

Abstract
Bordetella pertussis is a Gram-negative pathogen causing the human respiratory disease called pertussis or whooping cough. Here we examined the role of the RNA chaperone Hfq in B. pertussis virulence. Hfq mediates interactions between small regulatory RNAs and their mRNA targets and thus plays an important role in posttranscriptional regulation of many cellular processes in bacteria, including production of virulence factors. We characterized an hfq deletion mutant (Δhfq) of B. pertussis 18323 and show that the Δhfq strain produces decreased amounts of the adenylate cyclase toxin that plays a central role in B. pertussis virulence. Production of pertussis toxin and filamentous hemagglutinin was affected to a lesser extent. In vitro, the ability of the Δhfq strain to survive within macrophages was significantly reduced compared to that of the wild-type (wt) strain. The virulence of the Δhfq strain in the mouse respiratory model of infection was attenuated, with its capacity to colonize mouse lungs being strongly reduced and its 50% lethal dose value being increased by one order of magnitude over that of the wt strain. In mixed-infection experiments, the Δhfq strain was then clearly outcompeted by the wt strain. This requirement for Hfq suggests involvement of small noncoding RNA regulation in B. pertussis virulence.
INTRODUCTION
The RNA chaperone Hfq was discovered and characterized more than 40 years ago in Escherichia coli as a host factor required for replication of the bacteriophage Qβ (1). Biochemical and structural studies revealed that Hfq forms a hexameric ring-shaped doughnut structure and contains two distinct RNA binding surfaces on proximal and distal sites of the hexamer (2–4). More recently, Hfq has been recognized as a global posttranscriptional regulatory factor involved in numerous functions in bacteria (5). Several lines of evidence indicate that in its hexameric form Hfq binds cellular mRNAs and small noncoding RNAs (sRNAs) at its distal and proximal sites, respectively (2, 6). In addition, binding of Hfq was shown to induce conformational changes in mRNAs and sRNAs, as well as to modulate their stability (7–10). Bacterial trans-encoded sRNAs are transcribed in response to various stresses and regulate gene expression by base pairing with target mRNA, resulting in either translational activation or repression (11–13). Currently, the ability of the Hfq protein to facilitate interaction between sRNAs and their target mRNAs is considered to be its most prominent feature (14). Disruption of the hfq gene causes broadly pleiotropic effects in Escherichia coli, such as a decreased growth rate and increased sensitivity to stresses, such as UV or high osmolarity (15), showing that Hfq plays a general role in bacterial physiology. Indeed, Hfq-associated sRNAs were shown to be involved in regulation of a variety of cellular processes, including catabolite repression, iron homeostasis, cell envelope integrity, and pathogenesis (16–20). Accordingly, Hfq appears to be essential for the physiological fitness and virulence of a broad spectrum of bacterial pathogens (21). In one of the first studies of Hfq, Hfq was shown to be required for efficient translation of the σS factor of Salmonella enterica serovar Typhimurium, a factor that is crucial for virulence (22). Consequently, an hfq mutant of S. Typhimurium was found to be highly attenuated in a mouse infection model (23). Similarly, experiments employing murine or rat infection models revealed a role of Hfq in the virulence of several pathogenic bacteria (24–28).
Bordetella pertussis is the causative agent of human whooping cough (pertussis), a highly contagious disease that remains one of the 10 most common causes of death from infectious diseases worldwide (29). According to WHO, pertussis accounts for the death of almost 300,000 infants annually, predominantly in developing countries. Despite extensive vaccination programs, the incidence of pertussis is again on the rise even in industrialized countries (30–32). Therefore, there is an urgent need for a better understanding of the molecular mechanisms underlying the pathogenesis of B. pertussis infection. B. pertussis produces a complex array of virulence factors, including adhesins and toxins (33). Among the major adhesins are the filamentous hemagglutinin (FHA), fimbriae, and pertactin. These factors ensure adhesion of B. pertussis to human respiratory tract cells (34). Furthermore, B. pertussis produces two major toxins required for virulence, the pertussis toxin (PT) and the adenylate cyclase (AC) toxin (ACT). The first catalyzes transfer of an ADP-ribosyl group onto the Gαi subunit of the heterotrimeric guanine (G) nucleotide regulatory proteins that regulate endogenous adenylyl cyclase activity (35, 36). The second toxin carries out unregulated conversion of cytosolic ATP to the key second messenger signaling molecule cyclic AMP (cAMP) (37, 38). Both toxins, hence, manipulate cAMP levels in cells, and their cytotoxic enzymatic activities blunt the innate immunity functions of mammalian phagocytes by short-circuiting central signaling pathways. Together, these toxin activities constitute a strategy for B. pertussis to modulate the host immune system and evade its immune response (39–41).
Transcription of the majority of B. pertussis virulence genes, including adhesins and toxins, is controlled by a two-component system encoded by the bvg locus. This consists of a transmembrane sensor kinase (BvgS) and of a response regulator (BvgA), which in its phosphorylated form binds to promoter regions and activates transcription of dependent virulence genes (42). The activity of the BvgAS system can be modulated under laboratory conditions, as growth of B. pertussis cells at 37°C induces BvgAS activity, while growth at temperatures below 25°C or in the presence of millimolar amounts of sulfate or nicotinic acid renders the BvgAS system inactive (43, 44).
Posttranscriptional regulation of B. pertussis virulence has not been studied, and data on Hfq-mediated and sRNA-dependent posttranscriptional regulation of virulence in B. pertussis are lacking. Recently, several sRNAs were identified and characterized in B. pertussis (45), and the gene for an Hfq homologue was identified in the genomes of both fully sequenced B. pertussis strains, Tohama I and 18323 (http://www.sanger.ac.uk). However, its role in the virulence of B. pertussis has not yet been studied.
In this study, we examined the function of Hfq in B. pertussis virulence using an hfq deletion strain of B. pertussis 18323 (WHO reference strain). We show that compared to its parental strain, the Δhfq strain is impaired in growth and produces reduced amounts of one of the key virulence factors of B. pertussis, the adenylate cyclase toxin. Examination of the role of Hfq in B. pertussis virulence in vitro and in vivo revealed that the Δhfq strain is significantly attenuated. Moreover, in the mixed-infection experiment, the hfq mutant was clearly outcompeted by its parental strain.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
The WHO reference strain Bordetella pertussis 18323 (ATCC strain 97-97) and its derivatives were grown on Bordet-Gengou agar (BGA) supplemented with 15% defibrinated sheep blood for 3 to 4 days at 37°C. For liquid cultures, bacteria were grown in Stainer-Scholte (SS) medium (46) at 37°C. Samples for protein analyses were taken at early exponential phase (optical density at 600 nm [OD600], 0.7 and 0.4 for the wild-type [wt] and Δhfq strains, respectively), late exponential phase (OD600, ≈1.3 and 0.7, respectively), early stationary phase (OD600, ≈3.0 and 1.3, respectively), and late stationary phase (OD600 of >4.5 and >1.8, respectively) of growth. Escherichia coli strains were cultured on Luria-Bertani (LB) agar or in LB broth (47). When appropriate, culture media were supplemented with 5 μg/ml of chloramphenicol (Cm) to maintain the Δhfq::Cmr strain and 30 μg/ml of chloramphenicol to select for the pBBRhfq plasmid in the Δhfq strain.
Construction of hfq deletion in B. pertussis and its complementation.
To delete the hfq gene in B. pertussis 18323, two DNA fragments of approximately 750 bp corresponding to either the 5′ or the 3′ flanking region of the hfq gene were created using PCR, as follows: the region upstream of hfq was amplified using forward primer 5′-AAGGATCCCCAATCCCAATGCCCCGAC-3′ containing a BamHI site (underlined) and reverse primer 5′-AAAGCTAGCCATTGGCCAGGCTCCATTTGTTTGT-3′ containing an NheI site (underlined) and the ATG initiation codon of the hfq gene (bold). Similarly, the region downstream of hfq was amplified using forward primer 5′-AAAGCTAGCTAATCGCCTCGCTCCAGCATTACCCTTGA-3′ containing an NheI site (underlined) and the TAA stop codon of the hfq gene (bold) and reverse primer 5′-AAGGATCCCGACCGTGTCCGACAGCACCACCGA-3′ containing a BamHI site (underlined). Both PCR products were cleaved by NheI and ligated. The ligation mixture was then used as a template for the next PCR, together with BamHI site-containing primers. In the resulting product, the start and stop codons of hfq separated by an NheI restriction site create a markerless in-frame hfq deletion. The amplified PCR product was cleaved with BamHI and inserted into the corresponding site of allelic exchange plasmid pSS4245 (48) (kindly provided by Scott Stibitz). The resulting plasmid, pSS4245Δhfq, was transformed into the E. coli SM10 strain (donor strain) and transferred to B. pertussis 18323 (recipient strain) by conjugation, as described elsewhere (48). As a result of two subsequent recombination events, the strain carrying a chromosomal in-frame deletion of the hfq gene was obtained. The deletion of the hfq gene and the sequences of its 5′ and 3′ adjacent regions in the mutant strain were confirmed by DNA sequencing. To generate the Δhfq::Cmr strain, a chloramphenicol resistance cassette flanked by NheI sites was generated by PCR using the vector pBBR1MCS (49) as the template and was inserted into NheI site of plasmid pSS4245Δhfq. The resulting plasmid was used to construct the B. pertussis Δhfq strain resistant to chloramphenicol by homologous recombination, as described above.
To obtain a strain in which the hfq deletion is complemented, plasmid pBBRhfq carrying the hfq gene of the B. pertussis 18323 strain including its own promoter sequence was constructed as follows: the fragment containing the hfq gene was amplified by PCR using the forward primer 5′-CCCGTATCGATCAATTCCTGCGT-3′ and the reverse primer 5′-CTCCTCGACCTTGCCTGAACCG-3′. The blunt-ended PCR product was inserted into the EcoRV site of the cloning vector pBBR1MCS, which replicates in the Bordetellae. The resulting plasmid, pBBRhfq, was transformed into competent cells of E. coli SM10 and transferred into the B. pertussis 18323 Δhfq strain by conjugation. All plasmid constructs were confirmed by DNA sequence analysis.
Determination of enzymatic and biological activities of ACT.
For quantification of the amounts of produced and secreted ACT in cultures, the B. pertussis 18323 wt, Δhfq, and complemented strains were cultivated in SS medium. Samples were taken in duplicate at the times indicated in Fig. 1B and C. The first sample was directly mixed with 8 M urea (1:1) and served for determination of total AC enzyme activity (that secreted into medium plus that associated with cells). The second aliquot was first centrifuged at 16,000 × g to pellet the cells. The supernatant obtained was then mixed with 8 M urea (1:1) and was used to determine the AC activity of the toxin secreted into the medium. For determination of the toxin secretion efficiency, presented in Fig. 1E, the activity of the AC toxin attached to the bacterial surface was assayed in the pelleted fraction. Cell pellets were resuspended in 4 M urea, incubated for 20 min at room temperature to extract the toxin, and centrifuged for 10 min at 13,000 rpm. The soluble fraction was used for determination of extracytoplasmic toxin activity. Aliquots of extracts and of washed cells were loaded on SDS-polyacrylamide gels to check the integrity of the cells after extraction with 4 M urea. AC activities were measured in the presence of 1 μM calmodulin as described previously (50). One unit of AC activity corresponds to 1 μmol of cAMP formed per min at 30°C and pH 8.0. Enzyme activities in samples were normalized to the OD600 of the culture.
Fig 1.

Effect of hfq gene deletion on growth and ACT production. (A) Growth curves of the B. pertussis 18323 wt, Δhfq, and complemented strains. The strains were cultured in SS medium, and the OD600 was measured at the indicated times. (B) Total AC enzyme activities in cultures of the wt, Δhfq, and complemented strains. Culture samples were taken at the indicated times and mixed with 8 M urea (1:1), and cells were lysed by freezing and thawing and used to determine the total AC enzyme activity (that secreted into medium plus that associated with cells). (C) AC activities in culture supernatants of the wt, Δhfq, and complemented strains. (D) Western blot analysis of cell-associated ACT protein levels in both the wt and Δhfq strains. Samples were taken at the early exponential (EE), late exponential (LE), early stationary (ES), and late stationary (LS) phases of growth. Samples equivalent to 0.1 OD600 units of initial culture were separated on SDS-polyacrylamide gels and analyzed by immunoblotting using anti-ACT antibody. (E) The extent (percent) of ACT secretion by the wt and Δhfq strain was determined in samples from exponential (exp) and stationary (stat) phases of growth. The percentage of secreted AC was calculated as the ratio between the AC activity of the secreted toxin (the AC extractable from the bacterial cell surface in 4 M urea plus the AC released into the supernatant fraction) and the total AC activity. Total AC activity in the sample (determined as described for panel B) was set equal to 100%. Bars represent means ± standard deviations of three experiments. (F) Cell-associated activities of AC toxins produced by B. pertussis wt and Δhfq cells. Sheep erythrocytes (5 × 108/ml) were incubated at 37°C with 4 M urea extracts of wt and Δhfq bacteria adjusted to 5 mU/ml of input AC enzyme activity. After 30 min of incubation, aliquots of cell suspensions were repeatedly washed to remove unbound ACT and used to determine the amounts of cell-associated and cell-invasive AC enzyme activities. The activities of ACT produced by the wt strain were taken as 100%. Bars represent the means ± standard deviations of three experiments.
Cell binding and the invasive capacity of the ACT present in urea extracts of bacterial cells were determined as previously described (51). Briefly, sheep erythrocytes (5 × 108 cells/ml) were incubated at 37°C in TNC buffer (20 mM Tris at pH 8.0, 150 mM NaCl, 2 mM CaCl2) with urea extracts of wt and Δhfq cells adjusted to an 80 mM final urea concentration and equal AC enzyme activity. Mock urea buffer was diluted into control erythrocyte suspensions. After 30 min, cells were washed three times in TNE buffer (20 mM Tris at pH 8.0, 150 mM NaCl, 5 mM EDTA) to remove unbound ACT and divided into two aliquots. The first aliquot was directly used to determine the amount of cell-associated AC activity. The second aliquot was treated with 20 μg of trypsin for 15 min at 37°C in order to inactivate the extracellular AC toxin which did not translocate into cells. Fifty micrograms of soybean trypsin inhibitor was added to stop the reaction before the samples were washed three times in TNE buffer and used to determine the amount of cell-invasive AC activity.
Separation of total membrane fractions.
The B. pertussis 18323 wt and Δhfq strains were cultivated in SS medium at 37°C. Samples (≈20 OD600 units) were harvested at late exponential and stationary phases of growth, and cells were pelleted by centrifugation, washed in 20 ml of ice-cold Tris-NaCl (20 mM Tris-HCl, 200 mM NaCl, 1 mM EDTA), and resuspended in 10 ml of Tris-NaCl containing protease inhibitor cocktail (Complete Mini; Roche) and 0.1 μg/ml lysozyme. Cells were broken by sonication, and samples were cleared of unbroken cells by centrifugation at 5,000 × g for 10 min. Cleared lysates were centrifuged at 140,000 × g for 1 h to separate the membrane and soluble fractions. After centrifugation, the pellet (total membrane fraction) was resuspended in 1 ml of Tris-NaCl, divided into aliquots, and stored at −80°C. Membrane fractions were examined by immunoblotting using polyclonal antibodies raised against PtlH (52) and PtlI (kindly provided by C. Locht).
Immunoblotting.
The B. pertussis 18323 wt and Δhfq strains were cultivated in SS medium at 37°C, and cells from 1-ml aliquots were lysed and boiled in sample buffer. To analyze the supernatant fractions, 20-ml aliquots of cell-free supernatants were filtered through 0.22-μm-pore-size filters and precipitated with 10% (wt/vol) trichloroacetic acid, and the precipitates were washed with 80% acetone, dissolved in sample buffer, and boiled. Samples with ODs equivalent to 1 OD600 unit (supernatants) or 0.1 OD600 unit (whole-cell lysates) were separated on SDS-polyacrylamide gels and transferred onto a nitrocellulose membrane. Membranes were probed with mouse polyclonal antibodies raised against the ACT, FHA, and PT proteins (kindly provided by Nicole Guiso) and the BvgA protein (kindly provided by K. Keidel), followed by incubation with anti-mouse IgG conjugated with alkaline phosphatase. The antibody-antigen complexes were visualized using nitroblue tetrazolium– 5-bromo-4-chloro-3-indolylphosphate staining according to the standard protocol.
qPCR.
Bacterial pellets were lysed, and total RNA was extracted using TRI reagent solution (Molecular Research Center), including a treatment with DNase I (Promega, Madison, WI). RNA integrity was checked by electrophoresis. The concentration and purity of RNA were determined using a NanoDrop ND-1000 spectrophotometer (Thermo Fisher Scientific). To study the effect on gene expression, duplicates of 1 μg of total isolated RNA were reverse transcribed into cDNA following the manufacturer's instructions in a 25-μl reaction mixture using a reverse transcription (RT) system (Promega). All primers (Table 1) were designed to anneal at 60°C and were analyzed for secondary structures and for cross-dimers in primer pairs. Quantitative PCR (qPCR) was performed on a Bio-Rad CFX96 instrument using SYBR green JumpStart Taq ReadyMix (Sigma). Briefly, 200 nmol/liter of each primer together with 40 ng of reverse-transcribed RNA was used in a 20-μl qPCR mixture volume, with PCR consisting of an initial step at 95°C for 2 min, followed by 40 cycles of 95°C for 15 s, 60°C for 30 s, and 72°C for 30 s and then recording of the melting curve. The rpoB gene was used as the reference gene, and relative gene expression was quantified using amplification efficiency values (53).
Table 1.
PCR primers used for quantification of gene expression
| Protein | Gene | Orientationa | Oligonucleotide sequence (5′–3′) | Amplicon size (bp) |
|---|---|---|---|---|
| Hemolysin-adenylate cyclase | cyaA | F | CGAGGCGGTCAAGGTGAT | 93 |
| R | GCGGAAGTTGGACAGATGC | |||
| Virulence factor transcription regulator | bvgA | F | AGGTCATCAATGCCGCCA | 140 |
| R | GCAGGACGGTCAGTTCGC | |||
| Filamentous hemagglutinin | fhaB | F | CAAGGGCGGCAAGGTGA | 120 |
| R | ACAGGATGGCGAACAGGCT | |||
| RNA polymerase subunit beta | rpoB | F | GCTGGGACCCGAGGAAAT | 95 |
| R | CGCCAATGTAGACGATGCC | |||
| Pertussis toxin subunit 1 | ptxA | F | CCAGAACGGATTCACGGC | 112 |
| R | CTGCTGCTGGTGGAGACGA | |||
| Pertussis toxin transport protein A | ptlA | F | CCCTTGAAAGACTTGAGAGCATC | 154 |
| R | GCCGCGCAGTACGACCA | |||
| Pertussis toxin transport protein F | ptlF | F | TGTGCTGTATATCAAGGCCAAAT | 96 |
| R | GTTTTGACGAGCAGATTGGTGT | |||
| Pertussis toxin transport protein G | ptlG | F | CACGTTTATCGATTGCATCCTG | 140 |
| R | GTATTCGCCCACCACTGTCG | |||
| Pertussis toxin transport protein H | ptlH | F | ATATAGCGGAAATCTGCGTGAA | 150 |
| R | GGCAAATGTCGCGGTCG |
F, forward primer; R, reverse primer.
Macrophage infection assay.
B. pertussis cells were suspended in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% bovine serum albumin, and RAW 264.7 murine macrophages were infected at a multiplicity of infection (MOI) of 10 bacteria per cell. Bacterial inocula were quantified by plating appropriate dilutions on BGA plates. After addition of bacteria, tissue culture plates were centrifuged for 5 min at 300 × g to facilitate bacterial interaction with macrophage cells. After 60 min of incubation at 37°C with 5% CO2, nonadherent bacteria were removed by three washing steps in DMEM. Then, medium containing 100 μg/ml of polymyxin B sulfate was added for 60 min at 37°C with 5% CO2 to kill extracellular bacteria. Next, infected macrophages were washed with fresh medium containing 30 μg/ml of polymyxin B sulfate and further incubated at 37°C with 5% CO2. The number of viable intracellular bacteria was determined immediately after washing with fresh medium (2 h postinfection) and at 24 and 48 h postinfection. Infected macrophages were lysed with sterile water, and serial dilutions of lysates were rapidly plated in triplicate onto BGA plates to enumerate the CFU.
Mouse infection.
Four-week-old CD-1 Swiss outbred mice were obtained from Charles River Laboratories and maintained under specific-pathogen-free conditions. B. pertussis 18323 strains were grown to exponential phase of growth at 37°C. On the next day, the OD600 values of the cultures were measured and cells were resuspended in sterile phosphate-buffered saline (PBS; pH 7.4) to adjust the cells to the appropriate concentrations. Bacteria were administered intranasally in suspensions of 50 μl. In all experiments, the exact number of delivered bacteria was determined in parallel by plating serial dilutions of challenge suspensions on BGA plates. All experiments were performed at least twice and yielded consistent results.
To determine the 50% lethal dose (LD50) values, groups of five animals were infected with doses of 6 × 106 to 1.5 × 109 CFU/mouse. Mortality was recorded daily for 7 days.
To test the ability of the B. pertussis wt and Δhfq strains to colonize mouse lungs, cohorts of 15 animals were challenged with 2 × 105 CFU/mouse of the wt strain and with either 2 × 105 or 2 × 106 CFU/mouse of the Δhfq strain. Three mice per group were sacrificed at 2 h and 5, 8, 14, and 21 days postinfection. The lungs were removed aseptically and homogenized in 2 ml of PBS using a cell grinder (Heidolph RZR2020). The resulting homogenates were serially diluted with PBS and plated in duplicate on BGA plates, and colonies were counted to calculate the numbers of CFU per mouse.
In the mixed-infection competition experiments, a 1:1 mixture of both wt and Δhfq::Cmr strains (1 × 105 CFU/mouse of each strain) was used to infect mice. Serial dilutions of the infection mixture were plated to determine the input number of CFU (CFUin) for both strains. The bacterial colonization of the lungs was assayed as described above. The lung homogenates were plated in parallel on BGA plates with 5 μg/ml of chloramphenicol (BGACm) or without chloramphenicol to determine the output number of CFU (CFUout). The numbers of CFU of the Δhfq::Cmr strain were calculated directly from BGACm plates, and the numbers of CFU of the wt strain were calculated as the difference between the numbers of CFU obtained from BGA plates and the numbers obtained from BGACm plates. The competitive index (CI) was calculated by the following formula: (number of CFUout of Δhfq::Cmr/number of CFUout of wt)/(number of CFUin of Δhfq::Cmr/number of CFUin of wt) (54).
All animal experiments were approved by the Animal Welfare Committee of the Institute of Microbiology of the ASCR, v.v.i., in Prague, Czech Republic. Handling of the animals was performed according to the Guide for the Care and Use of Laboratory Animals (55); the Act of the Czech National Assembly, Collection of Laws No. 149/2004, inclusive of the amendments on the Protection of Animals against Cruelty; and the Public Notice of the Ministry of Agriculture of the Czech Republic, Collection of Laws No. 207/2004, on the care and use of experimental animals.
Statistical analysis.
When appropriate, the Student t test (SigmaPlot, version 1; Systat Software Inc., CA) was used to compare the mean values between two groups. Differences were considered significant at P values of ≤0.05.
RESULTS
The B. pertussis hfq mutant is impaired in growth and production of ACT.
To determine the impact of hfq gene deletion on B. pertussis physiology, we first compared the growth of the Δhfq mutant in SS medium at 37°C to that of parental strain 18323 (wt). As shown in Fig. 1A, the Δhfq strain grew slower (doubling time [Td], ∼6 h) and reached a lower final optical density than the wt or the complemented Δhfq strain (Td, ∼4 h) that carried the hfq gene on the pBBRhfq plasmid. To examine whether deletion of the hfq gene affected expression of important virulence factors of B. pertussis, we first analyzed the production of the adenylate cyclase (AC) toxin. Therefore, the amounts of the AC enzyme were determined and normalized to the OD600 values of the bacterial cultures, in order to compensate for the growth defect of the Δhfq mutant. As also shown in Fig. 1, in all phases of growth the hfq mutant produced significantly smaller total amounts of ACT (Fig. 1B) and it also released much less ACT into the culture supernatants (Fig. 1C) than the wt or the complemented strain. The amounts of cell-associated ACT detected by immunoblot analysis of whole-cell lysates then paralleled well the total amounts of cell-associated AC enzyme activity measured (cf. Fig. 1B and D).
Typically, most of the ACT secreted by B. pertussis stays firmly bound to the outer bacterial surface (56) and can be extracted from unbroken cells with 4 M urea (57). To test whether the hfq deletion did selectively impair ACT secretion to the bacterial cell surface, the amount of secreted AC activity (i.e., the amount surface bound plus the amount released into the medium) and the amount of total AC activity produced in the bacterial cultures were compared. As shown in Fig. 1E, however, no specific intracellular accumulation of ACT was observed for the Δhfq mutant, and for both the Δhfq and the wt strains, the secreted activity represented about 70 to 80% of the total amount of ACT activity produced per unit of bacterial culture. Apparently, the total level of production rather than the efficacy of ACT secretion specifically was reduced in the Δhfq strain. As further shown in Fig. 1F, the Δhfq strain still produced an active AC toxin that was properly activated for target cell interaction by the essential posttranslational fatty acylation by the CyaC protein, as the AC toxins produced by the Δhfq and wt strains exhibited the same specific erythrocyte binding and cell-invasive AC activities.
Expression and production of virulence factors in wt and Δhfq strains.
To assess if the production of other virulence factors of B. pertussis was affected in the Δhfq strain, we analyzed the production of pertussis toxin (i.e., production of its S1 subunit, PTS1) and of the filamentous hemagglutinin (FHA). As shown in Fig. 2A, the levels of PTS1 detected in whole-cell lysates of the hfq mutant (row P) by Western blotting were at least as high as or higher than those detected in whole-cell lysates of the parental strain. In culture supernatants (row S), however, lower PTS1 levels were observed, suggesting that the capacity of the Δhfq strain to secrete the toxin across the outer bacterial membrane was affected. This defect was hfq specific, as PT secretion was fully restored in the complemented Δhfq mutant (Fig. 2B).
Fig 2.

Effects of hfq deletion on expression and production of virulence factors in B. pertussis 18323. (A) Western blot analysis of pertussis toxin (PTS1 subunit), FHA, and BvgA protein levels in both wt and Δhfq strains. Samples were taken at early exponential (EE), late exponential (LE), early stationary (ES), and late stationary (LS) phases of growth. Samples equivalent to 0.1 OD600 units (pellets [rows P]) or 1 OD600 unit (supernatants [rows S]) of the initial culture were analyzed by immunoblotting using anti-ACT, anti-PTS1, anti-FHA, and anti-BvgA antibodies. Relevant parts of the membrane are shown. (B) Western blot analysis of pertussis toxin (PTS1 subunit) protein levels in the wt, Δhfq, and complemented strain. Samples were taken at late exponential (Ex) and late stationary (St) phases of growth. Samples equivalent to 0.1 OD600 units (pellets) or 1 OD600 unit (supernatants) of the initial culture were analyzed by immunoblotting using anti-PTS1 antibodies. Relevant parts of the membrane are shown. compl, complemented strain. (C) RT-qPCR analysis of ptxA, cyaA, fhaB, and bvgA transcript levels in wt and Δhfq bacteria. Total RNA was isolated from cells grown to late exponential (exp) and late stationary (stat) phases, and RT-qPCR was performed using primers specific for the ptxA (encoding PTS1), cyaA, fhaB, and bvgA genes. Target expression levels in the Δhfq and wt strains were normalized to the expression level of the reference gene rpoB, and the Δhfq strain/wt expression ratios were calculated (50). Bars represent means ± standard deviations from three experiments. (D) RT-qPCR analysis of ptlA, ptlF, ptlG, and ptlH transcript levels in both wt and Δhfq strains was performed as described for panel C. (E) Composition of membrane fractions of the wt and Δhfq strains (top) and Western blot analysis of PtlH and PtlI protein levels (middle and bottom). Samples of total membrane fractions (10 μg per lane) isolated from wt and Δhfq cells grown to late exponential or late stationary phases were loaded in duplicate onto SDS-polyacrylamide gels and either stained with Coomassie brilliant blue (top) or transferred onto a nitrocellulose membrane. Membranes were probed with anti-PtlH and anti-PtlI antibodies (middle and bottom; the relevant parts of the membrane are shown). Asterisks, major differences in membrane protein composition between wt and Δhfq samples. The positions of molecular size markers are indicated in the lane on the left.
Similar to PT secretion, FHA secretion was also altered in the Δhfq strain. When grown to exponential phase, the mutant released smaller amounts of FHA into the culture supernatants than the wt strain and the FHA produced accumulated in association with Δhfq cells (Fig. 2A). In the stationary phase, however, the levels of cell-associated and secreted FHA were comparable for both strains.
To analyze in more detail the potential differences in expression of the virulence factor genes between the wt and the Δhfq strains, total RNA was purified from bacterial cells grown to exponential and stationary phases and the relative levels of cyaA, ptxA, and fhaB mRNAs (encoding ACT, PTS1, and FHA, respectively) were analyzed by quantitative PCR (RT-qPCR). As shown in Fig. 2C, similar relative levels of cyaA gene expression were observed for the wt and the Δhfq strain, thus suggesting that posttranscriptional regulation may be accounting for the reduction of ACT production by the Δhfq strain (Fig. 1). Interestingly, in the stationary phase of growth, the relative amount of the ptxA transcript was even increased in the Δhfq strain, which was in good agreement with the increased levels of the PTS1 protein in extracts of the Δhfq cells. The relative levels of the fhaB transcript, then, did not differ importantly between the strains. To assess whether alteration of bvgAS expression might have accounted for the decrease of ACT production and decreased secretion of PT and FHA, the bvgA mRNA level and the level of BvgA protein production were examined. As shown in Fig. 2C and A, however, the levels of these proteins did not markedly differ between the wt and the Δhfq strain.
To corroborate these observations, we examined whether the decreased secretion of PT by the Δhfq strain could be due to altered expression of the ptl genes. Secretion of PT across the outer bacterial membrane depends on the function of a dedicated type IV transport system that is encoded by nine ptl genes located directly downstream of the ptx genes within a single ptx-ptl operon (58, 59). Therefore, we quantified by RT-qPCR the transcription levels of the ptlA, ptlF, ptlG, and ptlH genes, which are located at the 5′ and 3′ ends of the multicistronic ptl transcript. As shown in Fig. 2D, however, similar to ptxA, the relative expression level of the ptl genes was also increased severalfold in the hfq mutant. Nevertheless, this did not translate into increased Ptl protein incorporation into the bacterial cell envelope, since similar amounts of the PtlH and PtlI proteins were detected in total membrane fractions of the Δhfq and wt bacteria by Western blotting (Fig. 2E). As further indicated by asterisks in Fig. 2E, some differences in the protein compositions of the membrane fractions of the Δhfq and wt bacteria were already observed upon a simple one-dimensional SDS-PAGE analysis. This suggests that alterations of PT and FHA secretion may have been due, at least in part, to unspecific alterations of the cell envelope composition/functionality in the Δhfq bacteria.
The Δhfq strain is significantly attenuated in virulence.
Manipulation of cAMP signaling in immune cells by the AC toxin results in inhibition of the chemotactic, phagocytic, and oxidative burst capacities of host phagocytes and thereby contributes to the persistence of B. pertussis in the host (37, 57). Since the Δhfq strain produced smaller amounts of AC toxin, we predicted that its capacity to persist within immune cells could be compromised. Therefore, the intracellular survival of wt and Δhfq bacteria in RAW 264.7 macrophages was compared. As shown in Fig. 3, the relative survival capacity of the hfq mutant in macrophages was, indeed, significantly affected. Compared to the wt or the complemented strain, about 2-fold lower numbers of viable Δhfq bacteria were recovered from macrophages at 2 h postinfection. The difference was even more pronounced at later time points, and in contrast to the results for the wt strain, no viable Δhfq bacteria could be recovered from macrophage cells at 48 h postinfection. The numbers of complemented Δhfq bacteria recovered at 2, 24, and 48 h postinfection did not differ statistically significantly from the numbers determined for the wt strain. It can, hence, be concluded that Hfq is required for expression of factors involved in the intracellular persistence of B. pertussis in macrophages in vitro.
Fig 3.
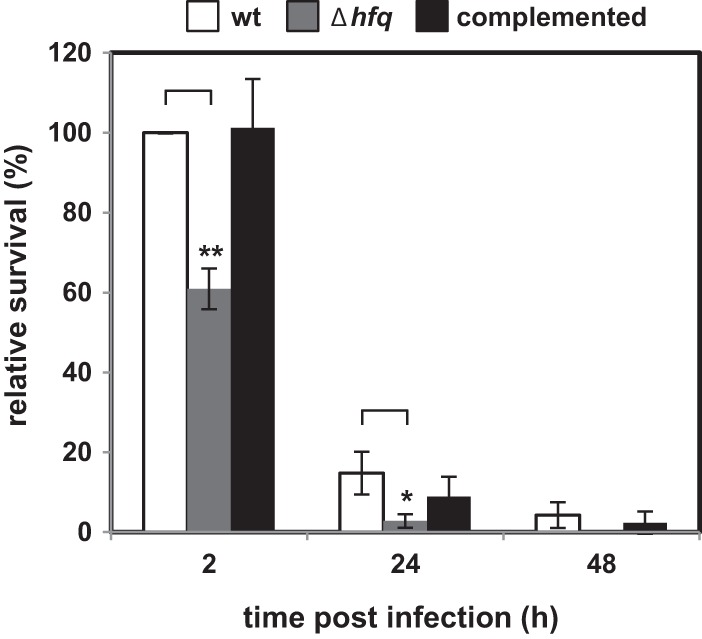
Effect of hfq deletion on B. pertussis survival within macrophages. Approximately 2 × 106 CFU of the wt, Δhfq, and complemented bacteria were incubated with 2 × 105 RAW 264.7 macrophage cells (MOI, 10) in DMEM at 37°C. After 1 h of incubation, noninternalized bacteria were repeatedly washed away and the remaining extracellular bacteria were killed by addition of polymyxin B sulfate to 100 μg/ml for 1 h. Macrophages were lysed at 2, 24, and 48 h postinfection, and the numbers of CFU of viable intracellular bacteria were determined by plating on BGA. Results are expressed as the percentage of the numbers of CFU of wt, Δhfq, and complemented bacteria and are representative of three independent experiments performed in triplicate. The numbers of CFU of surviving wt cells recovered at 2 h postinfection were taken as 100% survival. Bars represent means ± standard deviations. *, P < 0.05 for the Δhfq strain versus the wt strain; **, P < 0.002 for the Δhfq strain versus the wt strain.
To analyze the impact of hfq deletion on B. pertussis virulence in vivo, the mouse intranasal infection model was used. As shown in Fig. 4A, while administration of 1.5 × 108 CFU/mouse of the wt bacteria caused the death of all inoculated mice within 4 days, no lethality was observed with the same dose of Δhfq bacteria, which caused a comparably lethal infection only at a 10-fold increased challenge dose (1.5 × 109 CFU/mouse). When the doses causing the death of 50% of the infected animals (LD50s) were calculated from several infection experiments, values of 6 × 107, 7 × 107, and 8 × 108 CFU/mouse were found for the wt, the complemented, and the Δhfq strains, respectively. Clearly, a lack of Hfq function resulted in an important reduction of B. pertussis virulence.
Fig 4.

Effect of hfq deletion on B. pertussis 18323 virulence in mice. (A) Survival of mice after intranasal challenge with wt or Δhfq bacteria. Groups of five animals were infected with doses of 1.5 × 108 CFU/mouse of the wt strain and with either 1.5 × 108 or 1.5 × 109 CFU/mouse of the Δhfq strain. Mortality was recorded daily for 7 days. Results are representative of three experiments. (B) Colonization of mouse lungs after intranasal challenge with the wt or Δhfq strain. For each strain, 15 mice were infected intranasally with 2 × 105 CFU/mouse and groups of three animals were sacrificed at 2 h (day 0) and 5, 8, 14, and 21 days after infection. Lung homogenates were plated in duplicate on BGA plates, and the numbers of CFU of the wt and Δhfq strains were counted. The values are means ± standard deviations. *, P < 0.02 for the Δhfq strain versus the wt strain; **, P < 0.001 for the Δhfq strain versus the wt strain. Results are representative of three experiments. (C) Colonization of mouse lungs upon infection with an increased inoculum of the Δhfq strain. For each strain, 15 mice were infected intranasally either with 2 × 105 CFU/mouse of the wt strain or with 2 × 106 CFU/mouse of the Δhfq strain. The numbers of CFU in mouse lung homogenates were counted as described for panel B at 2 h (day 0) and 5, 8, 14, and 21 days after infection. The values are means ± standard deviations. *, P < 0.05 for the Δhfq strain versus the wt strain; **, P < 0.005 for the Δhfq strain versus the wt strain. (D) In vivo competition between the B. pertussis 18323 wt and Δhfq::Cmr strains during mixed infection of mice. Eighteen mice were challenged intranasally with a 1:1 mixture of the two strains (1 × 105 CFU/mouse of each strain), and groups of three animals were sacrificed at 2 h (day 0) and 5, 8, 12, 15, and 21 days after infection. Lung homogenates were plated in duplicate on BGA and BGACm plates, and the numbers of CFU of the wt and Δhfq::Cmr strains were counted. The values are means ± standard deviations. *, P < 0.003 for the Δhfq strain versus the wt strain in mixed culture; **, P < 0.001 for the Δhfq strain versus the wt strain in mixed culture. Results are representative of two experiments. Dashed lines, limit of detection.
Therefore, the capacity of the Δhfq strain to colonize the lungs of mice infected with sublethal doses of 2 × 105 CFU/mouse was assessed. As illustrated in Fig. 4B, at this inoculation dose, the wt strain was able to proliferate in lungs and its counts increased by almost 2 orders of magnitude within 8 days, with approximately 7 × 106 CFU being recovered from mouse lungs at days 5 and 8 postinfection. In contrast, the hfq mutant was unable to proliferate and colonized mouse lungs with a significantly lower efficacy at all times. Moreover, as shown in Fig. 4C, the Δhfq strain was unable to proliferate or colonize mouse lungs even when its infection dose was increased 10-fold (2 × 106 CFU/mouse), in order to compensate for the extended generation time of the hfq mutant. This suggests that the colonization defect of the hfq mutant was primarily due to reduced expression of virulence factors rather than due to its reduced growth rate.
The Δhfq strain is outcompeted in mixed-infection experiments.
To determine whether the Δhfq strain was at a competitive disadvantage when coinfecting mice together with the wt strain, we performed competitive infection experiments. Mice were infected intranasally with a sublethal dose of a 1:1 mixture of wt and Δhfq bacteria (1 × 105 CFU/mouse of each strain). In order to distinguish viable wt and Δhfq cells recovered from lungs, an isogenic hfq mutant carrying a chloramphenicol resistance cassette (Δhfq::Cmr) was used here. As a parallel noncompetitive condition control, both wt and Δhfq::Cmr strains were also administered separately (data not shown). As shown in Fig. 4D, in a mixed-infection experiment, the wt strain colonized lungs at levels which were significantly higher than those reached by the hfq::Cmr mutant. Similar to the findings for the single-strain infection (Fig. 4B and C), the wt bacteria exhibited a typical initial rise in numbers of CFU, peaking at days 5 and 8, while at between days 8 and 21, the wt bacteria were progressively cleared from the lungs. In contrast, the Δhfq bacteria already began to be cleared from day 0 in the mixed-infection experiment, and Δhfq cells were completely eliminated from mouse lungs by day 12. As expected, however, in the single-strain infection, the Δhfq::Cmr bacteria persisted in the lungs up to day 21 (data not shown), as did the markerless Δhfq strain (Fig. 4B). Hence, in the mixed-infection experiment, the hfq mutant was much less competitive than its parental strain, exhibiting a dramatically decreased persistence capacity and calculated competition indexes (CIs) of only 0.003 and 0.001 on days 5 and 8 postinfection, respectively.
DISCUSSION
We show here that the RNA chaperone Hfq is required for full expression of B. pertussis virulence and persistence in in vitro and in vivo models of infection. This indicates that expression of at least some of the virulence genes of B. pertussis might involve regulatory sRNA that depends on the function of Hfq.
In agreement with the phenotypes of the Δhfq derivatives of other bacteria (15, 25), the B. pertussis 18323 hfq mutant was moderately impaired in growth, exhibited a longer replication time, and reached a lower final culture density than the parental strain. This phenotype was expected, as pleiotropic effects of deletion of the hfq gene on the physiology of Gram-negative bacteria were previously reported (15, 21). Furthermore, Hfq was also previously implicated in the regulation of toxin production in several pathogenic bacteria (24, 60, 61). In line with this, we observed that Hfq function was required for the production and/or secretion of several key virulence factors of B. pertussis, such as the adenylate cyclase toxin, the pertussis toxin, and the filamentous hemagglutinin.
Compared to the parental strain on a per bacterial cell basis, the Δhfq strain produced about 5-fold lower total amounts of the AC toxin. The AC toxin, however, exhibited the same specific activity in binding and penetration of red blood cells as the toxin produced by the wt strain. Hence, the ACT produced by the Δhfq strain appeared to be properly activated through posttranslational palmitoylation by the CyaC expressed from the cya locus (62, 63). Moreover, the fraction (in percent) of the total amount of AC toxin produced that was secreted out of the bacterial cytosol to the outer bacterial surface and to the culture supernatants was similar for the parental and the Δhfq strain. The decreased production of ACT was not due to a reduced transcription of the cyaA gene, since normal levels of the cyaA transcript were detected in Δhfq cells (Fig. 2C) and the levels of transcription of the cyaBDE genes were also comparable for both the Δhfq and the wt strains (data not shown). It appears that the decrease of ACT secretion by the hfq mutant was primarily due to a reduction in the level of ACT protein synthesis. The possibility that the ACT protein may have been produced at a normal rate in Δhfq cells and was rapidly turned over intracellularly remains to be excluded. More plausibly, however, these results indicate that translation of cyaA mRNA may be subject to posttranscriptional regulation dependent on the function of the RNA chaperone Hfq, such as riboregulation by an Hfq-dependent sRNA.
A somewhat different picture was observed for production and secretion of PT and FHA. Secretion of FHA was reduced in the Δhfq strain, albeit only in the exponential phase, while expression of the fhaB gene was not affected. The Δhfq strain was then found to secrete smaller amounts of PTS1 into culture supernatants than the parental strain, suggesting that secretion of PT was affected in the hfq mutant. This, however, was not due to reduced expression of the ptx-ptl operon. Expression of the ptxA gene encoding the S1 subunit of PT (PTS1) was found to be increased, and increased amounts of the PTS1 protein accumulated in Δhfq cells. Similarly, expression of the ptl genes, encoding proteins involved in secretion of pertussis toxin, was elevated in the hfq mutant, while the levels of PtlH, a transport protein with ATPase activity (52), were comparable in both the Δhfq and the parental strains. Since the levels and membrane localization of the PtlH protein depend on the presence and localization of the PtlD, PtlE, PtlF, and PtlG proteins (52), it is plausible to assume that the levels of all of the Ptl proteins produced were similar for the hfq mutant and the parental strain. These results indicate that the observed reduction of PT secretion by the Δhfq strain was not due to a problem in expression of the type IV translocator of PT but, rather, that it was due to a more general alteration of cell envelope composition and function in the Δhfq bacteria. Similarly, secretion of FHA was reduced in the Δhfq strain as well; however, it was reduced only in the exponential phase (Fig. 2A), while there was no difference in expression of the corresponding gene, fhaB (Fig. 2C). Since the expression of virulence factors is under the control of the BvgAS two-component system, both the protein and transcript levels of the transcriptional regulator BvgA and its structural gene were analyzed as well. Compared to the wt strain, neither protein nor relative transcript levels were significantly changed in the hfq mutant (Fig. 2A and C). Although we did not analyze the phosphorylated form of BvgA, which is the only active form, we believe that the observed effects are linked to hfq.
Loss of Hfq was, indeed, previously reported to induce cell envelope stress and was shown to affect membrane homeostasis in several pathogenic bacteria (23, 27, 64). In line with that, we also observed differences in the protein compositions of membranes from wt and Δhfq bacteria (Fig. 2E). On the other hand, no difference in sensitivity to the membrane-disrupting activity of the antimicrobial compound polymyxin B was observed between wt and Δhfq cells (data not shown). Hence, the integrity of the outer membrane of the B. pertussis Δhfq strain was not as importantly compromised, as previously reported for the hfq mutant of a uropathogenic E. coli strain (65).
Besides impacting the growth rate, deletion of the hfq gene resulted in reduced production of both the adenylate cyclase toxin and the pertussis toxin, which are required for the efficient and successful establishment and persistence of B. pertussis infections (66, 67). Therefore, we further analyzed if reduced fitness and toxin production compromised the survival of the Δhfq strain within macrophage cells in vitro. While no significant difference in the ability to associate with cells was observed with the Δhfq strain (data not shown), its relative survival capacity in macrophage cells was significantly reduced compared to that of the wt or the complemented strain (Fig. 3). A similar phenotype was, indeed, also recently reported for hfq mutants of Yersinia pestis and S. Typhimurium (23, 25).
Not surprisingly, the reduced growth rate and altered production and secretion of such central virulence factors like ACT, PT, and FHA resulted in a decrease of virulence of the Δhfq strain in the in vivo challenge model of intranasal mouse infection. The hfq mutant was affected both in its capacity to cause a lethal infection (LD50) and in its ability to efficiently multiply and persist in mouse lungs. The LD50 value for the Δhfq strain was about 10-fold higher than that of the parental strain or of the complemented strain. These results go well with the previous observations obtained with ΔcyaA strains of the Tohama I and 18323 strains, which also exhibited a colonization defect in mice (67, 68). Importantly, the impairment of proliferation of the Δhfq strain in mouse lungs was not primarily due to slower growth of the mutant, since it could not be compensated for by a 10-fold increased infection dose (Fig. 4C), and even a 100-fold increase of the infection dose did not allow the colonization deficiency of the Δhfq strain to be overcome (data not shown).
Previous mixed-infection experiments demonstrated that in contrast to PT, which acts as a soluble factor benefiting the whole infecting bacterial population (69), ACT acts more as a cell-associated factor that benefits only the cells that produce it, and therefore, cells lacking ACT have been outcompeted in mixed infections (68). Therefore, we have asked whether the Δhfq strain would also show a competitive disadvantage in a mixed-infection experiment. The hfq mutant of B. pertussis exhibited, indeed, a striking colonization defect, when used in coinfection experiments with the wt strain. In contrast to single-strain infections, where Δhfq bacteria could be detected in lungs until day 21, in the mixed infection, the Δhfq strain was outcompeted by wt bacteria and was already completely eliminated from mouse lungs by day 12. The competitive index (CI) is considered a sensitive measure with which to assay virulence attenuation (54, 70). CI indexes calculated at days 5 and 8 postinfection were found to be only 0.003 and 0.001, respectively, which were values very similar to those obtained in mixed infections with wt and Δhfq strains of Y. pestis and S. Typhimurium (23, 25).
To conclude, the results reported here clearly point toward the requirement for Hfq function in posttranscriptional regulation of B. pertussis virulence. It will, hence, be of interest to use the Δhfq strain characterized here to unravel the sRNA-mediated regulation of B. pertussis virulence genes by employing genome-wide expression analysis methods, such as DNA microarray analysis and high-throughput sequencing.
ACKNOWLEDGMENTS
We thank H. Lukeová for excellent technical help and J. Držmíšek for help with gene cloning. The kind gifts of antibodies by Nicolle Guiso (Institute Pasteur, Paris, France), Drusilla Burns (FDA, Bethesda, MD), Camille Locht (Pasteur Institute, Lille, France), and Kristina Keidel (Julius Maximillian University, Wurzburg, Germany), as well as advice on the use of the pSS4245 plasmid and the helpful discussions with Scott Stibitz (FDA, Bethesda, MD), are gratefully acknowledged.
This work was supported by grants P302/11/1940 (to B.V.) and P302/12/0460 (to J.M.) from the Czech Science Foundation (www.gacr.cz) and by funding from RVO61388971.
I.B. and K.S. are doctoral students of the Institute of Chemical Technology in Prague, Czech Republic, and O.C. is a doctoral student of the Faculty of Science of the Charles University in Prague, Czech Republic.
Footnotes
Published ahead of print 26 August 2013
REFERENCES
- 1.Franze de Fernandez MT, Eoyang L, August JT. 1968. Factor fraction required for the synthesis of bacteriophage Qbeta-RNA. Nature 219:588–590 [DOI] [PubMed] [Google Scholar]
- 2.Mikulecky PJ, Kaw MK, Brescia CC, Takach JC, Sledjeski DD, Feig AL. 2004. Escherichia coli Hfq has distinct interaction surfaces for DsrA, rpoS and poly(A) RNAs. Nat. Struct. Mol. Biol. 11:1206–1214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Valentin-Hansen P, Eriksen M, Udesen C. 2004. The bacterial Sm-like protein Hfq: a key player in RNA transactions. Mol. Microbiol. 51:1525–1533 [DOI] [PubMed] [Google Scholar]
- 4.Schumacher MA, Pearson RF, Moller T, Valentin-Hansen P, Brennan RG. 2002. Structures of the pleiotropic translational regulator Hfq and an Hfq-RNA complex: a bacterial Sm-like protein. EMBO J. 21:3546–3556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brennan RG, Link TM. 2007. Hfq structure, function and ligand binding. Curr. Opin. Microbiol. 10:125–133 [DOI] [PubMed] [Google Scholar]
- 6.Link TM, Valentin-Hansen P, Brennan RG. 2009. Structure of Escherichia coli Hfq bound to polyriboadenylate RNA. Proc. Natl. Acad. Sci. U. S. A. 106:19292–19297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Geissmann TA, Touati D. 2004. Hfq, a new chaperoning role: binding to messenger RNA determines access for small RNA regulator. EMBO J. 23:396–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moll I, Afonyushkin T, Vytvytska O, Kaberdin VR, Blasi U. 2003. Coincident Hfq binding and RNase E cleavage sites on mRNA and small regulatory RNAs. RNA 9:1308–1314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sledjeski DD, Whitman C, Zhang A. 2001. Hfq is necessary for regulation by the untranslated RNA DsrA. J. Bacteriol. 183:1997–2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Afonyushkin T, Vecerek B, Moll I, Blasi U, Kaberdin VR. 2005. Both RNase E and RNase III control the stability of sodB mRNA upon translational inhibition by the small regulatory RNA RyhB. Nucleic Acids Res. 33:1678–1689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Waters LS, Storz G. 2009. Regulatory RNAs in bacteria. Cell 136:615–628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Majdalani N, Vanderpool CK, Gottesman S. 2005. Bacterial small RNA regulators. Crit. Rev. Biochem. Mol. Biol. 40:93–113 [DOI] [PubMed] [Google Scholar]
- 13.Gottesman S, McCullen CA, Guillier M, Vanderpool CK, Majdalani N, Benhammou J, Thompson KM, FitzGerald PC, Sowa NA, FitzGerald DJ. 2006. Small RNA regulators and the bacterial response to stress. Cold Spring Harbor Symp. Quant. Biol. 71:1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vogel J, Luisi BF. 2011. Hfq and its constellation of RNA. Nat. Rev. Microbiol. 9:578–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tsui HC, Leung HC, Winkler ME. 1994. Characterization of broadly pleiotropic phenotypes caused by an hfq insertion mutation in Escherichia coli K-12. Mol. Microbiol. 13:35–49 [DOI] [PubMed] [Google Scholar]
- 16.Masse E, Gottesman S. 2002. A small RNA regulates the expression of genes involved in iron metabolism in Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 99:4620–4625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang A, Altuvia S, Tiwari A, Argaman L, Hengge-Aronis R, Storz G. 1998. The OxyS regulatory RNA represses rpoS translation and binds the Hfq (HF-I) protein. EMBO J. 17:6061–6068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vecerek B, Moll I, Blasi U. 2007. Control of Fur synthesis by the non-coding RNA RyhB and iron-responsive decoding. EMBO J. 26:965–975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Udekwu KI, Darfeuille F, Vogel J, Reimegard J, Holmqvist E, Wagner EG. 2005. Hfq-dependent regulation of OmpA synthesis is mediated by an antisense RNA. Genes Dev. 19:2355–2366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sonnleitner E, Abdou L, Haas D. 2009. Small RNA as global regulator of carbon catabolite repression in Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. U. S. A. 106:21866–21871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chao Y, Vogel J. 2010. The role of Hfq in bacterial pathogens. Curr. Opin. Microbiol. 13:24–33 [DOI] [PubMed] [Google Scholar]
- 22.Brown L, Elliott T. 1996. Efficient translation of the RpoS sigma factor in Salmonella typhimurium requires host factor I, an RNA-binding protein encoded by the hfq gene. J. Bacteriol. 178:3763–3770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sittka A, Pfeiffer V, Tedin K, Vogel J. 2007. The RNA chaperone Hfq is essential for the virulence of Salmonella typhimurium. Mol. Microbiol. 63:193–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sonnleitner E, Hagens S, Rosenau F, Wilhelm S, Habel A, Jager KE, Blasi U. 2003. Reduced virulence of a hfq mutant of Pseudomonas aeruginosa O1. Microb. Pathog. 35:217–228 [DOI] [PubMed] [Google Scholar]
- 25.Geng J, Song Y, Yang L, Feng Y, Qiu Y, Li G, Guo J, Bi Y, Qu Y, Wang W, Wang X, Guo Z, Yang R, Han Y. 2009. Involvement of the post-transcriptional regulator Hfq in Yersinia pestis virulence. PLoS One 4:e6213. 10.1371/journal.pone.0006213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Christiansen JK, Larsen MH, Ingmer H, Sogaard-Andersen L, Kallipolitis BH. 2004. The RNA-binding protein Hfq of Listeria monocytogenes: role in stress tolerance and virulence. J. Bacteriol. 186:3355–3362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ding Y, Davis BM, Waldor MK. 2004. Hfq is essential for Vibrio cholerae virulence and downregulates sigma expression. Mol. Microbiol. 53:345–354 [DOI] [PubMed] [Google Scholar]
- 28.Dietrich M, Munke R, Gottschald M, Ziska E, Boettcher JP, Mollenkopf H, Friedrich A. 2009. The effect of hfq on global gene expression and virulence in Neisseria gonorrhoeae. FEBS J. 276:5507–5520 [DOI] [PubMed] [Google Scholar]
- 29.WHO 2006. Vaccine preventable deaths and the Global Immunization Vision and Strategy, 2006-2015. MMWR Morb. Mortal. Wkly. Rep. 55:511–515 [PubMed] [Google Scholar]
- 30.de Melker HE, Versteegh FG, Schellekens JF, Teunis PF, Kretzschmar M. 2006. The incidence of Bordetella pertussis infections estimated in the population from a combination of serological surveys. J. Infect. 53:106–113 [DOI] [PubMed] [Google Scholar]
- 31.Mooi FR. 2010. Bordetella pertussis and vaccination: the persistence of a genetically monomorphic pathogen. Infect. Genet. Evol. 10:36–49 [DOI] [PubMed] [Google Scholar]
- 32.Crowcroft NS, Stein C, Duclos P, Birmingham M. 2003. How best to estimate the global burden of pertussis? Lancet Infect. Dis. 3:413–418 [DOI] [PubMed] [Google Scholar]
- 33.Locht C. 1999. Molecular aspects of Bordetella pertussis pathogenesis. Int. Microbiol. 2:137–144 [PubMed] [Google Scholar]
- 34.van den Berg BM, Beekhuizen H, Willems RJ, Mooi FR, van Furth R. 1999. Role of Bordetella pertussis virulence factors in adherence to epithelial cell lines derived from the human respiratory tract. Infect. Immun. 67:1056–1062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Katada T, Tamura M, Ui M. 1983. The A protomer of islet-activating protein, pertussis toxin, as an active peptide catalyzing ADP-ribosylation of a membrane protein. Arch. Biochem. Biophys. 224:290–298 [DOI] [PubMed] [Google Scholar]
- 36.Moss J, Stanley SJ, Burns DL, Hsia JA, Yost DA, Myers GA, Hewlett EL. 1983. Activation by thiol of the latent NAD glycohydrolase and ADP-ribosyltransferase activities of Bordetella pertussis toxin (islet-activating protein). J. Biol. Chem. 258:11879–11882 [PubMed] [Google Scholar]
- 37.Confer DL, Eaton JW. 1982. Phagocyte impotence caused by an invasive bacterial adenylate cyclase. Science 217:948–950 [DOI] [PubMed] [Google Scholar]
- 38.Glaser P, Sakamoto H, Bellalou J, Ullmann A, Danchin A. 1988. Secretion of cyclolysin, the calmodulin-sensitive adenylate cyclase-haemolysin bifunctional protein of Bordetella pertussis. EMBO J. 7:3997–4004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vojtova J, Kamanova J, Sebo P. 2006. Bordetella adenylate cyclase toxin: a swift saboteur of host defense. Curr. Opin. Microbiol. 9:69–75 [DOI] [PubMed] [Google Scholar]
- 40.Khelef N, Zychlinsky A, Guiso N. 1993. Bordetella pertussis induces apoptosis in macrophages: role of adenylate cyclase-hemolysin. Infect. Immun. 61:4064–4071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Boyd AP, Ross PJ, Conroy H, Mahon N, Lavelle EC, Mills KH. 2005. Bordetella pertussis adenylate cyclase toxin modulates innate and adaptive immune responses: distinct roles for acylation and enzymatic activity in immunomodulation and cell death. J. Immunol. 175:730–738 [DOI] [PubMed] [Google Scholar]
- 42.Uhl MA, Miller JF. 1995. Bordetella pertussis BvgAS virulence control system, p 333–349 In Hoch J, Silhavy T. (ed), Two-component signal transduction. American Society for Microbiology, Washington, DC [Google Scholar]
- 43.Melton AR, Weiss AA. 1989. Environmental regulation of expression of virulence determinants in Bordetella pertussis. J. Bacteriol. 171:6206–6212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lacey BW. 1960. Antigenic modulation of Bordetella pertussis. J. Hyg. (Lond.) 58:57–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hot D, Slupek S, Wulbrecht B, D'Hondt A, Hubans C, Antoine R, Locht C, Lemoine Y. 2011. Detection of small RNAs in Bordetella pertussis and identification of a novel repeated genetic element. BMC Genomics 12:207. 10.1186/1471-2164-12-207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stainer DW, Scholte MJ. 1970. A simple chemically defined medium for the production of phase I Bordetella pertussis. J. Gen. Microbiol. 63:211–220 [DOI] [PubMed] [Google Scholar]
- 47.Bertani G. 1951. Studies on lysogenesis. I. The mode of phage liberation by lysogenic Escherichia coli. J. Bacteriol. 62:293–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Inatsuka CS, Xu Q, Vujkovic-Cvijin I, Wong S, Stibitz S, Miller JF, Cotter PA. 2010. Pertactin is required for Bordetella species to resist neutrophil-mediated clearance. Infect. Immun. 78:2901–2909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kovach ME, Phillips RW, Elzer PH, Roop RM, II, Peterson KM. 1994. pBBR1MCS: a broad-host-range cloning vector. Biotechniques 16:800–802 [PubMed] [Google Scholar]
- 50.Ladant D. 1988. Interaction of Bordetella pertussis adenylate cyclase with calmodulin. Identification of two separated calmodulin-binding domains. J. Biol. Chem. 263:2612–2618 [PubMed] [Google Scholar]
- 51.Masin J, Basler M, Knapp O, El-Azami-El-Idrissi M, Maier E, Konopasek I, Benz R, Leclerc C, Sebo P. 2005. Acylation of lysine 860 allows tight binding and cytotoxicity of Bordetella adenylate cyclase on CD11b-expressing cells. Biochemistry 44:12759–12766 [DOI] [PubMed] [Google Scholar]
- 52.Verma A, Burns DL. 2007. Requirements for assembly of PtlH with the pertussis toxin transporter apparatus of Bordetella pertussis. Infect. Immun. 75:2297–2306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pfaffl MW. 2001. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29:e45. 10.1093/nar/29.9.e45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Beuzon CR, Holden DW. 2001. Use of mixed infections with Salmonella strains to study virulence genes and their interactions in vivo. Microbes Infect. 3:1345–1352 [DOI] [PubMed] [Google Scholar]
- 55.National Research Council 2011. Guide for the care and use of laboratory animals, 8th ed. National Academies Press, Washington, DC [Google Scholar]
- 56.Hewlett EL, Urban MA, Manclark CR, Wolff J. 1976. Extracytoplasmic adenylate cyclase of Bordetella pertussis. Proc. Natl. Acad. Sci. U. S. A. 73:1926–1930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pearson RD, Symes P, Conboy M, Weiss AA, Hewlett EL. 1987. Inhibition of monocyte oxidative responses by Bordetella pertussis adenylate cyclase toxin. J. Immunol. 139:2749–2754 [PubMed] [Google Scholar]
- 58.Farizo KM, Cafarella TG, Burns DL. 1996. Evidence for a ninth gene, ptlI, in the locus encoding the pertussis toxin secretion system of Bordetella pertussis and formation of a PtlI-PtlF complex. J. Biol. Chem. 271:31643–31649 [DOI] [PubMed] [Google Scholar]
- 59.Weiss AA, Johnson FD, Burns DL. 1993. Molecular characterization of an operon required for pertussis toxin secretion. Proc. Natl. Acad. Sci. U. S. A. 90:2970–2974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nakao H, Watanabe H, Nakayama S, Takeda T. 1995. yst gene expression in Yersinia enterocolitica is positively regulated by a chromosomal region that is highly homologous to Escherichia coli host factor 1 gene (hfq). Mol. Microbiol. 18:859–865 [DOI] [PubMed] [Google Scholar]
- 61.Nakano M, Takahashi A, Su Z, Harada N, Mawatari K, Nakaya Y. 2008. Hfq regulates the expression of the thermostable direct hemolysin gene in Vibrio parahaemolyticus. BMC Microbiol. 8:155. 10.1186/1471-2180-8-155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hackett M, Guo L, Shabanowitz J, Hunt DF, Hewlett EL. 1994. Internal lysine palmitoylation in adenylate cyclase toxin from Bordetella pertussis. Science 266:433–435 [DOI] [PubMed] [Google Scholar]
- 63.Barry EM, Weiss AA, Ehrmann IE, Gray MC, Hewlett EL, Goodwin MS. 1991. Bordetella pertussis adenylate cyclase toxin and hemolytic activities require a second gene, cyaC, for activation. J. Bacteriol. 173:720–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Figueroa-Bossi N, Lemire S, Maloriol D, Balbontin R, Casadesus J, Bossi L. 2006. Loss of Hfq activates the sigmaE-dependent envelope stress response in Salmonella enterica. Mol. Microbiol. 62:838–852 [DOI] [PubMed] [Google Scholar]
- 65.Kulesus RR, Diaz-Perez K, Slechta ES, Eto DS, Mulvey MA. 2008. Impact of the RNA chaperone Hfq on the fitness and virulence potential of uropathogenic Escherichia coli. Infect. Immun. 76:3019–3026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Weiss AA, Hewlett EL, Myers GA, Falkow S. 1984. Pertussis toxin and extracytoplasmic adenylate cyclase as virulence factors of Bordetella pertussis. J. Infect. Dis. 150:219–222 [DOI] [PubMed] [Google Scholar]
- 67.Khelef N, Sakamoto H, Guiso N. 1992. Both adenylate cyclase and hemolytic activities are required by Bordetella pertussis to initiate infection. Microb. Pathog. 12:227–235 [DOI] [PubMed] [Google Scholar]
- 68.Carbonetti NH, Artamonova GV, Andreasen C, Bushar N. 2005. Pertussis toxin and adenylate cyclase toxin provide a one-two punch for establishment of Bordetella pertussis infection of the respiratory tract. Infect. Immun. 73:2698–2703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Carbonetti NH, Artamonova GV, Mays RM, Worthington ZE. 2003. Pertussis toxin plays an early role in respiratory tract colonization by Bordetella pertussis. Infect. Immun. 71:6358–6366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Monk IR, Casey PG, Cronin M, Gahan CG, Hill C. 2008. Development of multiple strain competitive index assays for Listeria monocytogenes using pIMC; a new site-specific integrative vector. BMC Microbiol. 8:96. 10.1186/1471-2180-8-96 [DOI] [PMC free article] [PubMed] [Google Scholar]