs were synthesized using the different capping systems and delivered into KB cells, and protein expression was detected by Western blot analysis. Eight, 24, and 48 h after transfection with ARCA-capped mRNAs, KB cells expressed more CAMP protein than that after transfection with CE cap 1, which was higher than that after transfection with CE cap 0 (see Fig. S1 in the supplemental material). ARCA capping was then used for all experiments. To determine whether the lengths of 3′ poly(A) tails increased mRNA stability, we compared mRNAs with 64-A and 150-A 3′ extensions. The 150-A mRNAs showed a slightly longer half-life (not significant; data not shown). We used 150-A mRNA for all subsequent experiments. CAMP protein levels peaked at about 16 h and decreased by about 50% 40 to 48 h after ARCA-capped mRNA delivery (Fig. 1). CAMP was hydrolyzed to LL-37, and levels peaked from 24 to 48 h after mRNA transfection (Fig. 1). LL-37 appeared as the presence of CAMP was reduced.
Fig 1.
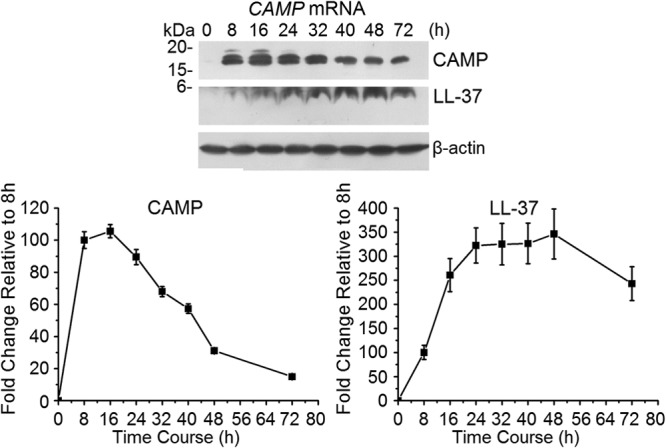
Relative CAMP protein and LL-37 expression over time after transfection with ARCA-capped CAMP. CAMP protein and LL-37 expression after ARCA-capped CAMP mRNA transfection was detected using Western blotting (upper panel) and is presented in the lower panel relative to the level at 8 h, which was set to 100. Intensity levels of each band are normalized to that of β-actin. The error bars show the means ± standard deviations (SD) for three to six independent experiments.
Stability of mRNAs capped with ARCA.
KB cells were used because they express CAMP, S100A8, and S100A9 mRNAs below the level of detection (data not shown). Therefore, endogenous S100A8, S100A9, and CAMP mRNAs expressed in KB cells contributed little to levels after transfection with ARCA-capped mRNAs. To synthesize a fusion mRNA containing S100A8 and S100A9, the S100A8 ORF containing a Kozak sequence was cloned into pIRES MCS A, and S100A9 was cloned into MCS B. To eliminate the attenuation of downstream translation with pIRES, native IRES (nIRES) was constructed and inserted between the S100A8 and S100A9 ORFs. Sequences containing all necessary elements were cloned into pGEM4Z.2bgUTR.150A, and then A8-IRES-A9 and A8-nIRES-A9 mRNAs were synthesized. Transfection of in vitro-transcribed ARCA-capped CAMP, S100A8, S100A9, A8-IRES-A9, and A8-nIRES-A9 mRNAs was analyzed using qRT-PCR (see Fig. S2 in the supplemental material). Four hours after each mRNA transfection, we first measured the intracellular mRNA levels. Sixteen to 20 h following transfection, the CAMP mRNA levels were approximately half that at 4 h, indicating that the half-life in KB cells was between 12 and 16 h (see Fig. S2A). In contrast, cotransfection of S100A8 and S100A9 mRNAs resulted in mRNA half-lives of between 12 and 16 h and 8 to 12 h, respectively (see Fig. S2B). When the tandem mRNAs were transfected, the half-life of A8-IRES-A9 was 12 to 16 h, whereas that of A8-nIRES-A9 mRNA was between 8 and 12 h (see Fig. S2C and D).
Cotransfection of S100A8 and S100A9 increases stability of each protein.
After delivery into KB cells, ARCA-capped mRNAs for S100A8 (Fig. 2A), S100A9 (Fig. 2B), and calprotectin (1:1 [mol/mol] S100A8 plus S100A9) (Fig. 2C) were compared for protein expression. Expression of S100A8 protein alone was maximal at 16 h and decreased by about half at about 28 h (Fig. 2A), whereas expression of S100A9 protein was maximized at 16 h and decreased to half at about 40 to 48 h (Fig. 2B). The stability of each peptide appeared to increase after S100A8 and S100A9 mRNAs (half the amount of each compared to transfections with either S100A8 or S100A9 mRNA) were cotransfected. Compared to S100A8 protein after transfection with S100A8 mRNA, for example (Fig. 2D), S100A8/A9 was more sustained over time after cotransfection of S100A8 and S100A9 mRNAs (Fig. 2C). When S100A8 mRNA was cotransfected with S100A9 mRNA, S100A8 protein expression was maximized at 24 h and decreased by half at about 72 h (Fig. 2C). Under the same conditions, S100A9 protein expression was also maximized at 24 h and decreased to half at about 48 to 72 h (Fig. 2D). Cotransfection of S100A8 and S100A9 mRNAs led to increased stability of each peptide, suggesting that the formation of spontaneous calprotectin heterodimers promotes resistance to degradation.
Fig 2.

S100A8 and S100A9 protein expression by KB cells after transfection with ARCA-capped S100A8, S100A9, A8-IRES-A9, or A8-nIRES-A9 mRNA. (A) S100A8 protein expression after KB cells were transfected with S100A8 mRNA, as detected using Western blot analysis. Identical experiments were performed to determine the time course of expression of S100A9 protein (B); S100A8 and S100A9 protein levels after KB cells were cotransfected with S100A8 and S100A9 mRNAs (C); S100A8 and S100A9 protein levels after cells were transfected with S100A8, S100A9, or S100A8/S100A9 mRNA (D); S100A8 and S100A9 protein levels after KB cells were transfected with A8-IRES-A9 mRNA (E); and S100A8 and S100A9 protein levels after KB cells were transfected with A8-nIRES-A9 mRNA (F). (G) S100A8 and S100A9 proteins were quantitated after cells were transfected with A8-IRES-A9 and A8-nIRES-A9 mRNAs, respectively. Intensity levels of each band are normalized to that of β-actin. Protein expression 8 h after mRNA transfection was set to 100. Representative images are shown. Error bars show the means ± SD for three to six independent experiments.
S100A8/S100A9 protein expression using tandem constructs.
After A8-IRES-A9 mRNA transfection, S100A8 protein expression was maximal at 16 h and decreased to half at 24 to 32 h (Fig. 2E), whereas S100A9 protein was maximal at 32 h and decreased to half at about 48 to 72 h (Fig. 2E). For A8-nIRES-A9 mRNA transfection, expression of S100A8 protein was maximal at 16 h and decreased to half at 24 to 32 h (Fig. 2F and G), whereas S100A9 protein expression was maximal at 40 h and decreased to half at about 40 to 48 h (Fig. 2F and G). As when S100A8 was coexpressed with S100A9 (Fig. 2C), use of the tandem mRNA constructs generally appeared to stabilize the protein subunits compared with just S100A8 mRNA transfection (Fig. 2A).
Intracellular presentation of CAMP and calprotectin.
Following mRNA transfections, CAMP and calprotectin (neoproteins) were also detected in KB cells by use of immunofluorescence (see Fig. S3 in the supplemental material). Endogenous or background CAMP, S100A8, and S100A9 protein levels were at or below the limits of detection, and fluorescence above background was not detected with mock transfections (see Fig. S3). Sixteen hours after delivery of mRNAs for EGFP, CAMP, or calprotectin (S100A8/S100A9), significant fluorescence was detected, suggesting that mRNA delivery increases enhanced green fluorescent protein (EGFP), CAMP, and calprotectin protein expression (see Fig. S3A to C). Sixteen hours after transfection with A8-IRES-A9 and A8-nIRES-A9 mRNAs, no significant fluorescence was detected (see Fig. S3D). Forty hours following mRNA transfection, however, fluorescence signals for calprotectin were detected (see Fig. S3E). These results are consistent with the results of Western blot analysis (Fig. 1 and 2E and F). The tandem constructs contained fewer molar equivalents of S100A8 and S100A9 mRNAs than those with S100A8 and S100A9 mRNA cotransfection, but they may be as effective.
CAMP or calprotectin mRNA transfection increases resistance of KB cells to bacterial invasion.
We determined whether select antimicrobial proteins produced after mRNA transfections into epithelial cells affected resistance to invasion by bacterial pathogens. We determined the numbers of intracellular CFU of Listeria and Salmonella that invaded untreated KB cells (vehicle control) and cells treated by delivery of S100A8, S100A9, or CAMP mRNA (Fig. 3A). We then expressed the data as percentages of vehicle control levels at 8 (Fig. 3B), 24 (Fig. 3C), and 48 (Fig. 3D) h posttransfection to determine the effect of each mRNA treatment over time. When intracellular antimicrobial proteins/peptides were present, invading bacteria were significantly reduced at each time point. Following transfection with all CAMP and calprotectin mRNA constructs, epithelial cell resistance to invasion significantly increased for Listeria and Salmonella at 8 h (Fig. 3A and B), 24 h (Fig. 3C), and 48 h (Fig. 3D). Resistance seemed to be greatest 8 h after transfection. The CAMP and S100A8-plus-S100A9 mRNA transfection schemes appeared to increase resistance more effectively than the tandem mRNA constructs did.
Fig 3.

CAMP and calprotectin mRNAs increase resistance to invasion by Listeria and Salmonella. Monolayers were transfected with mRNAs for 8, 24, or 48 h and then incubated for 2 h with L. monocytogenes ATCC 10403S at an MOI of 100:1 or S. Typhimurium ATCC 14028 at an MOI of 1:1. Cells were split 8 and 32 h after transfection, and then cells were incubated for another 16 h. Intracellular bacteria were recovered as described in Materials and Methods, and an antibiotic protection assay was used to determine the numbers of viable intracellular CFU. (A) Raw numbers of CFU per well after correcting for dilutions during the experimental protocol are shown as means ± SD for three to six replicates at 8 h. Numbers of CFU were normalized to the number of CFU in KB cells mock transfected with vehicle (set to 100) and are reported as means ± SD for three to six replicates at 8 (B), 24 (C), or 48 (D) h. Statistical analysis was performed using one-way ANOVA with Bonferroni's test. *, significantly decreased compared to vehicle control (P < 0.05).
Effects of cotransfection with calprotectin (S100A8/S100A9) and CAMP mRNAs.
To determine whether S100A8/A9 and CAMP (LL-37) function cooperatively and additively, S100A8 and S100A9 mRNAs (1:1 molar ratio) were cotransfected at different molar ratios with CAMP mRNA. Compared to the vehicle control, CAMP and S100A8/A9 in all combinations caused a reduction in viable intracellular Listeria (Fig. 4A) and Salmonella (Fig. 4B). When CAMP and S100A8/S100A9 mRNAs were transfected at a 1:1 molar ratio (S100A8/S100A9 at 0.5:0.5 relative to CAMP), resistance to invasion by Listeria (Fig. 4A) and Salmonella (Fig. 4B) was significantly increased compared to that with S100A8/A9 or CAMP alone or at great excess (5:1 or 1:5 molar ratio).
Fig 4.
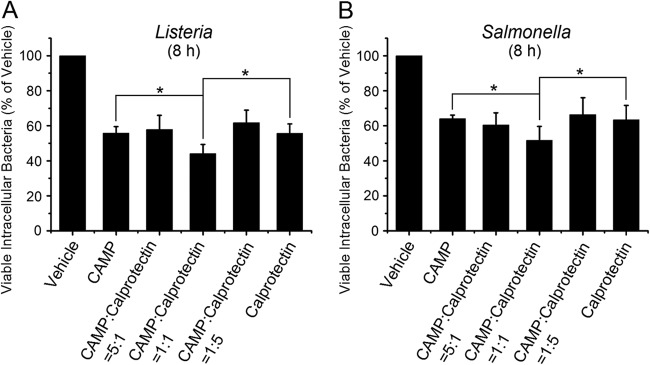
Cotransfection of CAMP and calprotectin mRNAs increases resistance to invasion by Listeria and Salmonella. S100A8 and S100A9 mRNAs were transfected into cell monolayers at various molar ratios for 8 h to optimize production of S100A8/A9. Monolayers were then incubated for 2 h with L. monocytogenes ATCC 10403S at an MOI of 100:1 (A) or S. Typhimurium ATCC 14028 at an MOI of 1:1 (B). Viable intracellular bacteria were enumerated using the antibiotic protection assay, and data are reported as mean numbers of CFU ± SD relative to the number of CFU in KB cells mock transfected with vehicle, which was set to 100. Results represent three to six independent experiments. Statistical analysis was performed using Student's t test. *, P < 0.05.
The TransIT-mRNA delivery system reduces cell viability without triggering apoptosis.
To determine the cytolethality of the TransIT-mRNA transfection reagent with and without mRNA cargoes, changes in cell viability were measured using an MTT assay and by quantifying apoptotic cells using flow cytometry and PARP cleavage. Compared with untransfected cells, transfection with vehicle alone caused reductions in cell viability over time similar to those with vehicle packaged with CAMP, S100A8/S100A9, A8-IRES-A9, or A8-nIRES-A9 mRNA (Fig. 5A). Cell viability was significantly reduced 48 and 72 h after transfection (Fig. 5A). Approximately 2% of the cells showing loss of viability had undergone apoptosis. Apoptosis, however, was unaffected by the vehicle, with or without cargoes, compared to that of untransfected cells (Fig. 5B). Even at 72 h, the percentage of apoptotic cells after each of the transfections was indistinguishable from that for untransfected cells. The apoptotic status of the KB cells was confirmed using Western blot analysis for PARP cleavage, an indicator of caspase activation (28). PARP cleavage was elevated 32 h after mRNA transfection (Fig. 5C). Compared to the untransfected cells, the pattern of cleaved PARP was similar after delivery of the mRNAs (Fig. 5C).
Fig 5.

mRNA delivery reduces cell viability without triggering apoptosis. (A) Cell viability was determined using the MTT assay at the indicated times for 3 days after mRNA delivery of mRNA constructs to KB cells. Mock (PBS)-transfected cells were used as a control, with their viability set at 100%. Each experiment was done in triplicate and repeated at least twice. Statistical analysis was performed using Student's t test. *, significantly decreased compared to untransfected cells (P < 0.05). (B) Analysis of apoptotic cells by flow cytometry after delivery of mRNA constructs. Cells that were positively stained by annexin V-FITC by flow cytometry were considered apoptotic. In panels A and B, error bars show the means ± SD for three to six independent experiments. (C) Western blot analysis of PARP cleavage after delivery of mRNA constructs. Mock (PBS)-transfected (vehicle) cells and untransfected cells were used as controls.
DISCUSSION
In health, mucosal epithelial cells contain putative commensals and endogenous pathogens (29). Since the interiors of mucosal epithelial cells are not sterile, mechanisms must exist to control the growth of these bacteria. The best-studied innate immune effector molecules, antimicrobial peptides (AMPs), include the defensins, CAMP (LL-37), and S100A8/A9. S100A8/A9 is expressed in the cytoplasm, and LL-37 in the endosomes, in association with Toll-like receptor 9 (TLR9) (6). As we have reported (5, 6, 30), S100A8/A9 promotes functional innate immunity in the cytoplasm of epithelial cells by increasing resistance against invading bacteria, including Listeria and Salmonella. Functioning in the cytoplasm, S100A8/A9 has no signal sequence for export and generally appears extracellularly when cells lyse. CAMP contains a signal peptide, but the active LL-37 fragment does not. Hence, we focused on whether S100A8/A9 and LL-37 function cooperatively within epithelial cells to attenuate the effects of invasive bacteria (3, 6).
The fact that these innate immune effector molecules function intracellularly distinguishes S100A8/A9 and LL-37 from the defensins and other AMPs that are packaged in granules and contain signals for export and secretion. We show for the first time that epithelial cell resistance to invasive pathogens in vitro can be augmented by transient transfections using mRNAs encoding antimicrobial CAMP and calprotectin (S100A8/S100A9, A8-IRES-A9, and A8-nIRES-A9). S100A8/A9 and LL-37 or CAMP function independently and additively in controlling intracellular bacteria. Importantly, stable presentation of S100A8/A9 and LL-37 neoproteins continued for at least 72 h, with corresponding protection against bacterial invasion.
The experimental approach using transient transfections of mRNAs to obtain neoprotein production is somewhat unusual, and this report appears to be the first in which epithelial cells are targeted. We recognized that the stability of the neoproteins depended on the stability of the transfected mRNAs. To optimize for stability, the mRNA cargo synthesized by in vitro translation was modified through 5′ capping with ARCA and by 3′ mRNA capping using poly(A) chains in cis and in trans (31). After transfection with ARCA-A100 Luc-mRNA, CD34-derived mouse dendritic cells (JAWSII cells) showed detectable luciferase activity within 1 h, which maximized at 8 h and declined 20-fold at 30 h (31), whereas transfection of primary human mesenchymal stem cells with a CXCR4-GFP fusion mRNA resulted in CXCR4 expression for up to 72 h and in efficient cell migration (32). To increase mRNA stability (33), our constructs included two sequential human beta-globin untranslated regions (UTRs). Following transfection into KB cells, ARCA-capped CAMP, S100A8/S100A9, A8-IRES-A9, or A8-nIRES-A9 each showed mRNA half-lives of 8 to 16 h (see Fig. S2A to D in the supplemental material). Our transfections used a commercially available nonliposomal, cationic polymer-lipid formulation (TransIT-mRNA transfection kit). Clearly, certain mRNA modifications improve the stability of the mRNA, but the specific mRNA cargo, the stability of the translated protein, and the target cell all affect the kinetics and efficiency of new protein expression.
After cotransfection with S100A8 and S100A9 mRNAs, S100A8/A9 forms spontaneously, as seen in Fig. S3 in the supplemental material. The stability of the S100A8/A9 heterodimer is greater than that of the individual subunit proteins (34). Use of tandem constructs was similarly successful. S100A8/A9 protein expression was detectable after cotransfection with S100A8 and S100A9 mRNAs (see Fig. S3C) and with the A8-IRES-A9 and A8-nIRES-A9 bicistronic transcripts (see Fig. S3E). The several constructs for S100A8/S100A9, A8-IRES-A9, and A8-nIRES-A9 mRNAs did result in different kinetics of translation. Translation from the MCS B site insert proximal to pIRES (Fig. 2E and G) and nIRES (Fig. 2F and G) showed a delay. We speculate that the mRNAs in the MCS B site were scanned and that translation was initiated by a low-efficiency, cap-independent IRES mechanism in KB cells.
CAMP, LL-37, and calprotectin produced as neoproteins after mRNA transfections are expressed inside the cell (see Fig. S3 in the supplemental material). S100A8, S100A9, and LL-37 all lack signal sequences, a membrane anchor sequence, and N-linked glycosylation consensus sequences, so they are unlikely to be secreted from the cell by a canonical pathway. We checked for release of S100A8/A9 and LL-37 from our cells, and the levels were below the limits of detection (data not shown). Being slightly antimicrobial, CAMP has a signal peptide and can be secreted from the cell. We did show, however, that innate intraepithelial immunity could be augmented through concurrent delivery of mRNAs encoding antimicrobial proteins, and we did not rigorously exclude the possibility that the effect is extracellular.
To perform our experiments, we reconciled differences in optimal cell confluence needed for the TransIT-mRNA transfection reagent (60 to 90%) and the antibiotic protection assay (40 to 60%) (4, 6). For the antibiotic protection assay, cells were transfected, split 8 and 32 h later, and then incubated for another 16 h to reach the 40 to 60% confluence needed for bacterial invasion. As expected, the apparent antimicrobial activity at 24 and 48 h was lower than that at 8 h following mRNA transfection (Fig. 3B to D); cell division over time (and subculturing) decreased intracellular CAMP and calprotectin protein levels. Nonetheless, the residual LL-37 and calprotectin levels up to 48 h after transfection were sufficient to provide statistically significant increases in resistance to the invading pathogens.
Cells in culture undergo apoptosis, but we found no evidence that mRNA delivery of any of the cargo mRNAs or their translated neoprotein products, S100A8, S100A9, S100A8/A9, CAMP, and LL-37, affected apoptosis or cell viability. Apoptosis in untransfected and transfected cells was indistinguishable. Our observations are consistent with previous reports using many different cell types and mRNA packaging systems (35, 36). To minimize the loss of cell viability, the delivery system may need to be modified to reduce epithelial cytotoxicity. During the last decade, investigators have explored the utility of nonviral packaging for mRNA delivery, and systems have been developed for production of HBD-2 in a macrophage line (37) and of luciferase mRNA in several normal and cancer cell lines (38). While alternatives will need to be developed, the modest loss of cell viability resulting from transfection in our experiments did not affect resistance to invasion (Fig. 3 and 5A).
How S100A8/A9 and LL-37 function within eukaryotic cells to increase resistance to bacterial invasion is the subject of current investigations. Both are directly antibacterial (3, 5, 6). S100A9 E36Q and E78Q mutations in S100A8/A9 result in diminished resistance to bacterial invasion (6). S100A8/A9 also interacts with NADPH oxidase to activate oxidative killing in epithelial cells (HaCaT) (39) and neutrophils (40, 41). LL-37 also appears to function as an endosomal chaperone for bacteria and bacterial DNA (3), also functioning in cooperation with NADPH oxidase to effect intracellular killing. Since we now show that LL-37 and S100A8/A9 function cooperatively, we must learn how these various mechanisms are coordinated with the production of antimicrobial oxygen metabolites to control the microbial colonization of epithelia without causing tissue damage.
Since this report shows that mRNA transfections can be used to augment innate epithelial immunity, this approach may be the basis for development of therapeutic agents on mucosal or epidermal surfaces. We appreciate that only moderate increases in resistance to bacterial invasion were noted in our experiments. Yet these moderate effects may be of considerable biological importance. For example, when keratinocytes were transfected using a lentivirus to overexpress cathelicidin (42), the antimicrobial effects (<0.2 log) in vitro were more modest than what we report. When the anatomy of the squamous mucosa is considered, with expressing cells stacked on expressing cells, the moderate effect within individual cells is multiplied geometrically into the antimicrobial protection of the tissue. When cathelicidin was augmented in mice, a profound epithelial resistance to group A streptococci developed (42).
mRNA delivery has several advantages over DNA gene transfer techniques for the treatment of disease (33), despite persistent concerns that the ease of synthesis and stability of transfected mRNA would prove insufficient to yield a useful protein product (23). Since mRNA does not integrate into the genome and the transfection remains transient, this transfection approach avoids imprecise, mutagenic insertion into the host cells and offers greater pharmaceutical safety (34). mRNA delivery facilitates simultaneous expression of all epitopes of an antigen, and manipulation and purification are rather simple (33). mRNA transfections have been used to express polyepitopes (43), to target dendritic cells ex vivo, and as anticancer vaccines in patients (44, 45). Similarly, mRNA delivery systems are in development for T cell-based immunotherapy of certain tumors (46) and to increase erythropoiesis (47). With a view toward augmenting innate immunity, HBD-2 mRNA transfection into monocyte-derived macrophages was shown to be effective against Mycobacterium tuberculosis, where the AMP and bacterium inexplicably colocalized in phagosomes (37). Similarly, insect-derived short proline-rich antimicrobial peptides (48) linked to a cell-penetrating peptide, penetratin, entered the cytoplasm of two mammalian cell lines without marked toxicity; intracellular resistance was increased against Micrococcus luteus but not against Escherichia coli (48). The insect-derived peptides show homology to mammalian heat shock proteins, but concern about potential cross-reactivity with host proteins and an autoimmune response may limit their clinical utility. Augmentation of intracellular innate immunity using native, endogenous antimicrobial peptides/proteins, however, could supplement or supplant the use of antibiotics, avoiding the clinical challenge of emergence of antibiotic resistance.
The present data show proof of principle that an mRNA delivery system could be an effective surrogate for a gene therapy approach to augment innate mucosal epithelial immunity and for treatment or control of mucosal infections. Use of this approach for therapeutic purposes in mucosal infections would be enhanced with an mRNA packaging system that would target epithelial cells more specifically and increase mRNA stability and minimize immune activation, as suggested by Weissman and colleagues (35). Ultimately, the test for mRNA transfection efficiency is a new or augmented function and minimal associated cytotoxicity or induction of apoptosis. Our data show that CAMP and calprotectin mRNA delivery could significantly increase resistance of epithelial cells to invasion by Listeria and Salmonella.
Supplementary Material
ACKNOWLEDGMENTS
We thank Lori A. Fischer for helpful suggestions about our flow cytometry analysis.
This work was supported by NIH/NIDCR grant R01DE021206 to M.C.H.
Footnotes
Published ahead of print 12 August 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.00539-13.
REFERENCES
- 1.Bando M, Hiroshima Y, Kataoka M, Shinohara Y, Herzberg MC, Ross KF, Nagata T, Kido J. 2007. Interleukin-1alpha regulates antimicrobial peptide expression in human keratinocytes. Immunol. Cell Biol. 85:532–537 [DOI] [PubMed] [Google Scholar]
- 2.Pasupuleti M, Schmidtchen A, Malmsten M. 2012. Antimicrobial peptides: key components of the innate immune system. Crit. Rev. Biotechnol. 32:143–171 [DOI] [PubMed] [Google Scholar]
- 3.Lee HM, Shin DM, Choi DK, Lee ZW, Kim KH, Yuk JM, Kim CD, Lee JH, Jo EK. 2009. Innate immune responses to Mycobacterium ulcerans via Toll-like receptors and dectin-1 in human keratinocytes. Cell. Microbiol. 11:678–692 [DOI] [PubMed] [Google Scholar]
- 4.Nisapakultorn K, Ross KF, Herzberg MC. 2001. Calprotectin expression in vitro by oral epithelial cells confers resistance to infection by Porphyromonas gingivalis. Infect. Immun. 69:4242–4247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zaia AA, Sappington KJ, Nisapakultorn K, Chazin WJ, Dietrich EA, Ross KF, Herzberg MC. 2009. Subversion of antimicrobial calprotectin (S100A8/S100A9 complex) in the cytoplasm of TR146 epithelial cells after invasion by Listeria monocytogenes. Mucosal Immunol. 2:43–53 [DOI] [PubMed] [Google Scholar]
- 6.Champaiboon C, Sappington KJ, Guenther BD, Ross KF, Herzberg MC. 2009. Calprotectin S100A9 calcium-binding loops I and II are essential for keratinocyte resistance to bacterial invasion. J. Biol. Chem. 284:7078–7090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burton MF, Steel PG. 2009. The chemistry and biology of LL-37. Nat. Prod. Rep. 26:1572–1584 [DOI] [PubMed] [Google Scholar]
- 8.Yamasaki K, Schauber J, Coda A, Lin H, Dorschner RA, Schechter NM, Bonnart C, Descargues P, Hovnanian A, Gallo RL. 2006. Kallikrein-mediated proteolysis regulates the antimicrobial effects of cathelicidins in skin. FASEB J. 20:2068–2080 [DOI] [PubMed] [Google Scholar]
- 9.Nijnik A, Hancock RE. 2009. The roles of cathelicidin LL-37 in immune defences and novel clinical applications. Curr. Opin. Hematol. 16:41–47 [DOI] [PubMed] [Google Scholar]
- 10.Turner J, Cho Y, Dinh NN, Waring AJ, Lehrer RI. 1998. Activities of LL-37, a cathelicidin-associated antimicrobial peptide of human neutrophils. Antimicrob. Agents Chemother. 42:2206–2214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Larrick JW, Hirata M, Zhong J, Wright SC. 1995. Antimicrobial activity of human CAP18 peptides. Immunotechnology 1:65–72 [DOI] [PubMed] [Google Scholar]
- 12.Dorschner RA, Pestonjamasp VK, Tamakuwala S, Ohtake T, Rudisill J, Nizet V, Agerberth B, Gudmundsson GH, Gallo RL. 2001. Cutaneous injury induces the release of cathelicidin antimicrobial peptides active against group A Streptococcus. J. Invest. Dermatol. 117:91–97 [DOI] [PubMed] [Google Scholar]
- 13.Wang TT, Nestel FP, Bourdeau V, Nagai Y, Wang Q, Liao J, Tavera-Mendoza L, Lin R, Hanrahan JW, Mader S, White JH. 2004. Cutting edge: 1,25-dihydroxyvitamin D3 is a direct inducer of antimicrobial peptide gene expression. J. Immunol. 173:2909–2912 [DOI] [PubMed] [Google Scholar]
- 14.Nell MJ, Tjabringa GS, Vonk MJ, Hiemstra PS, Grote JJ. 2004. Bacterial products increase expression of the human cathelicidin hCAP-18/LL-37 in cultured human sinus epithelial cells. FEMS Immunol. Med. Microbiol. 42:225–231 [DOI] [PubMed] [Google Scholar]
- 15.Carlsson G, Wahlin YB, Johansson A, Olsson A, Eriksson T, Claesson R, Hänström L, Henter JI. 2006. Periodontal disease in patients from the original Kostmann family with severe congenital neutropenia. J. Periodontol. 77:744–751 [DOI] [PubMed] [Google Scholar]
- 16.Ong PY, Ohtake T, Brandt C, Strickland I, Boguniewicz M, Ganz T, Gallo RL, Leung DY. 2002. Endogenous antimicrobial peptides and skin infections in atopic dermatitis. N. Engl. J. Med. 347:1151–1160 [DOI] [PubMed] [Google Scholar]
- 17.Marenholz I, Heizmann CW, Fritz G. 2004. S100 proteins in mouse and man: from evolution to function and pathology (including an update of the nomenclature). Biochem. Biophys. Res. Commun. 322:1111–1122 [DOI] [PubMed] [Google Scholar]
- 18.Santamaria-Kisiel L, Rintala-Dempsey AC, Shaw GS. 2006. Calcium-dependent and -independent interactions of the S100 protein family. Biochem. J. 396:201–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miyasaki KT, Bodeau AL, Murthy AR, Lehrer RI. 1993. In vitro antimicrobial activity of the human neutrophil cytosolic S-100 protein complex, calprotectin, against Capnocytophaga sputigena. J. Dent. Res. 72:517–523 [DOI] [PubMed] [Google Scholar]
- 20.Sohnle PG, Collins-Lech C, Wiessner JH. 1991. Antimicrobial activity of an abundant calcium-binding protein in the cytoplasm of human neutrophils. J. Infect. Dis. 163:187–192 [DOI] [PubMed] [Google Scholar]
- 21.Steinbakk M, Naess-Andresen CF, Lingaas E, Dale I, Brandtzaeg P, Fagerhol MK. 1990. Antimicrobial actions of calcium binding leucocyte L1 protein, calprotectin. Lancet 336:763–765 [DOI] [PubMed] [Google Scholar]
- 22.Chamilos G, Gregorio J, Meller S, Lande R, Kontoyiannis DP, Modlin RL, Gilliet M. 2012. Cytosolic sensing of extracellular self-DNA transported into monocytes by the antimicrobial peptide LL37. Blood 120:3699–3707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tavernier G, Andries O, Demeester J, Sanders NN, De Smedt SC, Rejman J. 2011. mRNA as gene therapeutic: how to control protein expression. J. Control. Release 150:238–247 [DOI] [PubMed] [Google Scholar]
- 24.Zhao Y, Moon E, Carpenito C, Paulos CM, Liu X, Brennan AL, Chew A, Carroll RG, Scholler J, Levine BL, Albelda SM, June CH. 2010. Multiple injections of electroporated autologous T cells expressing a chimeric antigen receptor mediate regression of human disseminated tumor. Cancer Res. 70:9053–9061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rees S, Coote J, Stables J, Goodson S, Harris S, Lee MG. 1996. Bicistronic vector for the creation of stable mammalian cell lines that predisposes all antibiotic-resistant cells to express recombinant protein. Biotechniques 20:102–110 [DOI] [PubMed] [Google Scholar]
- 26.Bochkov YA, Palmenberg AC. 2006. Translational efficiency of EMCV IRES in bicistronic vectors is dependent upon IRES sequence and gene location. Biotechniques 41:283, 284, 286, 288 [DOI] [PubMed] [Google Scholar]
- 27.Pelicano H, Feng L, Zhou Y, Carew JS, Hileman EO, Plunkett W, Keating MJ, Huang P. 2003. Inhibition of mitochondrial respiration: a novel strategy to enhance drug-induced apoptosis in human leukemia cells by a reactive oxygen species-mediated mechanism. J. Biol. Chem. 278:37832–37839 [DOI] [PubMed] [Google Scholar]
- 28.Taylor RC, Cullen SP, Martin SJ. 2008. Apoptosis: controlled demolition at the cellular level. Nat. Rev. Mol. Cell. Biol. 9:231–241 [DOI] [PubMed] [Google Scholar]
- 29.Rudney JD, Chen R, Zhang G. 2005. Streptococci dominate the diverse flora within buccal cells. J. Dent. Res. 84:1165–1171 [DOI] [PubMed] [Google Scholar]
- 30.Sorenson BS, Khammanivong A, Guenther BD, Ross KF, Herzberg MC. 2012. IL-1 receptor regulates S100A8/A9-dependent keratinocyte resistance to bacterial invasion. Mucosal Immunol. 5:66–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mockey M, Bourseau E, Chandrashekhar V, Chaudhuri A, Lafosse S, Le Cam E, Quesniaux VF, Ryffel B, Pichon C, Midoux P. 2007. mRNA-based cancer vaccine: prevention of B16 melanoma progression and metastasis by systemic injection of MART1 mRNA histidylated lipopolyplexes. Cancer Gene Ther. 14:802–814 [DOI] [PubMed] [Google Scholar]
- 32.Ryser MF, Ugarte F, Thieme S, Bornhäuser M, Roesen-Wolff A, Brenner S. 2008. mRNA transfection of CXCR4-GFP fusion—simply generated by PCR—results in efficient migration of primary human mesenchymal stem cells. Tissue Eng. Part C Methods 14:179–184 [DOI] [PubMed] [Google Scholar]
- 33.Yamamoto A, Kormann M, Rosenecker J, Rudolph C. 2009. Current prospects for mRNA gene delivery. Eur. J. Pharm. Biopharm. 71:484–489 [DOI] [PubMed] [Google Scholar]
- 34.Hsu K, Champaiboon C, Guenther BD, Sorenson BS, Khammanivong A, Ross KF, Geczy CL, Herzberg MC. 2009. Anti-infective protective properties of S100 calgranulins. Antiinflamm. Antiallergy Agents Med. Chem. 8:290–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Karikó K, Muramatsu H, Ludwig J, Weissman D. 2011. Generating the optimal mRNA for therapy: HPLC purification eliminates immune activation and improves translation of nucleoside-modified, protein-encoding mRNA. Nucleic Acids Res. 39:e142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gonzalez G, Pfannes L, Brazas R, Striker R. 2007. Selection of an optimal RNA transfection reagent and comparison to electroporation for the delivery of viral RNA. J. Virol. Methods 145:14–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kisich KO, Heifets L, Higgins M, Diamond G. 2001. Antimycobacterial agent based on mRNA encoding human beta-defensin 2 enables primary macrophages to restrict growth of Mycobacterium tuberculosis. Infect. Immun. 69:2692–2699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bettinger T, Carlisle RC, Read ML, Ogris M, Seymour LW. 2001. Peptide-mediated RNA delivery: a novel approach for enhanced transfection of primary and post-mitotic cells. Nucleic Acids Res. 29:3882–3891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Benedyk M, Sopalla C, Nacken W, Bode G, Melkonyan H, Banfi B, Kerkhoff C. 2007. HaCaT keratinocytes overexpressing the S100 proteins S100A8 and S100A9 show increased NADPH oxidase and NF-kappaB activities. J. Invest. Dermatol. 127:2001–2011 [DOI] [PubMed] [Google Scholar]
- 40.Lim SY, Raftery MJ, Goyette J, Hsu K, Geczy CL. 2009. Oxidative modifications of S100 proteins: functional regulation by redox. J. Leukoc. Biol. 86:577–587 [DOI] [PubMed] [Google Scholar]
- 41.Simard JC, Simon MM, Tessier PA, Girard D. 2011. Damage-associated molecular pattern S100A9 increases bactericidal activity of human neutrophils by enhancing phagocytosis. J. Immunol. 186:3622–3631 [DOI] [PubMed] [Google Scholar]
- 42.Braff MH, Zaiou M, Fierer J, Nizet V, Gallo RL. 2005. Keratinocyte production of cathelicidin provides direct activity against bacterial skin pathogens. Infect. Immun. 73:6771–6781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nielsen JS, Wick DA, Tran E, Nelson BH, Webb JR. 2010. An in vitro-transcribed-mRNA polyepitope construct encoding 32 distinct HLA class I-restricted epitopes from CMV, EBV, and influenza for use as a functional control in human immune monitoring studies. J. Immunol. Methods 360:149–156 [DOI] [PubMed] [Google Scholar]
- 44.Lesterhuis WJ, De Vries IJ, Schreibelt G, Schuurhuis DH, Aarntzen EH, De Boer A, Scharenborg NM, Van De Rakt M, Hesselink EJ, Figdor CG, Adema GJ, Punt CJ. 2010. Immunogenicity of dendritic cells pulsed with CEA peptide or transfected with CEA mRNA for vaccination of colorectal cancer patients. Anticancer Res. 30:5091–5097 [PubMed] [Google Scholar]
- 45.Suso EM, Dueland S, Rasmussen AM, Vetrhus T, Aamdal S, Kvalheim G, Gaudernack G. 2011. hTERT mRNA dendritic cell vaccination: complete response in a pancreatic cancer patient associated with response against several hTERT epitopes. Cancer Immunol. Immunother. 60:809–818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lehner M, Götz G, Proff J, Schaft N, Dörrie J, Full F, Ensser A, Muller YA, Cerwenka A, Abken H, Parolini O, Ambros PF, Kovar H, Holter W. 2012. Redirecting T cells to Ewing's sarcoma family of tumors by a chimeric NKG2D receptor expressed by lentiviral transduction or mRNA transfection. PLoS One 7:e31210. 10.1371/journal.pone.0031210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Karikó K, Muramatsu H, Keller JM, Weissman D. 2012. Increased erythropoiesis in mice injected with submicrogram quantities of pseudouridine-containing mRNA encoding erythropoietin. Mol. Ther. 20:948–953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hansen A, Schäfer I, Knappe D, Seibel P, Hoffmann R. 2012. Intracellular toxicity of proline-rich antimicrobial peptides shuttled into mammalian cells by the cell-penetrating peptide penetratin. Antimicrob. Agents Chemother. 56:5194–5201 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.