

Abstract
Spt2/Sin1 is a DNA binding protein with HMG-like domains. It plays a role in chromatin modulations associated with transcription elongation in Saccharomyces cerevisiae. Spt2 maintains the nucleosome level in coding regions and is important for the inhibition of spurious transcription in yeast. In this work, we undertook a biochemical approach to identify Spt2-interacting partners. Interestingly, casein kinase 2 (CK2) interacts with Spt2 and phosphorylates it in vitro as well as in vivo on two small regions, region I (RI) (amino acids 226 to 230) and RII (amino acids 277 to 281), located in its essential C-terminal domain. Mutation of the phosphorylation sites in RI and RII to acidic residues, thereby mimicking CK2 phosphorylation, leads to the inhibition of Spt2 function in the repression of spurious transcription and to a loss of its recruitment to coding regions. Inversely, depleting cells of CK2 activity leads to an increased Spt2 association with genes. We further show that Spt2 physically interacts with the essential histone chaperone Spt6 and that this association is inhibited in vitro and in vivo by CK2-dependent phosphorylation. Taken together, our data suggest that CK2 regulates the function of Spt2 by modulating its interaction with chromatin and the histone chaperone Spt6.
INTRODUCTION
The genome of the eukaryotic cell is packaged in a topologically complex structure known as chromatin. The nucleosome is the basic fundamental unit of chromatin. It is composed of a core histone octamer formed by two molecules of H2A, H2B, H3, and H4, wrapped around by 146 bp of DNA (1). Chromatin represents an additional level of regulation for DNA-mediated processes, such as transcription, replication, repair, and recombination, by acting as a platform where biological signals are integrated and molecular responses take place. In addition to histones, several nonhistone proteins are important for defining chromatin structure. These include DNA binding proteins and chromatin-associated factors that alter nucleosome structure and function. Spt2/Sin1 is a DNA binding protein with HMG-like domains that is associated with yeast chromatin (2–4). It was originally identified by mutations that suppress the insertion of the transposon Ty in the promoter of the HIS4 gene (suppressor of Ty2) (5). Spt2 is also called Sin1 (Swi-independent 1) because a mutation in SPT2 suppresses the effects of the loss of the Swi/Snf chromatin-remodeling complex (2). In addition, mutations in SPT2 suppress the mutant phenotypes associated with mutations in the SAGA complex and partial deletions in the C-terminal domain of the largest subunit of RNA polymerase II (RNAP II), Rpb1 (2–4). Taken together, these reports strongly suggested a negative role of Spt2 in transcription initiation.
In recent work, we have shown that the Spt2 function is also tightly linked to nucleosome reassembly in the wake of transcription (6, 7). Defects in this function lead to spurious transcription from thousands of cryptic promoters located within the coding regions of genes (8, 9). Although the exact role of Spt2 remains unknown, there is now clear evidence that associates this protein with chromatin modulations linked to transcription elongation and repression of spurious transcription (6, 7). First, Spt2 travels with RNAP II during transcription elongation, and its recruitment to transcribed regions is dependent on the essential histone chaperone Spt6 (6, 7). Second, SPT2 interacts genetically with several factors, such as the Paf and HIR/HPC complexes, that are involved directly in chromatin modulation associated with transcription elongation (6). Third, Spt2 is required for the maintenance of chromatin structure in the open reading frames (ORFs) of transcribed genes, participates in the reassembly of nucleosomes at these locations, and is required for the repression of spurious transcription (6, 7). Fourth, Spt2 suppresses the histone exchange in coding regions (7). Fifth, similar to the histone chaperones Spt6 and yFACT, this factor is involved in the transcription-dependent deposition of nucleosomes at the transcribed intergenic region of SRG1-SER3 (7, 10–12). This deposition is a crucial event in the regulation of the SER3 gene (7, 10–12). Taken together, these molecular and genetic observations indicate an important role of Spt2 in chromatin reassembly associated with transcription elongation. Other Spt2 roles have been reported, specifically in the maintenance of genome stability. Indeed, overexpression of this factor leads to transcription-dependent gross chromosomal rearrangement (GCR), while the deletion of its gene is associated with transcription-dependent hyperrecombination (6, 13). Spt2 overexpression was associated with high levels of DNA/RNA hybrids that were linked to increases in the number of GCR events and suggested that Spt2 association with transcription sites must be tightly regulated (6, 13).
The genetic and biochemical evidence regarding Spt2 points clearly toward an important role of this protein during transcription elongation. However, despite its functional importance, the exact mechanistic details of Spt2 function and regulation are still not well understood. In this work, we show that CK2 phosphorylates Spt2 and modulates its function in the repression of spurious transcription. In addition, we provide evidence that Spt2 interacts directly with Spt6, and we further show that this interaction is inhibited in vitro and in vivo by CK2 phosphorylation. Taken together, our findings indicate that CK2 regulates the function of the chromatin reassembly factor Spt2 through the modulation of its interaction with Spt6 and the chromatin of coding regions.
MATERIALS AND METHODS
Saccharomyces cerevisiae strains and plasmids.
Strains were constructed by using standard genetic methods and are isogenic to S288C (14) (see Table 1 posted at http://www.crc.ulaval.ca/nourani/supplemental_data). The strains containing the alleles encoding the epitope-tagged proteins were constructed by integrating the DNA encoding the epitope at the 3′ end of the corresponding gene (15–17). The cka1Δ::KANMX6 allele was constructed by replacing the open reading frame with the KANMX6 marker (16). The point mutation in the CKA2 allele (D225N) was introduced as described previously (18), with NatMX6 as the selection marker. Plasmids were constructed by using standard molecular biology techniques. Glutathione S-transferase (GST) fusion and 6His fusion plasmids were constructed by the insertion of PCR-amplified fragments into the appropriate sites of pGEX-4T3 (GE Healthcare, Life Sciences) and pet15b (Novagen). Plasmids pRS415 and pRS416 were described previously (19). Spt2 and Spt2-Flag along with the promoter (900 bp upstream of start codon to 500 bp downstream of stop codon) were amplified from the genome and inserted into plasmids pRS415 and pRS416 (19). Mutagenesis was done by using the QuikChange mutagenesis kit from Stratagene and the instructions therein. The constructions used in this study were checked by sequencing.
Protein purification.
Recombinant 6His-tagged and GST-tagged proteins were expressed in Escherichia coli BL21 bacteria and purified with Ni2+-nitrilotriacetic acid (NTA)-agarose (Qiagen) or glutathione-Sepharose (GE Healthcare) according to the manufacturer's protocol. Tandem affinity purification (TAP) of Spt2 and Ckb2 was done as described previously (17).
GST pulldown assays and coimmunoprecipitation.
GST-Spt2, GST–Spt2-5A, or GST–Spt2-acidic coupled to beads (500 ng) was incubated or not with 500 ng of His-Spt6 in pulldown buffer (20 mM HEPES [pH 7.5], 150 mM NaCl, 10% glycerol, 100 μg/ml bovine serum albumin [BSA], 0.5 mM dithiothreitol [DTT], 0.1% NP-40, 2 μg/ml leupeptin and pepstatin, 5 μg/ml aprotinin) for 3 h at 4°C. Beads were washed three times with pulldown buffer, and bound proteins were then analyzed by Western blotting with anti-His tag antibodies. To study the Spt2-Spt6 interaction, the GST-Spt2/His-Spt6 complex was assembled by the GST pulldown assay as described above. After the pulldown, the beads were washed three times with phosphorylation buffer (80 mM NaCl-KCl, 25 mM Tris-HCl [pH 8], 10 mM MgCl2, 1 mM DTT) and suspended in phosphorylation buffer to make a 50% slurry. One microliter of the TAP-Ckb2 purification fraction was then added to the slurry in the presence or absence of 50 μM ATP. After incubation at 30°C for 30 min on a rotating wheel, the reactions were stopped by centrifugation at 3,000 rpm for 2 min. Finally, 15% of the beads and a large fraction of the supernatant were resolved by 12% SDS-polyacrylamide gel electrophoresis (PAGE) and analyzed by Western blotting with anti-His tag and anti-GST antibodies. Coimmunoprecipitation assays were performed as described previously (20). Briefly, 10 μl of anti-Flag M2 agarose beads were incubated overnight at 4°C with yeast whole-cell extract (WCE) (5 mg of total protein) in binding buffer (20 mM HEPES [pH 7.5], 300 mM NaCl, 10% glycerol, 0.1% NP-40, 2 μg/ml of leupeptin and pepstatin, 5 μg/ml of aprotinin, 1 mM phenylmethylsulfonyl fluoride [PMSF]). Beads were washed three times with binding buffer. Bound proteins were eluted with the 3×Flag peptide and analyzed by Western blotting.
In vitro phosphorylation.
In vitro phosphorylation of recombinant proteins was done as described previously (21, 22), with slight modifications. GST- or His-tagged recombinant proteins (1 μg) were incubated for 30 min at 30°C with CK2 purified from yeast in kinase buffer (final concentration of 80 mM NaCl-KCl, 25 mM Tris-HCl [pH 8], 10 mM MgCl2, 1 mM DTT, 50 μM cold ATP, and 1 μCi [γ-32P]ATP). Samples were run on 12% SDS-PAGE gels, blotted onto nitrocellulose, dried, and exposed to film. For the in vitro phosphorylation experiments done with yeast whole-cell extracts, 1 μg of GST-tagged proteins coupled to beads was incubated with yeast whole-cell extract (5 mg total proteins) at 4°C for 3 h in binding buffer (20 mM HEPES [pH 7.5], 150 mM NaCl, 10% glycerol, 0.1% NP-40, 2 μg/ml of leupeptin and pepstatin, 5 μg/ml of aprotinin, 1 mM PMSF). Beads were washed three times with binding buffer and once with kinase buffer (without [γ-32P]ATP), and phosphorylation was carried by the addition of kinase buffer with [γ-32P]ATP.
Phosphatase treatment and two-dimensional electrophoresis.
For dephosphorylation with alkaline phosphatase, cells were lysed in 1× NEB buffer 3. Cell lysates were cleared by centrifugation at 15,000 × g for 30 min. Total protein (10 μg) was incubated with 10 units of alkaline phosphatase at 37°C for 1 h (calf intestinal alkaline phosphatase; NEB). The reactions were stopped by trichloroacetic acid (TCA) precipitation, and proteins were then analyzed by two-dimensional PAGE (2D-PAGE), as indicated below. For 2D-PAGE, soluble proteins were precipitated by 20% TCA followed by an acetone wash. Precipitated proteins were then solubilized in isoelectric focusing (IEF) buffer {8 M urea, 2% 3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate (CHAPS), 18.5 mM DTT, and 2% IPG buffer (pH 7 to 11)} (GE Healthcare) and separated in the first dimension by a nonlinear pH gradient (Immobiline DryStrip NL 7-11; GE Healthcare) at 90 kV/h. After isoelectric focusing in the first dimension, proteins were resolved in the second dimension by standard SDS-PAGE and analyzed by Western blotting.
RNA extraction and Northern blotting.
Total RNA was isolated by using the hot-phenol method (23). Northern blot analysis was done as described previously (6). RNA (40 μg) was separated on a 1% agarose formaldehyde morpholinepropanesulfonic acid (MOPS) gel and transferred onto a nylon membrane. The FLO8, SCR1, and SER3 probes were amplified by PCR and radiolabeled by random priming.
Chromatin immunoprecipitation.
Chromatin immunoprecipitation (ChIP) experiments were performed as described previously (6).
RESULTS
CK2 subunits copurify with Spt2.
To further understand the role of Spt2 during transcription elongation, we searched for proteins that interact in vivo with this factor. For this, we constructed a yeast strain expressing a TAP-tagged Spt2 protein, performed TAP sequentially on IgG and calmodulin columns, and analyzed the proteins by SDS-PAGE followed by silver staining (Fig. 1A). In addition to Spt2, we observed that three other proteins of 45 kDa, 40 kDa, and 30 kDa copurified consistently with this factor. To identify these proteins, we excised all bands, eluted the proteins, and analyzed them by mass spectrometry. The number of peptides identified and the corresponding proteins are reported in Fig. 1B (see also Fig. S1 posted at http://www.crc.ulaval.ca/nourani/supplemental_data). Interestingly, in addition to other proteins, we identified peptides of all subunits of the casein kinase 2 (CK2) complex. These results suggest that the multisubunit kinase CK2 interacts in vivo with Spt2.
Fig 1.

Spt2 associates stably with CK2. (A) Silver-stained gel of a TAP-purified Spt2 preparation. The right lane of the silver-stained gel is a mock purification performed with a whole-cell extract (WCE) of an untagged strain. (B) Protein bands excised from the Spt2 TAP purification gel were subjected to mass spectrometry, and the peptides identified are shown. (C and D) TAP of Spt2 from a strain expressing Spt2-TAP and Cka1-Myc or Cka2-Myc. A mock purification was performed by using a WCE of an untagged strain expressing either Cka1-Myc or Cka2-Myc. Different fractions were analyzed by Western blotting using anti-TAP and anti-Myc antibodies. (E) WCEs from cells expressing Spt2-Flag and Cka1-Myc or Cka2-Myc were immunoprecipitated with an anti-Flag antibody. The bound proteins were subsequently eluted from the anti-Flag beads with Flag peptide. A mock purification was also performed by using a WCE of an untagged strain expressing either Cka1-Myc or Cka2-Myc. The proteins eluted by the Flag peptide were analyzed by Western blotting using anti-Myc and anti-Flag antibodies. (F) Diagram illustrating the purification protocol where TAP of Ckb2 from a strain expressing Spt2-Flag and Ckb2-TAP was followed by Flag bead purification and a final Flag peptide elution. A mock purification was performed by using a WCE of an untagged strain expressing only Spt2-Flag. FT, flowthrough. (G) Different fractions from the Ckb2-TAP/Flag purification and mock purification were analyzed by Western blotting using anti-TAP and anti-Flag antibodies.
CK2 has a broad range of targets and interacts with many proteins in vivo in yeast, including elongation factors (24, 25). To confirm the mass spectrometry data, we constructed yeast strains expressing TAP-tagged Spt2 and Myc-tagged versions of two different CK2 subunits, Cka1 and Cka2. We purified Spt2 by TAP and found that both Cka1-Myc and Cka2-Myc coeluted with Spt2 after the purification steps (Fig. 1C and D). Importantly, no such coelution was observed for the control strains that do not express an Spt2-TAP-tagged version. This suggests that Cka1-Myc and Cka2-Myc signals observed after TAP are specific to an association with Spt2. To test whether this coelution is dependent on the purification protocol, we Flag purified Spt2 from a strain expressing Spt2-Flag, Cka1-Myc, or Cka2-Myc. After immunopurification with anti-Flag beads followed by elution with Flag peptides, Cka1-Myc and Cka2-Myc coeluted with Spt2-Flag (Fig. 1E). These data show that CK2 and Spt2 copurify with each other independently of the purification procedure.
Finally, we tested the stability and strength of the Spt2-CK2 complex. For this, we used a multistep purification protocol by combining TAP steps with other affinity purification steps at a high salt concentration (see purification protocol in Fig. 1F). In these experiments, we purified another CK2 subunit by TAP, Ckb2, from a strain expressing Spt2-Flag. The fractions containing the purified CK2 complex were then pooled and submitted to Flag immunoprecipitation followed by Flag peptide elution. The two latter steps were done at a high salt concentration (300 mM NaCl). As shown in Fig. 1G, after being submitted to multiple purification steps, Spt2 copurified with Ckb2, clearly indicating a strong and stable association between the elongation factor and the kinase. Finally, we tested in vitro the association between Spt2 and CK2 by GST pulldown assays and found a direct interaction between Spt2 and Ckb2 (see Fig. S2 posted at http://www.crc.ulaval.ca/nourani/supplemental_data). Taken together, our data indicate a specific and direct interaction between the elongation factor Spt2 and the essential kinase CK2.
Spt2 is phosphorylated in vitro by CK2 on its C-terminal domain.
The association of Spt2 with CK2 led us to think that Spt2 could be a substrate of this kinase. To address this possibility directly, we first asked if Spt2 could be phosphorylated by CK2 in vitro. We purified 6His-Spt2 from bacteria and tested this recombinant protein in an in vitro kinase assay. As indicated in Fig. 2A, 6His-Spt2 was incubated with or without the CK2 complex purified from yeast in the presence of [γ-32P]ATP. The proteins were then analyzed by SDS-PAGE followed by autoradiography or Western blotting using an anti-His tag antibody. As shown in Fig. 2A, 6His-Spt2 incorporated labeled phosphate only in the presence of purified CK2, indicating that it is phosphorylated by this kinase. Interestingly, a lower band was observed in the presence or absence of Spt2, suggesting that there is autophosphorylation of CK2. We conclude that Spt2 is a direct target of CK2. Next, we wanted to determine which Spt2 domain is phosphorylated by CK2. For this, we purified different recombinant domains of Spt2 fused to a His tag or GST (described in the legend of Fig. 2B) and tested the ability of purified yeast CK2 to phosphorylate them. As shown in Fig. 2C, we tested the full-length fusion protein His-Spt2, two versions deleted in the C-terminal domain (His–Spt2-213 or GST–Spt2-179), and the GST-Spt2 C-terminal domain (GST–Spt2-Ct [amino acids {aa} 200 to 333]). Full-length Spt2 and the C-terminal domain of Spt2 fused to GST were the only versions phosphorylated under these conditions, indicating that CK2 phosphorylates the C-terminal domain of Spt2 directly. Thus, our data indicate that the phosphorylation site(s) is located in the region between amino acids 214 and 333. We next analyzed the phosphorylation of the different Spt2 domains by a wild-type or CK2-depleted yeast whole-cell extract (Fig. 2D). The C-terminal domain of Spt2 was the only domain phosphorylated in vitro by yeast whole-cell extract (Fig. 2D, top). Interestingly, when we used a whole-cell extract from a ck2ts strain, the phosphorylation of the GST–Spt2-Ct fusion protein was significantly reduced (Fig. 2D, middle). Therefore, we conclude that CK2 is the major yeast kinase that interacts with Spt2 and directly phosphorylates this factor on its C-terminal domain (aa 214 to 333).
Fig 2.
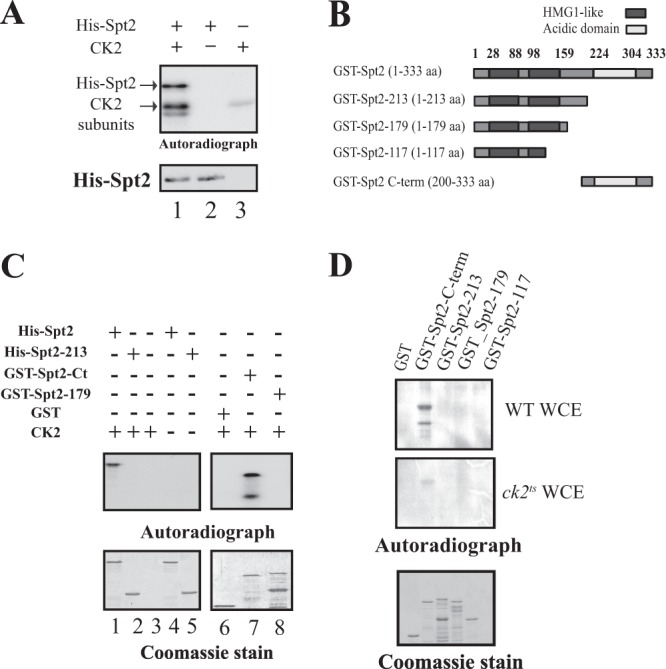
CK2 phosphorylates Spt2 in vitro on the essential C-terminal domain. (A) Yeast CK2 phosphorylates Spt2 in vitro. Recombinant His-Spt2 was incubated with TAP-purified CK2 (lane 1) or with a mock TAP purification in the presence of radiolabeled ATP (lane 2). Lane 3 shows a control kinase reaction without recombinant His-Spt2. The samples were resolved by SDS-PAGE followed by a transfer onto nitrocellulose membranes for antihistidine Western blotting or autoradiography. (B) Diagram indicating the different parts of Spt2 that were fused to GST and used in the kinase assay shown in subsequent panels. (C) Yeast CK2 modifies the Spt2 C-terminal domain in vitro. Kinase assays were performed by using TAP-purified CK2, radiolabeled ATP, and an equal amount of recombinant His-Spt2 (aa 1 to 333) (lanes 1 and 4) and the His-Spt2 N-terminal domain (aa 1 to 213) (lanes 2 and 5) or GST fused to the Spt2 N-terminal domain (aa 1 to 179) (lane 8) and to the Spt2 C-terminal domain (aa 200 to 300) (lane 7). Control reactions were carried out without recombinant proteins (lane 3), in the absence of CK2 (lanes 4 and 5), or by using purified recombinant GST (lane 6). The samples were then resolved by SDS-PAGE followed by Coomassie staining (bottom) or autoradiography (top). (D) CK2 is the major yeast Spt2 C-terminal kinase. Wild-type (WT) and ck2ts cells were grown at the permissive temperature (30°C) to mid-log phase and subsequently shifted to the restrictive temperature (37°C) for 2 h. Equal amounts of WCEs prepared from these cells were incubated with 1 μg of recombinant GST or GST fused to the indicated portion of Spt2. After GST pulldowns were performed, the beads were incubated with radiolabeled ATP. The different samples were then resolved by SDS-PAGE followed by Coomassie staining (bottom) or autoradiography (top).
Spt2 is a phosphoprotein in vivo.
To test whether Spt2 is a phosphoprotein in vivo, we submitted WCEs from wild-type cells expressing Spt2-Flag to alkaline phosphatase treatment followed by migration on SDS-PAGE gels. This treatment did not change the migration of Spt2 appreciably (see Fig. S3 posted at http://www.crc.ulaval.ca/nourani/supplemental_data). However, we also observed that in vitro phosphorylation of Spt2 was not associated with modifications in the electrophoresis mobility of Spt2, as shown by the anti-His Western analysis (Fig. 2A, bottom, compare lanes 1 and 2). As an alternative way to test the possibility that Spt2 is a phosphoprotein in vivo, we separated the different Spt2 species by two-dimensional electrophoresis and observed two major forms of Spt2 (Fig. 3A and B, arrows), one of which was much more acidic than the other, suggesting a possible incorporation of phosphogroups in the former. Importantly, while the acidic species of Spt2 seemed to represent the major form in vivo, alkaline phosphatase treatment of the yeast extracts had a dramatic effect on the ratio of Spt2 species and shifted the major part of the protein toward the less acidic form (Fig. 3B). These observations strongly suggest that Spt2 is a phosphoprotein in vivo.
Fig 3.
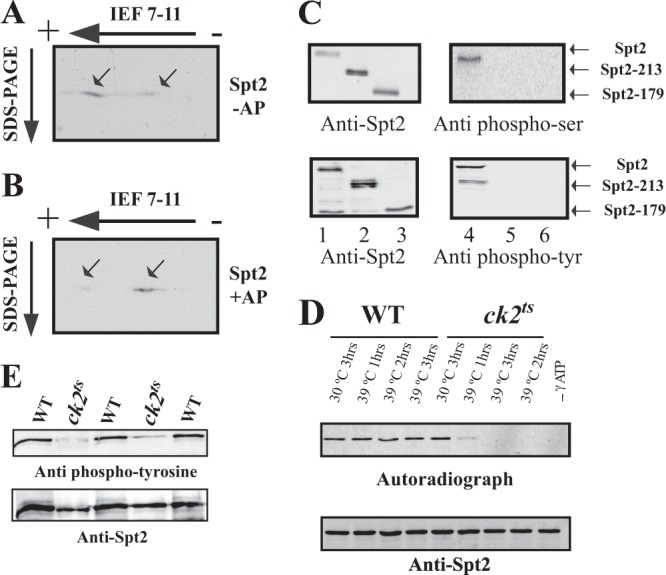
CK2 is required for the in vivo phosphorylation of Spt2. (A and B) Identification of Spt2 isoforms by 2D gel electrophoresis. WCE from a strain expressing Spt2-Flag was subjected (+AP) (B) or not subjected (−AP) (A) to alkaline phosphatase treatment followed by 2D gel electrophoresis (isoelectric focusing [IEF] followed by SDS-PAGE) and then analyzed by anti-Flag Western blotting. The arrows indicate the major Spt2 isoforms. Panel B shows that, after alkaline phosphatase, the migration of the most acidic form of Spt2 is shifted to that of a less acidic isoform(s). (C) Spt2 is phosphorylated in vivo on serine and tyrosine residues located in the C-terminal domain. In vivo phosphorylation of Spt2 domains was assessed by Western blotting using antiphosphoserine (top right), antiphosphotyrosine (bottom right), and anti-Flag (left) antibodies. The WCEs tested were prepared from yeast cells expressing the indicated Spt2-Flag version, subjected to anti-Flag immunoprecipitation, and then analyzed by Western blotting with the indicated antibody. (D) CK2 depletion affects the phosphorylation of Spt2. WCEs were prepared from wild-type or ck2ts cells expressing Spt2-13Myc. These cells were grown at the permissive (30°C) or restrictive (39°C) temperature for the indicated times. Equal amount of WCEs were incubated with radiolabeled ATP. The proteins were immunoprecipitated with anti-Myc antibodies and resolved by SDS-PAGE followed by transfer onto a nitrocellulose membrane for anti-Myc Western blotting (bottom) or autoradiography (top). (E) Spt2 tyrosine phosphorylation is dependent on CK2. In vivo phosphorylation of Spt2 was assessed by antiphosphotyrosine (top) and anti-Myc (bottom) Western blotting. The cells from wild-type or ck2ts strains expressing Spt2-13Myc were grown to mid-log phase at the permissive temperature (30°C) and then shifted to the nonpermissive temperature (37°C) for 2 h. The WCEs from these cells were subjected to anti-Myc immunoprecipitation and then analyzed by Western blotting with the indicated antibody. Three wild-type and two ck2ts mutant clones were analyzed.
Spt2 is phosphorylated in vivo on serine and tyrosine residues.
We wished to use another approach first to confirm that Spt2 is a phosphoprotein in vivo and second to identify the Spt2 residues targeted for phosphorylation. Whereas CK2 has been shown to be a serine/threonine kinase in various organisms (26), several studies have indicated that in yeast, it can also function as a tyrosine kinase (27–30). Therefore, we asked whether Spt2 is phosphorylated in vivo on serine or tyrosine residues. To address this, we immunopurified Spt2 from yeast cells and analyzed the proteins by Western blotting with antibodies raised against phosphoserine or phosphotyrosine residues (Fig. 3C). Interestingly, a protein the size of Spt2 was recognized by both antibodies, suggesting that Spt2 is phosphorylated on serine and tyrosine residues (Fig. 3C). The deletion of the Spt2 C-terminal domain leads to the loss of phosphotyrosine and phosphoserine signals. This indicates that Spt2 is phosphorylated in vivo on serine and tyrosine residues located in its C-terminal domain.
CK2 kinase activity is essential for Spt2 phosphorylation in vivo.
We next asked if Spt2 is phosphorylated in vivo by CK2. For this, we analyzed the role of CK2 in the incorporation of radiolabeled phosphate into the yeast Spt2. This was done by testing the incorporation of radioactive phosphate into the Spt2 protein that was purified from the wild type or a ck2ts mutant grown at a permissive (30°C) or restrictive (39°C) temperature (Fig. 3D). As expected, Spt2 purified from wild-type yeast incorporated radioactive phosphate under permissive or restrictive conditions at all times. In contrast to this, Spt2 purified from ck2ts cells grown at the restrictive temperature for 1, 2, or 3 h did not incorporate detectable radioactive phosphate. This result indicates that depletion of CK2 from yeast cells affects the phosphorylation of Spt2. In a second approach, we analyzed the role of CK2 in the in vivo phosphorylation of Spt2 by Western blotting using antiphosphotyrosine antibodies. Cells from the wild-type or ck2ts mutant strain were grown to mid-logarithmic phase and then shifted to the restrictive temperature for 2 h. We then analyzed the phosphorylation of immunopurified Spt2 by antiphosphotyrosine Western blotting (Fig. 3E). The depletion of CK2 activity in the ck2ts mutant was associated with a clear loss of the Spt2-specific phosphotyrosine signal. Thus, we conclude that the in vivo phosphorylation of Spt2, including that of tyrosine residues, requires CK2 activity.
CK2 phosphorylates multiple sites in Spt2.
Our analyses of recombinant or purified yeast Spt2 led us to believe that the phosphorylation sites are localized within its C-terminal domain (aa 213 to 333). To identify the phosphorylated residues, we submitted recombinant Spt2 to in vitro phosphorylation using purified yeast CK2 followed by mass spectrometry. Unfortunately, we identified only a few peptides from the C-terminal domain (see Fig. S4 posted at http://www.crc.ulaval.ca/nourani/supplemental_data). The highly charged nature of the Spt2 C-terminal domain and the specificity of trypsin could explain why only a part of the C-terminal domain was identified. Few Spt2 C-terminal peptides were recovered by mass spectrometry, and none of these contained a phosphorylated residue (see Fig. S4 posted at http://www.crc.ulaval.ca/nourani/supplemental_data). Based on these results, we decided to attempt to identify the phosphorylated residue(s) in Spt2 by searching for serine and tyrosine residues within CK2 consensus sites in peptides other than those recovered by mass spectrometry. Using this approach, we identified five potential residues (Fig. 4A) in the C-terminal domain of Spt2: four tyrosines (Y226, Y230, Y279, and Y281) and one serine (S277). To test directly if these residues are phosphorylated by CK2, we decided to change them to alanine (S to A) or phenylalanine (Y to F) and tested the phosphorylation of the corresponding recombinant Spt2. None of these single mutations affected the level of Spt2 in vitro phosphorylation by CK2 (data not shown). Careful examination of the potential phosphorylation sites showed that these residues are located in two distinct short stretches of amino acids (region I [RI] at aa 226 to 230 and RII at aa 277 to 281). It is possible that CK2 phosphorylates all sites located in RI or in RII. We combined mutations in each region and performed kinase assays. As shown in Fig. 4B, the modification of RI had no effect, while RII mutations reduced the phosphorylation by CK2 without completely abolishing it (compare lanes 3, 4, and 5). We next combined mutations in RI and RII and assayed phosphorylation by CK2. Importantly, Spt2 protein that is mutated in RI and RII was no longer phosphorylated by CK2, indicating that the RI and RII sites are targeted by CK2 (Fig. 4B). We next wanted to know whether these sites are the in vivo targets of Spt2 phosphorylation. To address this, we analyzed the Spt2 RI- and RII (RI+RII)-mutated proteins by 2D electrophoresis and compared their migration to that of the wild-type forms. As shown in Fig. 4C and D, the levels of acidic Spt2 species were significantly reduced in the whole-cell extract from the strain expressing the RI+RII-mutated Spt2 compared to that derived from Spt2 wild-type cells (Fig. 4D). Our results indicate that the RI+RII mutations decrease the in vivo phosphorylation of Spt2 and show that these sites are the major CK2 targets in vivo.
Fig 4.

CK2 phosphorylates five residues on two short regions (RI and RII) in the C-terminal domain of Spt2. (A) Diagram showing the potential CK2 phosphorylation sites on Spt2. (B) Mutations of the RI and RII sites abolish Spt2 phosphorylation by CK2. Kinase assays were performed by using equal amounts of recombinant wild-type His-Spt2 or the indicated mutated version. The samples were analyzed by antihistidine Western blotting (bottom) and autoradiography (top). (C and D) Mutations of RI and RII affect the in vivo phosphorylation of Spt2. Shown are data for 2D-PAGE analysis of Spt2 (wild type [WT]) and Spt2 (RI+RII mutated to alanine). WCEs from a strain expressing Spt2-Flag (C) or a nonphosphorylatable Spt2-Flag version (RI+RII mutated to alanine) (D) were resolved by 2D-PAGE and analyzed by anti-Flag Western blotting.
Phosphorylation of Spt2 RI and RII sites by CK2 modulates its function.
After mapping the phosphorylated residues in Spt2, we sought to determine the functional significance of this modification. Although CK2 phosphorylates five sites in the Spt2 C terminus, it is possible that only one has functional significance. For this, we constructed different yeast strains, each one expressing an Spt2 protein containing one substitution at a single phosphorylation site. Each of these mutations replaced phosphorylatable residues by nonphosphorylatable amino acids. To analyze the function of these alleles, we tested for a specific phenotype associated with Spt2 function. The deletion of the SPT2 gene suppresses Ty and δ insertion mutations in the HIS4 or LYS2 promoter (this is known as an Spt− phenotype [5, 31]). In a wild-type strain containing the lys2-128δ allele, the LYS2 gene is not expressed, and the cells are not able to grow in medium lacking lysine. The deletion of SPT2 suppresses this phenotype, and spt2Δ cells are able to express LYS2 and grow in medium lacking lysine (31). As expected, in the absence of Spt2 (spt2Δ strain), the cells were able to grow in medium lacking lysine (Fig. 5A and B). Adding back wild-type SPT2 or any single mutants restored the wild-type phenotype. This shows that mutation of a single RI or RII site does not affect Spt2 function as it relates to the Spt phenotype. We next combined mutations of CK2 sites in RI, in RII, or in all sites (RI+RII) to alanine or phenylalanine and observed the same results (Fig. 5A to C). Taken together, these data show that the absence of phosphorylation by CK2 in RI or/and RII has no effect on Spt2 function.
Fig 5.
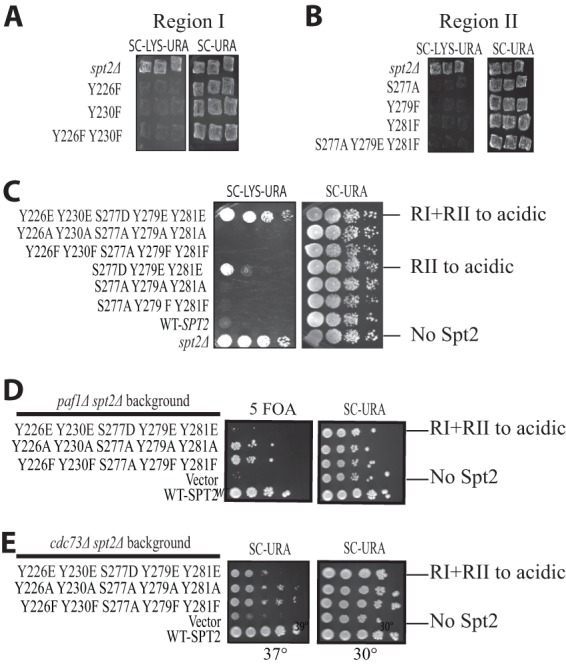
Phosphorylation by CK2 negatively regulates Spt2 chromatin function. (A and B) Mutations of CK2 sites to nonphosphorylatable residues do not result in an Spt− phenotype. Patches show the growth of an spt2Δ mutant containing the lys2-128δ Spt reporter and transformed with a plasmid carrying either the wild type or a mutated version of SPT2, as indicated. The photographs were taken after 1 or 2 days of incubation at 30°C. (C) Mimicking phosphorylation of Spt2 at CK2 RII or RI/RII leads to an Spt− phenotype. Serial dilutions of the spt2Δ mutant culture containing the lys2-128δ Spt reporter and transformed with a plasmid carrying either wild-type SPT2 or the indicated mutated version of SPT2 were grown on synthetic complete medium lacking uracil (SC−URA) or on synthetic complete medium lacking both uracil and lysine (SC−URA−LYS). The photographs were taken after 2 days of incubation at 30°C. (D) An spt2 mutant mimicking RI/II CK2 phosphorylation is synthetically lethal with the paf1Δ mutant. An spt2Δ mutant containing a CEN URA3 SPT2 vector was transformed with a second CEN LEU2 plasmid containing wild-type SPT2 or the indicated mutated version of SPT2. This strain was then mated with a paf1Δ mutant, diploids were sporulated and dissected, and representative progeny containing both plasmids were spotted onto medium lacking uracil or medium containing 5-fluoroorotic acid (5 FOA). The photograph was taken after 2 days of incubation at 30°C. (E) An spt2 mutant mimicking RI/II CK2 permanent phosphorylation has synthetic interactions with a cdc73Δ mutant. An spt2Δ cdc73Δ mutant transformed with a CEN URA3 plasmid containing wild-type SPT2 or the indicated mutated version of SPT2 was spotted onto medium lacking uracil at 30°C or 37°C for 2 days.
Next, we reasoned that phosphorylation by CK2 could negatively regulate the function of Spt2. In this case, mimicking a permanent phosphorylation state could result in the inhibition of Spt2 function. To test this hypothesis, we replaced each residue in RI or RII with an acidic amino acid, thereby mimicking CK2 phosphorylation. Interestingly, while mutation of RI to acidic residues had no visible effect on growth in medium lacking lysine, the strain expressing the Spt2 acidic mutant in RII grew on this same medium (Fig. 5C). Moreover, the strain expressing an Spt2 protein where RI and RII residues were changed to acidic amino acids grew better than the RII mutant. In fact, the acidic RI+RII double mutant grew as well as the strain deleted in the SPT2 gene. This indicates that mimicking of phosphorylation of RI and RII by CK2 inhibits the Spt2 function in the suppression of δ insertion mutations. We next wanted to determine if the mutations in RI and RII affect Spt2 function in chromatin modulation associated with transcription elongation. The PAF complex is known to be involved in chromatin modulation associated with transcription elongation (32, 33). Spt2 has a functional association with the PAF complex, and a spt2Δ mutation shows synthetic phenotypes with mutations in the PAF complex (6). We therefore tested whether phosphorylation of RI and RII has a functional link with Paf complex components. Since the Paf complex subunits Cdc73 and Paf1 are necessary for the normal growth of cells lacking Spt2 (the spt2Δ cdc73Δ strain is thermosensitive, while the spt2Δ paf1Δ strain is not viable [6]), we asked whether different Spt2 mutants are able to complement the loss of Spt2 in spt2Δ paf1Δ and spt2Δ cdc73Δ strains. As shown in Fig. 5, cells containing the RI and RII sites mutated to acidic residues were not capable of compensating for the loss of the SPT2 gene in either the paf1Δ or cdc73Δ background. Together, our observations strongly suggest that phosphorylation of RI/RII inhibits the function of Spt2 in the chromatin modulations associated with elongation.
Spt2 phosphorylation sites play an important role in the inhibition of spurious transcription from a cryptic promoter.
A defect in the reassembly of nucleosomes leads to the loss of transcriptional inhibition of cryptic promoters that are located within coding regions (6, 9, 34–39). We wanted to know if the phosphorylation of Spt2 RI/RII regions by CK2 plays any role in the nucleosome reassembly at coding regions and therefore in the repression of spurious transcription. For this, we used a reporter system for FLO8 cryptic initiation in which the 3′ coding region of FLO8 has been replaced with the HIS3 coding region (6, 37) (Fig. 6A). In this construct, HIS3 is expressed only when the FLO8 cryptic promoter is active, indicating a significant defect in the refolding of nucleosomes after the passage of RNAP II (37). In addition, the FLO8 promoter was replaced with the GAL1 promoter to allow regulation and modulation of FLO8 transcription levels. Using this reporter, we then tested the expression of GAL1-FLO8-HIS3 in different mutants by assaying growth on medium lacking histidine, using either glucose or galactose as the carbon source. In these tests, growth on galactose is a more permissive condition to detect cryptic initiation than is growth on glucose. Our results (Fig. 6B) show that, similar to the spt2Δ mutant, mutation of RI/II to acidic residues allows expression from the FLO8 cryptic promoter when cells are grown on galactose. Spt2 cooperates with the PAF and HIR/HPC complexes to inhibit the initiation of transcription from the FLO8 cryptic promoter and therefore represses spurious transcription (6). In the cdc73Δ mutant deleted in SPT2 and transformed with an empty vector or a plasmid encoding the Spt2 acidic version, we observed growth in glucose medium lacking histidine, indicating stronger spurious transcription (Fig. 6C). This growth was suppressed when cdc73Δ spt2Δ cells were transformed with a plasmid containing wild-type SPT2 or the nonphosphorylatable mutant (Fig. 6C). Next, to test cryptic initiation at FLO8 more directly, we performed Northern hybridization analysis of the wild-type FLO8 gene. These experiments were done with an spt2Δ cdc73Δ thermosensitive strain transformed with a plasmid containing either wild-type SPT2 or the RI/II acidic or nonphosphorylatable mutants. RNA samples were prepared from cells grown at a permissive temperature or 2 h after shifting to a nonpermissive temperature, as described previously (6). As shown in Fig. 6D, strains harboring either wild-type SPT2 or the RI/II nonphosphorylatable mutant expressed only the full-length FLO8 transcript. In contrast, in either the absence of SPT2 (empty vector) or the presence of the RI/II acidic mutant, the FLO8 short transcript was produced at a significant level. This conclusion is further supported by quantification of the data (see Fig. S5 posted at http://www.crc.ulaval.ca/nourani/supplemental_data). Thus, mimicking phosphorylation of Spt2 RI/II inhibits the function of Spt2 in the regulation of nucleosome refolding during transcription elongation at FLO8.
Fig 6.

Mimicking CK2 phosphorylation of Spt2 leads to spurious transcription. (A) Diagram of the pGAL1::FLO8::HIS3 reporter construct. (B and C) The RI/II spt2 acidic mutant is not able to inhibit spurious transcription from the cryptic promoter of the pGAL1::FLO8::HIS3 construct. Below are serial dilutions of cultures of an spt2Δ (B) or spt2Δ cdc73Δ (C) strain containing the pGAL1::FLO8::HIS3 reporter and transformed with a plasmid carrying either the wild type or a mutated version of SPT2, as indicated. The cells were spotted onto synthetic complete medium lacking uracil (SC−URA) or lacking both uracil and histidine (SC−URA−HIS) and containing galactose (B) or glucose (C) as the carbon source. The photographs were taken after 1 or 2 days of incubation at 30°C. (D) The RI/II spt2 acidic mutant is not able to suppress spurious transcription from the cryptic promoter of the FLO8 gene. The cdc73Δ spt2Δ strain transformed with a plasmid carrying either the wild type or a mutated version of SPT2, as indicated, was grown in yeast extract-peptone-dextrose at 30°C and then shifted for the indicated times to 39°C. Total RNA was extracted and analyzed by Northern blotting with a probe for FLO8. SCR1 served as a loading control. The FLO8 probe identifies the full-length FLO8 mRNA and the FLO8 short RNA, which was shown previously to initiate from a cryptic promoter (34). (E) Mimicking permanent Spt2 phosphorylation does not affect Spt2 function at the SRG1/SER3 locus. The diagram explains the complex regulation of SER3 by the intergenic transcription of the noncoding DNA of SRG1. In wild-type cells, noncoding DNA of SRG1 is actively transcribed, and nucleosomes are refolded in the wake of transcription, resulting in the repression of SER3. In the absence of Spt2, nucleosomes are not refolded at noncoding DNA of SRG1, and SER3 is derepressed. (F) Wild-type (WT) or spt2Δ strains were transformed with an empty plasmid, a plasmid containing wild-type SPT2 (WT-SPT2), or nonphosphorylatable or acidic spt2 mutants. These cells were grown in yeast extract-peptone-dextrose at 30°C. Total RNA was extracted and analyzed by Northern blotting with a probe against SER3 and SCR1.
We have recently shown that Spt2 regulates the SER3 gene by modulating the chromatin structure of the intergenic region of SRG1 (7). In the absence of Spt2, because the transcription of SRG1 does not deposit nucleosomes at the SER3 promoter, the transcription of this gene is significantly induced (7). To determine whether Spt2 phosphorylation by CK2 plays any role in this function, we performed Northern analysis of SER3 RNA levels in wild-type and spt2Δ strains transformed with plasmids containing wild-type or different spt2 phosphorylation mutant alleles. Our results (Fig. 6E) showed that, similar to the wild type, all phosphorylation mutants inhibited the transcription of SER3. We also found that the spt2 acidic mutant, similar to wild-type SPT2, was able to repress histone H3 exchange at the SRG1/SER3 locus (see Fig. S6 posted at http://www.crc.ulaval.ca/nourani/supplemental_data). These observations indicate that mutation of the CK2 consensus RI/II to acidic residues does not render Spt2 totally nonfunctional. Rather, it shows that it inhibits specifically chromatin reassembly at defined locations.
Phosphorylation by CK2 regulates Spt2 recruitment to the coding regions of different genes.
Phosphorylation by CK2 may inhibit directly the activity of Spt2 at the sites of transcription. It could modulate the interaction with partners involved in the same function and therefore inhibit the reassembly of nucleosomes indirectly. Alternatively, CK2 phosphorylation could simply regulate the recruitment of Spt2 to the sites of transcription. To test this, we analyzed the association of wild-type and RI/RII acidic Spt2 with the transcribed regions of different active genes. As shown in Fig. 7A, and as expected, wild-type Spt2 was recruited to coding regions of PMA1, ACT1, RPL5, RPL10, RPL25, PDC1, and POL1. Interestingly, a change of RI/II sites to acidic amino acids led to a drastic reduction of Spt2 recruitment to the open reading frames of these genes. This loss of Spt2 recruitment was not associated with an appreciable change in the level of the protein (Fig. 7B). This observation suggests that phosphorylation of Spt2 by CK2 regulates its function through an inhibition of its recruitment to active sites of transcription. Previous reports showed that a loss of elongation factors or chromatin marks sometimes reflects changes in distribution over these regions rather than a total loss (40). We therefore asked whether the association of an Spt2 version mimicking phosphorylation is similar in all regions of a specific gene. We performed Spt2 ChIP assays at different regions of the highly active PMA1 gene (Fig. 7C). The Spt2 acidic mutant was not associated with all PMA1 regions tested (promoter, 5′ ORF, mid-ORF, and 3′ ORF). Thus, mimicking CK2 phosphorylation does not lead to the inhibition of Spt2 recruitment to one specific region in the gene tested but affects instead the Spt2 association with the entire coding region.
Fig 7.
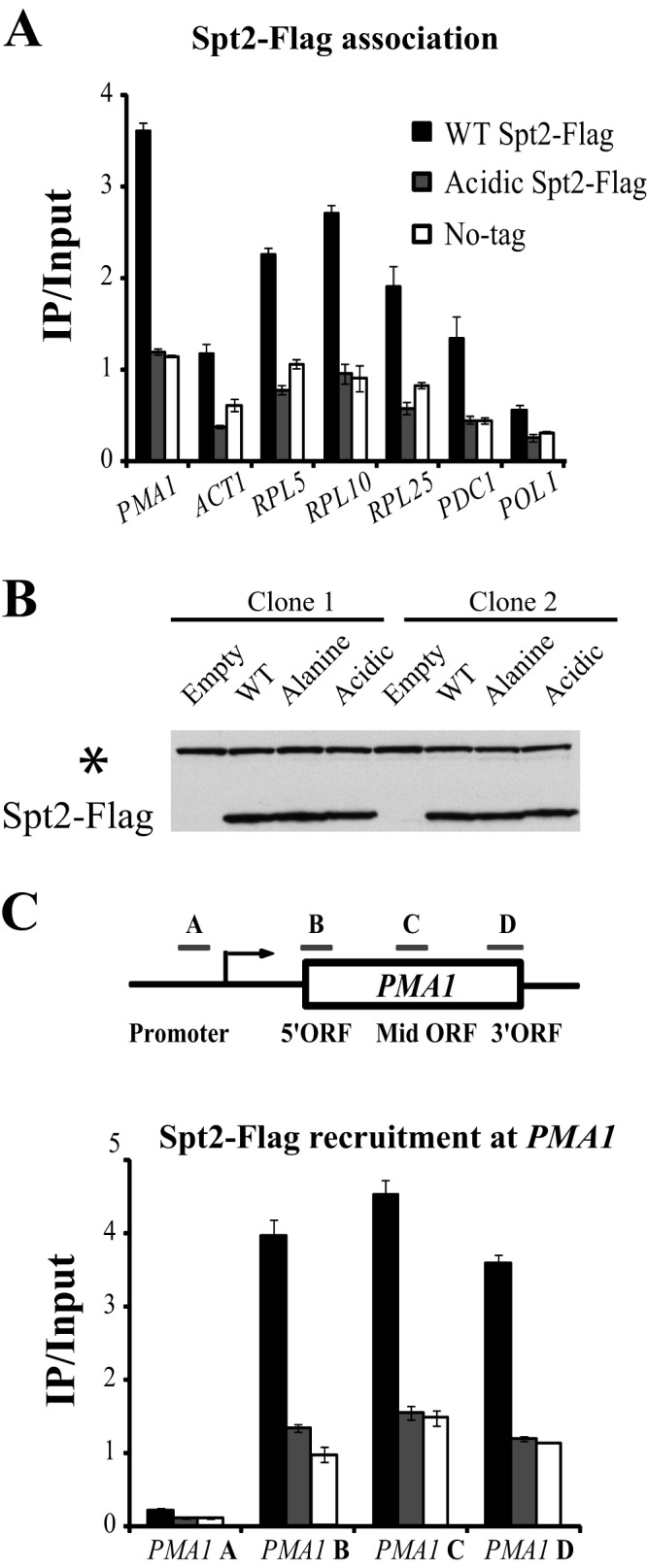
Mimicking CK2 phosphorylation results in the loss of Spt2 from the transcribed regions of different genes. (A) Mutation of Spt2 RI/II to acidic residues results in a dramatic decrease of the Spt2-Flag association with coding regions of different genes. Yeast cells from spt2Δ strains containing a plasmid expressing Flag-tagged wild-type Spt2 or the RI/II acidic version of Spt2 were grown in yeast extract-peptone-dextrose to mid-log phase prior to formaldehyde treatment. Chromatin immunoprecipitations were then performed by using the anti-Flag antibody. The values shown (IP [immunoprecipitated]/Input) represent the averages and standard errors of two to three independent experiments. For each gene, one region located generally near the middle of the open reading frame was tested. (B) Mutation in RI/II sites does not affect the protein level of Spt2 in yeast cells. Yeast cells from the spt2Δ strains were transformed with an empty vector or a plasmid encoding wild-type, acidic, or nonphosphorylatable Spt2 fused to a Flag tag. The WCEs of these cells were subjected to Western blotting with anti-Flag antibodies. The asterisk indicates a protein that was recognized nonspecifically by the antibody and could be used as a loading control. (C) The spt2 mutant mimicking CK2 phosphorylation is lost from all transcribed regions of the active gene PMA1. The chromatin used in this experiment was prepared from the same strains as those in panel A and treated as described above for panel A. The bars labeled A to D represent the PMA1 regions assayed by quantitative PCR. The values shown (IP/Input) represent the averages and standard errors of two to three independent experiments.
CK2 kinase activity is required for the regulation of Spt2 targeting to chromatin.
We next asked if the loss of CK2 activity has an impact on the recruitment of Spt2 to coding regions. To address this question, we performed Spt2 ChIP assays (Fig. 8A) using chromatin extracted from wild-type or ck2ts cells grown at the permissive temperature (30°C) or following a shift to the restrictive temperature (37°C) for 1 or 2 h. Interestingly, in CK2-depleted cells, we observed a higher level of association of Spt2 with most of the genes tested (Fig. 8A; see also Fig. S8 posted at http://www.crc.ulaval.ca/nourani/supplemental_data). This higher level of association was observed with both highly active genes (ACT1 and PMA1) and infrequently transcribed genes (PDC1, POLI, and AZR1) and does not appear to be a result of elevated levels of RNAP II occupancy at these genes (Fig. 8B). This indicates that CK2 activity is required for the regulation of Spt2 recruitment to coding regions of many genes. We next examined the effect of CK2 depletion on the relative distribution of Spt2 across two different coding regions, those of PMA1 and FKS1 (Fig. 8C and D). Interestingly, the loss of CK2 was associated with a greater increase of the Spt2 association at the 3′ ends of PMA1 and FKS1 (Fig. 8C and D). This is particularly clear for the long gene FKS1 and suggests that CK2 phosphorylation could control not only the recruitment but also the distribution of Spt2 on coding regions of specific genes. Taken together, our data indicate that CK2 has a negative effect on the association of Spt2 with coding regions.
Fig 8.
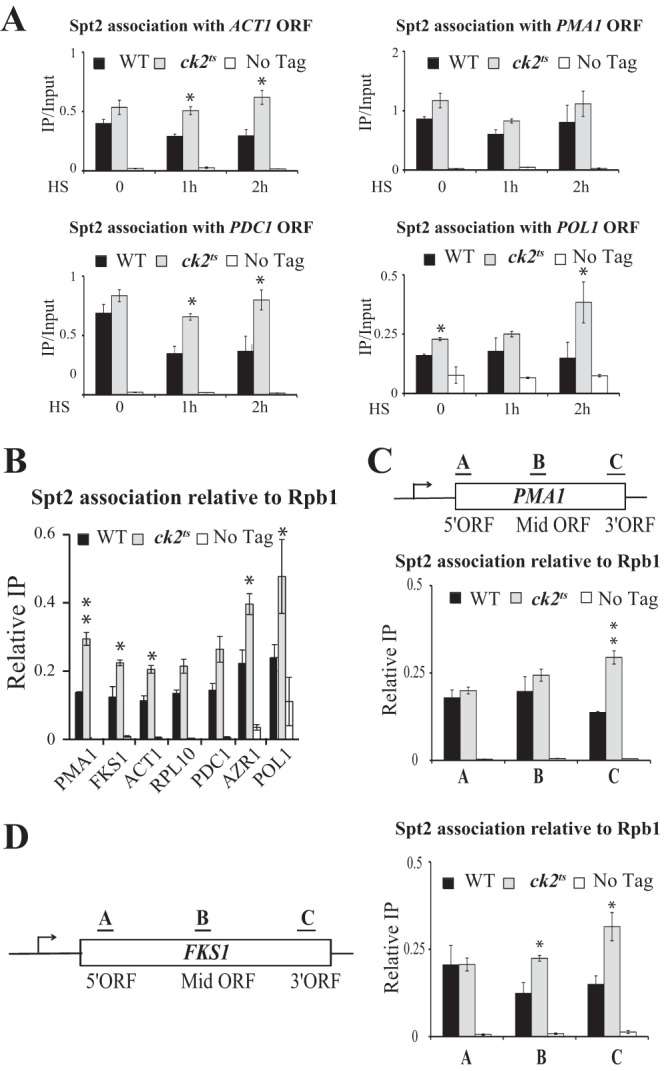
Phosphorylation by CK2 inhibits Spt2 association with chromatin. (A) CK2 depletion is associated with an increased association of Spt2 with coding regions of genes. Shown are data for ChIP assays for Spt2-13Myc. The coding regions of ACT1, PMA1, PDC1, and POL1 were analyzed by quantitative PCR. The values shown (IP/Input) represent the averages and standard errors of three independent experiments. HS, heat shock. (B) CK2 controls Spt2 recruitment to coding regions of genes independently of the transcription level. ChIP assays of Spt2-13Myc and Rpb1 were performed with chromatin extracted from wild-type, ck2ts, or untagged cells grown in yeast extract-peptone-dextrose to mid-log phase at 30°C. For each strain, the value shown represents the ratio of the percent immunoprecipitated with anti-Myc relative to the percent immunoprecipitated with anti-Rpb1 for three independent experiments. The asterisks indicate P values of <0.05. (C and D) CK2 inhibits the association of Spt2 with the 3′ ends of genes. ChIP assays of Spt2-13Myc and Rpb1 were performed as described above for panel B. The bars labeled A to C in each diagram represent the regions assayed by PCR for each gene. As in panel B, the relative Spt2 association (Relative IP) represents the ratio of the percent immunoprecipitated with anti-Myc relative to the percent immunoprecipitated with anti-Rpb1 for three independent experiments.
Phosphorylation of Spt2 by CK2 partially inhibits the Spt2-Spt6 interaction.
Spt2 is recruited to coding regions of transcriptionally active genes in an Spt6-dependent manner, while Spt6 recruitment is partially dependent on Spt2 (6, 7). This suggested a mechanism in which Spt6 directly recruits Spt2 to sites of transcriptional elongation (6, 7). However, whether Spt6 directly recruits Spt2 or indirectly stabilizes its interaction with chromatin postrecruitment is not clear. In order to examine this question, we asked whether Spt2 and Spt6 interact directly. For this, recombinant GST, GST-Spt2, and His-Spt6 were produced, purified, and used in GST pulldown assays (Fig. 9A). Our results show that GST-Spt2 interacted with Spt6, while no such interaction was observed with the GST control (Fig. 9A). These data indicate that Spt6 interacts directly with Spt2 and suggest that it could recruit Spt2 directly to chromatin. We next asked which Spt2 domain interacts with Spt6. We produced N- and C-terminal domains of Spt2 fused to GST (GST–Spt2-Nt and GST–Spt2-Ct) and performed pulldowns with purified His-Spt6. As shown in Fig. 9A, full-length Spt2 and the N-terminal domain of Spt2 were able to interact with Spt6, whereas no such interaction was observed with the Spt2 C-terminal domain. This observation indicates that Spt6 interacts with the N-terminal (aa 1 to 213) part of Spt2 and suggests that it targets Spt2 to transcribed regions. We performed ChIP assays with the N-terminal version of Spt2 (aa 1 to 213) tested in these in vitro experiments and found that it was still recruited in vivo to transcribed regions of active genes (data not shown). This finding indicates that the Spt2 domain that is required for the interaction with Spt6 is sufficient to direct Spt2 to transcribed genes.
Fig 9.
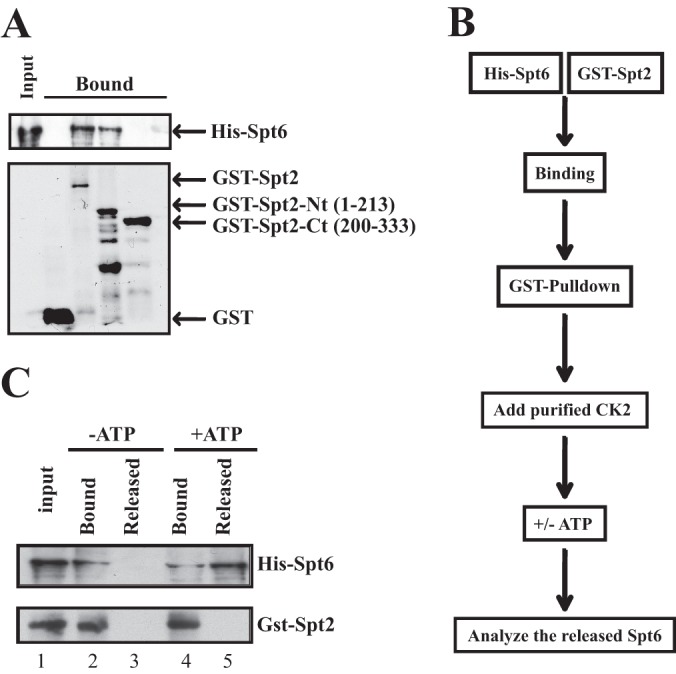
Phosphorylation by CK2 regulates the Spt2-Spt6 interaction. (A) Spt2 interacts directly with Spt6. GST pulldown experiments were conducted by using recombinant GST, GST-Spt2, GST–Spt2-Nt (aa 1 to 213), GST–Spt2-Ct (aa 200 to 333), and His-Spt6. After washes, input (20%) and bound proteins were analyzed by anti-His Western blotting or by Coomassie staining. (B) Diagram summarizing the experimental procedure. (C) The Spt2-Spt6 interaction is regulated by CK2 phosphorylation. The His-Spt6/GST-Spt2 complex was bound on glutathione beads. After washes, the complex was incubated with purified yeast CK2 in the presence of ATP (+ATP) or in the absence of ATP (−ATP). The Spt6 released was assessed in the soluble fraction. The input (15%), the bound fraction (15%), and a large fraction of the supernatant were analyzed by anti-His and anti-GST Western blotting.
CK2 could regulate Spt2 recruitment by modulating the Spt2-Spt6 interaction. To test this, we conducted an experiment where we analyzed the effect of CK2 phosphorylation on the Spt2-Spt6 interaction. As indicated in Fig. 9B, we first assembled the Spt2-Spt6 complex and added the purified yeast CK2 complex. After incubation in the presence or absence of ATP, we assessed the disruption of the His-Spt6/GST-Spt2 complex by analyzing the Spt6 proteins that were released from the GST-Spt2 beads (Fig. 9C). Interestingly, after phosphorylation of the complex, we observed significant levels of Spt6 proteins that were released from the GST-Spt2 beads, whereas no Spt6 was detected in the absence of ATP. Taken together, our data suggest that CK2 phosphorylation may regulate the binding of Spt2 to coding regions through the modulation of the Spt2-Spt6 complex. This is also supported by the fact that an spt6-140 mutation does not further decrease the level of Spt2 phosphomimic association with active genes (see Fig. S11 posted at http://www.crc.ulaval.ca/nourani/supplemental_data). To confirm that the disruption of the Spt2-Spt6 complex is mediated by the modification of CK2 sites in Spt2, we conducted the same experiment described above using the recombinant nonphosphorylatable Spt2 mutant (GST–Spt2-5A). As shown in Fig. 10A, when GST-Spt2 was used, phosphorylation by CK2 led to Spt6 release from GST beads. This effect was not observed when Spt2-Spt6 complexes were formed with nonphosphorylatable Spt2 (GST–Spt2-5A). Thus, our data strongly suggest that direct phosphorylation of Spt2 by CK2 destabilizes the Spt2-Spt6 complex.
Fig 10.

CK2-dependent phosphorylation of Spt2 RI/RII sites regulates Spt2-Spt6 interactions in vitro and in vivo. (A) Mutation of RI/RII sites to alanine abolishes the disruption of the Spt2-Spt6 complex by CK2 phosphorylation. The His-Spt6/GST-Spt2 or GST–Spt2-5A complex was bound on glutathione beads. After washes, the complex was incubated with or without purified yeast CK2 in presence or absence of ATP. The Spt6 released was assessed in the soluble fraction. As described in the legend of Fig. 9C, proteins were analyzed by anti-His and anti-GST Western blotting. (B) Mutation of RI/RII sites to acidic amino acids decreases the interaction between Spt2 and Spt6. The His-Spt6/GST-Spt2 or GST–Spt2-acidic complex was bound on glutathione beads. After washes, the level of each protein was assessed by anti-His and anti-GST Western blotting. (C) Quantification of the amount of Spt6 that is pulled down by GST-Spt2 and the GST–Spt2-acidic version. The values on the histogram represent the averages and standard errors of two independent experiments. The Western blots were quantified by using a Fluor-S-MultiImager. (D) Depletion of CK2 activity increases the level of Spt2 that interacts with Spt6 in vivo. Shown are coimmunoprecipitation experiments using WCEs of wild-type or ck2ts cells grown to mid-log phase and shifted to a nonpermissive temperature for 2 h. These cells express Flag-Spt6 and Spt2-13Myc, and immunoprecipitation was conducted with Flag antibodies coupled to agarose beads. The immunoprecipitated proteins were analyzed by anti-Flag and anti-Myc Western blotting. The relative amount of each protein was quantified and is reported. (E) Model of Spt2 regulation by CK2-dependent phosphorylation.
To further confirm the role of Spt2 phosphorylation in the Spt2-Spt6 interaction, we asked if mimicking CK2 phosphorylation in Spt2 would have an effect on the complex. We produced and purified the recombinant GST–Spt2-acidic version that mimics CK2 phosphorylation. Using the wild type or phosphomimic GST-Spt2 fusions, we performed GST pulldown assays and assessed the level of Spt6 by Western blotting (Fig. 10B). Interestingly, we found that the level of Spt6 interacting with the phosphomimic Spt2 version was consistently lower than the one observed with wild-type Spt2. Quantification (Fig. 10C) showed that mimicking phosphorylation of Spt2 decreased the Spt6 level associated with Spt2 by 50%. Together, these observations indicate that the phosphorylation of the Spt2 C-terminal domain by CK2 inhibits the interaction in vitro of this factor with Spt6.
Finally, to test if CK2 activity has any role in the Spt2-Spt6 interaction in vivo, we performed coimmunoprecipitation experiments in the presence or absence of CK2. Whole-cell extracts were prepared from wild-type or ck2ts cells grown to mid-log phase and shifted for 2 h to the nonpermissive temperature. We immunoprecipitated Flag-Spt6 from these extracts and analyzed the immunoprecipitated proteins by Western blotting (Fig. 10C). Importantly, the Spt2 level associated with Spt6 was clearly higher in CK2-depleted cells. This indicates that CK2 activity has a negative role in the in vivo interaction between Spt2 and Spt6. Taken together, our data led us to conclude that CK2 plays an important role in the regulation of the Spt2-Spt6 interaction.
DISCUSSION
Nucleosomes represent a major obstacle to RNAP II transcription elongation. To efficiently transcribe through chromatin, RNAP II is assisted by various proteins that collaborate in a dynamic process. These factors unfold repressive nucleosomes and rapidly refold them following the passage of RNAP II (8, 9). Beyond efficient production of a specific transcript, these chromatin modulations also appear to be important for the maintenance of a repressive structure that counteracts spurious transcription (8, 9, 41). A defect in the refolding of nucleosomes results in the production of thousands of spurious transcripts (37, 42). Interestingly, recent observations suggested that spurious transcription interferes with the maintenance of human stem cell pluripotency (43–45). Thus, refolding of nucleosomes to control cryptic transcription appears to be an important conserved cellular function. The present study is focused on one factor, Spt2, involved in refolding of nucleosomes and repression of spurious transcription. We provide new evidence showing that Spt2 function is regulated directly by CK2-dependent phosphorylation. Thus, in addition to the role of histone posttranslational modifications (PTMs) such as H3K36me3, our work suggests that nucleosome reassembly factors are the targets of dynamic PTMs that have a direct impact on spurious transcription. It also opens an intriguing possibility that transcription from many cryptic sites is controlled by specific stimuli and triggered to produce specific sets of transcripts, as previously suggested (37). CK2 is localized in the nucleus and copurifies with many other nuclear proteins, including elongation factors involved in chromatin reassembly such as yFACT, Chd1, Elf1, and the Spt4/Spt5 complex (24, 46, 47). Remarkably, while CK2 associates with these different factors, none of them were shown to be phosphorylated by this kinase in vivo (24, 46, 47). In fact, the functional relevance and the biochemical consequences of such associations have rarely been investigated. Here, going beyond previous studies linking one or more subunits of CK2 and an elongation factor, we provide strong evidence of a direct functional association between Spt2 and CK2.
CK2 modifies two small regions (RI and RII) in the essential C-terminal domain of Spt2 and modulates Spt2 function.
Combining mutagenesis and functional studies, we were able to identify the Spt2 phosphorylation sites and test their function. Several reasons could explain why these sites were not identified by mass spectrometry. The main one is the poor coverage of the Spt2 C-terminal region when trypsin is used to produce the peptides to be analyzed by mass spectrometry. The mutation of each site by itself had no effect on the total phosphorylation of the protein, nor did it affect its function, suggesting that none of these sites is playing a major role by itself. It is rather the multiple phosphorylations that are involved in proper regulation of the protein. Indeed, our analyses show that mutation of all sites to residues mimicking phosphorylation has dramatic functional consequences for Spt2. The RI/II mutations to acidic residues are associated with a strong Spt− phenotype and significant cryptic activation of spurious transcription from the FLO8 gene. It is, however, conceivable that a change of these amino acids affects the folding of Spt2, making the mutated form nonfunctional. Much evidence leads us to rule out such possibilities. First, mutation of RI, RII, or all sites to alanine and phenylalanine had no effect on Spt2 function. Second, changing of RI/RII sites to alanine or acidic amino acids did not interfere with the important function of Spt2 in the complex regulation of SER3 (Fig. 6D). Third, we found that, similar to wild-type Spt2, the phosphomimic version favors the use of old histones and lowers the deposition of new histones at this locus (see Fig. S6 posted at http://www.crc.ulaval.ca/nourani/supplemental_data). Fourth, DNA and nucleosome binding activities of the phosphomimic version do not seem to be different form those of the wild type (see Fig. S9 at the website above). Fifth, overexpression of the Spt2 acidic version suppresses the Spt− phenotype of the Δspt2 strain (see Fig. S10 at the website above). Taken together, these observations exclude the possibility that the effects associated with the acidic version of Spt2 are the consequence of misfolding affecting the function of Spt2.
Role(s) of Spt2 regulation by CK2.
Our data suggest a new model of nucleosome reassembly in the wake of the transcription machinery by the Spt2/Spt6 pathway (Fig. 10D). Without excluding other possibilities, we propose that following RNAP II passage, Spt2 and Spt6 contribute to the redeposition of nucleosomes. Once this has been done, CK2 phosphorylates Spt2, disrupts the Spt2-Spt6 complex, and releases Spt2 from coding regions of many genes. The precise role of CK2 regulation remains an interesting question. In other words, why would CK2 stimulate the dissociation of Spt2 from coding regions? Two models could be envisioned. First, the Spt2 level at coding regions must be precisely controlled, and CK2-dependent phosphorylations could play a key role in this process. Interestingly, other work, done by us and others, clearly indicates that the level of Spt2 association with chromatin must be regulated. Overexpression of Spt2, which is associated with a high level of recruitment to chromatin, reduces cell growth and affects the progression through S phase (3, 13) (data not shown). Furthermore, high levels of Spt2 associated with transcription sites, a situation reminiscent of CK2 depletion, lead to hyperrecombination and gross chromosomal rearrangements (GCRs) at coding regions (13). Thus, Spt2 has a positive role in nucleosome reassembly in the wake of transcription, but it seems to have negative impacts on the genome stability at those sites (13). In fact, it appears that in addition to its role in nucleosome reassembly, Spt2 stabilizes long DNA-RNA duplexes in transcription sites, and presumably, these structures stimulate GCR (13). Therefore, it is likely that after the completion of nucleosome reassembly, CK2 phosphorylations trigger the dissociation of Spt2 from coding regions and thereby maintain an appropriate level of this protein at coding regions. Hence, CK2 may protect genome integrity by disrupting the Spt2-Spt6 interaction and releasing Spt2 from chromatin upon completion of chromatin reassembly.
Alternatively, CK2 regulation may play a direct positive role in the reassembly reaction. Interestingly, structural and biochemical studies on Spt6 suggested that following the reassembly of nucleosomes in the wake of transcription, Spt6-nucleosome complexes needed to be disrupted, thereby allowing the release of a mature nucleosome with repressive properties toward spurious transcription (48). CK2 may play a role in this process through the regulation of the Spt2-Sp6 interaction. Indeed, Spt2 is recruited by Spt6 to active sites of transcription and may assist this histone chaperone during the reassembly of nucleosomes (7). This is likely because Spt6, similar to FACT, needs HMG boxes to interact with DNA and nucleosomes (48). In this work, we show new data indicating that the HMG-like protein Spt2 interacts directly with Spt6 and that phosphorylation by CK2 disrupts this interaction. As suggested by our model (Fig. 10), Spt2 could stabilize the Spt6-nucleosome interaction, and CK2 phosphorylation could destabilize these complexes after the completion of nucleosome reassembly. This is consistent with our data showing that the Spt6 association with chromatin is dependent on Spt2 (7). As discussed above, Spt6 is not able to bind nucleosomes by itself and needs HMG boxes for that. Spt2 has two HMG-like boxes in its N-terminal domain, and at least one of these boxes is capable of interacting with four-way DNA junctions, which is similar to the DNA structure at nucleosome entry/exit points. Furthermore, we have evidence that Spt2 is able to bind mononucleosomes (data not shown; see also Fig. S9 posted at http://www.crc.ulaval.ca/nourani/supplemental_data). These biochemical and functional observations suggest that Spt2 could facilitate in vivo the interaction of Spt6 with nucleosomes. Therefore, CK2 phosphorylation, by disrupting the Spt6-Spt2 interaction, could enhance the disruption of Spt6-nucleosome complexes at the final stages of refolding. Thus, upon completion of nucleosome deposition, Spt2 RI/RII sites may become accessible to CK2, which phosphorylates them. This may contribute directly or indirectly to the release of Spt2 and the destabilization of Spt6-nucleosome complexes that would be destabilized further by the other partner of Spt6, Iws1/Spn1 (48).
Finally, our work sheds light on an intriguing new form of regulation involving CK2 and the elongation factor Spt2. The function of this factor in the repression of cryptic transcription is inhibited by CK2-dependent phosphorylation, and this is associated with regulation of the Spt2-chromatin and Spt2-Spt6 interactions. Future work should establish the exact role of CK2 in the regulation of chromatin modulations at coding regions and in the repression of spurious transcription.
ACKNOWLEDGMENTS
We thank Alan Anderson, Nicolas Bisson, Andrea Duina, and Jacques Côté for helpful comments. We also thank Marie-Michelle Genois and Jean-Yves Masson for the technical assistance with electrophoretic mobility shift assays.
This work was supported by CIHR grant MOP81245 to A.N. G.B. holds an NSERC Ph.D. fellowship, and A.N. holds a Canada research chair.
Footnotes
Published ahead of print 26 August 2013
REFERENCES
- 1.Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. 1997. Crystal structure of the nucleosome core particle at 2.8A resolution. Nature 389:251–260 [DOI] [PubMed] [Google Scholar]
- 2.Pollard KJ, Peterson CL. 1997. Role for ADA/GCN5 products in antagonizing chromatin-mediated transcriptional repression. Mol. Cell. Biol. 17:6212–6222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Perez-Martin J, Johnson AD. 1998. The C-terminal domain of Sin1 interacts with the SWI-SNF complex in yeast. Mol. Cell. Biol. 18:4157–4164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Peterson CL, Kruger W, Herskowitz I. 1991. A functional interaction between the C-terminal domain of RNA polymerase II and the negative regulator SIN1. Cell 64:1135–1143 [DOI] [PubMed] [Google Scholar]
- 5.Winston F, Chaleff DT, Valent B, Fink GR. 1984. Mutations affecting Ty-mediated expression of the HIS4 gene of Saccharomyces cerevisiae. Genetics 107:179–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nourani A, Robert F, Winston F. 2006. Evidence that Spt2/Sin1, an HMG-like factor, plays roles in transcription elongation, chromatin structure, and genome stability in Saccharomyces cerevisiae. Mol. Cell. Biol. 26:1496–1509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thebault P, Boutin G, Bhat W, Rufiange A, Martens J, Nourani A. 2011. Transcription regulation by the noncoding RNA SRG1 requires Spt2-dependent chromatin deposition in the wake of RNA polymerase II. Mol. Cell. Biol. 31:1288–1300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Smolle M, Workman JL. 2013. Transcription-associated histone modifications and cryptic transcription. Biochim. Biophys. Acta 1829:84–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Smolle M, Workman JL, Venkatesh S. 2013. reSETting chromatin during transcription elongation. Epigenetics 8:10–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hainer SJ, Pruneski JA, Mitchell RD, Monteverde RM, Martens JA. 2011. Intergenic transcription causes repression by directing nucleosome assembly. Genes Dev. 25:29–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pruneski JA, Hainer SJ, Petrov KO, Martens JA. 2011. The Paf1 complex represses SER3 transcription in Saccharomyces cerevisiae by facilitating intergenic transcription-dependent nucleosome occupancy of the SER3 promoter. Eukaryot. Cell 10:1283–1294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hainer SJ, Charsar BA, Cohen SB, Martens JA. 2012. Identification of mutant versions of the Spt16 histone chaperone that are defective for transcription-coupled nucleosome occupancy in Saccharomyces cerevisiae. G3 2:555–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sikdar N, Banerjee S, Zhang H, Smith S, Myung K. 2008. Spt2p defines a new transcription-dependent gross chromosomal rearrangement pathway. PLoS Genet. 4:e1000290. 10.1371/journal.pgen.1000290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Winston F, Dollard C, Ricupero-Hovasse SL. 1995. Construction of a set of convenient Saccharomyces cerevisiae strains that are isogenic to S288C. Yeast 11:53–55 [DOI] [PubMed] [Google Scholar]
- 15.Gelbart ME, Rechsteiner T, Richmond TJ, Tsukiyama T. 2001. Interactions of Isw2 chromatin remodeling complex with nucleosomal arrays: analyses using recombinant yeast histones and immobilized templates. Mol. Cell. Biol. 21:2098–2106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Longtine MS, McKenzie A, III, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pringle JR. 1998. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14:953–961 [DOI] [PubMed] [Google Scholar]
- 17.Puig O, Caspary F, Rigaut G, Rutz B, Bouveret E, Bragado-Nilsson E, Wilm M, Seraphin B. 2001. The tandem affinity purification (TAP) method: a general procedure of protein complex purification. Methods 24:218–229 [DOI] [PubMed] [Google Scholar]
- 18.Kitazono AA, Tobe BT, Kalton H, Diamant N, Kron SJ. 2002. Marker-fusion PCR for one-step mutagenesis of essential genes in yeast. Yeast 19:141–149 [DOI] [PubMed] [Google Scholar]
- 19.Sikorski RS, Hieter P. 1989. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122:19–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nourani A, Doyon Y, Utley RT, Allard S, Lane WS, Cote J. 2001. Role of an ING1 growth regulator in transcriptional activation and targeted histone acetylation by the NuA4 complex. Mol. Cell. Biol. 21:7629–7640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Utley RT, Lacoste N, Jobin-Robitaille O, Allard S, Cote J. 2005. Regulation of NuA4 histone acetyltransferase activity in transcription and DNA repair by phosphorylation of histone H4. Mol. Cell. Biol. 25:8179–8190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sawa C, Nedea E, Krogan N, Wada T, Handa H, Greenblatt J, Buratowski S. 2004. Bromodomain factor 1 (Bdf1) is phosphorylated by protein kinase CK2. Mol. Cell. Biol. 24:4734–4742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schmitt ME, Brown TA, Trumpower BL. 1990. A rapid and simple method for preparation of RNA from Saccharomyces cerevisiae. Nucleic Acids Res. 18:3091–3092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krogan NJ, Kim M, Ahn SH, Zhong G, Kobor MS, Cagney G, Emili A, Shilatifard A, Buratowski S, Greenblatt JF. 2002. RNA polymerase II elongation factors of Saccharomyces cerevisiae: a targeted proteomics approach. Mol. Cell. Biol. 22:6979–6992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gavin AC, Bosche M, Krause R, Grandi P, Marzioch M, Bauer A, Schultz J, Rick JM, Michon AM, Cruciat CM, Remor M, Hofert C, Schelder M, Brajenovic M, Ruffner H, Merino A, Klein K, Hudak M, Dickson D, Rudi T, Gnau V, Bauch A, Bastuck S, Huhse B, Leutwein C, Heurtier MA, Copley RR, Edelmann A, Querfurth E, Rybin V, Drewes G, Raida M, Bouwmeester T, Bork P, Seraphin B, Kuster B, Neubauer G, Superti-Furga G. 2002. Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature 415:141–147 [DOI] [PubMed] [Google Scholar]
- 26.Meggio F, Pinna LA. 2003. One-thousand-and-one substrates of protein kinase CK2? FASEB J. 17:349–368 [DOI] [PubMed] [Google Scholar]
- 27.Wilson LK, Dhillon N, Thorner J, Martin GS. 1997. Casein kinase II catalyzes tyrosine phosphorylation of the yeast nucleolar immunophilin Fpr3. J. Biol. Chem. 272:12961–12967 [DOI] [PubMed] [Google Scholar]
- 28.Vilk G, Weber JE, Turowec JP, Duncan JS, Wu C, Derksen DR, Zien P, Sarno S, Donella-Deana A, Lajoie G, Pinna LA, Li SS, Litchfield DW. 2008. Protein kinase CK2 catalyzes tyrosine phosphorylation in mammalian cells. Cell. Signal. 20:1942–1951 [DOI] [PubMed] [Google Scholar]
- 29.Marin O, Meggio F, Sarno S, Cesaro L, Pagano MA, Pinna LA. 1999. Tyrosine versus serine/threonine phosphorylation by protein kinase casein kinase-2. A study with peptide substrates derived from immunophilin Fpr3. J. Biol. Chem. 274:29260–29265 [DOI] [PubMed] [Google Scholar]
- 30.Zhu H, Klemic JF, Chang S, Bertone P, Casamayor A, Klemic KG, Smith D, Gerstein M, Reed MA, Snyder M. 2000. Analysis of yeast protein kinases using protein chips. Nat. Genet. 26:283–289 [DOI] [PubMed] [Google Scholar]
- 31.Simchen G, Winston F, Styles CA, Fink GR. 1984. Ty-mediated gene expression of the LYS2 and HIS4 genes of Saccharomyces cerevisiae is controlled by the same SPT genes. Proc. Natl. Acad. Sci. U. S. A. 81:2431–2434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jaehning JA. 2010. The Paf1 complex: platform or player in RNA polymerase II transcription? Biochim. Biophys. Acta 1799:379–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rondon AG, Gallardo M, Garcia-Rubio M, Aguilera A. 2004. Molecular evidence indicating that the yeast PAF complex is required for transcription elongation. EMBO Rep. 5:47–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kaplan CD, Laprade L, Winston F. 2003. Transcription elongation factors repress transcription initiation from cryptic sites. Science 301:1096–1099 [DOI] [PubMed] [Google Scholar]
- 35.Keogh MC, Kurdistani SK, Morris SA, Ahn SH, Podolny V, Collins SR, Schuldiner M, Chin K, Punna T, Thompson NJ, Boone C, Emili A, Weissman JS, Hughes TR, Strahl BD, Grunstein M, Greenblatt JF, Buratowski S, Krogan NJ. 2005. Cotranscriptional set2 methylation of histone H3 lysine 36 recruits a repressive Rpd3 complex. Cell 123:593–605 [DOI] [PubMed] [Google Scholar]
- 36.Carrozza MJ, Li B, Florens L, Suganuma T, Swanson SK, Lee KK, Shia WJ, Anderson S, Yates J, Washburn MP, Workman JL. 2005. Histone H3 methylation by Set2 directs deacetylation of coding regions by Rpd3S to suppress spurious intragenic transcription. Cell 123:581–592 [DOI] [PubMed] [Google Scholar]
- 37.Cheung V, Chua G, Batada NN, Landry CR, Michnick SW, Hughes TR, Winston F. 2008. Chromatin- and transcription-related factors repress transcription from within coding regions throughout the Saccharomyces cerevisiae genome. PLoS Biol. 6:e277. 10.1371/journal.pbio.0060277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Venkatesh S, Smolle M, Li H, Gogol MM, Saint M, Kumar S, Natarajan K, Workman JL. 2012. Set2 methylation of histone H3 lysine 36 suppresses histone exchange on transcribed genes. Nature 489:452–455 [DOI] [PubMed] [Google Scholar]
- 39.Smolle M, Venkatesh S, Gogol MM, Li H, Zhang Y, Florens L, Washburn MP, Workman JL. 2012. Chromatin remodelers Isw1 and Chd1 maintain chromatin structure during transcription by preventing histone exchange. Nat. Struct. Mol. Biol. 19:884–892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chu Y, Simic R, Warner MH, Arndt KM, Prelich G. 2007. Regulation of histone modification and cryptic transcription by the Bur1 and Paf1 complexes. EMBO J. 26:4646–4656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Avvakumov N, Nourani A, Cote J. 2011. Histone chaperones: modulators of chromatin marks. Mol. Cell 41:502–514 [DOI] [PubMed] [Google Scholar]
- 42.Li B, Gogol M, Carey M, Pattenden SG, Seidel C, Workman JL. 2007. Infrequently transcribed long genes depend on the Set2/Rpd3S pathway for accurate transcription. Genes Dev. 21:1422–1430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lin CH, Workman JL. 2011. Suppression of cryptic intragenic transcripts is required for embryonic stem cell self-renewal. EMBO J. 30:1420–1421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xie L, Pelz C, Wang W, Bashar A, Varlamova O, Shadle S, Impey S. 2011. KDM5B regulates embryonic stem cell self-renewal and represses cryptic intragenic transcription. EMBO J. 30:1473–1484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Carvalho S, Raposo AC, Martins FB, Grosso AR, Sridhara SC, Rino J, Carmo-Fonseca M, de Almeida SF. 2013. Histone methyltransferase SETD2 coordinates FACT recruitment with nucleosome dynamics during transcription. Nucleic Acids Res. 41:2881–2893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Prather D, Krogan NJ, Emili A, Greenblatt JF, Winston F. 2005. Identification and characterization of Elf1, a conserved transcription elongation factor in Saccharomyces cerevisiae. Mol. Cell. Biol. 25:10122–10135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lindstrom DL, Squazzo SL, Muster N, Burckin TA, Wachter KC, Emigh CA, McCleery JA, Yates JR, III, Hartzog GA. 2003. Dual roles for Spt5 in pre-mRNA processing and transcription elongation revealed by identification of Spt5-associated proteins. Mol. Cell. Biol. 23:1368–1378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McDonald SM, Close D, Xin H, Formosa T, Hill CP. 2010. Structure and biological importance of the Spn1-Spt6 interaction, and its regulatory role in nucleosome binding. Mol. Cell 40:725–735 [DOI] [PMC free article] [PubMed] [Google Scholar]