

Abstract
Pneumocystis species are opportunistic fungal pathogens that induce tumor necrosis factor (TNF) production by alveolar macrophages. Here we report that B cells from the draining lymph nodes as well as lung CD4+ T cells are important producers of TNF upon Pneumocystis murina infection. To determine the importance of B cell-derived TNF in the primary response to P. murina, we generated bone marrow chimeras whose B cells were unable to produce TNF. The lung P. murina burden at 10 days postinfection in TNF knockout (TNFKO) chimeras was significantly higher than that in wild-type (WT) chimeras, which corresponded to reduced numbers of activated CD4+ T cells in the lungs at this early time point. Furthermore, CD4+ T cells isolated from P. murina-infected TNFKO chimeras were unable to stimulate clearance of P. murina upon adoptive transfer to recombinase-deficient (RAG1KO) hosts. Together, these data indicate that B cell-derived TNF plays an important function in promoting CD4+ T cell expansion and production of TNF and facilitating protection against P. murina infection.
INTRODUCTION
Pneumocystis species are opportunistic fungal pathogens that cause Pneumocystis pneumonia (PCP), especially in immunocompromised patients such as those with AIDS (1, 2). Many studies have demonstrated that both CD4+ T cells and B cells contribute to protection against Pneumocystis infections (3–10), but the interactive roles of these lymphocytes in host defense have not been fully defined. Previous studies from our laboratory and others have shown that reconstitution of SCID mice with primed wild-type (WT) CD4+ T cells was sufficient to facilitate Pneumocystis murina (the species found in mice) clearance from their lungs (5, 6, 8). However, reconstitution with CD4+ T cells primed in a B cell-deficient environment failed to clear P. murina (6). This suggests that CD4+ T cells primed in a wild-type environment have the ability to produce important cytokines that activate macrophages to eliminate the organisms. In addition, T cells have a survival advantage when primed in WT mice, since they are able to expand in both the tracheobronchial lymph node (TBLN) and the alveolar spaces, as opposed to those primed in a B cell-deficient environment (6).
P. murina-specific antibodies (Abs) are important in protecting experimental animals from infection (3, 8); however, we have demonstrated that mice containing CD40-deficient B cells, unable to produce class-switched antibodies, cleared P. murina infection, suggesting that B cells have important functions other than antibody secretion (7). Consistent with this, we have also shown that mice with targeted mutations in Fcγ and ϵ receptors cleared P. murina organisms (7). Other laboratories have also demonstrated that B cells are essential in T cell-mediated protection of hosts against various infectious pathogens (11–15). It is believed that naive CD4+ T cells respond to some soluble antigens when processed by B cells (16, 17). Furthermore, the proximity of B cells to CD4+ T cells in the lymph node (LN) could enable them to continuously present antigen to maintain CD4+ T cell effector or memory function (15, 18).
Protection from P. murina is highly dependent on proinflammatory cytokines, such as gamma interferon (IFN-γ) and tumor necrosis factor (TNF), produced by effector cells (19–25). Neutralization of TNF with monoclonal Abs (MAbs) resulted in persistent P. murina pneumonia in infected SCID mice that were reconstituted with splenocytes (22). Recently, it was reported that some individuals on monoclonal antibody therapy targeting TNF have developed PCP (26, 27). It is well documented that TNF can be produced by a number of cell types, including B cells and T cells (24–26, 28–30). However, TNF produced primarily by macrophages in response to P. murina is thought to be important for clearance of infection (31–34). There is also evidence that TNF derived from lung epithelial cells contributes to the immune response to P. murina (35).
Given that B cell-deficient mice are susceptible to PCP (36) even though they have functional CD4+ T cells, we addressed the importance of B cells in promoting CD4+ T cell activation in response to P. murina infection. We demonstrate that B cells produce TNF in the draining lymph node, impacting CD4+ T cell expansion in response to the pathogen. Importantly, we show that in the absence of B cell-derived TNF, CD4+ T cells are unable to clear P. murina upon adoptive transfer to lymphocyte-deficient RAG1KO mice.
MATERIALS AND METHODS
Mice.
Adult C57BL/6, B6.129S2-Ighmtm1Cgn/J (μMT), B6.129S-Tnftm1Gk1/J (TNFKO), and B6.129S7-Rag1tm1Mom/J (RAG1KO) mice on a C57BL/6 background were purchased from The Jackson Laboratory (Bar Harbor, ME). Adult BALB/c mice were obtained from Taconic Farms. B cell-deficient mice on a BALB/c background (Igh-Jtm1Dhu [JhKO]) (14) were obtained from our breeding colony, initially obtained from Taconic Farms. A colony of C.129S6(B6)-Rag2tm1Fwa (RAG2KO) mice on a BALB/c background, originally obtained from Taconic Farms, was used to maintain a source of P. murina for infection of experimental mice. Severe combined immunodeficient (SCID) mice on a BALB/c background (C.B-17) were obtained from The Jackson Laboratory. All experimental mice were housed in the Lexington, KY, Veterans Administration (VA) Medical Center veterinary medical unit or the University of Kentucky Division of Laboratory Animal Resources in sterile, filter-topped cages and were given sterile food and water. All animal procedures were approved by the Lexington VA or University of Kentucky Animal Institutional Care and Use Committee.
Enumeration and inoculation of Pneumocystis organisms.
For isolation of organisms for inoculation, lungs were excised from P. murina-infected RAG2KO mice and pushed through stainless steel mesh in Hanks' balanced salt solution (HBSS). Cell debris was removed by centrifugation at 200 × g for 2 min, and organisms were pelleted at 1,300 × g for 15 min. Aliquots were spun onto glass slides, fixed in methanol, and stained with Diff-Quik (Siemens Healthcare Diagnostics, Deerfield, IL). P. murina nuclei were enumerated by microscopy. Mice to be infected were anesthetized lightly with isoflurane gas, and 105 to 107 P. murina organisms were injected intratracheally (i.t.) in 100 μl of HBSS supplemented with 1 μM penicillin-streptomycin and 0.1 μM gentamicin. For determination of lung P. murina burden, the lung lobes were excised, minced, and digested in RPMI 1640 supplemented with 2% fetal calf serum (FCS), 1 mg/ml collagenase A, and 50 U/ml DNase for 1 h at 37°C. Digested lung fragments were pushed through mesh screens, and aliquots were spun onto glass slides and stained with Diff-Quik for microscopic enumeration. Lung burden is expressed as log10 P. murina nuclei per lung. For these experiments, the limit of detection was 2.93 log10 P. murina nuclei per lung.
In vivo proliferation of lymphocytes.
Adult BALB/c or JhKO mice were infected i.t. with 107 P. murina organisms. Approximately 12 h before a specified time point, the mice were injected intraperitoneally (i.p.) with 100 μl of 10 ng/ml bromodeoxyuridine (BrdU) from BD Biosciences (San Jose, CA). Cells were harvested from the TBLN and lung and stained with fluorescently labeled anti-CD4, -CD19, and -BrdU antibodies according to the manufacturer's instructions (BD Biosciences). BrdU incorporation into DNA of T and B cells was determined by flow cytometry (37, 38).
Adoptive transfer of B cells and CD4 T cells.
Donor WT C57BL/6 or BALB/c, TNFKO chimera, and B cell-deficient μMT or JhKO mice were given intratracheal inoculations of 107 P. murina organisms. The draining LNs were collected at day 14 postinfection. CD4 T cells were isolated from single-cell suspensions of TBLN using negative-selection columns from R&D Systems (Minneapolis, MN). B cells were isolated from the spleens of either WT or TNFKO mice by positive selection using anti-CD19-coated magnetic beads and columns from Miltenyi Biotec Inc. (Auburn, CA) or StemCell magnet negative-selection kits, according to the manufacturer's protocol. C.B-17 SCID or RAG1KO mice were injected intravenously (i.v.) with 105 purified CD4+ T cells either alone or together with 107 purified B cells, and 4 days later, the adoptive recipients were given intratracheal inoculations of 105 to 106 P. murina organisms.
Generation of mixed chimeras and adoptive transfer of B cells and CD4+ T cells.
Mixed chimeric mice in which B cells were derived from TNFKO mice were generated as previously described (6, 7). Briefly, recipient μMT mice were lethally irradiated with 9.5 Gy from a 137Cs source and reconstituted with bone marrow cells either from μMT mice or from a mix of cells composed of 75% μMT plus 25% TNFKO or C57BL/6 mouse cells. Chimerism was confirmed after 10 weeks by flow cytometry using peripheral blood samples and again at individual time points.
Donor C57BL/6, TNFKO, μMT, or mixed chimeric mice were given i.t. inoculations of 107 P. murina organisms. The draining TBLNs were isolated at day 14 postinfection, and CD4+ T cells were isolated from single-cell suspensions by using negative-selection columns from R&D Systems (Minneapolis, MN) according to the manufacturer's protocol. B cells were isolated from the spleens of either WT or TNFKO mice by positive selection using anti-CD19-coated magnetic beads and columns from Miltenyi Biotec Inc. (Auburn, CA). We routinely obtained ∼95% pure CD4+ T cells and ∼90% pure B cells. RAG1KO mice were injected i.v. with 105 purified CD4+ T cells either alone or together with 107 purified B cells, and 4 days later, these mice were given i.t. inoculations of 105 to 106 P. murina organisms.
Isolation of cells from alveolar spaces, lungs, and LNs.
Lungs were lavaged with 5 1-ml washes of HBSS containing 3 mM EDTA to obtain alveolar cells. The fluid from the first wash was retained for cytokine analysis, and the cells were pooled with cells collected from the subsequent washes. Lungs were minced and digested as described above, and after removal of an aliquot for enumeration of P. murina organisms, erythrocytes were removed from the lung digest cells by using a hypotonic lysing buffer. Cells were washed, and single-cell suspensions were enumerated. TBLNs were pushed through mesh screens in HBSS to obtain single-cell suspensions, erythrocytes were removed, and cells were enumerated.
Determination of cytokine concentrations in serum and BALF.
Serum and bronchial alveolar lavage fluid (BALF) collected from the first lung lavage wash was spun down, and supernatants were frozen for subsequent determination of cytokine levels by using an enzyme-linked immunosorbent assay (ELISA) according to the manufacturer's directions (eBioscience, San Diego, CA).
Flow cytometric analysis of lung and LN lymphocytes.
Lung lavage, lung digest, and TBLN cells were washed in phosphate-buffered saline (PBS) containing 0.1% bovine serum albumin (BSA) and 0.02% NaN3 (PBA) and stained with appropriate concentrations of fluorochrome-conjugated antibodies specific for murine CD4, CD8, CD44, and CD62L. Antibodies were purchased from BD Biosciences and eBioscience. Expression of these molecules on the surface of lymphocytes was determined by multiparameter flow cytometry using a FACSCalibur cytofluorimeter (BD Biosciences). Fifty thousand events were routinely collected. Unless otherwise stated, either the proportion of cells or total numbers of cells per tissue are reported.
Intracellular cytokine staining.
Single-cell suspensions were obtained as described above. The cells were incubated for 2 h in 50 ng/ml phorbol myristate acetate (PMA) and 1 μg/ml ionomycin to increase production of cytokines (39). Brefeldin A (10 μg/ml) was added to inhibit secretion, followed by a further 2-h incubation. Cells were surface stained in PBA with brefeldin A with fluorescently labeled anti-CD4 and -CD19 antibodies, followed by fixation in 10% formalin. Cells were permeabilized with PBA–0.5% saponin, FcRs were blocked with anti-CD16/CD32, and cells were stained with allophycocyanin (APC)-conjugated anti-TNF or isotype control antibody and analyzed by flow cytometry.
Pneumocystis-specific ELISA.
Blood was collected from the abdominal aorta under isoflurane anesthesia, and sera were frozen at −80°C. A crude sonicate of P. murina protein (10 μg/ml) was coated onto microtiter plates for 2 h, and coated wells were blocked with 5% dry milk in HBSS containing 0.05% Tween 20 for 1 h. Test sera were serially diluted and incubated in plates overnight (4°C). Plates were washed extensively, and bound Ab was detected by using alkaline phosphatase-conjugated specific Abs (anti-IgM and -IgG). After 4 h at 37°C, plates were washed and developed by using p-nitrophenylphosphate at 1 mg/ml in diethanolamine buffer. The optical density was read at 405 nm.
Statistical analysis.
Differences between experimental groups were determined by using analysis of variance (ANOVA), followed by Student-Newman-Keuls or Dunn's post hoc tests where appropriate. Differences were considered statistically significant when the P value was <0.05. SigmaPlot 11.1 statistical software (Systat, San Jose, CA) was used for all analyses.
RESULTS
B cells enhance CD4+ T cell activation in response to P. murina infection.
We previously reported that B cell-deficient mice have reduced numbers of activated CD4+ T cells in the alveolar spaces in response to P. murina and that these cells fail to expand when transferred to immunodeficient SCID mice infected with P. murina (6, 7). To determine whether this failure to expand is due to a problem with proliferation, T cells from B cell-deficient mice were examined by using our adoptive transfer model. CD4+ T cells from P. murina-infected wild-type (WT) or JhKO mice (on a BALB/c background) were purified and transferred to C.B-17 SCID mice on a BALB/c background, followed by P. murina infection 4 days later. Twelve hours prior to each time point, BrdU was injected into the mice, and cells that had recently proliferated were examined by using flow cytometry. There was no difference in the number of CD4+ BrdU-positive (BrdU+) cells in the draining lymph nodes (TBLN) of mice that received T cells from WT compared to JhKO donors at 14 days postinfection, but there were significantly more proliferating cells in the airways of mice that received WT T cells than in those of mice that received T cells from JhKO donors (Fig. 1A). This reduced number of proliferating cells in the alveolar spaces of the JhKO CD4+ T cell recipients raised the question of whether B cells could affect activation of T cells primed in JhKO mice. To test this, CD4+ T cells from P. murina-infected JhKO mice were adoptively transferred alone or together with B cells from P. murina-infected WT donors into SCID mice. T cell activation was determined by flow cytometry. As shown in Fig. 1B to D, there was an increase in the number of activated (CD44hi CD62Llo) CD4+ T cells in the lungs, bronchoalveolar lavage fluid (BALF), and TBLN of the recipient mice that received both T cells from JhKO mice and WT B cells as opposed to those that received JhKO T cells alone. The number of activated T cells decreased by day 29, which correlated with a lower lung burden of P. murina organisms (Fig. 1E). Consistent with our previous findings (6), adoptive transfer of WT CD4+ T cells alone cleared the organisms. Interestingly, the group of mice that received both JhKO T cells and B cells had a faster kinetics of accumulation of activated T cells and cleared the pathogen faster than the group that received WT T cells only (Fig. 1B to E). Notably, we were able to detect P. murina-specific IgM in the BALF of mice that received both JhKO T cells and B cells (data not shown), and so we cannot rule out the possibility that the antibody made a contribution to the reduction in organism burden in these mice. However, these data do suggest that B cells play a critical role in CD4+ T cell-mediated host protection against P. murina. This could be through antigen presentation or local cytokine secretion.
Fig 1.
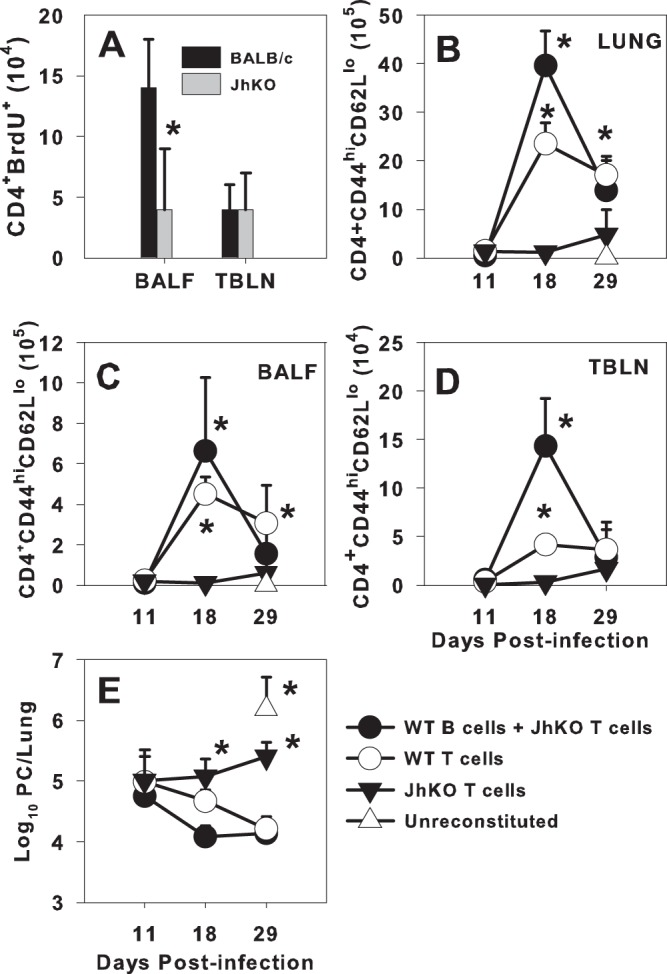
B cells promote CD4+ T cell proliferation in BALF and activation in the TBLN and lung digest. BALB/c WT or JhKO mice were infected with 107 P. murina organisms, and 14 days thereafter, CD4+ T cells were purified from the TBLN of the infected donors and transferred to SCID mice. (A) The recipients were infected with 106 P. murina organisms, and proliferation was determined by using BrdU at day 14 postinfection. CD4+ T cells from the TBLN of P. murina-infected JhKO mice were transferred alone or together with B cells purified from the spleens of WT P. murina-primed donors into SCID mice. (B to D) Control recipient mice received WT T cells from P. murina-infected TBLNs, and activation in lung digest (B), BALF (C), or TBLN (D) was determined on days 11, 18, and 29 postinfection by using flow cytometry. (E) P. murina lung burden was determined microscopically. Data represent the means ± standard deviations for 4 mice per group and are representative of two separate experiments. ∗, P < 0.05 compared to JhKO mouse CD4+ T cells alone at the same time point, using ANOVA and Student-Newman-Keuls post hoc tests.
T cell proliferation in B cell-deficient mice is not impaired.
Because of the inability of JhKO CD4+ T cells to expand in the alveolar spaces upon adoptive transfer (6) (Fig. 1), we next determined whether they proliferated as well as cells from WT mice during the primary response to the pathogen. Adult BALB/c or JhKO mice were infected with P. murina, and approximately 12 h before each time point, the mice were injected with BrdU. As shown in Fig. 2A, there were no differences in CD4 T cell proliferation in the TBLNs between the two groups of mice. In contrast, there were significantly higher numbers of proliferating CD4+ T cells in the lungs of the JhKO mice at days 20 and 27 postinfection than in the WT mice (Fig. 2B). This corresponded to higher numbers of P. murina organisms in the lungs of the JhKO mice at these time points (Fig. 2E). Although the JhKO mice were not able to eliminate P. murina organisms from the lungs, the CD4+ T cell-proliferative response to P. murina infection was robust. Additionally, we found similar proportions of IFN-γ- and interleukin-17 (IL-17)-producing CD4+ T cells in the TBLNs of JhKO and wild-type mice as well as similar proportions of regulatory T cells (Treg) (data not shown), indicating that the failure to expand in adoptive hosts was not due to inhibition by Tregs.
Fig 2.
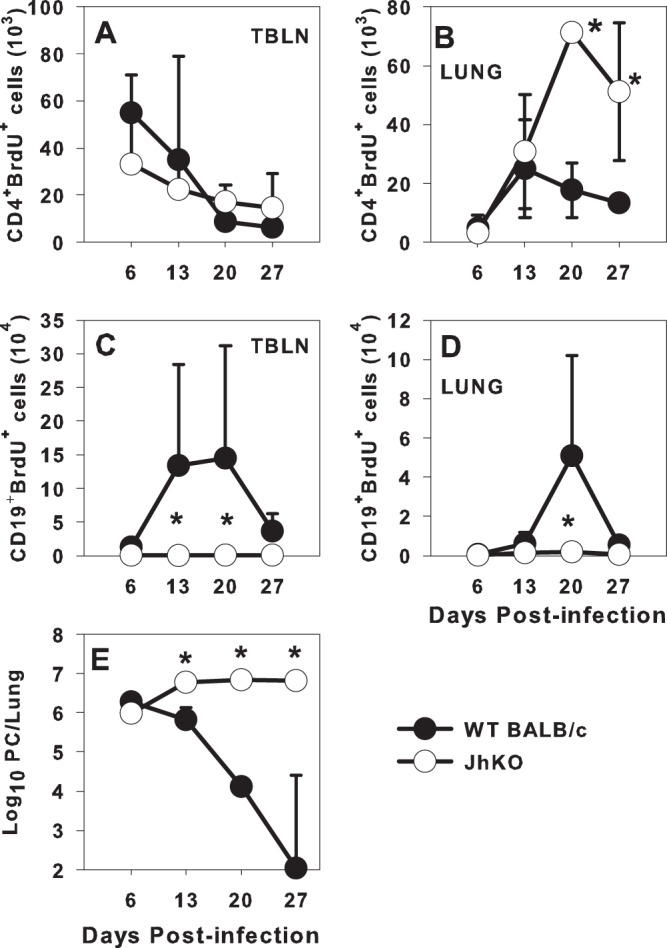
CD4+ T cells proliferate in the lungs of Pneumocystis-infected wild-type and JhKO mice. WT and JhKO mice were infected with P. murina organisms and injected with BrdU 12 h before the indicated time points. (A to D) Cell proliferation of CD4+ T cells (A and B) and CD19+ B cells (C and D) was determined by staining for uptake of BrdU in TBLN (A and C) and lung (B and D). (E) P. murina lung burden was enumerated microscopically. Data represent the means ± standard deviations for 5 mice per group and are representative of two separate experiments. ∗, P < 0.05 compared to WT mice at the indicated time point, using ANOVA and Dunn's post hoc test.
Examination of B cell proliferation in P. murina-infected WT mice showed a small increase in the number of CD19+ BrdU+ cells in the TBLN starting at day 6 postinfection (Fig. 2C). The proportion of proliferating CD19+ cells increased significantly through day 13 postinfection (Fig. 2C). Levels of CD19+ BrdU+ cells remained high through day 20 and then decreased by day 27, which corresponded with P. murina clearance (Fig. 2E). In the lungs, the number of CD19+ cells that had proliferated increased from day 13 through day 20 postinfection (Fig. 2D), suggesting that B cells that were proliferating in the TBLN started moving to the lungs at around day 13 postinfection. Proliferation of both lung and TBLN B cells decreased by day 27, which corresponded with decreased P. murina lung burden (Fig. 2E). Taken together with the data described above, these data indicate that T cell proliferation takes place early in the TBLN, followed by B cell proliferation, and that both T cells and B cells then migrate to the lungs after proliferation. Moreover, B cells do not appear to be critical for proliferation of T cells during primary infection.
B cells produce TNF upon P. murina infection.
Although there is evidence that B cells are able to produce TNF in response to some stimuli (28), it has not been shown whether B cells are able to produce TNF in response to P. murina infection. We next determined whether B cells contribute to the production of TNF in this infection model. B cells were harvested from the TBLN of P. murina-infected BALB/c mice at day 14 postinfection and restimulated in vitro with sonicated P. murina protein, whole P. murina cells, or lipopolysaccharide (LPS). B cells produced elevated TNF levels only when restimulated with sonicated P. murina (Fig. 3A). There was a small increase in the level of TNF secretion upon restimulation with whole P. murina, but there was no detectable TNF secretion by the cells that were stimulated with LPS compared to unstimulated B cells (Fig. 3A).
Fig 3.
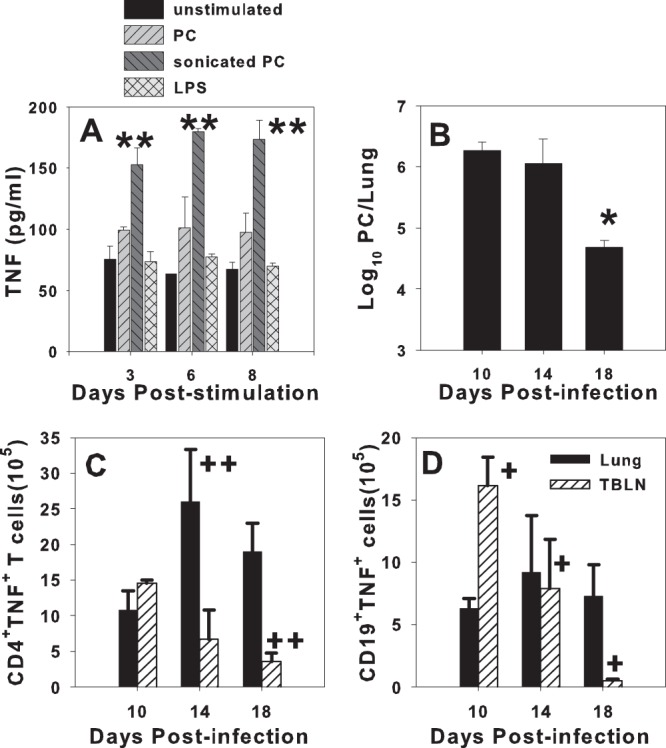
B cells produce TNF in response to P. murina infection. B cells were purified from the TBLN of P. murina-infected BALB/c mice at day 10 postinfection and restimulated in vitro. (A) At the indicated days poststimulation, the TNF-α concentration in the supernatants was determined by ELISA. (B) BALB/c mice were infected with 107 P. murina organisms, and P. murina lung burden was determined microscopically over time. (C and D) TBLN and lung digest CD4+ T cells (C) and CD19+ B cells (D) were analyzed for the ability to secrete TNF by intracellular cytokine staining at days 10, 14, and 17 postinfection. Data represent the means ± standard deviations for 3 to 4 mice per group and are representative of three separate experiments. ∗∗, P < 0.05 among groups at indicated time points by Kruskal-Wallis one-way ANOVA on ranks (A); ∗, P < 0.05 compared to the other time points by one-way ANOVA (B); ++, P < 0.05 compared to the same tissue at day 10 by one-way ANOVA or ANOVA on ranks (C); +, P < 0.05 compared to the same tissue at other time points by using one-way ANOVA (D). PC, Pneumocystis.
The ability of B cells to facilitate T cell activation (Fig. 1) and produce TNF in the in vitro system (Fig. 3A) raised the question of when and where during the infection B cell-derived TNF might be important for defense against the pathogen. The kinetics of TNF secretion by CD4+ T and B cells in vivo was therefore determined by using intracellular staining (Fig. 3C and D). The number of CD4+ T cells that produced TNF was highest in the lungs of BALB/c mice at day 14 during the peak of infection (Fig. 3C). In contrast, the number of TNF-producing CD4+ T cells peaked in the lymph node at day 10 postinfection (Fig. 3C). B cell-derived TNF was predominantly produced by the cells in the lymph node at days 10 and 14 postinfection (Fig. 3D). TNF-producing B cells were also found in the lungs, although there was less fluctuation in CD19+ TNF+ cells in the lungs and no real peak seen over the course of the experiment (Fig. 3D). We did not observe significant TNF production by either CD4+ T cells or B cells in the TBLN at day 18 postinfection. This correlated with a significant decrease in the number of P. murina organisms in the lungs (Fig. 3B). These data suggest that CD4+ T and possibly B cells produce TNF in the lymph node and then move to the lungs, where they continue to produce TNF.
Chimeric mice with B cells that are unable to produce TNF clear P. murina with delayed kinetics.
In order to determine whether B cell-derived TNF is important for a primary immune response and clearance of P. murina, we generated mixed bone marrow chimeras with B cells derived from TNFKO mice (TNFKO chimeras). Control chimeric mice included mice without B cells (μMT chimeras) and mice with B cells derived from wild-type C57BL/6 donors (WT chimeras). Figure 4A demonstrates that CD19+ cells from the draining TBLNs of TNFKO chimeras at day 10 postinfection produce background levels of TNF compared to WT chimeras, while the proportion of CD4+ T cells that produce TNF is similar for all three groups. As we have previously reported (6, 7), μMT chimeras failed to control P. murina lung burden over the course of the experiment, while WT chimeras cleared P. murina by day 30 postinfection (Fig. 4B). TNFKO chimeras also cleared P. murina by day 30 postinfection but had about a 10-fold-higher lung burden at day 10 postinfection than did the WT chimeras (Fig. 4B). This elevated lung burden at day 10 was not due to the activation state of CD19+ B cells in the TBLN or lungs or to P. murina-specific IgG in the BALF, since there were no differences between the WT chimeras and TNFKO chimeras (Fig. 4E to G and data not shown).
Fig 4.

Chimeric mice with B cells deficient in TNF have slower P. murina clearance kinetics than wild-type chimeras. Mixed bone marrow chimeras were generated by reconstituting lethally irradiated μMT mice with 100% μMT bone marrow or a mix of 75% μMT plus 25% TNFKO or WT bone marrow. (A) Twelve weeks after transplantation, chimeric mice were infected with P. murina, and TBLN cells were examined for chimerism at day 10 postinfection. Flow cytometry dot plots show TBLN CD19+ B cells (top) and CD4+ T cells (bottom) that produced TNF from WT (left), TNFKO (center), and μMT (right) chimeras. (B) Lung P. murina burdens were determined by microscopy. (C to E) The numbers of activated CD4+ CD44hi CD62Llo cells in the lungs (C) and TBLN (D) and the numbers of activated CD19+ CD86hi Iab+ B cells in the TBLN (E) were determined by flow cytometry. (F and G) The level of P. murina-specific IgG in BALF (F) and sera (G) was determined by ELISA. (H and I) TNF (H) and IFN-γ (I) concentrations in the BALF were determined by ELISA. Data represent the means ± standard deviations for 4 mice per group per time point and are representative of two separate experiments. ∗, P < 0.05 compared to WT chimeras; +, P < 0.05 compared to other groups at the same time point, using ANOVA and Student-Newman-Keuls post hoc tests.
There were no significant differences in the number or activation state of CD4+ T cells isolated from the BALF of the different chimeras (data not shown). However, at day 10 postinfection, there were fewer activated CD4+ CD44hi CD62Llo cells in both the TNFKO and μMT chimeras than in the WT chimeras (Fig. 4C), which corresponded to differences in lung P. murina burden (Fig. 4B). Although there were no differences in the number of CD4+ T cells in the TBLN between the groups, the μMT chimeras had significantly fewer activated T cells than did the other two groups (Fig. 4D). Interestingly, there was no difference between WT and TNFKO chimeras in the concentration of TNF or IFN-γ in the BALF; however, μMT chimeras had significantly less TNF at day 10 and more IFN-γ at each time point postinfection (Fig. 4H and I). Together, these data indicate that B cell-derived TNF is important for the early control of P. murina infection. However, it was not immediately obvious from this model what the mechanism of B cell-derived TNF is in contributing to control of P. murina infection.
CD4+ T cells primed in the absence of B cell-derived TNF fail to stimulate clearance of P. murina.
To determine the functional consequences of initial priming of CD4+ T cells in an environment lacking B cell-derived TNF, we adoptively transferred CD4+ cells from the TBLNs of P. murina-infected chimeric mice into RAG1KO mice 4 days prior to infection with P. murina. RAG1KO recipients that received CD4+ T cells from WT chimeras were able to significantly reduce lung P. murina burden by day 30 postinfection, while the mice that received T cells from either μMT or TNFKO chimeras were unable to control P. murina through day 30 postinfection (Fig. 5A). Numbers of activated and TNF-producing CD4+ T cells from WT chimeras were slightly elevated at day 10 postinfection in the lungs, while they remained near the limit of detection in mice reconstituted with cells from TNFKO or μMT chimeras (Fig. 5B and D). The reduction in P. murina lung burden in mice that received T cells from WT chimeras followed the appearance of elevated numbers of activated CD4+ and TNF-producing CD4+ cells in the lung at day 21 postinfection (Fig. 5B and D). In contrast, very few activated or TNF-producing CD4+ T cells were found in the BALF or TBLN of mice reconstituted with T cells from μMT chimeras (Fig. 5C and data not shown). Reduced numbers of activated and TNF-producing T cells from TNFKO chimeras were found in the lung digests at day 21 postinfection, although the numbers of these cells increased through day 30, while the number of T cells from WT chimera recipients decreased over this time frame as P. murina lung burden was reduced (Fig. 5A, B, and D). IFN-γ concentrations in the BALF were similar in the wild-type and TNFKO chimeras, although the peak was earlier in the TNFKO chimeras than in the other two groups (Fig. 5F). The μMT chimeras had significantly higher levels of IFN-γ in the BALF at day 20 postinfection than the other groups (Fig. 5F), but the ability to produce IFN-γ did not correspond to an ability to control infection in this experiment. These data demonstrate that B cell-derived TNF is important for the initial priming of CD4+ T cells in response to P. murina and clearance of infection.
Fig 5.
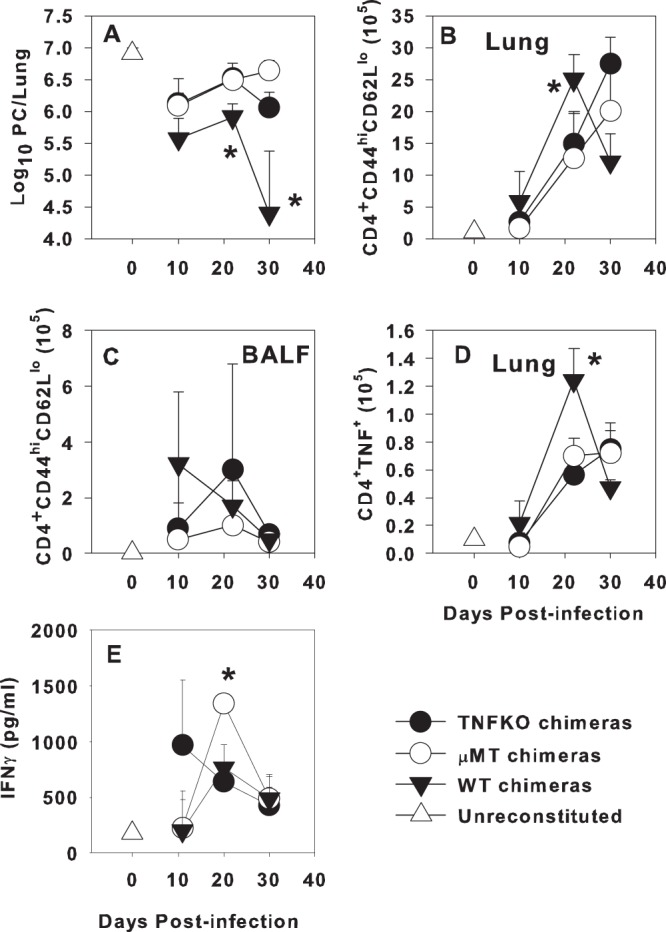
Adoptively transferred CD4+ T cells from μMT or TNFKO B cell chimeras fail to induce clearance of P. murina in adoptive hosts. WT, μMT, and TNFKO mixed bone marrow chimeras were generated as described in Materials and Methods and infected with P. murina, and TBLN CD4+ T cells were isolated at day 14 postinfection for transfer into RAGKO hosts. (A) RAGKO mice were infected with P. murina 4 days after transfer of T cells, and the lung P. murina burden was determined microscopically. Cellular infiltration into the lungs and BALF was determined by flow cytometry. (B to D) The number of activated CD4+ CD44hi CD62Llo cells in the lungs (B) and BALF (C) and the number of CD4+ cells that produce TNF in the lungs (D) are shown. (E) IFN-γ concentrations in BALF were determined by ELISA. Data represent the means ± standard deviations for 4 mice per group per time point. ∗, P < 0.05 compared to WT chimeras at the same time point, as determined by ANOVA and Student-Newman-Keuls post hoc tests.
DISCUSSION
The work presented here confirms our previous finding that B cells promote CD4+ T cell activation in response to P. murina organisms. An important new finding is that B cells produce TNF in the tracheobronchial lymph node as well as in the lungs in response to P. murina stimulation. The data indicate that B cells participate in CD4+ T cell expansion and secretion of proinflammatory cytokines in the course of infection with the fungal pathogen. Work by Whitmire et al. showed that B cells play an important function in sustaining CD4+ T cell memory (40). In their studies, they found that CD4+ T cells form a normal primary response to lymphocytic choriomeningitis virus (LCMV) in the absence of B cells but that this CD4+ T cell response disintegrates quickly and cannot be detected 1 month later. They further observed that the role of B cells in their system was not dependent on antibody secretion. Consistent with our data in this report, they speculate that this novel role of B cells could be a result of B cell-specific local cytokines or direct contact through costimulatory molecules involved in the second signal for T cell activation (40).
It is well established that activated B cells are competent at presenting antigens (16, 17). Previous data from our laboratory showed that major histocompatibility complex class II (MHCII) B cell chimeric mice in which MHC was present on other antigen-presenting cells but not on B cells were not able to clear P. murina infection (6). Moreover, chimeric mice whose B cells were deficient in CD40 cleared P. murina infection but with delayed kinetics compared to wild-type mice (7). Those studies and the current data are consistent with our hypothesis that B cells provide important signals to stimulate CD4+ T cell effector function during P. murina infection. Other investigators have reported that cytokines produced by B cells increase the function of effector T cells (12, 15, 28, 41, 42). In addition, it has been shown that TNF and lymphotoxin produced by B cells are important in the development of lymphoid follicles in Peyer's patches (43) and in the development of T helper responses in lymph nodes (15). It is possible that the TNF produced by B cells in our model could be important in lymphoid architecture, but this cannot be concluded from these studies. Rather, we report that priming of CD4+ T cells in the presence of TNF-producing B cells is important for T cell activation and expansion.
We normally find the same numbers of CD44hi CD62Llo CD4+ T cells in the TBLNs of both WT and B cell-deficient mice, suggesting that the T cells are able to be activated initially, likely by dendritic cell (DC) interactions. This confirms previous studies where we demonstrated that the CD4+ T cells isolated from the TBLNs of B cell-deficient mice responded to in vitro restimulation by proliferating and producing both IFN-γ and IL-2 as well as their counterparts from WT mice (6). Here we report significantly increased numbers of proliferating CD4+ T cells in B cell-deficient mice at later time points postinfection compared to the numbers in WT mice. This could be a result of an increased lung P. murina burden in the B cell-deficient mice. We clearly show no functional defect in the ability of JhKO T cells to proliferate in response to the organisms. However, we have preliminary data suggesting that T cells from infected JhKO mice are also undergoing apoptosis at a higher rate than in WT mice, suggesting that B cells may be providing survival signals for T cells during infection. Indeed, our adoptive transfer model, in which T cells from B cell-deficient mice do not expand as readily as T cells from WT donors, was highly suggestive that B cells are needed for expansion of CD4+ T cells in adoptive hosts. We were able to induce expansion of these cells by transferring B cells along with CD4+ T cells primed in B cell-deficient mice, resulting in the rescue of T cells and control of infection.
Although the role of TNF in host defense against Pneumocystis has been extensively studied, it had been presumed that alveolar macrophages were the most important source of TNF (34, 44). We demonstrated here that B cells were able to produce TNF in the TBLN and lungs in response to P. murina and that CD4+ T cells produced TNF in the TBLN only at early time points. TNF production by B cells corresponded to the proliferation of B cells in the TBLN, which started at day 6 postinfection. There was a significant change in TNF production by CD4+ T cells at time points corresponding to the peak of infection, suggesting that the TNF-producing T cells in the TBLN had moved to the lung after being activated. During germinal center formation, cognate interaction between the B and T cells in the T cell zone leads to activation of B cells. Ngo and colleagues demonstrated that B cells influence splenic T cell zone development by providing signals that promote T cell accumulation (45). Our data suggest that this B cell-T cell interaction could have a beneficial effect by facilitating CD4+ T cell activation or survival (Fig. 4). Consistent with this idea, we found that B cells from wild-type mice were able to rescue the expansion of CD4+ T cells primed in B cell-deficient donors upon transfer to SCID mice (Fig. 1). This could in part be through TNF secretion enabling the CD4+ T cells to expand and secrete proinflammatory cytokines essential for P. murina elimination.
Utilizing mixed bone marrow chimeric mice, we were able to demonstrate for a primary immune response that in the absence of B cell-derived TNF, there was about a 10-fold-higher lung P. murina burden than in wild-type chimeras at day 10 postinfection (Fig. 4). By day 22, there was no difference between the WT and TNFKO chimeras in lung P. murina burden, indicating that B cell-derived TNF is important during the early phase of infection. The increased P. murina burden in the TNFKO chimeras at day 10 corresponded to reduced numbers of activated CD4+ T cells in the lungs but not in the BALF. Transferred CD4+ T cells primed in the TNFKO chimeras were unable to stimulate clearance of P. murina in RAGKO hosts, indicating that B cell-derived TNF is important for the effector function, including TNF production, of T cells. These data suggest that B cells are important for stimulating TNF production in T cells, and it may be that T cell-derived TNF is critical for stimulating the phagocytosis and killing of P. murina by lung phagocytes.
A number of recent reports have demonstrated the importance of B cells and/or TNF in host defense against Pneumocystis jirovecii in humans. Case reports have been sprinkled in the literature, showing that treatment of patients with rituximab, anti-CD20, as a part of chemotherapy for B cell lymphoma can result in severe PCP (46, 47), confirming the importance of B cell function in controlling Pneumocystis infection. There have also been reports that TNF blockers (infliximab, adalimumab, and etanercept) used as therapy for Crohn's disease or rheumatoid arthritis have resulted in PCP in some individuals (26, 27, 48). This confirms the strong animal data demonstrating an important role of TNF in mediating the immune response to Pneumocystis. Together, the data presented here demonstrate a role for B cell-derived TNF in the expansion and function of CD4+ T cells in controlling and clearing lung infection with Pneumocystis. In addition to the well-known role of alveolar macrophages in secreting TNF in response to Pneumocystis, presumably for initiation of an innate inflammatory response in the lungs, we show that B cells produce TNF in the draining lymph nodes during the initiation of adaptive immune responses, and activated CD4+ T cells elaborate TNF in the lungs. B cell-derived TNF appears to provide an expansion or survival signal for CD4+ T cells. These data provide important evidence that TNF is vital during multiple phases of the immune response and is produced locally by multiple cell types to stimulate different immune effector functions.
ACKNOWLEDGMENTS
This work was supported by grant HL088989 from the National Heart, Lung, and Blood Institute to B.A.G. and by resources provided by the Veteran's Affairs Medical Center, Lexington, KY.
We thank J. Louise Lines and Weihua Jiang for technical support.
None of the authors have a financial or other conflict of interest with findings reported in the manuscript.
Footnotes
Published ahead of print 3 September 2013
REFERENCES
- 1.Gamsu G, Hecht ST, Birnberg FA, Coleman DL, Golden JA. 1982. Pneumocystis carinii pneumonia in homosexual men. Am. J. Roentgenol. 139:647–651 [DOI] [PubMed] [Google Scholar]
- 2.Gottlieb MS, Schroff R, Schanker HM, Weisman JD, Fan PT, Wolf RA, Saxon A. 1981. Pneumocystis carinii pneumonitis and mucosal candidiasis in previously healthy homosexual men: evidence of a new acquired cellular immunodeficiency. N. Engl. J. Med. 305:1424–1431 [DOI] [PubMed] [Google Scholar]
- 3.Garvy BA, Wiley JA, Gigliotti F, Harmsen AH. 1997. Protection against Pneumocystis carinii pneumonia by antibodies generated from either T helper 1 or T helper 2 responses. Infect. Immun. 65:5052–5056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Harmsen AG, Chen W. 1992. Resolution of Pneumocystis carinii pneumonia in CD4+ lymphocyte-depleted mice given aerosols of heat-treated Escherichia coli. J. Exp. Med. 176:881–886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harmsen AG, Stankiewicz M. 1990. Requirement for CD4+ cells in resistance to Pneumocystis carinii pneumonia in mice. J. Exp. Med. 172:937–945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lund FE, Hollifield M, Schuer K, Lines JL, Randall TD, Garvy BA. 2006. B cells are required for generation of protective effector and memory CD4 cells in response to Pneumocystis lung infection. J. Immunol. 176:6147–6154 [DOI] [PubMed] [Google Scholar]
- 7.Lund FE, Schuer K, Hollifield M, Randall TD, Garvy BA. 2003. Clearance of Pneumocystis carinii in mice is dependent on B cells but not on P. carinii-specific antibody. J. Immunol. 171:1423–1430 [DOI] [PubMed] [Google Scholar]
- 8.Roths JB, Sidman CL. 1992. Both immunity and hyperresponsiveness to Pneumocystis carinii result from transfer of CD4+ but not CD8+ T cells into severe combined immunodeficiency mice. J. Clin. Invest. 90:673–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roths JB, Sidman CL. 1993. Single and combined humoral and cell-mediated immunotherapy of Pneumocystis carinii pneumonia in immunodeficient scid mice. Infect. Immun. 61:1641–1649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shellito J, Tate C, Ruan S, Kolls J. 2000. Murine CD4+ T lymphocyte subsets and host defense against Pneumocystis carinii. J. Infect. Dis. 181:2011–2017 [DOI] [PubMed] [Google Scholar]
- 11.Rangel-Moreno J, Carragher DM, Misra RS, Kusser K, Hartson L, Moquin A, Lund FE, Randall TD. 2008. B cells promote resistance to heterosubtypic strains of influenza via multiple mechanisms. J. Immunol. 180:454–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wojciechowski W, Harris D, Sprague F, Mousseau B, Makris M, Kusser K, Honjo T, Mohrs K, Mohrs M, Randall T, Lund F. 2009. Cytokine-producing effector B cells regulate type 2 immunity to H. polygyrus. Immunity 30:421–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Matsuzaki G, Vordermeier HM, Hashimoto A, Nomoto K, Ivanyi J. 1999. The role of B cells in the establishment of T cell response in mice infected with an intracellular bacteria, Listeria monocytogenes. Cell. Immunol. 194:178–185 [DOI] [PubMed] [Google Scholar]
- 14.Yang X, Brunham RC. 1998. Gene knockout B cell-deficient mice demonstrate that B cells play an important role in the initiation of T cell responses to Chlamydia trachomatis (mouse pneumonitis) lung infection. J. Immunol. 161:1439–1446 [PubMed] [Google Scholar]
- 15.Leon B, Ballesteros-Tato A, Browning JL, Dunn R, Randall TD, Lund FE. 2012. Regulation of Th2 development by CXCR5+ dendritic cells and lymphotoxin-expressing B cells. Nat. Immunol. 13:681–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Constant SL. 1999. B lymphocytes as antigen-presenting cells for CD4+ T cell priming in vivo. J. Immunol. 162:5695–5703 [PubMed] [Google Scholar]
- 17.Constant S, Schweitzer N, West J, Ranney P, Bottomly K. 1995. B lymphocytes can be competent antigen-presenting cells for priming CD4+ T cells to protein antigens in vivo. J. Immunol. 155:3734–3741 [PubMed] [Google Scholar]
- 18.Gray D, Kosco M, Stockinger B. 1991. Novel pathways of antigen presentation for the maintenance of memory. Int. Immunol. 3:141–148 [DOI] [PubMed] [Google Scholar]
- 19.Meissner N, Rutkowski M, Harmsen AL, Han S, Harmsen AG. 2007. Type I interferon signaling and B cells maintain hemopoiesis during Pneumocystis infection of the lung. J. Immunol. 178:6604–6615 [DOI] [PubMed] [Google Scholar]
- 20.Garvy BA, Gigliotti F, Harmsen AG. 1997. Neutralization of IFNγ exacerbates Pneumocystis-driven interstitial pneumonitis following bone marrow transplantation in mice. J. Clin. Invest. 99:1637–1644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Garvy BA, Ezekowitz RAB, Harmsen AG. 1997. Role of gamma interferon in the host immune and inflammatory responses to Pneumocystis carinii infection. Infect. Immun. 65:373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen W, Havell EA, Harmsen AG. 1992. Importance of endogenous tumor necrosis factor alpha and gamma interferon in host resistance against Pneumocystis carinii infection. Infect. Immun. 60:1279–1284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beck JM, Liggitt HD, Brunette EN, Fuchs HJ, Shellito JE, Debs RJ. 1991. Reduction in intensity of Pneumocystis carinii pneumonia in mice by aerosol administration of gamma interferon. Infect. Immun. 59:3859–3862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rudmann DG, Preston AM, Moore MW, Beck JM. 1998. Susceptibility to Pneumocystis carinii in mice is dependent on simultaneous deletion of IFN-γ and type 1 and 2 TNF receptor genes. J. Immunol. 161:360–366 [PubMed] [Google Scholar]
- 25.Downing J, Kachel D, Pasula R, Martin WJ., II 1999. Gamma interferon stimulates rat alveolar macrophages to kill Pneumocystis carinii by L-arginine- and tumor necrosis factor-dependent mechanisms. Infect. Immun. 67:1347–1352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Seddik M, Meliez H, Seguy D, Viget N, Cortot A, Colombel JF. 2004. Pneumocystis jiroveci (carinii) pneumonia following initiation of infliximab and azathiprine therapy in a patient with Crohn's disease. Inflamm. Bowel Dis. 10:436–437 [DOI] [PubMed] [Google Scholar]
- 27.Itaba S, Iwasa T, Sadamoto Y, Nasu T, Misawa T, Inoue K, Shimokawa H, Nakamura K, Takayanagi R. 2007. Pneumocystis pneumonia during combined therapy of infliximab, corticosteroid, and azathioprine in a patient with Crohn's disease. Dig. Dis. Sci. 52:1438–1441 [DOI] [PubMed] [Google Scholar]
- 28.Harris DP, Haynes L, Sayles PC, Duso DK, Eaton SM, Lepak NM, Johnson LL, Swain SL, Lund FE. 2000. Reciprocal regulation of polarized cytokine production by effector B and T cells. Nat. Immunol. 1:475–482 [DOI] [PubMed] [Google Scholar]
- 29.Agrawal S, Gupta S. 2011. TLR1/2, TLR7, and TLR9 signals directly activate human peripheral blood naive and memory B cell subsets to produce cytokines, chemokines, and hematopoietic growth factors. J. Clin. Immunol. 31:89–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yu M, Wen S, Wang M, Liang W, Li H-H, Long Q, Guo H-P, Liao Y-H, Yuan J. 2013. TNF-α-secreting B cells contribute to myocardial fibrosis in dilated cardiomyopathy. J. Clin. Immunol. 33:1002–1008 [DOI] [PubMed] [Google Scholar]
- 31.Tamburrini E, De Luca A, Ventura G, Maiuro G, Siracusano A, Ortona E, Antinori A. 1991. Pneumocystis carinii stimulates in vitro production of tumor necrosis factor-alpha by human macrophages. Med. Microbiol. Immunol. 180:15–20 [DOI] [PubMed] [Google Scholar]
- 32.Krishnan V, Meager A, Mitchell D, Pinching A. 1990. Alveolar macrophages in AIDS patients: increased spontaneous tumour necrosis factor-alpha production in Pneumocystis carinii pneumonia. Clin. Exp. Immunol. 80:156–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hoffman OA, Standing JE, Limper AH. 1993. Pneumocystis carinii stimulates tumor necrosis factor-alpha release from alveolar macrophages through a beta-glucan-mediated mechanism. J. Immunol. 150:3932–3940 [PubMed] [Google Scholar]
- 34.Kolls JK, Beck JM, Nelson S, Summer WR, Shellito J. 1993. Alveolar macrophage release of tumor necrosis factor during murine Pneumocystis carinii pneumonia. Am. J. Respir. Cell Mol. Biol. 8:370–376 [DOI] [PubMed] [Google Scholar]
- 35.Evans SE, Hahn PY, McCann F, Kottom TJ, Pavlovic ZV, Limper AH. 2005. Pneumocystis cell wall β-glucans stimulate alveolar epithelial cell chemokine generation through nuclear factor-κB-dependent mechanisms. Am. J. Respir. Cell Mol. Biol. 32:490–497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marcotte H, Levesque D, Delanay K, Bourgeault A, de la Durantaye R, Brochu S, Lavoie MC. 1996. Pneumocystis carinii infection in transgenic B cell-deficient mice. J. Infect. Dis. 173:1034–1037 [DOI] [PubMed] [Google Scholar]
- 37.Tough DF, Sprent J, Stephens GL. 2007. Measurement of T and B cell turnover with bromodeoxyuridine. Curr. Protoc. Immunol. 77:4.7.1–4.7.9. 10.1002/0471142735.im0407s77 [DOI] [PubMed] [Google Scholar]
- 38.Carayon P, Bord A. 1992. Identification of DNA-replicating lymphocyte subsets using a new method to label the bromo-doxyuridine incorporated into the DNA. J. Immunol. Methods 147:225–230 [DOI] [PubMed] [Google Scholar]
- 39.Openshaw P, Murphy EE, Hosken NA, Maino V, Davis K, Murphy K, O'Garra A. 1995. Heterogeneity of intracellular cytokine synthesis at the single-cell level in polarized T helper 1 and T helper 2 populations. J. Exp. Med. 182:1357–1367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Whitmire JK, Asano MS, Kaech SM, Sarkar S, Hannum LG, Shlomchik MJ, Ahmed R. 2009. Requirement of B cells for generating CD4+ T cell memory. J. Immunol. 182:1868–1876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Menard LC, Minns LA, Darche S, Mielcarz DW, Foureau DM, Roos D, Dzierszinski F, Kasper LH, Buzoni-Gatel D. 2007. B cells amplify IFN-γ production by T cells via a TNF-α-mediated mechanism. J. Immunol. 179:4857–4866 [DOI] [PubMed] [Google Scholar]
- 42.Wagner M, Poeck H, Jahrsdoerfer B, Rothenfusser S, Prell D, Bohle B, Tuma E, Giese T, Ellwart JW, Endres S, Hartmann G. 2004. IL-12p70-dependent Th1 induction by human B cells requires combined activation with CD40 ligand and CpG DNA. J. Immunol. 172:954–963 [DOI] [PubMed] [Google Scholar]
- 43.Tumanov A, Kuprash D, Lagarkova M, Grivennikov S, Abe K, Shakhov A, Druskaya L, Stewart C, Chervonsky A, Nedospasov S. 2002. Distinct role of surface lymphotoxin expressed by B cell in the organization of secondary lymphoid tissues. Immunity 17:239–250 [DOI] [PubMed] [Google Scholar]
- 44.Vassallo R, Thomas J, Vuk-Pavlovic CFZ, Limper AH. 1999. Alveolar macrophage interactions with Pneumocystis carinii. J. Lab. Clin. Med. 133:535–540 [DOI] [PubMed] [Google Scholar]
- 45.Ngo VN, Cornall RJ, Cyster JG. 2001. Splenic T zone development is B cell dependent. J. Exp. Med. 194:1649–1660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hashimoto K, Kobayashi Y, Asakura Y, Mori M, Azuma T, Maruyama D, Kim SW, Watanabe T, Tobinai K. 2010. Pneumocystis jiroveci pneumonia in relation to CD4+ lymphocyte count in patients with B-cell non-Hodgkin lymphoma treated with chemotherapy. Leuk. Lymphoma 51:1816–1821 [DOI] [PubMed] [Google Scholar]
- 47.Martin-Garrido I, Camona EM, Specks U, Limper AH. 2013. Pneumocystis pneumonia in patients treated with rituximab. Chest 144:258–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kalyoncu U, Karadag O, Akdogan A, Kisacik B, Erman M, Erguven S, Ertenli A. 2007. Pneumocystis carinii pneumonia in a rheumatoid arthritis patient treated with adalimumab. Scand. J. Infect. Dis. 39:475–478 [DOI] [PubMed] [Google Scholar]