

Abstract
Oncogene-induced senescence is a stable proliferative arrest that serves as a tumor-suppressing defense mechanism. p38 mitogen-activated protein kinase (MAPK) has been implicated in oncogene-induced senescence and tumor suppression. However, the specific role of each of the four p38 isoforms in oncogene-induced senescence is not fully understood. Here, we demonstrate that p38δ mediates oncogene-induced senescence through a p53- and p16INK4A-independent mechanism. Instead, evidence suggests a link between p38δ and the DNA damage pathways. Moreover, we have discovered a novel mechanism that enhances the expression of p38δ during senescence. In this mechanism, oncogenic ras induces the Raf-1–MEK–extracellular signal-regulated kinase (ERK) pathway, which, in turn, activates the AP-1 and Ets transcription factors that are bound to the p38δ promoter, leading to increased transcription of p38δ. These findings indicate that induction of the prosenescent function of p38δ by oncogenic ras is achieved through 2 mechanisms, transcriptional activation by the Raf-1–MEK–ERK–AP-1/Ets pathway, which increases the cellular concentration of the p38δ protein, and posttranslational modification by MKK3/6, which stimulates the enzymatic activity of p38δ. In addition, these studies identify the AP-1 and Ets transcription factors as novel signaling components in the senescence-inducing pathway.
INTRODUCTION
Although aberrant activation of Ras is associated with human tumors, activated ras in early-passage primary human and rodent cells causes permanent growth arrest known as oncogene-induced senescence (OIS) (1–4). Like apoptosis, OIS is a tumor-suppressing defense mechanism, the disruption of which leads to tumorigenesis (5–10).
Multiple signaling intermediates have been identified that play critical roles in the pathways mediating oncogene-induced senescence. The ability of ras to induce senescence depends on activation of the Raf–MEK–extracellular signal-regulated kinase (ERK) mitogen-activated protein kinase (MAPK) pathway (4, 11) and is accompanied by upregulation of p16INK4A, p53, p14/p19ARF, and p21WAF1 (3, 12) and silencing of E2F target genes (13). We previously showed that ras-induced senescence relies on activation of p38, a MAPK previously identified as a major mediator of inflammation and stress responses (14). p38 and its upstream MAPK kinases MKK3 and MKK6 are activated by oncogenic ras as a result of persistent MEK/ERK activation in senescent cells. Constitutive activation of p38 causes premature senescence, whereas inhibition of p38 prevents ras-induced senescence (14). Consistent with the important role of p38 in oncogene-induced senescence and tumor suppression, targeted deletion of p38α or PRAK, a downstream substrate kinase of p38, accelerates cancer development in mouse models (10, 15, 16).
p38 MAK has four mammalian isoforms, α, β, δ, and γ, which are encoded by different genes and differ in tissue-specific expression, substrate spectrum, and affinity for upstream MAPK activators (17–23). Our previous data indicated that p38α and p38γ contribute to oncogenic ras-induced senescence by upregulating p16INK4A and p53, respectively, while p38β is not essential for senescence induction (27). In the current study, we found that p38δ also regulates ras-induced senescence, yet through a p53- and p16INK4A-independent mechanism. More importantly, in addition to the induction of p38δ enzymatic activity through phosphorylation by MKK3/6, the transcription of the p38δ gene is greatly increased by oncogenic ras through the AP-1 and Ets transcriptional factors upon their activation by the Ras–Raf-1–MEK–ERK signaling pathway. These findings reveal that in response to activation of ras, the prosenescent function of p38δ is induced at the levels of both gene transcription and posttranslational modification through multiple signaling cascades downstream of ras.
MATERIALS AND METHODS
Cell culture.
BJ human foreskin fibroblasts were maintained in minimum essential medium supplemented with 10% fetal calf serum, nonessential amino acids, glutamine, and antibiotics. WI38 and IMR90 human fibroblasts, 293T cells, mouse embryonic fibroblasts (MEFs), and LinX-A retroviral packaging cells were grown in Dulbecco's modified Eagle medium supplemented with 10% fetal calf serum, glutamine, and antibiotics. MEFs were maintained in a 3% O2 incubator.
Plasmids.
BabePuro-Ha-rasV12, -MEK1Q56P, -MKK3E, -MKK6E, -MKK4E, and -MKK7D expression vectors were described previously (14). cDNA for Raf-1 CAAX was obtained from Scott Lowe (Cold Spring Harbor Laboratory); hemagglutinin (HA)-tagged wild-type and intrinsically active mutant (F324S) forms of p38δ were from David Engelberg and Oded Livnah (The Hebrew University of Jerusalem, Israel); MEK1E(+) and MEK1-AA (a rabbit cDNA containing the Ser218/Ser222→Ala mutations) were from Shuang Huang (Medical College of Georgia); and myristoylated p110α and AKT were gifts from Peter Vogt (the Scripps Research Institute). These cDNAs were subcloned into pBabePuro, pWZLHygro, or pWZLNeo to generate the retroviral expression vectors.
Oligonucleotides for small hairpin RNA (shRNA) targeting p38δ-386 (CTCCAGTACCTGGTGTATCAGA), p38δ-695 (AACCAGCTGACCCAGATCCTGA), and green fluorescent protein (GFP) (28) were cloned into pSUPER.reto according to a published protocol (29). Targeted sequences for c-Jun and Ets1 shRNAs are the following: shc-Jun-1555 (GCAAACCTCAGCAACTTCAA), shEts1-247 (CCAAGCAGCAAAGAAATGA), and shEts1-257 (GCAACTCAGGAAGTTCCTA). Appropriate small hairpins were generated by PCR and cloned into the lentiviral pLV-RNAi vector (Biosettia), according to the manufacturer's protocol.
cDNA encoding TAM67, a dominant negative c-Jun mutant lacking the transactivation domain, was amplified by PCR from pIRES-Tam67puro (a gift from Robert Hennigan, University of Cincinnati College of Medicine) (30) and cloned between the BamHI and EcoRI sites in pBabePuro.
Retrovirus- and lentivirus-based gene transduction.
Retroviruses were packaged in LinX-A cells using calcium phosphate transfection and transduced into primary human cells as previously described (31). Lentiviruses were packaged in 293T cells using a Lipofectamine-based transfection procedure and transduced into primary human cells as previously described (32). Transduced cells were selected with 120 (BJ) or 50 (WI38 and IMR90) μg/ml of hygromycin B, 1.2 μg/ml of puromycin, 600 μg/ml of G418, or 5 μg/ml of blasticidin.
Analysis of senescence.
Senescence analysis in cell culture was performed by measuring the rate of proliferation and the expression of the senescence-associated β-galactosidase (SA-β-Gal) senescence marker, as described previously (14). Population doublings (PD) were calculated with the formula PD = log(N2/N1)/log2, where N1 is the number of cells seeded and N2 is the number of cells recovered (33). To quantify SA-β-Gal positives, at least 200 cells were counted in random fields in each well. Each experiment was performed in triplicates or duplicates.
Western blot analysis.
Western blotting was performed with lysates prepared 7 to 10 days after transduction of Ras or MKK3/6E from subconfluent cells as described previously (14). Primary antibodies were from Abcam (c-Fos, phospho-c-Fos-S374, ETS-1, phospho-ETS-1-T38, and tubulin), Cell Signaling (phospho-p53-Ser33, phospho-p53-Ser15, phospho-p38-Thr180/Tyr182, ERK2, phospho-p44/42 MAPK-Thr202/Tyr 204, phosphor-Chk1-Ser345, Chk1, phosphor-Chk2-Thr68, Chk2, MKK7, and AKT), Santa Cruz [Ras C-20, p21 c-19, p53 FL-393, c-Jun H-79, c-Jun (D), phospho-c-Jun-S73, p14ARF-C18, cyclin A-H432, cyclin E-C19, Cdk2-M2, Cdk4-H22, Cdk6-C21, MEK4-C20, Raf-1-C20, p110α, and Myc N-262], BD Pharmingen (cyclin D1), and Sigma (actin and p16INK4A DCS-50). Antibodies against p38α,- β, -γ, and -δ were described previously (27). Antibodies against MKK3 (no. 1330), MKK6, and MEK1 (no. 1329) were gifts from Jiahuai Han (Xiamen University, China). Signals were detected using enhanced chemiluminescence and captured by the FluorChem HD2 imaging system (Cell Biosciences).
Quantitative real-time PCR.
RNA was isolated from BJ cells using TRIzol reagent (Life Technologies). cDNA was synthesized from 100 ng of RNA using iScript Reverse Transcription Supermix (Bio-Rad) according to the manufacturer's protocol. Quantitative real-time reverse transcription-PCR (RT-PCR) was formed using SsoAdvanced SYBR green Supermix (Bio-Rad) on a CFX96 real-time system (Bio-Rad). Signals were normalized to that of a housekeeping gene, the porphobilinogen deaminase (PBGD) or glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene. The primers used were 5′-ATAATGGCCGAGCTGTTGACTGG-3′ and 5′-TTTCTTGCCTCATGGCTTGGCATC-3′ for p38α, 5′-ACTACATTGACCAGCTGAAGCGCA-3′ and 5′-ACTGGATATATGTCCGGGCGTGTT-3′ for p38β, 5′-AGATGATCACAGGCAAGACGCTGT-3′ and 5′-AGGGCTTGCATTGGTCAGGATAGA-3′ for p38γ, 5′-ACGAAACTTTGCCCATAGCA-3′ and 5′-GCAAGGAGAGCCTTTCAGAG-3′ for c-Myc, 5′-TCCAGTCCAATTATCACCAGC-3′ and 5′-TGCTTGGAGTTAATAGTGGGAC-3′ for Ets1, 5′-TCCAAGCGGAGCCATGTCTG-3′ and 5′-AGAATCTTGTCCCCTGTGGTGGA-3′ for PBGD, and 5′-CCCTTCATTGACCTCAACTA-3′ and 5′-CCTTCTCCATGGTGGTGAA-3′ for GAPDH. Primers for p38δ was purchased from Qiagen.
mRNA stability assay.
RNA was prepared from BJ cells treated with 5 μg/ml of actinomycin D for 0, 0.5, 1, 1.5, and 2 h using the RNeasy Plus minikit (Qiagen). Fifty nanograms of each RNA sample was converted into cDNA by reverse transcription and analyzed by quantitative real-time PCR for p38δ and c-Myc levels. Signals for p38δ and c-Myc were normalized to that of a housekeeping gene, the PBGD gene.
Protein stability assay.
Protein lysates were prepared from BJ cells treated with 20 μg/ml of cycloheximide for 0, 1, 2, 6, 9, and 15 h and analyzed by Western blotting. Signals for p38δ and c-Myc were quantified by AlphaView Imaging Software (Cell Biosciences) and normalized to that of actin.
Luciferase reporter assay.
The retroviral p53 reporter PG-Luc and its non-p53-binding mutant control MG-Luc have been previously reported (27).
To construct the 3kb-Luc reporter for p38δ, a 3-kb p38δ promoter fragment (nucleotide −2970 to +20 relative to the transcriptional start site) was amplified by PCR from genomic DNA of BJ cells and cloned between BglII and EcoRI sites upstream of a firefly luciferase reporter in a self-inactivating retroviral vector (pBabe-PGK-Blast-SIN-Luc) (34). To generate reporters containing the 2-kb, 1-kb, 2.8-kb, 2.6-kb, and 2.4-kb p38δ promoter region upstream of the transcriptional start site, and those containing the 3-kb p38δ promoter lacking the AP-1 binding sites within the region from kb −3 to −2.8 (see Fig. 6A), appropriate fragments were amplified by PCR from the 3kb-Luc and cloned between BglII and EcoRI sites upstream of a firefly luciferase reporter in pBabe-PGK-Blast-SIN-Luc. The reporter containing the 3-kb p38δ promoter with a mutant Ets binding site within the region from kb −3 to −2.8 (Ets Mut-Luc [see Fig. 6A]) was generated by ligating a 5′ BglII-XhoI fragment generated by primers 5′-GGAATTCCTCCCTTTCATGACTCCCCAACTGGATCCTC-3′ and 5′-GGCCTCGAGTGACTCAGGCCTCCGACTCTGCTGACACTG-3′ and a 3′ XhoI-EcoRI fragment generated by primers 5′-CCGCTCGAGGACCCTTGCTCAAGAGCATCCGGAGCTACAG-3′ and 5′-GAAGATCTCCAACGGGCTCCCAGCGCCCCGACCCCCGC-3′ into the BglII and EcoRI sites of pBabe-PGK-Blast-SIN-Luc.
Fig 6.

Induction of p38δ transcription by oncogenic ras requires the AP-1 and Ets transcription factor binding sites on the p38δ promoter. (A) Schematic diagram of the p38δ promoter reporter constructs carrying deletion or mutation of the AP-1 and Ets transcription factor binding sites within the upstream region from kb −3 to −2.8 of the transcription start site. (B) BJ cells stably transduced with a retroviral luciferase reporter containing a 3-kb, 2-kb, or 1-kb p38δ promoter sequence (3kb, 2kb, or 1kb, respectively) upstream of the transcription start site was transduced with Ha-rasV12 (Ras) or vector (WH). *, P < 0.001 versus 3kb by Student's t test. (C) BJ cells stably transduced with a retroviral luciferase reporter containing a 3-kb, 2.8-kb, 2.6-kb, or 2.4-kb p38δ promoter sequence upstream of the transcription start site was transduced with Ha-rasV12 (Ras) or vector (WH). *, P < 0.01 versus 3kb by Student's t test. (D) BJ cells stably transduced with a retroviral luciferase reporter containing a 3-kb or 2.8-kb p38δ promoter sequence upstream of the transcription start site was transduced with Ha-rasV12 (Ras), MEK1Q56P, or vector (WH). *, P < 0.005 versus 3kb by Student's t test. (E) BJ cells stably transduced with a retroviral luciferase reporter containing a 2.95-kb wild-type p38δ promoter sequence upstream of the transcription start site (2.95kb) or that harboring deletion of one (ΔAPx1), two (ΔAPx2), or all three (ΔAPx3) AP-1 binding sites and/or mutation of the Ets binding site within the region from kb −3 to −2.8 (Ets Mut) was transduced with Ha-rasV12 (Ras) or vector (WH). *, P < 0.05; **, P > 0.5 (versus 2.95kb; by Student's t test). (F) BJ cells stably transduced with a retroviral luciferase reporter containing a 3-kb p38δ promoter sequence upstream of the transcription start site was transduced first with a dominant negative mutant of MEK1 (MEK1-AA) or vector (WH) and then with Ha-rasV12 (Ras) or vector (BP). *, P < 0.005 versus WH by Student's t test. For panels B to F, cells were lysed on day 6 to 8 after Ras transduction. Luciferase activity was measured and normalized to protein concentration. Fold induction by ras/MEK1Q56P was calculated by dividing the luciferase activity in ras/MEK1Q56P-expressing cells by that in control cells. Values are means ± SDs for triplicates.
The retroviral reporter constructs were transduced into BJ cells to create stable reporter cells, which were in some cases transduced with p38δ shRNA, MEK1-AA, or TAM67 or proper controls. The resulting cell lines were then transduced with Ha-rasV12, MEK1Q56P, or vector controls at PD28-34. Cells were split into 12-well plates on day 7 or 8 after ras/MEK1Q56P transduction and lysed on day 8 or 9. Luciferase activity was determined using a luciferase assay system (Promega) according to the manufacturer's instructions and normalized to protein concentrations as determined by Bradford assays. Each experiment was performed in triplicates or duplicates.
Kinase assay with recombinant p38.
Recombinant glutathione S-transferase (GST)–MKK6E, His-p38δ, His-p38γ, and wild-type and mutant human p53 (1-61) were prepared as described previously (27).
His-p38 (0.4 μg) was first incubated with 50 ng of GST-MKK6E at 30°C for 10 min in 14 μl of 1× kinase buffer (20 mM Tris-HCl [pH 7.5], 20 mM NaCl, 10 mM MgCl2, 1 mM dithiothreitol [DTT], 20 μM cold ATP, and 1 mM NaF). Subsequently, 6 μl of substrate mix in 1× kinase buffer (same as above) containing 20 μg of human p53 (1-61) (wild type or S33A or S46A mutant) and 2 μCi of [γ-32P]ATP was added to each reaction mixture. The resulting 20 μl of reaction mixture was incubated at 30°C for 30 min, the reaction was stopped by addition of 7 μl of 4× Laemmli buffer, and the mixture was heated at 95°C for 10 min. The reactions were separated on 4 to 20% gradient SDS-PAGE gels. Radioactive signals were detected by a phosphorimager.
ChIP assays.
A total of 2 × 106 of BJ cells/15-cm plate were cross-linked with 1% formaldehyde for 10 min at room temperature. The cross-linking was stopped by incubation in 0.125 M glycine for 5 min at room temperature. Cells were washed with phosphate-buffered saline (PBS), dislodged from plates in 1 ml of hypotonic buffer (25 mM HEPES [pH 7.8], 1.5 mM MgCl2, 10 mM KCl, and 1 mM DTT), and passed 5 times through a 25.5-gauge needle. Nuclei were spun down at 5,000 rpm for 5 min and lysed with 1 ml of lysis buffer (50 mM HEPES [pH 7.9], 140 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% sodium deoxycholate [NaDOC], 0.1% SDS, 0.5 mM phenylmethylsulfonyl fluoride [PMSF], and Roche Complete protease inhibitor cocktail with no EDTA). Chromatin was sonicated to 0.5 to 1 kb in size, at an output of 4 with 5 15-s pulses and 1 min 15 s of cooldown between each pulse (Branson Sonifier 150). Samples of the same cell line from different plates were pooled, and protein concentrations were measured by the Bradford assay. Ten percent of each sample was saved as total input. A 0.8- to 1-mg quantity of chromatin was incubated with 5 μg of an anti-c-Jun (H-79X; Santa Cruz), anti-c-Fos (4X, Santa Cruz, or 9F6, Cell Signaling), or anti-Ets1 (C-20X; Santa Cruz) antibody or normal rabbit or goat IgG (Santa Cruz) at 4°C overnight and then with 50 μl (bead volume) of PureProteome protein A or G magnetic beads (Millipore) at 4°C for up to 4 h. The beads were washed three times sequentially with lysis buffer, buffer 2 (50 mM HEPES [pH 7.9], 500 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% NaDOC, and 0.1% SDS), buffer 3 (20 mM Tris-HCl [pH 8], 250 mM LiCl, 1 mM EDTA, 1% NP-40, and 1% NaDOC), and Tris-EDTA (TE; pH 8). After the final wash, protein-DNA-bead complexes were resuspended in 300 μl of TE and treated with 10 μg/ml of RNase A at 37°C for 15 min. Cross-linking was reversed by addition of NaCl to 300 mM and incubation at 65°C overnight, after which 2.5 μl of 20% SDS and 36 μg of proteinase K were added and the reaction mixtures were incubated at 37°C for an additional 2 to 4 h. After pelleting of the beads, DNA in the supernatant was extracted with phenol-chloroform, precipitated with ethanol, and dissolved in diethyl pyrocarbonate (DEPC)-treated water. DNA precipitated by chromatin immunoprecipitation (ChIP) was quantified by real-time PCR using primers amplifying the region from −3086 to −2759 of the p38δ promoter (5′-CCTGCCTCCATGCTCCATGCTCATA-3′ and 5′-TGGCTGTAGCTCCGGATGCTCTTGA-3′) or those amplifying the region from −7007 to −6723 of the p38δ promoter (5′-CTCGGTCCCACCATTTGGGTCTTCA-3′ and 5′-CCAGGCCTCCGCTCTGTTTGATCTC-3′).
RESULTS
p38δ mediates oncogenic ras-induced senescence.
In light of the critical role of p38α and p38γ in senescence, we investigated whether p38δ was also important for oncogenic ras-induced senescence. Two independent p38δ shRNAs (shδ386 and shδ695) were constructed. These shRNAs effectively knocked down p38δ expression without affecting the expression of the other p38 isoforms when stably transduced into primary BJ human fibroblasts via retroviruses (Fig. 1A). The p38δ knockdown cell lines, shδ386 and shδ695, and shGFP control cell line were then transduced with an activated ras allele, Ha-rasV12. While the control cells transduced with Ha-rasV12 became growth arrested, BJ cells expressing the p38δ shRNAs continued to proliferate in the presence of activated ras (Fig. 1B). In addition, the p38δ shRNAs greatly decreased ras-induced accumulation of cells positive for senescence-associated β-galactosidase (SA-β-Gal), a biomarker for senescence (Fig. 1C). These results demonstrate that like p38α and p38γ, p38δ also plays an essential role in oncogenic ras-induced senescence. Further supporting the notion that p38δ is required for senescence induction, oncogenic ras-induced senescence was impaired in p38δ−/− mouse embryonic fibroblasts (MEFs) (Fig. 1G).
Fig 1.

p38δ mediates oncogenic ras-induced senescence. (A) Western blot analysis of BJ cells transduced with shRNA for GFP (shGFP) or p38δ (shδ386 and shδ695) and Ha-rasV12 (Ras) or vector (WH) on day 8 after ras transduction. Numbers represent relative levels of protein. (B) Population doublings of BJ cells transduced with shRNA for GFP (shGFP) or p38δ (shδ386 and shδ695) and Ha-rasV12 (Ras) or vector (WH) over 19 days starting at PD 30. Values are means ± SDs for triplicates. *, P < 0.05 versus shGFP by Student's t test. (C) Percentage of SA-β-Gal-positive cells in BJ populations transduced with shRNA for GFP (shGFP) or p38δ (shδ386 and shδ695) and Ha-rasV12 (Ras) or vector (WH). Values are means ± SDs for triplicates. *, P < 0.005 versus shGFP by Student's t test. (D) Western blot analysis of BJ cells transduced with Ha-rasV12 (Ras), wild-type p38δ, the p38δ-F324S mutant, or vector (BP) on day 8 posttransduction. (E) Population doublings of BJ cells transduced with Ha-rasV12 (Ras), wild-type p38δ, the p38δ-F324S mutant, or vector (BP) over 9 days starting at PD 30. Values are means ± SDs for triplicates. *, P < 0.05 versus BP by Student's t test. (F) Percentage of SA-β-Gal-positive cells in BJ populations transduced with Ha-rasV12 (Ras), wild-type p38δ, the p38δ-F324S mutant, or vector (BP). Values are means ± SDs for triplicates. *, P < 0.05 versus BP by Student's t test. (G) p38δ deficiency disrupts ras-induced senescence in MEFs. p38δ+/+ and p38δ−/− MEFs were transduced with Ha-rasV12 or vector (WH). A total of 2 × 104 cells were seeded into 12-well plates on day 5 after ras transduction, after selection of transduced cells. Cells were counted 4 days after seeding, and PD was calculated (top graph). Cells were stained for SA-β-Gal on day 10 after ras transduction. The percentage of cells positive for SA-β-Gal was quantified (bottom graph). Values are means ± SDs for duplicates. *, P < 0.05 versus p38δ+/+; **, P < 0.005 versus p38δ+/+ (Student's t test).
To further show the importance of p38δ in senescence, we explored the possibility that the constitutively active form of p38δ is sufficient to induce senescence in the absence of Ras. We utilized an intrinsically active mutant (F324S) of p38δ that had acquired spontaneous protein kinase activity in vitro and in vivo, while preserving the substrate and inhibitor specificities of the wild-type protein (35, 36). The wild type and the constitutively active F324S mutant of p38δ were transduced into BJ cells via retrovirus. As reported previously, p38δ-F324S displayed a higher level of autophosphorylation in the activation loop than wild-type p38δ when transduced into BJ cells, indicating that the mutant is indeed constitutively active (Fig. 1D). Moreover, p38δ-F324S reduced cell proliferation rate (Fig. 1E) and induced an increase in the percentage of cells positive for SA-β-Gal (Fig. 1F) compared to those with wild-type p38δ. Thus, constitutively activated p38δ is capable of inducing senescence, although its effect is not as robust as that of oncogenic ras. Taken together, our results demonstrate that p38δ plays an essential role in senescence induction by oncogenic ras.
p38δ mediates oncogenic ras-induced senescence in a p53/p21WAF1- and p16INK4A-independent manner.
Since p38α and p38γ mediate oncogenic ras-induced senescence by upregulating p16INK4A and p53, respectively (27), we investigated the impact of p38δ on these pathways in senescence.
p53 is one of the crucial mediators of ras-induced senescence. The activity of p53 is regulated through phosphorylation of its N-terminal transactivation domain. We showed previously that p38γ mediates ras-induced p53 activation by directly phosphorylating p53 at Ser33 (27). Like p38γ, recombinant p38δ phosphorylated p53, and the phosphorylation was abolished when Ser33 of p53 was mutated to Ala (Fig. 2A), suggesting that p53-Ser33 is also a p38δ substrate in vitro. Nevertheless, while the p38δ shRNAs effectively knocked down p38δ expression and disrupted ras-induced senescence (Fig. 1A to C), they failed to reduce ras-induced phosphorylation of p53 at Ser33 in BJ cells (Fig. 2B). This finding suggests that although recombinant p38δ can phosphorylate p53-S33 in vitro, p38δ is not the major p38 isoform phosphorylating this site during senescence induction in cells. Instead, our previous study identified p38γ as the main p53-S33 kinase in senescent cells (27). Supporting the notion that p38δ does not play a key role in p53 activation, silencing of p38δ expression did not abrogate ras-induced expression of p21WAF1, a transcriptional target of p53 and a critical effector of senescence (Fig. 2B), and had no effect on the induction of p53 transcriptional activity by ras, as determined by a reporter assay using a luciferase gene driven by a p53-dependent promoter (PG14-Luc) (Fig. 2C). Moreover, although the constitutively active mutant of p38δ (F324S) induced a modest level of senescence (Fig. 1E and F), it failed to increase p53-S33 phosphorylation and p21WAF1 expression (Fig. 2E). These results indicate that p38δ mediates ras-induced senescence without contributing to p53 activation.
Fig 2.

p38δ mediates oncogenic ras-induced senescence through a p53/p21WAF1- and p16INK4A-independent mechanism. (A) Like p38γ, recombinant p38δ phosphorylates p53 at Ser33. His-p38γ or -p38δ was incubated first with GST-MKK6E and cold ATP and then with wild-type p53 (1-61) (WT) or p53 (1-61) carrying the S33A (A33) or S46A (A46) mutation in the presence of [γ-32P]ATP. The reactions were separated by SDS-PAGE. Phosphorylated p53 was detected using a phosphorimager. The input of substrate was determined by staining with Coomassie brilliant blue R. (B) Western blot analysis of BJ cells transduced with shRNA for GFP (shGFP) or p38δ (shδ386 and shδ695) and Ha-rasV12 (Ras) or vector (WH) on day 8 after ras transduction. Numbers represent relative levels of proteins. (C) BJ cells stably transduced with a retroviral luciferase reporter driven by a promoter containing multiple copies of a functional p53-binding site (PG-Luc, top) or a mutant p53-binding site (MG-Luc, bottom) were transduced with retroviruses encoding shRNA for GFP (shGFP) or p38δ (shp38δ-386 or -695) at population doubling 33 and with Ha-rasV12 (Ras) or vector (WH) at population doubling 35. Cells were lysed on day 8 after Ras transduction. Luciferase activity was measured and normalized to protein concentration. Fold induction by ras was calculated by dividing the luciferase activity in ras-expressing cells by that in control cells. Values are means ± SDs for triplicates. *, P > 0.1 versus shGFP by Student's t test. (D and E) Western blot analysis of BJ cells transduced with Ha-rasV12 (Ras), wild-type p38δ, the p38δ-F324S mutant, or vector (BP) on day 8 posttransduction, detecting indicated proteins. (F) Western blot analysis of BJ cells transduced with shRNA for GFP (shGFP) or p38δ (shδ386 and shδ695) and Ha-rasV12 (Ras) or vector (WH) on day 8 after ras transduction.
Besides p53 activation, another hallmark of senescence is the induction of expression of p16INK4A, a key effector of stable proliferative arrest associated with senescence. We found that shRNA-mediated silencing of p38δ did not alter ras-induced p16INK4A expression (Fig. 2B) and that the constitutively active mutant of p38δ failed to increase the p16INK4A protein level (Fig. 2D). Taken together, our data demonstrate that p38δ mediates ras-induced senescence through a p53/p21WAF1- and p16INK4A-independent mechanism. Moreover, p38δ knockdown had no effect on some of the other cell cycle regulators, including p14ARF, cyclin A, cyclin D1, cyclin E, cyclin-dependent kinase 2 (CDK2), CDK4, and CDK6 (Fig. 2F). In addition, neither p38δ knockdown (Fig. 1A) nor overexpression of the wild type or active mutant of p38δ (Fig. 2D) had any effect on the activation of ERK, another MAPK involved in senescence.
Oncogenic ras-induced senescence is mediated by DNA damage responses caused by DNA hyperreplication (37, 38). We found that p38δ shRNAs reduced the ras-induced activating phosphorylation of Chk1 and Chk2, two DNA damage checkpoint kinases (Fig. 2B). This finding suggests that p38δ may participate in the activation of DNA damage pathways in response to oncogenic ras. How p38δ regulates DNA damage responses in a p53- and p16INK4A-indepdendent fashion is currently unclear.
Oncogenic ras induces both activity and expression of p38δ in senescent cells.
As detected by Western blotting using an anti-phospho-p38 antibody, oncogenic ras induced a phospho-p38 signal that comigrated with p38δ (Fig. 3A). Moreover, we immunoprecipitated an equal amount of p38δ from BJ cells transduced with Ha-rasV12 or vector control, after adjusting the input, using an antibody specific for p38δ, and the p38δ from ras cells displayed an increased protein kinase activity toward ATF2 compared to that from control cells (Fig. 3B). These results indicate that oncogenic ras induces the phosphorylation and activation of p38δ.
Fig 3.

Oncogenic ras induces p38δ activity and expression. (A) Western blot analysis of BJ cells transduced with Ha-rasV12 (Ras) or vector (WH). Identical sets of lysates were resolved side by side on the same SDS-PAGE gel and transferred to a nitrocellulose membrane. The membrane was cut into pieces, each containing one set of lysates, which were then hybridized to the antibody against phospho-p38, p38α, p38δ, and p38β, respectively. The enhanced chemiluminscence signals were captured after the membranes were realigned into the original position. The positions of p38α, p38δ,and p38β are marked by arrows. (B) Oncogenic ras induces the kinase activity of p38δ. An equal amount of p38δ was immunoprecipitated from BJ cells transduced with Ha-rasV12 (Ras) or vector (WH) after adjustment of the input and assayed for kinase activity toward ATF2. Part of the IPs was subjected to Western blotting to ensure an equal amount of p38δ IP. (C) Western blot analysis of the protein levels of p38 isoforms in BJ cells transduced with Ha-rasV12, MKK6E, or vector (WH) on day 8 after ras transduction. (D) Analysis of the mRNA levels of p38 isoforms in BJ cells transduced with Ha-rasV12, MKK6E, or vector (WH) on day 8 after ras transduction, by quantitative real-time RT-PCR. Signals for p38 were normalized to that of PBGD. Values are means ± SDs for triplicates. *, P < 0.01 versus WH by Student's t test. (E) Time course analysis of the p38δ mRNA (bar graph) and protein (inset) levels in BJ cells transduced with Ha-rasV12, MKK6E, or vector (BP) on day 4 through day 8 after ras/MKK6E transduction. Bar graph, signals for p38δ mRNA were detected by quantitative real-time RT-PCR and normalized first to that of PBGD and then to that from cells transduced with vector control (BP). Values are means ± SDs for triplicates. (Inset) The p38δ and actin protein levels were detected by Western blotting. *, P < 0.05 versus BP by Student's t test. (F) Western blot analysis of p38 isoforms in IMR90 and WI38 cells transduced with Ha-rasV12, MKK3E, MKK6E, or vector (WH or BP) on day 8 after ras/MKK3/6E transduction. (G) Western blot analysis of p38 isoforms in p38δ+/+, p38δ+/−, and p38δ−/− MEFs transduced with Ha-rasV12 (Ras) or vector (WH) on day 8 after ras transduction.
In the course of analyzing the role of p38δ in senescence, we observed that while the basal level of p38δ is very low in the absence of ras, the p38δ protein level was significantly increased by oncogenic ras but not by MKK6E, a constitutively active mutant of the p38 upstream activator MKK6 (Fig. 1A, 2B, and 3C). The protein level of p38α was also increased by ras in BJ cells. However, neither ras nor MKK6E altered the levels of p38β and p38γ (Fig. 1A and 3C). We also examined the induction of p38δ at the mRNA level. Quantitative real-time PCR assays revealed that oncogenic ras, but not MKK6E, also increased the mRNA level of p38δ in senescent BJ cells while having no significant effect of the mRNA expression of p38α, p38β, or p38γ (Fig. 3D). In time course analyses of the effect of ras on p38δ expression, we found that p38δ was induced at both mRNA (Fig. 3E, bar graph) and protein (Fig. 3E, inset) levels on as early as day 4 after transduction of oncogenic ras and that the induction persisted at least into day 8.
Furthermore, the induction of p38δ expression by oncogenic ras was also observed in other primary human fibroblast lines such as IMR90 and WI38 and mouse embryonic fibroblasts (Fig. 3F and G). Again in these additional cell lines, constitutively active MKK3 or MKK6 failed to induced p38δ, and the expression of p38β and p38γ was not significantly increased by ras.
These findings therefore indicate that p38δ is unique among all the p38 isoforms in that its activity is induced in senescent cells not only through phosphorylation of the activation loop by its upstream kinases MKK3 and MKK6 (27) but also through upregulation of its expression at protein and mRNA levels by oncogenic ras.
Oncogenic ras induces p38δ expression through the Raf-1–MEK1–ERK pathway.
Oncogenic ras activates multiple downstream effector pathways, including Raf-1–MEK–ERK, MKK3/6-p38, MKK4/7–Jun N-terminal protein kinase (JNK), and phosphatidylinositol 3-kinase (PI3K)–AKT. Since the constitutively active MKK3 and MKK6 failed to increase the p38δ level, it is likely that oncogenic ras induces p38δ expression through another downstream effector pathway. To identify the ras effector pathway that mediates the induction of p38δ expression in senescent cells, we examined the effects of constitutively active forms of Raf-1 (hRaf-1 CAAX, membrane bound), MEK1 (MEK1E+ and MEK1Q56P), MKK3 (MKK3E), MKK4 (MKK4E), MKK7 (MKK7D), PI3K (MF-p110α, myristoylated), and AKT (MF-AKT, myristoylated) (Fig. 4). The results indicate that the active forms of Raf-1 and MEK1 induced p38δ expression at both protein (Fig. 4A) and mRNA (Fig. 4B) levels, while the others failed to do so. Moreover, a dominant negative mutant of MEK1 (MEK1-AA) (see Fig. 8B) and a chemical inhibitor of MEK1 (U0126) (see Fig. 8C) reduced the induction of p38δ expression by ras. These findings indicate that oncogenic ras induces p38δ expression through the Raf-1–MEK–ERK pathway.
Fig 4.
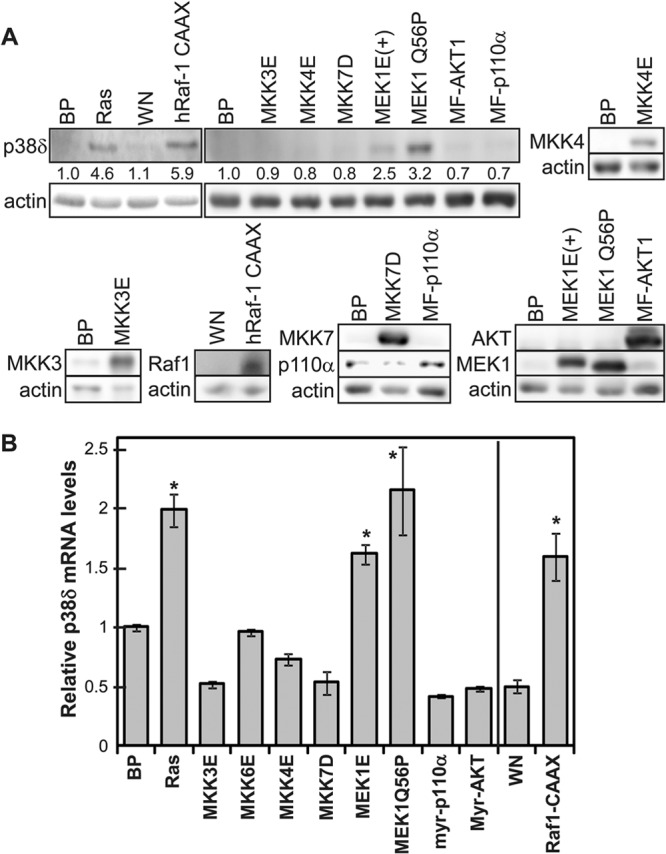
Induction of p38δ expression by the Ras–Raf-1–MEK pathway. (A) Western blot analysis of the p38δ protein levels in BJ cells on day 8 after transduction of Ha-rasV12 (Ras), the constitutively active forms of the indicated Ras downstream effectors, or vector controls (BP or WN). Numbers represent relative levels of the protein. (B) Analysis of the p38δ mRNA levels by quantitative real-time RT-PCR in BJ cells on day 8 after transduction of Ha-rasV12 (Ras), the constitutively active forms of the indicated Ras downstream effectors, or vector controls (BP or WN). Signals for p38δ mRNA were normalized first to that of PBGD and then to that from cells transduced with vector control (BP or WN). Values are means ± SDs for triplicates. *, P < 0.05 versus BP or WN by Student's t test.
Fig 8.

c-Jun, c-Fos, and Ets1 are activated by oncogenic ras through the MEK-ERK pathway during senescence induction. Numbers represent relative levels of proteins. (A) Western blot analysis of BJ cells transduced with Ha-rasV12 (Ras), constitutively active MEK1 (MEK1Q56P and MEK1E+), or vector (BP) on day 8 after ras transduction. (B) Western blot analysis of BJ cells transduced first with a dominant negative mutant of MEK1 (MEK1-AA) or vector (WH) and then with Ha-rasV12 (Ras) or vector (BP) on day 8 after ras transduction. (C) Western blot analysis of BJ cells transduced with Ha-rasV12 (Ras) or vector (WH) and treated with 5 μM U0126 or vehicle (Ctrl) on day 8 after ras transduction.
Oncogenic ras induces p38δ expression by stimulating transcription from the p38δ promoter through the AP-1 and Ets transcription factor binding sites.
To investigate the mechanism for the induction of p38δ level by ras, we explored several possible modes of regulation of p38δ expression.
By Western blotting, we first compared the stabilities of the p38δ protein in control and ras-expressing BJ cells after inhibition of novel protein synthesis by cycloheximide. p38δ appeared to be a quite stable protein, and its half-life was unaltered in senescent cells compared to control cells (Fig. 5A and B). Furthermore, the stability of p38δ mRNA was determined in control and senescent cells that had been treated with actinomycin D, an inhibitor of novel DNA transcription. Again, no difference was observed in p38δ mRNA stability between control cells and those undergoing ras-induced senescence (Fig. 5E). As a positive control for these experiments, c-Myc decayed rapidly at both protein (Fig. 5C and D) and mRNA (Fig. 5F) levels in the protein stability and mRNA stability assays, respectively. These results indicate that ras-induced p38δ expression does not occur through stabilization of p38δ protein or mRNA.
Fig 5.

Oncogenic ras does not stabilize p38δ protein or mRNA. (A) Western blot analysis of p38δ protein levels in BJ cells transduced with Ha-rasV12 (Ras) or vector (WH) after treatment with cycloheximide (Cycl) for 0, 1, 2, 9, or 15 h on day 8 after ras transduction. (B) Quantification of relative p38δ protein levels in panel A. The relative p38δ protein level was calculated by dividing the p38δ signal at each time point after cycloheximide treatment by that at 0 h, after normalization to the actin signal. (C) Western blot analysis of c-Myc protein levels in BJ cells transduced with Ha-rasV12 (Ras) or vector (WH) after treatment with cycloheximide for 0, 1, 2, 6, 9, or 15 h on day 8 after ras transduction. (D) Quantification of relative c-Myc protein levels in panel C. The relative c-Myc protein level was calculated by dividing the c-Myc signal at each time point after cycloheximide treatment by that at 0 h, after normalization to the actin signal. (E) Quantification of p38δ mRNA levels by real-time RT-PCR in BJ cells transduced with Ha-rasV12 or vector (WH), after treatment with actinomycin D for 0, 0.5, 1, 1.5, or 2 h on day 8 after ras transduction. The relative p38δ mRNA level was calculated by dividing the p38δ signal at each time point after actinomycin D treatment by that at 0 h, after normalization to the PBGD signal. Values are means ± SDs for triplicates. (F) Quantification of c-Myc mRNA levels by real-time RT-PCR in BJ cells transduced with Ha-rasV12 or vector (WH) after treatment with actinomycin D for 0, 0.5, 1, 1.5, or 2 h on day 8 after ras transduction. The relative c-Myc mRNA level was calculated by dividing the c-Myc signal at each time point after actinomycin D treatment by that at 0 h, after normalization to the PBGD signal. Values are means ± SDs for triplicates.
We next investigated whether oncogenic ras stimulates p38δ gene transcription in senescent cells. To this end, a 3-kb p38δ promoter (a region from nucleotide −2970 to +20 relative to the transcriptional start site) was cloned upstream of a firefly luciferase reporter in a self-inactivating retroviral vector (34) (Fig. 6A). This construct (3kb-Luc) was transduced into BJ cells to generate a stable reporter cell line for p38δ promoter activity. Transduction of oncogenic ras into this cell line led to increased transcription from the 3-kb p38δ promoter (Fig. 6B), indicating that ras induces p38δ expression by stimulating its transcription. Moreover, constitutively active MEK1 (MEK1Q56P) also induced the p38δ promoter activity (Fig. 6D), while a dominant negative mutant of MEK1 (MEK1-AA) abrogated ras-induced transcription from the p38δ promoter (Fig. 6F). These findings indicate that oncogenic ras stimulates p38δ transcription through MEK1, consistent with our prior finding that p38δ expression is induced by the Ras–Raf-1-MEK–ERK pathway (Fig. 4).
To identify the cis-regulatory elements that mediate induction by ras on the p38δ promoter, luciferase reporters containing 2-kb (2kb-Luc) or 1-kb (1kb-Luc) promoter sequences upstream of the transcriptional start site were generated and tested for ras responsiveness. In contrast to findings with 3kb-Luc, transcription from 2kb-Luc and 1kb-Luc could no longer be induced by oncogenic ras (Fig. 6B), suggesting that the ras-responsive element resides in the region between kb −3 and −2 upstream of the transcriptional start site. A series of 200-bp deletion mutants were made within this region and analyzed by the luciferase reporter assay. Deletion of the region from kb −3 to −2.8 (2.8kb-Luc) essentially abolished ras-induced transcription (Fig. 6C). Moreover, deletion of this region also disrupted transcriptional stimulation by constitutively active MEK1 (Fig. 6D). These data indicate that the region from kb −3 to −2.8 contains cis elements required for the induction of p38δ transcription by the ras–Raf-1–MEK–ERK pathway.
We searched for potential transcription factor binding sites within the region from kb −3 to −2.8 using bioinformatic tools offered by Transcription Element Search System (TESS) and Searching Transcription Factor Binding Sites (TFSEARCH). The analysis revealed 3 potential binding sites for AP-1 followed by 1 potential binding site for Ets (Fig. 6A); AP-1 and Ets are transcription factors previously shown to be activated by the MEK-ERK cascade. In fact, this is the only region with clustered AP-1/Ets binding sites along the whole 3-kb promoter. We deleted one, two, or all three AP-1 binding sites and mutated the Ets binding sites in the context of 3kb-Luc (Fig. 6A) and tested the effects of these mutations on transcriptional stimulation by ras. While deletion of the first AP-1 site (ΔAPx1-Luc) had no effect on reporter expression compared to the control reporter (2.95kb-Luc), deletion of two (ΔAPx2-Luc) or all three (ΔAPx3-Luc) AP-1 sites or mutation of the Ets site (Ets Mut-Luc) partially reduced transcriptional stimulation of the p38δ promoter by ras (Fig. 6E). Furthermore, combination of deletion of all the AP-1 sites and mutation of the Ets site (ΔAPx3/Ets-Luc) led to complete elimination of ras induction (Fig. 6E). These findings indicate that the binding sites for AP-1 and Ets work cooperatively to mediate the induction of p38δ transcription.
c-Jun and Ets1 transcription factors directly bind to the ras-responsive element on the p38δ promoter.
AP-1 transcription factors are dimers mainly composed of members of the Jun (c-Jun, JunB, and JunD) and Fos (c-Fos, FosB, Fra-1, and Fra-2) families of proteins (39). While c-Jun is the most potent transcriptional activator in the Jun family, c-Fos and FosB are the only Fos proteins containing transcriptional activation domains. Chromatin immunoprecipitation (ChIP) was performed to determine whether c-Jun, c-Fos, and Ets1 directly bind to the region from kb −3 to −2.8 of the p38δ promoter (Fig. 7). Chromatin DNA associated with c-Jun, c-Fos, and Ets1 was immunoprecipitated by appropriate antibodies and quantified by real-time PCR using primers amplifying the region from −3086 to −2759 of the p38δ promoter, which encompasses the predicted AP-1 and Ets binding sites. The results showed that c-Jun and Ets1 constitutively bound to this region in BJ cells, although their binding increased modestly in the presence of oncogenic ras (Fig. 7A). In contrast, c-Fos failed to bind to this region with or without ras. We failed to detect enrichment of the region from −3086 to −2759 of the p38δ promoter after ChIP using 2 independent ChIP-grade anti-c-fos antibodies, 4X from Santa Cruz (Fig. 7A) and 9F6 from Cell Signaling (data not shown). As a negative control, none of these transcription factors bound to a distal region (−7007 to −6723) on the p38δ promoter (Fig. 7B). These results indicate that c-Jun and Ets1 directly bind to the ras-responsive element on the p38δ promoter in cells. The lack of c-Fos binding suggests that c-Jun may bind to this region either as homodimers or as heterodimers with another Fos family member.
Fig 7.
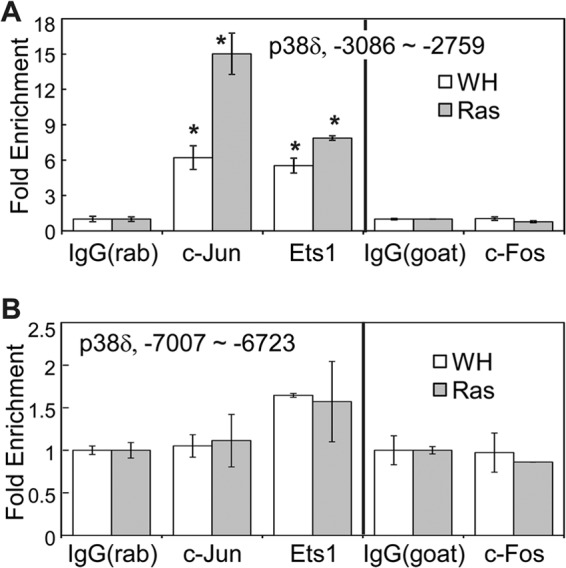
c-Jun and Ets1 directly bind to the ras-responsive element on the p38δ promoter. Chromatin was immunoprecipitated from BJ cells transduced with Ha-rasV12 (Ras) or vector (WH) using an anti-c-Jun, anti-Ets1, or anti-c-Fos antibody or control IgG (rabbit IgG for the anti-c-Jun and anti-Ets1 antibodies and goat IgG for the anti-c-Fos antibody). DNA present in the immunoprecipitated complexes was quantified by real-time PCR using primers that amplify the region from −3086 to −2759 (A) or that from −7007 to −6723 (B) of the p38δ promoter. Fold enrichment was calculated by dividing signals from the antibody immunoprecipitates by that from the IgG immunoprecipitates. Values are means ± SDs for triplicates. *, P < 0.01 versus IgG by Student's t test.
AP-1 and Ets transcription factors are activated by oncogenic ras through the MEK-ERK pathway during senescence induction.
The observation that MEK1 mediates ras-induced transcription from the p38δ promoter region from kb −3 to −2.8 containing the AP-1 and Ets binding sites (Fig. 6D and F) raises the possibility that the Ras–Raf-1–MEK–ERK pathway induces p38δ transcription by activating the AP-1 and Ets transcription factors. Indeed, previous studies showed that oncogenic Ha-rasV12 stimulates the transcriptional activity of c-Jun through phosphorylation at Ser63 and Ser73 within its transactivation domain, which can be mediated at least partly by ERK (40, 41). The Ets transcription factors, Ets1 and Ets2, contain conserved ERK phosphorylation sites (Thr38 in Ets1 and Thr72 in Ets2) that are critical for the induction of transcriptional activity by oncogenic ras signaling (42–44). ERK also phosphorylates c-Fos on multiple sites, including Ser374, in its transactivation domain, leading to activation of c-Fos (45, 46).
We thus investigated the contribution of the MEK-ERK pathway to AP-1 and Ets activation during ras-induced senescence. We found that like oncogenic ras, constitutively active MEK1 (MEK1Q56P and MEK1E+) induced phosphorylation of c-Jun-S73, c-Fos-S374, and Ets1-T38, which are critical for the induction of transcriptional activity of these transcription factors (Fig. 8A). Activated ras and MEK1 also increased the total c-Jun protein level in BJ cells, consistent with a previous report that Ha-ras stimulates c-Jun gene expression in NIH 3T3 fibroblasts (47). As expected, ras and active MEK1 induced activating phosphorylation of ERK and increased the expression of p38δ (Fig. 8A). Furthermore, a dominant negative mutant of MEK1 (MEK1-AA) abrogated ras-induced activating phosphorylation of c-Jun, Ets1, and c-Fos and, at the same time, greatly reduced the induction of p38δ expression by ras (Fig. 8B). Treatment of BJ cells with a MEK inhibitor, U0126, essentially abolished ras-induced ERK phosphorylation as well as the induction of p38δ expression (Fig. 8C). Our results thus demonstrate that oncogenic ras activates the AP-1 and Ets transcription factors through phosphorylation mediated by the MEK-ERK pathway, leading to induction of p38δ expression associated with ras-induced senescence. Notably, the MEK1-AA mutant greatly reduced, but did not completely abolish, ras-induced p38δ expression and c-Jun and Ets1 phosphorylation, likely because the MEK1-AA mutant did not completely eradicate the activity of the endogenous MEK1 when acting in a dominant negative fashion.
AP-1 and Ets1 are required for ras-induced p38δ transcription and ras-induced senescence.
The requirement of the AP-1 binding sites for ras-induced p38δ transcription (Fig. 6E) suggests that the AP-1 transcription factors may play an essential role in the induction of p38δ in senescence. To investigate the role of AP-1 in p38δ expression and senescence, we utilized a dominant negative c-Jun mutant, TAM67 (30, 48). TAM67 lacks the transactivation domain (amino acid 3 to 122) but retains the DNA binding domain of c-Jun. As a result, ectopic expression of TAM67 dominant negatively inhibits AP-1 activity by blocking the DNA binding of endogenous Jun-Jun homodimers and Jun-Fos heterodimers. When stably transduced into BJ primary fibroblasts, TAM67 greatly reduced the induction of p38δ mRNA (Fig. 9A) and protein (Fig. 9B) levels by oncogenic ras. In addition, while ras induced transcription from the luciferase reporter for the p38δ promoter (3kb-Luc), this induction was abolished by TAM67 (Fig. 9C). Together, these results indicate that the AP1 transcription factor is essential for the induction of p38δ gene transcription by ras in senescent cells.
Fig 9.

Activity of the AP-1 transcription factor is required for oncogenic ras-induced p38δ expression and senescence. (A) Analysis of p38δ mRNA levels by quantitative real-time RT-PCR in BJ cells transduced with a dominant negative mutant of c-Jun (TAM67) or vector (BP); and Ha-rasV12 (Ras) or vector (WH) on day 8 after ras transduction. The signals for p38δ mRNA were normalized to that of PBGD. Values are means ± SDs for triplicates. *, P < 0.05 versus BP by Student's t test. (B) Western blot analysis of p38δ protein levels in BJ cells transduced with a dominant negative mutant of c-Jun (TAM67) or vector (BP) and Ha-rasV12 (Ras) or vector (WH) on day 8 after ras transduction. (C) BJ cells stably transduced with a retroviral luciferase reporter containing a 3-kb p38δ promoter sequence upstream of the transcription start site were transduced with Ha-rasV12 (Ras) or vector (WH). Cells were lysed on day 8 after Ras transduction. Luciferase activity was measured and normalized to protein concentration. Fold induction by ras was calculated by dividing the luciferase activity in ras-expressing cells by that in control cells. Values are means ± SDs for triplicates. *, P < 0.005 versus BP by Student's t test. (D) Percentage of SA-β-Gal-positive cells in BJ cell populations transduced with a dominant negative mutant of c-Jun (TAM67) or vector (BP) and Ha-rasV12 (Ras) or vector (WH). *, P < 0.05 versus BP by Student's t test. (E) Morphology of BJ cells transduced with a dominant negative mutant of c-Jun (TAM67) or vector (BP) and Ha-rasV12 (Ras) or vector (WH). (F) Growth curves of BJ cells transduced with a scrambled shRNA (shSC) or c-Jun shRNA (shJun1555) and Ha-rasV12 (Ras) or vector (WH) over 21 days starting at PD32. Values are means ± SDs for triplicates. *, P < 0.01 versus shSC by Student's t test. (G) Percentage of SA-β-Gal-positive cells in BJ populations transduced with a scrambled shRNA (shSC) or c-Jun shRNA (shJun1555) and Ha-rasV12 (Ras) or vector (WH). Values are means ± SDs for triplicates. *, P < 0.01 versus shSC by Student's t test. (H) Western blot analysis of BJ populations transduced with a scrambled shRNA (shSC) or c-Jun shRNA (shJun1555) and Ha-rasV12 (Ras) or vector (WH). Numbers represent relative levels of proteins.
Moreover, we investigated the effect of TAM67-mediated AP-1 inhibition on ras-induced senescence. As reported previously (30), ectopic expression of TAM67 led to a proliferative arrest in BJ cells, which precluded the analysis of the effect of TAM67 on cell proliferation. However, ras-induced accumulation of SA-β-Gal-positive cells was essentially abolished in cells expressing TAM67 compared to the control cells (Fig. 9D), indicating disruption of ras-induced senescence upon AP-1 inhibition by TAM67. TAM67 also prevented the induction of the enlarged and flattened morphology characteristic of senescence in ras-expressing cells (Fig. 9E). In addition, a shRNA (shJun1555) that silenced c-Jun expression (Fig. 9H) disrupted ras-induced proliferative arrest (Fig. 9F) and accumulation of SA-β-Gal (Fig. 9G) and greatly reduced induction of p38δ expression by ras (Fig. 9H). These findings indicate that c-Jun is essential for ras-induced senescence and p38δ expression.
We further investigated the role of Ets1 in ras-induced senescence and p38δ expression. Two shRNAs were constructed that knocked down Ets1 expression at both mRNA and protein levels when stably transduced into BJ cells (shEts247 and shEts257) (Fig. 10A). These Ets1 shRNAs disrupted oncogenic ras-induced proliferative arrest (Fig. 10C) and accumulation of SA-β-Gal-positive cells (Fig. 10D) and concurrently abrogated induction of p38δ mRNA and protein expression by ras (Fig. 10B). Moreover, these Ets1 shRNAs greatly reduced ras-induced transcription from the 3-kb p38δ promoter reporter (Fig. 10E and F). These results indicate that Ets1 is required for the induction of senescence and p38δ expression by oncogenic ras.
Fig 10.

Ets1 is essential for oncogenic ras-induced p38δ expression and senescence. (A) Relative levels of Ets1 mRNA in BJ cells transduced with a scrambled shRNA (shSC) or Ets1 shRNA (shEts247 and shEts257) and Ha-rasV12 (Ras) or vector (WH), as detected by quantitative real-time RT-PCR on day 8 after ras transduction. Signals were normalized first to that of PBGD and then to that from cells transduced with scrambled shRNA control (shSC). Values are means ± SDs for triplicates. *, P < 0.05 versus shSC by Student's t test. (Inset) Western blot analysis of BJ cells transduced with a scrambled shRNA (shSC) or Ets1 shRNA (shEts247 and shEts257). (B) Relative levels of p38δ mRNA in BJ cells transduced with a scrambled shRNA (shSC) or Ets1 shRNA (shEts247 and shEts257) and Ha-rasV12 (Ras) or vector (WH), as detected by quantitative real-time RT-PCR on day 8 after ras transduction. Signals were normalized first to that of PBGD and then to that from cells transduced with vector control (WH). Values are means ± SDs for triplicates. *, P < 0.05 versus shSC by Student's t test. (Inset) Western blot analysis of the same cell lines. (C) Growth curve of BJ cells transduced with a scrambled shRNA (shSC) or Ets1 shRNA (shEts247 and shEts257) and Ha-rasV12 (Ras) or vector (WH) over 21 days starting on day 5 after ras transduction at PD 29. Values are means ± SDs for duplicates. *, P < 0.05 versus shSC by Student's t test. (D) Percentage of SA-β-Gal-positive cells in BJ populations transduced with a scrambled shRNA (shSC) or Ets1 shRNA (shEts247 and shEts257) and Ha-rasV12 (Ras) or vector (WH) on day 12 after ras transduction. Values are means ± SDs for duplicates. *, P < 0.01 versus shSC by Student's t test. (E and F) BJ cells stably transduced with the 3-kb p38δ promoter luciferase reporter were transduced first with a scrambled shRNA (shSC) or Ets1 shRNA (shEts247 in panel E and shEts257 in panel F) and then with Ha-rasV12 (Ras) or vector (WH). Cells were lysed on day 8 after Ras transduction. Luciferase activity was measured and normalized to protein concentration. Fold induction by ras was calculated by dividing the luciferase activity in ras-expressing cells by that in control cells. Values are means ± SDs for triplicates. *, P < 0.005 versus shSC by Student's t test.
Moreover, we investigated the epistatic relationship among p38δ, c-Jun, and Ets1 in the senescence pathway. While c-Jun and Ets1 shRNAs disrupted ras-induced senescence, they failed to abrogate senescence induced by constitutively active p38δ (Fig. 11A). In addition, wild-type p38δ restored ras-induced senescence in BJ cells expressing c-Jun or Ets1 shRNA (Fig. 11B). These results indicate that p38δ acts downstream of c-Jun and Ets1 to mediate senescence.
Fig 11.

p38δ acts downstream of c-Jun and Ets1 to mediate senescence. (A) BJ cells transduced with a scrambled shRNA (shSC) or shRNA for c-Jun (shJun1555) or Ets1 (shEts257) and a constitutively active mutant of p38δ (p38δF324S) or vector (WH) were seeded into 12-well plates on day 5 after p38δF324S transduction, after selection of transduced cells. Cell numbers (top graph) were counted 6 days after seeding. Cells were stained for SA-β-Gal on day 10 after p38δF324S transduction. The percentage of cells positive for SA-β-Gal was quantified (bottom graph). Values are means ± SDs for duplicates. *, P < 0.05 versus WH by Student's t test. (B) BJ cells transduced with a scrambled shRNA (shSC) or shRNA for c-Jun (shJun1555) or Ets1 (shEts257), wild-type p38δ (p38δWT) or vector (WN), and Ha-rasV12 (Ras) or vector (WH) were seeded into 12-well plates on day 5 after ras transduction, after selection of transduced cells. Cells (top graph) were counted 3 days after seeding. Cells were stained for SA-β-Gal on day 10 after ras transduction. The percentage of cells positive for SA-β-Gal was quantified (bottom graph). Values are means ± SDs for duplicates. *, P < 0.05 versus WN; **, P < 0.01 versus WN (by Student's t test).
Taken together, our results demonstrate that by stimulating the expression of p38δ, AP-1 and Ets1 play an essential role in oncogenic ras-induced senescence.
DISCUSSION
In the current study, we identified p38δ as an important mediator of oncogenic ras-induced senescence. Combined with our previous report (27), our results indicate that 3 out of the 4 p38 isoforms, p38α, p38γ, and p38δ, play an essential role in senescence induction by ras. Interestingly, these p38 isoforms seem to contribute to senescence induction by activating distinct downstream signaling pathways. While p38α and p38γ induce p16INK4A and p53/p21WAF1, respectively, p38δ mediates senescence through a mechanism that is independent of these senescence effectors. As previously reported for p38α and p38γ (27), the constitutively active mutant of p38δ only induced a partial senescence phenotype compared to oncogenic ras, as manifested by reduced, rather than arrested, cell proliferation and a lower percentage of SA-β-Gal-positive cells (Fig. 1E and F). This finding is consistent with the notion that each of these p38 isoforms is able to activate only some, and not all, of the senescence effectors required for the full induction of senescence.
Our findings demonstrate that activation of p38δ in the course of ras-induced senescence occurs at 2 levels. Oncogenic ras induces the Raf-1–MEK–ERK pathway, which, in turn, activates AP-1 and Ets transcription factors, leading to increased p38δ transcription. Once expressed at elevated levels, p38δ serves as the substrate for MKK3 and MKK6, which, upon activation by ras, induce p38δ activity through direct phosphorylation in its activation loop. It is important to note that the basal level of p38δ expression is very low and barely detectable by Western blotting in various primary human fibroblasts. A mechanism for the enhancement of p38δ expression may therefore be essential, in order to provide MKK3/6 with a sufficient amount of substrates and to ensure that the p38δ signaling is strong enough for senescence induction. Supporting the notion that ras-induced senescence requires a high level of p38δ protein, shRNA-mediated reduction in p38δ expression led to abrogation of senescence. In addition to p38δ, the expression of p38α was also induced by oncogenic ras in BJ cells; however, the induction of p38α occurs without an increase in mRNA (Fig. 3A and B). The mechanism and functional significance of ras-induced p38α expression are under investigation.
The current study identified the AP-1 and Ets transcription factors as novel signaling components in the pathway that mediates oncogene-induced senescence. We show that upon activation by oncogenic ras through the MEK-ERK effector pathway, AP-1 and Ets stimulate p38δ gene transcription on the p38δ promoter. Most importantly, inhibition of AP-1 or Ets activity leads to disruption of oncogenic ras-induced senescence. These findings demonstrate that AP-1 and Ets transcription factors are integral components of the senescence-inducing pathway, which play an essential role in senescence by mediating the induction of p38δ expression.
It has been reported previously that Ets binding sites and AP-1 binding sites often reside next to each other and that Ets and AP-1 cooperate in transcriptional activation (49, 50). Consistent with these reports, we found that Ets and AP-1 indeed bind to sites immediately adjacent to each other in the ras-responsive element of the p38δ promoter and cooperate to mediate p38δ induction by ras. However, the exact identities of the Ets and AP-1 transcription factors mediating the ras induction on the p38δ promoter remain to be fully elucidated. While our data indicate that Ets1 directly binds to the ras-responsive element of the p38δ promoter, the role of Ets2, the other member of the Ets family of transcription factors, is currently unknown. Moreover, ChIP analysis detected direct binding of c-Jun but not c-Fos to the ras-responsive element. Thus, it remains to be determined whether c-Jun stimulates transcription on the p38δ promoter as a homodimer or a heterodimer with a Fos family member other than c-Fos.
p53 is a key senescence effector and a known substrate of p38 MAPK. In fact, our previous study indicated that p38γ contributes to ras-induced senescence by mediating p53 activation through direct phosphorylation of p53 at Ser33 (27). However, despite the ability of recombinant p38δ to phosphorylate p53 at Ser33 in vitro, p38δ is not essential for ras-induced p53-Ser33 phosphorylation or p53 transcriptional activity in cells, and a constitutively active mutant of p38δ also fails to induce p53-Ser33 phosphorylation or a p53 transcriptional target, p21WAF1. Therefore, similar to p38α but unlike p38γ, p38δ is not the major protein kinase for p53-Ser33 phosphorylation during senescence induction. It is possible that in cells, the kinase activity of different p38 isoforms toward p53 is differentially regulated by posttranslational modifications, subcellular localization, or interactions with inhibitory or stimulatory factors.
ACKNOWLEDGMENTS
We thank Scott Lowe, Peter Vogt, Jiahuai Han, Shuang Huang, David Engelberg, Oded Livnah, and Robert Hennigan for providing reagents.
Research reported in this publication was supported by the National Cancer Institute of the National Institutes of Health under award numbers CA106768 and CA131231 (P.S.) and the 973 Program in China (2013CB957201) (R.X.).
Footnotes
Published ahead of print 22 July 2013
The Scripps manuscript number for this article is 22004.
REFERENCES
- 1.Dimri GP, Itahana K, Acosta M, Campisi J. 2000. Regulation of a senescence checkpoint response by the E2F1 transcription factor and p14(ARF) tumor suppressor. Mol. Cell. Biol. 20:273–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Olsen CL, Gardie B, Yaswen P, Stampfer MR. 2002. Raf-1-induced growth arrest in human mammary epithelial cells is p16-independent and is overcome in immortal cells during conversion. Oncogene 21:6328–6339 [DOI] [PubMed] [Google Scholar]
- 3.Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. 1997. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 88:593–602 [DOI] [PubMed] [Google Scholar]
- 4.Zhu J, Woods D, McMahon M, Bishop JM. 1998. Senescence of human fibroblasts induced by oncogenic Raf. Genes Dev. 12:2997–3007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Braig M, Lee S, Loddenkemper C, Rudolph C, Peters AH, Schlegelberger B, Stein H, Dorken B, Jenuwein T, Schmitt CA. 2005. Oncogene-induced senescence as an initial barrier in lymphoma development. Nature 436:660–665 [DOI] [PubMed] [Google Scholar]
- 6.Chen Z, Trotman LC, Shaffer D, Lin HK, Dotan ZA, Niki M, Koutcher JA, Scher HI, Ludwig T, Gerald W, Cordon-Cardo C, Pandolfi PP. 2005. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature 436:725–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Collado M, Gil J, Efeyan A, Guerra C, Schuhmacher AJ, Barradas M, Benguria A, Zaballos A, Flores JM, Barbacid M, Beach D, Serrano M. 2005. Tumour biology: senescence in premalignant tumours. Nature 436:642. [DOI] [PubMed] [Google Scholar]
- 8.Michaloglou C, Vredeveld LC, Soengas MS, Denoyelle C, Kuilman T, van der Horst CM, Majoor DM, Shay JW, Mooi WJ, Peeper DS. 2005. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature 436:720–724 [DOI] [PubMed] [Google Scholar]
- 9.Narita M, Lowe SW. 2005. Senescence comes of age. Nat. Med. 11:920–922 [DOI] [PubMed] [Google Scholar]
- 10.Sun P, Yoshizuka N, New L, Moser BA, Li Y, Liao R, Xie C, Chen J, Deng Q, Yamout M, Dong MQ, Frangou CG, Yates JRIII, Wright PE, Han J. 2007. PRAK is essential for ras-induced senescence and tumor suppression. Cell 128:295–308 [DOI] [PubMed] [Google Scholar]
- 11.Lin AW, Barradas M, Stone JC, Van Aelst L, Serrano M, Lowe SW. 1998. Premature senescence involving p53 and p16 is activated in response to constitutive MEK/MAPK mitogenic signaling. Genes Dev. 12:3008–3019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ferbeyre G, de Stanchina E, Lin AW, Querido E, McCurrach ME, Hannon GJ, Lowe SW. 2002. Oncogenic ras and p53 cooperate to induce cellular senescence. Mol. Cell. Biol. 22:3497–3508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Narita M, Nunez S, Heard E, Narita M, Lin AW, Hearn SA, Spector DL, Hannon GJ, Lowe SW. 2003. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 113:703–716 [DOI] [PubMed] [Google Scholar]
- 14.Wang W, Chen JX, Liao R, Deng Q, Zhou JJ, Huang S, Sun P. 2002. Sequential activation of the MEK-extracellular signal-regulated kinase and MKK3/6-p38 mitogen-activated protein kinase pathways mediates oncogenic ras-induced premature senescence. Mol. Cell. Biol. 22:3389–3403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hui L, Bakiri L, Mairhorfer A, Schweifer N, Haslinger C, Kenner L, Komnenovic V, Scheuch H, Beug H, Wagner EF. 2007. p38alpha suppresses normal and cancer cell proliferation by antagonizing the JNK-c-Jun pathway. Nat. Genet. 39:741–749 [DOI] [PubMed] [Google Scholar]
- 16.Ventura JJ, Tenbaum S, Perdiguero E, Huth M, Guerra C, Barbacid M, Pasparakis M, Nebreda AR. 2007. p38alpha MAP kinase is essential in lung stem and progenitor cell proliferation and differentiation. Nat. Genet. 39:750–758 [DOI] [PubMed] [Google Scholar]
- 17.Enslen H, Brancho DM, Davis RJ. 2000. Molecular determinants that mediate selective activation of p38 MAP kinase isoforms. EMBO J. 19:1301–1311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Han J, Lee JD, Bibbs L, Ulevitch RJ. 1994. A MAP kinase targeted by endotoxin and hyperosmolarity in mammalian cells. Science 265:808–811 [DOI] [PubMed] [Google Scholar]
- 19.Jiang Y, Chen C, Li Z, Guo W, Gegner JA, Lin S, Han J. 1996. Characterization of the structure and function of a new mitogen-activated protein kinase (p38beta). J. Biol. Chem. 271:17920–17926 [DOI] [PubMed] [Google Scholar]
- 20.Jiang Y, Gram H, Zhao M, New L, Gu J, Feng L, Di Padova F, Ulevitch RJ, Han J, Han J, Lee JD, Bibbs L, Ulevitch RJ. 1997. Characterization of the structure and function of the fourth member of p38 group mitogen-activated protein kinases, p38delta A MAP kinase targeted by endotoxin and hyperosmolarity in mammalian cells. J. Biol. Chem. 272:30122–30128 [DOI] [PubMed] [Google Scholar]
- 21.Li Z, Jiang Y, Ulevitch RJ, Han J. 1996. The primary structure of p38 gamma: a new member of p38 group of MAP kinases. Biochem. Biophys. Res. Commun. 228:334–340 [DOI] [PubMed] [Google Scholar]
- 22.Shi Y, Gaestel M. 2002. In the cellular garden of forking paths: how p38 MAPKs signal for downstream assistance. Biol. Chem. 383:1519–1536 [DOI] [PubMed] [Google Scholar]
- 23.Tanoue T, Yamamoto T, Maeda R, Nishida E. 2001. A novel MAPK phosphatase MKP-7 acts preferentially on JNK/SAPK and p38 alpha and beta MAPKs. J. Biol. Chem. 276:26629–26639 [DOI] [PubMed] [Google Scholar]
- 24.Reference deleted.
- 25.Reference deleted.
- 26.Reference deleted.
- 27.Kwong J, Hong L, Liao R, Deng Q, Han J, Sun P. 2009. p38alpha and p38gamma mediate oncogenic ras-induced senescence through differential mechanisms. J. Biol. Chem. 284:11237–11246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brummelkamp TR, Bernards R, Agami R. 2002. A system for stable expression of short interfering RNAs in mammalian cells. Science 296:550–553 [DOI] [PubMed] [Google Scholar]
- 29.Brummelkamp TR, Bernards R, Agami R. 2002. Stable suppression of tumorigenicity by virus-mediated RNA interference. Cancer Cell 2:243–247 [DOI] [PubMed] [Google Scholar]
- 30.Hennigan RF, Stambrook PJ. 2001. Dominant negative c-jun inhibits activation of the cyclin D1 and cyclin E kinase complexes. Mol. Biol. Cell 12:2352–2363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sun P, Dong P, Dai K, Hannon GJ, Beach D. 1998. p53-independent role of MDM2 in TGF-beta1 resistance. Science 282:2270–2272 [DOI] [PubMed] [Google Scholar]
- 32.Hong L, Lai M, Chen M, Xie C, Liao R, Kang YJ, Xiao C, Hu WY, Han J, Sun P. 2010. The miR-17-92 cluster of microRNAs confers tumorigenicity by inhibiting oncogene-induced senescence. Cancer Res. 70:8547–8557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shay JW, Wright WE. 1989. Quantitation of the frequency of immortalization of normal human diploid fibroblasts by SV40 large T-antigen. Exp. Cell Res. 184:109–118 [DOI] [PubMed] [Google Scholar]
- 34.Deng Q, Li Y, Tedesco D, Liao R, Fuhrmann G, Sun P. 2005. The ability of E1A to rescue ras-induced premature senescence and confer transformation relies on inactivation of both p300/CBP and Rb family proteins. Cancer Res. 65:8298–8307 [DOI] [PubMed] [Google Scholar]
- 35.Askari N, Diskin R, Avitzour M, Capone R, Livnah O, Engelberg D. 2007. Hyperactive variants of p38alpha induce, whereas hyperactive variants of p38gamma suppress, activating protein 1-mediated transcription. J. Biol. Chem. 282:91–99 [DOI] [PubMed] [Google Scholar]
- 36.Avitzour M, Diskin R, Raboy B, Askari N, Engelberg D, Livnah O. 2007. Intrinsically active variants of all human p38 isoforms. FEBS J. 274:963–975 [DOI] [PubMed] [Google Scholar]
- 37.Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, Issaeva N, Vassiliou LV, Kolettas E, Niforou K, Zoumpourlis VC, Takaoka M, Nakagawa H, Tort F, Fugger K, Johansson F, Sehested M, Andersen CL, Dyrskjot L, Orntoft T, Lukas J, Kittas C, Helleday T, Halazonetis TD, Bartek J, Gorgoulis VG. 2006. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 444:633–637 [DOI] [PubMed] [Google Scholar]
- 38.Di Micco R, Fumagalli M, Cicalese A, Piccinin S, Gasparini P, Luise C, Schurra C, Garre' M, Nuciforo PG, Bensimon A, Maestro R, Pelicci PG, d'Adda di Fagagna F. 2006. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 444:638–642 [DOI] [PubMed] [Google Scholar]
- 39.Shaulian E, Karin M. 2002. AP-1 as a regulator of cell life and death. Nat. Cell Biol. 4:E131–E136. 10.1038/ncb0502-e131 [DOI] [PubMed] [Google Scholar]
- 40.Pulverer BJ, Kyriakis JM, Avruch J, Nikolakaki E, Woodgett JR. 1991. Phosphorylation of c-jun mediated by MAP kinases. Nature 353:670–674 [DOI] [PubMed] [Google Scholar]
- 41.Smeal T, Binetruy B, Mercola DA, Birrer M, Karin M. 1991. Oncogenic and transcriptional cooperation with Ha-Ras requires phosphorylation of c-Jun on serines 63 and 73. Nature 354:494–496 [DOI] [PubMed] [Google Scholar]
- 42.McCarthy SA, Chen D, Yang BS, Garcia Ramirez JJ, Cherwinski H, Chen XR, Klagsbrun M, Hauser CA, Ostrowski MC, McMahon M. 1997. Rapid phosphorylation of Ets-2 accompanies mitogen-activated protein kinase activation and the induction of heparin-binding epidermal growth factor gene expression by oncogenic Raf-1. Mol. Cell. Biol. 17:2401–2412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Paumelle R, Tulasne D, Kherrouche Z, Plaza S, Leroy C, Reveneau S, Vandenbunder B, Fafeur V. 2002. Hepatocyte growth factor/scatter factor activates the ETS1 transcription factor by a RAS-RAF-MEK-ERK signaling pathway. Oncogene 21:2309–2319 [DOI] [PubMed] [Google Scholar]
- 44.Yang BS, Hauser CA, Henkel G, Colman MS, Van BC, Stacey KJ, Hume DA, Maki RA, Ostrowski MC. 1996. Ras-mediated phosphorylation of a conserved threonine residue enhances the transactivation activities of c-Ets1 and c-Ets2. Mol. Cell. Biol. 16:538–547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen RH, Abate C, Blenis J. 1993. Phosphorylation of the c-Fos transrepression domain by mitogen-activated protein kinase and 90-kDa ribosomal S6 kinase. Proc. Natl. Acad. Sci. U. S. A. 90:10952–10956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Monje P, Marinissen MJ, Gutkind JS. 2003. Phosphorylation of the carboxyl-terminal transactivation domain of c-Fos by extracellular signal-regulated kinase mediates the transcriptional activation of AP-1 and cellular transformation induced by platelet-derived growth factor. Mol. Cell. Biol. 23:7030–7043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sistonen L, Holtta E, Makela TP, Keski-Oja J, Alitalo K. 1989. The cellular response to induction of the p21 c-Ha-ras oncoprotein includes stimulation of jun gene expression. EMBO J. 8:815–822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brown PH, Alani R, Preis LH, Szabo E, Birrer MJ. 1993. Suppression of oncogene-induced transformation by a deletion mutant of c-jun. Oncogene 8:877–886 [PubMed] [Google Scholar]
- 49.Wasylyk B, Wasylyk C, Flores P, Begue A, Leprince D, Stehelin D. 1990. The c-ets proto-oncogenes encode transcription factors that cooperate with c-Fos and c-Jun for transcriptional activation. Nature 346:191–193 [DOI] [PubMed] [Google Scholar]
- 50.Wasylyk C, Flores P, Gutman A, Wasylyk B. 1989. PEA3 is a nuclear target for transcription activation by non-nuclear oncogenes. EMBO J. 8:3371–3378 [DOI] [PMC free article] [PubMed] [Google Scholar]