

Abstract
The adapter molecules SLP-76 and LAT play central roles in T cell activation by recruiting enzymes and other adapters into multiprotein complexes that coordinate highly regulated signal transduction pathways. While many of the associated proteins have been characterized, less is known concerning the mechanisms of assembly for these dynamic and potentially heterogeneous signaling complexes. Following T cell receptor (TCR) stimulation, SLP-76 is found in structures called microclusters, which contain many signaling complexes. Previous studies showed that a mutation to the SLP-76 C-terminal SH2 domain nearly abolished SLP-76 microclusters, suggesting that the SH2 domain facilitates incorporation of signaling complexes into microclusters. S. C. Bunnell, A. L. Singer, D. I. Hong, B. H. Jacque, M. S. Jordan, M. C. Seminario, V. A. Barr, G. A. Koretzky, and L. E. Samelson, Mol. Cell. Biol., 26:7155–7166, 2006). Using biophysical methods, we demonstrate that the adapter, ADAP, contains three binding sites for SLP-76, and that multipoint binding to ADAP fragments oligomerizes the SLP-76 SH2 domain in vitro. These results were complemented with confocal imaging and functional studies of cells expressing ADAP with various mutations. Our results demonstrate that all three binding sites are critical for SLP-76 microcluster assembly, but any combination of two sites will partially induce microclusters. These data support a model whereby multipoint binding of SLP-76 to ADAP facilitates the assembly of SLP-76 microclusters. This model has implications for the regulation of SLP-76 and LAT microclusters and, as a result, T cell signaling.
INTRODUCTION
Engagement of a T cell antigen receptor (TCR) by a cognate peptide-major histocompatibility complex (MHC) on an antigen-presenting cell triggers complex molecular cascades that control signaling pathways crucial for gene transcription, cytokine production, cell adhesion, mobility, proliferation, and differentiation (1). Appropriate T cell responses depend on the formation of multiprotein complexes at the plasma membrane that regulate efficient signal transduction via multiple pathways. TCR stimulation facilitates phosphorylation of the TCRζ chains by the Src family kinase Lck, which allows for the recruitment and activation of the protein tyrosine kinase Zap-70. Phosphorylation of the essential adapter proteins LAT and SLP-76 by Zap-70 creates docking sites for SH2 domain-containing adapter and effector proteins. The adapter, Grb2, binds LAT and recruits the guanine exchange factor, Sos, while the Grb2-related adapter, Gads, recruits the adapter SLP-76 (1). LAT and SLP-76 recruit other signaling proteins, thereby inducing the assembly of multiprotein complexes. Molecular associations within the multiprotein complexes are dynamic and highly cooperative, which might allow tight regulation of signal transduction pathways in T cells (2–5, 9, 32).
In confocal imaging studies of T cell lines and peripheral blood lymphocytes stimulated on anti-CD3-coated glass coverslips, within seconds of TCR engagement LAT and SLP-76 are visualized in microclusters, which contain many protein complexes (6, 7). Numerous reports indicate that effective TCR signaling depends on the assembly and persistence of the microclusters that contain LAT and SLP-76 (8–13). Microclusters containing LAT are abolished by tyrosine-to-phenylalanine (Y-F) changes of critical Gads and Grb2 binding sites on LAT (8, 9, 14). Microclusters containing SLP-76 are abolished by a mutation to the Gads binding motif of SLP-76, which prevents recruitment to LAT. In both cases, losses of microclusters are associated with defective signaling. A mutation of the C-terminal SH2 domain of SLP-76 also resulted in a similar loss of SLP-76 microclusters along with severely reduced CD69 upregulation and NFAT activity (8). This result was completely unanticipated, because the mutation is not expected to prevent recruitment to LAT. Instead, the result indicated that the SH2 domain plays a central and essential role in the stabilization of SLP-76 microclusters and signal transduction in T cells.
The adapter protein, ADAP, binds the SLP-76 SH2 domain, and the interaction appears to affect TCR signaling (15, 16). SLP-76-deficient J14 cells transfected with SLP-76 containing an SH2 mutation (SLP-76-SH2*) showed reduced NFAT activity, CD69 upregulation, and phospholipase C-γ1 (PLC-γ1) phosphorylation (8, 17). Recently, studies of T cells from conditional knockout mice expressing SLP-76-SH2* were described (18). Similarities to T cells from ADAP knockout mice were observed, including reduced thymocyte positive selection, cytokine production and proliferation, and defective peripheral T cell activation (19, 20).
Data in the literature suggest that ADAP has up to five binding sites for SLP-76 (21–24). The presence of multiple SLP-76 binding sites raises the question of whether these sites could promote the assembly of SLP-76 microclusters through multipoint binding of SLP-76 to ADAP. In this study, we demonstrated SLP-76 oligomerization mediated by the SH2 domain binding to multiple ADAP sites and examined a role for these interactions in TCR signaling. We used biophysical methods to demonstrate direct and multipoint binding of the SLP-76 SH2 domain to fragments of ADAP containing multiple phosphorylation sites in vitro. The significance of these results was confirmed by multicolor confocal imaging, signaling, and adhesion assays, which demonstrated that multipoint binding of SLP-76 to ADAP has functional consequences and controls the assembly of SLP-76 into microclusters in stimulated T cells.
MATERIALS AND METHODS
Reagents.
The anti-CD3ε antibodies UCHT1 (BD Pharmingen) and OKT3 were used for T cell stimulation. TGA resin and N-α-Fmoc-protected amino acids, including phosphorylated Tyr and the pseudoproline (oxazolidine) dipeptides Fmoc-Gly-Ser(ΨMe,Mepro)-OH and Fmoc-Ile-Ser(ΨMe,Mepro)-OH, were purchased from Novabiochem. Solvents were obtained from Sigma-Aldrich. All short interfering RNA (siRNA) reagents were purchased from Dharmacon, including three specific human Fyb duplexes targeting the untranslated region (UTR) of endogenous ADAP, GCUUUGGCUACAAUUAUGA, UCUCUCUGCUUAAUUGUUA, and GUGAGAUGUAAGAAUUAUU, as well as siGENOME nontargeting siRNA pool 1 (D-001206-13-20).
Peptide synthesis.
Peptides were synthesized by the solid-phase method, utilizing 9-fluorenylmethoxycarbonyl (Fmoc)–tert-butyl chemistry, and performed with an Applied Biosystems 431A synthesizer. Peptides were assembled on Fmoc-Asp(tBu) NovaSyn TGA resin (0.24 mmol/g substitution). The coupling reactions were conducted by the 1-hydroxybenzotriazole–2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HOBt–HBTU) method. Cleavage of the peptide from resin was achieved with a trifluoroacetic acid-water-triisopropylsilane mixture (92.5:5:2.5, vol/vol/vol) for 6 h at room temperature. After the resin had been removed by filtration, the filtrate was concentrated by flushing with nitrogen gas and crude peptides were precipitated with diethyl ether. Crude peptides were purified using reverse-phase high-performance liquid chromatography (RP-HPLC) on a BioAdvantage preparative C4 column with a water-acetonitrile solvent system containing trifluoroacetic acid. Purified peptides were characterized by matrix-associated laser desorption ionization time-of-flight mass spectrometry (MALDI micro MX; Waters) and RP-HPLC on a Vaydac analytical C4 column.
Protein expression and purification.
The SH2 domain of human SLP-76, residues 421 to 533, was expressed with an N-terminal polyhistidine (6-His) tag in BL21(DE3) cells (Invitrogen). Cells were grown at 37°C in LB medium, and protein overexpression was induced with 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) (MP Biomedicals, LLC) for 4 h. Harvested cells were resuspended in buffer A (phosphate-buffered saline [PBS; Quality Biologicals, Inc.], 5 mM imidazole, pH 7.4) and Complete EDTA-free protease inhibitor (Roche) and lysed using a French press. The supernatant was loaded onto a HisTrap FF 1-ml column (GE Healthcare), and the protein was eluted with a linear gradient of 5 to 500 mM imidazole. The protein was further purified with a Superdex 75 16/60 gel filtration column equilibrated in PBS, pH 7.4. Purity of SLP-76 SH2 was confirmed by SDS-PAGE and sedimentation velocity analytical ultracentrifugation.
Analytical ultracentrifugation.
Sedimentation velocity experiments were executed according to a standard protocol described previously (25). Briefly, 400 or 100 μl of protein sample in PBS buffer was loaded into 12- or 3-mm centerpieces, respectively. The sedimentation boundary was recorded at a rotor speed of 50,000 or 59,000 rpm and temperature of 20.0°C using both interference detection and absorbance at 280 nm and/or 230 nm in an analytical ultracentrifuge (Beckman Coulter, Indianapolis, IN) equipped with data acquisition software, version 5.7. SV experiments were carried out on the individual peptides and were performed as a titration series with constant ADAP peptide and variable SLP-76 SH2 concentrations. The buffer density (1.0051 g/ml) and viscosity (1.0100 cP) were measured using an Anton Paar DMA5000M density meter and an AMVn automated microvisometer (Graz, Austria).
The raw sedimentation velocity profiles of the individual components and their mixtures were analyzed by the software SEDFIT (version 12.5), using a continuous sedimentation coefficient [c(s)] distribution model (26) and allowing for the refinement of the meniscus position and average frictional ratio. In order to test whether peak positions in the sedimentation coefficient distribution were dependent on the extent of diffusional deconvolution, some c(s) analyses were carried out by fixing the weighted-average frictional ratio (f/f0,w) to values ranging between 1.25 to 1.80.
The molar interference fringe increment (at 655 nm) was calculated using SEDFIT for SLP-76 SH2 (45,873 M−1 cm−1), ADAP-70 (20,963 M−1 cm−1), ADAP-70-pY595 (21,182 M−1 cm−1), ADAP-70-pY651 (21,182 M−1 cm−1), and ADAP-70-pY595-pY651 (21,402 M−1 cm−1) (27). Because of the effects of phosphorylation on the predicted tyrosine contributions to the measured absorbance at 250 and 280 nm, the molar extinction coefficients for protein and adapter peptides at UV-visible wavelengths were calculated based on the concentrations determined by the number of interference fringes. Unexpectedly, the 280/250 extinction ratios for the ADAP-70-pY595 and ADAP-70-pY651 peptides were not identical despite the symmetry of the two phosphorylated tyrosine sites, although, as would be expected intuitively, they were bracketed by the values for the peptides with 0 or 2 phosphorylated tyrosine residues. The partial specific volumes for SLP-76 SH2 (0.7288 ml/g) and ADAP-70 (0.6902 ml/g) were calculated based on the amino acid composition using SEDFIT (28).
For the determination of binding constants, weighted-average sedimentation coefficient (sw) isotherms of weighted-average sedimentation coefficients were constructed by integration of the c(s) distributions. The isotherms were globally modeled for all peptides using custom-written MATLAB scripts (Mathworks, Natick, MA). The sedimentation coefficients of the individual components were fixed at values obtained from the respective c(s) analyses, whereas the s values of the complexes were refined within the constraints of 1.9 to 2.4S for the 1:1 complexes and 2.6 to 3.3S for the 1:2 complex, which were derived from hydrodynamic considerations. To account for binding to nonphosphorylated sites, for all peptides, binding to nonphosphorylated sites was included and, in the absence of contradictory information, assumed to be of the same average affinity. The Kd for the high-affinity binding to pY651 was constrained to be within the range of uncertainty of this parameter derived from isothermal titration calorimetry (ITC) experiments. Hydrodynamic interactions were approximated with a nonideality coefficient (ks) of 0.01 ml/mg.
Direct Lamm equation global modeling of the absorbance and interference profiles of ADAP-70-pY595-pY651 with various concentrations of SLP-76-SH2 was carried out using SEDPHAT (29, 30). In this analysis, the s values of individual free components were fixed, while the binding constant, cooperativity factor, and s values of the 2:1 and 1:1 complexes were fitted parameters, as were the total loading concentration and dissociation rate constant. Plots of the direct boundary modeling were created in the software GUSSI (http://biophysics.swmed.edu/MBR/software.html).
Isothermal titration calorimetry.
Samples were prepared by dilution from concentrated stocks using dialysate from exhaustive dialysis against PBS. Concentrations of the protein and peptide solutions were determined spectrophotometrically using experimentally determined molar extinction coefficients: SLP-76 SH2, ε280 = 20,400 M−1 cm−1; ADAP-70, ε280 = 3,566 M−1 cm−1; ADAP-70-pY595, ε280 = 2,742 M−1 cm−1; ADAP-70-pY651, ε280 = 2,567 M−1 cm−1; ADAP-70-pY595-pY651, ε280 = 2,016 M−1 cm−1; ADAP-14-pY559, ADAP-14-pY595, ADAP-14-pY625, ADAP-14-pY651, and ADAP-14-pY771, ε276 = 505 M−1 cm−1. Titrations were carried out using a MicroCal VP-ITC or ITC200 titration microcalorimeter (Northampton, MA). Raw thermograms were integrated with automated shape analysis using NITPIC (31) and then imported into the software SEDPHAT (32) for individual analysis or global analysis of multiple titrations, using models for 1:1 and 2:1 association schemes and nonlinear least-squares fitting. In addition to parameters for binding constants, change in enthalpy (ΔH), and baseline offsets, where necessary, a parameter to account for local incompetent fractions was included and refined. Statistical uncertainties for best-fit estimates of affinity and ΔH were calculated using standard error surface projection methods built into SEDPHAT.
Expression vectors and mutations.
All point mutations were introduced with the QuikChange II XL site-directed mutagenesis kit (Stratagene). All construct sequences were verified by DNA sequencing. A DNA sequence encoding the SH2 domain of SLP-76 from residues 421 to 533 was cloned into a pET28 plasmid (Novagen) using the restriction sites BamHI and HindIII. The SLP-76-YFP construct has been described previously (6); however, a monomeric mutation, A206K, was introduced into yellow fluorescent protein (YFP) as previously described (33). Also, an S342F mutation was introduced in order to make the sequence identical to the published sequence (NCBI reference sequence NM_005565.3). An additional construct with the SLP-76 SH2 domain mutation R448K was also made.
A plasmid for retroviral expression of wild-type ADAP was a gift from Mira Barda-Saad. In this plasmid, the cDNA sequence encoding ADAP amino acids 1 to 783, followed by a C-terminal Cerulean tag with the monomeric mutation A206K, had been cloned into the pMSCVhyg vector (Clontech). Additionally, the tyrosine-to-phenylalanine mutations Y595F, Y651F, and Y771F were introduced into the wild-type ADAP sequence in different combinations for this study.
Cell culture, transfection, and generation of stable Jurkat T cell lines.
SLP-76-deficient J14 Jurkat cells were a gift from Arthur Weiss and have been described previously (34). Jurkat cells were cultured under standard conditions in RPMI 1640. Stable J14 clones expressing SLP-76-mYFP or SLP-76-SH2*-mYFP were generated as described previously (7). For generation of stable cell lines expressing ADAP constructs (described above), retroviral expression plasmids were transfected into Phoenix-A packaging cells by the calcium phosphate method. After 48 and 72 h, the virus-containing medium was removed and concentrated with Retro-Concentin (System Biosciences) according to the manufacturer's instructions. J14 cells stably expressing either wild-type or R448K SLP-76 were infected with the concentrated retroviral particles. Drug selection medium was added at 72 h postinfection, and the cells were sorted for similar levels of Cerulean fluorescence. At 48 h prior to experiments, stable cells were transfected with siRNA reagents at 2 μM per 3.5 × 106 cells using AMAXA electroporation. For imaging experiments, cells were also transfected with an mKate reporter plasmid (1.58 μg per 3.5 × 106 cells).
Fixed- and live-cell imaging.
The cell-spreading assay has been described previously (35). Briefly, chambered coverslips (LabTek) were coated with the stimulatory antibody in PBS overnight at 4°C. Cells were plated onto anti-CD3ε-coated (UCHT1; 10 μg/ml) coverslips containing imaging buffer (RPMI 1640 without phenol red, 10% fetal calf serum, 20 mM HEPES) and fixed at 3 min with 2.4% paraformaldehyde. Images from live and fixed samples were collected with a Zeiss Axiovert 200 microscope, equipped with a PerkinElmer Ultraview spinning-disk confocal system, using a 63× objective (numeric aperture, 1.4). Images were captured with an Orca-ERII charge-coupled device camera (Hamamatsu). A hot air blower and an objective warmer were used to maintain live samples at 37°C.
Image processing and quantitation.
Imaris 7.4.0 (Bitplane; Andor) was used for image processing of fixed cells and colocalization analysis. Cells were excluded from the analysis if expression of the mKate transfection reporter was not observed. Briefly, single z-slices with the highest intensity SLP-76 punctae at the cell surface were chosen. The 514 channel was used to generate surfaces for analyses of all punctae, including calculation of the microcluster area. For colocalization, the surfaces were used to make a channel with all pixels outside the surfaces assigned a value of 0. This new channel was used to define a region of interest for the Imaris colocalization analysis. All pixels above background levels within the channel were included in the analysis. Average protein recruitment to punctae was determined by calculating the average YFP or Cerulean fluorescence on the surfaces and normalizing data to the average YFP or Cerulean fluorescence in a cytoplasmic region without punctae from the same z-slice of the cell. Movies were prepared from z-stacks by making a maximum-intensity projection of each time point and assembling a sequence of all projections. The particle-tracking feature of Slidebook 5.0 software (Intelligent Imaging Innovations, Inc.) was used for determination of metrics related to SLP-76 microcluster movement from live-cell imaging data.
Adhesion assay.
Wells of 96-well plates (Costar) were coated with anti-CD3 (OKT3; 10 μg/ml) in PBS overnight at 4°C. The wells were washed 3 times with PBS and then coated with VCAM-1 (1 μg/ml; R&D Systems) in PBS for 3 h at 25°C. The antibody solution was aspirated, and the wells were washed 3 times with PBS. Cells (1 × 106) were resuspended in 1 ml of PBS with 10% fetal bovine serum (FBS), dispensed at 100 μl per well, and allowed to adhere at 37°C for 1 h. Images of cells were collected from 4 nonoverlapping fields with a 10× objective using an IN Cell Analyzer 2000 (GE Healthcare) based on expression of the SLP-76-mYFP or SLP-76-SH2*-mYFP chimera. Cells were counted using IN Cell Analyzer software. The percentage of cells remaining was determined after stepwise 30-s bursts with a Vortex-Genie2 plate shaker set to 6 as described previously (10).
Flow-cytometric analysis of cytosolic Ca2+.
Cells (1 × 106) were incubated with 5 μM indo-1-acetoxymethyl-ester (Indo-1-AM; Molecular Probes) and 0.5 mM probenecid (Sigma) in RPMI 1640 without supplements for 45 min at 37°C. The cells were washed and resuspended in RPMI 1640 without phenol red, 20 mM HEPES, and 0.5 mM probenecid. The cells were maintained at room temperature for 30 min and incubated at 37°C for at least 10 min before measurements. The cells were stimulated with 6.25 ng/ml OKT3, and Ca2+ flux was measured by flow cytometry using an LSR II cytometer (BD Biosciences). All fluorescence-activated cell sorter (FACS) data were analyzed using FlowJo software.
Statistical analysis.
Data are presented as means ± standard errors of the mean (SEM). The statistical significance of differences between the means of two data sets was determined with a two-tailed Student's t test for unpaired samples using GraphPad Prism software. A P value of <0.05 was considered to be statistically significant.
RESULTS
Characterization of multiple SLP-76 binding sites in ADAP.
Several ADAP phosphotyrosine residues, pY559, pY595, and pY651, have been reported to bind the SLP-76 SH2 domain. One study indicated that pY595 and pY651 bound SLP-76 (21), while a second study implicated pY559 (22). Recent mass spectrometry studies identified SLP-76 among a list of proteins affinity precipitated from Jurkat lysates by ADAP-based phosphopeptides containing pY625 and pY771 (23, 24). However, the latter two associations could result from either direct or indirect interactions. To evaluate direct binding of SLP-76 to these sites, we used isothermal titration calorimetry (ITC) to characterize the association between the purified SLP-76 SH2 domain and individual 14-amino-acid phosphorylated and nonphosphorylated peptides with ADAP sequences (ADAP-14). Although we did not observe binding by ITC to either the ADAP-14-pY559 or ADAP-14-pY625 peptide, our ITC studies demonstrated that the SLP-76 SH2 domain binds to three ADAP phosphopeptides centered around pY595, pY651, and pY771 with strong affinities (Table 1). Therefore, our results confirm that SLP-76 binds two of the previously proposed sites and one new site. Because multiple binding sites might contribute to interactions between SLP-76 and ADAP, our results raised the possibility that multipoint binding of SLP-76 to ADAP oligomerizes SLP-76.
Table 1.
Thermodynamic binding parameters for the association of 14-amino-acid ADAP phosphopeptides to the SLP-76 SH2 domain as determined by ITCa
| Peptide | Apparent Kd (nM) | Apparent ΔH (kcal/mol) |
|---|---|---|
| ADAP-14-pY559 | ND | ND |
| ADAP-14-Y595 | 180,426b (59,744–787,589) | −3.30 (−1.51 to −13.17) |
| ADAP-14-pY595 | 45 (16–99) | −5.37 (−4.99 to −5.8) |
| ADAP-14-pY625 | ND | ND |
| ADAP-14-Y651 | Weak-affinity binding | ND |
| ADAP-14-pY651 | 59 (41–84) | −6.07 (−5.85 to −6.32) |
| ADAP-14-pY771 | 242 (90–542) | −3.92 (−3.41 to −4.68) |
Values in parentheses represent the 95% confidence intervals as determined by standard error surface projection methods from a global analysis of two or more experiments using SEDPHAT. ND, not determined.
This value is from a single experiment, and the values in parentheses represent the 68.3% confidence intervals.
Oligomerization of the SLP-76 SH2 domain by multipoint binding to ADAP.
We next investigated multipoint binding using analytical ultracentrifugation (AUC) and ITC. For these studies, we synthesized 70-amino-acid peptides corresponding to ADAP residues E589 to D658 (ADAP-70) specifically and differentially phosphorylated at Y595 and Y651 (Fig. 1, top). Peptides including either all three binding sites together or the Y651 and Y771 pair would require substantially longer polypeptide chains; therefore, they posed significant technical challenges. We examined associations between the purified SLP-76 SH2 domain and the nonphosphorylated, singly phosphorylated, and doubly phosphorylated peptides by AUC and ITC. The continuous sedimentation coefficient (c[s]) distributions indicated that individual components are monomeric and do not self-associate (Fig. 1, dotted and dashed lines). Unexpectedly, we observed binding of the SLP-76 SH2 domain to the nonphosphorylated peptide, ADAP-70, by AUC (Fig. 2A). We used a global analysis procedure to fit a model to data from multiple independent AUC experiments (see Materials and Methods). In the global analysis of all weighted-average sedimentation coefficient (sw) data, the data fit well to a model that allows weak binding to the nonphosphorylated Y595 and Y651 binding sites. The average affinity of binding (Kd) to nonphosphorylated sites was estimated to be 120 μM (range, 80 to 200 μM) (Table 2). Conversely, the sw data did not fit well to a model that does not allow binding to the nonphosphorylated sites (Fig. 2A), which caused a 1.6-fold increase in the root mean square deviation (RMSD) for the fit (data not shown). We also observed weak affinity binding to the nonphosphorylated 14-amino-acid peptides ADAP-14-Y595 and ADAP-14-Y651 by ITC (Table 1). Because of the weak affinities, the ITC experiments required concentrations near the solubility limit of both components; therefore, the data quality is limited. Nevertheless, the results suggest that binding to the nonphosphorylated sites of the 14-amino-acid peptides is sufficient to account for the detected binding to the 70-amino-acid peptide. For all ADAP peptides, nonphosphorylated or phosphorylated, mixed with the SLP-76 SH2 domain, there was a concentration-dependent increase in the sw with increasing loading concentrations, indicating interactions between the ADAP peptides and the SLP-76 SH2 domain (Fig. 2A to D).
Fig 1.

Sedimentation coefficient distributions of 30 μM equimolar mixtures of SLP-76 SH2 with phosphorylated and nonphosphorylated 70-amino-acid ADAP fragments (solid lines) or individual components (broken lines). The primary sequence of the 70-amino-acid peptides is shown at the top, with the positions of Y595 and Y651 indicated.
Fig 2.

Global analysis of weighted-average sedimentation coefficients (sw) as a function of SLP-76 SH2 concentration in mixtures with ADAP peptides. Symbols indicate experimentally determined sw values, with red circles for interference optical detection and triangles and blue circles for absorbance detection. Lines represent different best-fit models for the experimental data as described below. (A) Mixtures of nonphosphorylated ADAP-70 peptide (at concentrations ranging from 2.5 to 25 μM) and a 5-fold molar excess of SLP-76 SH2. Thick lines represent a model allowing for binding to both nonphosphorylated sites, while the thin lines represent a model lacking binding to the nonphosphorylated sites (leading to a 1.6-fold increase in the RMSD). (B to D) A global analysis of the sw data for all singly and doubly phosphorylated peptides. This model assumes that the affinity of SLP-76 SH2 to each site is equivalent among different peptides and only dependent on the phosphorylation state. The affinity of SLP-76 SH2 to pY651, as determined by ITC, was used as a constraint in this model. Thick lines are the global best fit to a model accounting for weak binding to nonphosphorylated sites and allowing for simultaneous binding to both phosphorylated sites in ADAP-70-pY595-pY651. The thin lines are the global best fit to a model lacking the potential for simultaneous occupancy of both sites (leading to a 2.1-fold increase in RMSD of the fit). (B) ADAP-70-pY595 at 9.1 μM (circles). The thick and thin lines superimpose exactly, so the thin lines are not visible (indicating a similar fit to both models). (C) ADAP-70-pY651 at 10 μM (circles). (D) ADAP-70-pY595-pY651 at 2.3 μM (circles and triangles).
Table 2.
Binding parameters for the association of the SLP-76 SH2 domain with 70-amino-acid ADAP peptidesa
| Peptide | Apparent Kd1/Kd2 (nM) | Apparent ΔH (kcal/mol) |
|---|---|---|
| ADAP-70 | 120,000/120,000 | ND |
| ADAP-70-pY595 | 5,400/120,000 | ND |
| ADAP-70-pY651 | 120,000/137 (103–171)* | −6.53 (−6.33 to −6.72)* |
| ADAP-70-pY595-pY651 | 3,900/513 | ND |
Values in parentheses represent the 95% confidence intervals as determined by standard error surface projection methods from a global analysis of two or more experiments using SEDPHAT. The values marked with an asterisk were determined from ITC studies; the others were from the global fit of AUC sw isotherms shown in Fig. 2. Dissociation constants for the two binding sites of the 70-amino-acid peptides are given as Kd1 and Kd2. ND, not determined.
An equimolar mixture of SLP-76 SH2 and ADAP-70-pY595 resulted in a bimodal c(s) distribution (1.6S and 2.25S), consistent with the sedimentation pattern of a mixture with reversibly forming complex, which undergoes slow dissociation on the time scale of sedimentation, and unbound SLP-76 SH2 (Fig. 1, blue line). We were unable to extract reliable estimates of binding parameters for SLP-76 SH2 and ADAP-70-pY595 by ITC due to low heats of reaction, despite numerous attempts to optimize the conditions. The global analysis of sw isotherms from sedimentation experiments at a range of concentrations fit well to a two-site model with a best-fit moderate affinity (Kd) of 5.4 μM for binding to pY595, in addition to weak binding (Kd = 120 μM) that was assumed to reflect interactions with nonphosphorylated Y651 at the same magnitude as that measured for the average binding to the nonphosphorylated peptide (Table 2 and Fig. 2B). The sw data fit equally well to a one-site binding model, which is seen as an overlap between the thick and thin lines that represent the two models in Fig. 2B. However, we favor the two-site binding model, because data from our studies of all the long peptides were well described by two-site binding models with affinities that vary according to the phosphorylation status of the sites, as described below. Interestingly, in comparing the ADAP-14-pY595 and ADAP-70-pY595 peptides, the affinities to SLP-76 SH2 are substantially different (Tables 1 and 2). We do not know the reason for this difference, but it is possible that the larger peptides are more representative of the full-length protein.
The association between SLP-76 SH2 and ADAP-70-pY651 is of stronger affinity than that between SLP-76 SH2 and ADAP-70-pY595 (Table 2). The c(s) distribution of an equimolar mixture of SLP-76 SH2 and ADAP-70-pY651 shows a broad monomodal peak with a weighted-average s value that is larger than that of any of the individual reactants alone. The peak shape is consistent with a reaction boundary of a reversible interaction that reflects the time-averaged state of complex formation between SLP-76 SH2 and the pY651 site (Fig. 1, black line). A global analysis of four ITC titration experiments was consistent with a 1:1 association model with a strong affinity (Kd) of 137 nM (Table 2). AUC experiments confirm a strong interaction, with a Kd too high to be independently determined. However, a two-site model of the sw isotherm with an estimate for the strong-affinity site taken from ITC, again superimposed with weak binding (Kd = 120 μM) to the nonphosphorylated Y595, can describe the data well (Fig. 2C). In comparing the thick and thin lines in Fig. 2C, it is clear that the two-site binding model is a better fit to the data than a one-site binding model.
Mixing equimolar mixtures of SLP-76 SH2 with the doubly phosphorylated ADAP-70-pY595-pY651 peptide resulted in the appearance of two major peaks in the c(s) distribution (Fig. 1, red line). The peak representing slower-sedimenting material (1.9S) presumably represents some time-averaged distribution of free SLP-76 SH2 and a 1:1 complex with the ADAP-70-pY595-pY651 peptide. The peak representing faster-sedimenting material (2.75S) presumably corresponds to components involved in a 2:1 association. Upon direct boundary modeling with Lamm equation solutions for reactive systems, the two-site binding model provides a significantly better description of the association between SLP-76 SH2 and the doubly phosphorylated peptide than a single-site binding model as judged by the global reduced chi-squared values (1.19 versus 1.40) (data not shown). The two-site binding model indicates a stronger-affinity site (best-fit Kd of 0.5 μM) (Table 2) and an s value for the 1:1 complex of 2.36S (not shown). The second site is described by moderate affinity (Kd = 3.9 μM) (Table 2) and an s value for the 2:1 complex of 2.95S (not shown). A global analysis of four ITC titration experiments of ADAP-70-pY595-pY651 and SLP-76 SH2 indicated strong-affinity binding (i.e., nM levels) (data not shown). From the ITC data, it could not be definitively discerned whether there is a single strong-affinity site or two sites where the second site has little heat associated with binding. Together, our analyses suggested that the doubly phosphorylated peptide contains strong- and moderate-affinity binding sites; however, it remained formally possible that the moderate affinity resulted from negative cooperativity between the two sites.
In order to determine whether negative cooperativity accounts for the observed moderate-affinity binding site in the doubly phosphorylated peptide, a global analysis of the sw isotherms from the singly and doubly phosphorylated peptides together was performed. The model assumed that the intrinsic affinity of each site is equivalent among different peptides and only dependent on the phosphorylation state, but it allowed for site-site interactions (cooperativity) in the doubly phosphorylated peptide. This model also incorporated the strong-affinity Kd, determined by ITC for binding to pY651. The data fit well to that model (Fig. 2D, thick lines), with a best-fit cooperativity factor of 1.8 (0.2 to 5.8). Within error, this is consistent with independent binding to both sites, and it excludes the possibility of strong, negative cooperativity (Fig. 2D, thin lines). The data did not fit well to a one-site binding model, which led to a 2.1-fold increase in the RMSD of the fit (data not shown). Thus, the doubly phosphorylated peptide contains the strong-affinity site characterized for pY651 and the moderate-affinity site characterized for pY595. Together, the AUC and ITC observations provide experimental evidence that ADAP can oligomerize SLP-76 by multipoint binding.
SLP-76 microclusters translocate with ADAP in T cells.
Recent imaging studies by Pauker and colleagues demonstrated that ADAP is present in SLP-76 microclusters of fixed Jurkat T cells (36). Moreover, the authors reported that knocking down the levels of ADAP by siRNA reduced the persistence of SLP-76 microclusters in live cells. These results indicate an association of ADAP with SLP-76 in microclusters and suggest a role for ADAP in the assembly and stabilization of these microclusters. We investigated the association between SLP-76-mYFP and ADAP-mCerulean in live cells by multicolor imaging. Our results indicate that ADAP continues to associate with SLP-76 microclusters throughout their lifetime (Fig. 3). Together, these results are consistent with a model in which ADAP stabilizes SLP-76 microclusters by continuous association with SLP-76.
Fig 3.
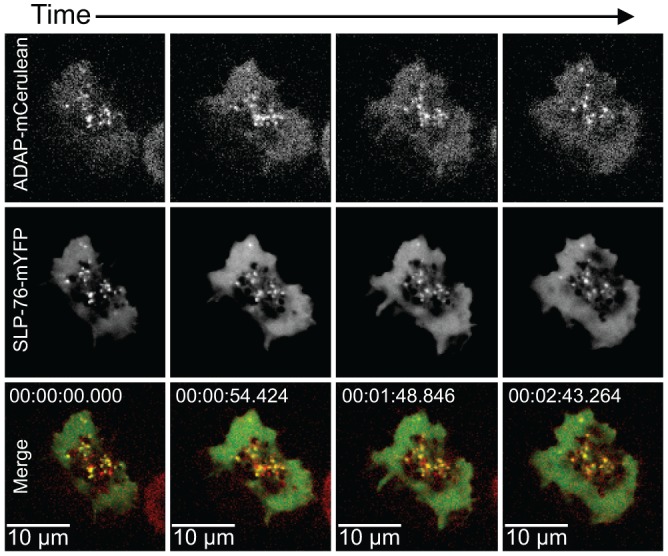
SLP-76 and ADAP translocate together in punctae. SLP-76-deficient Jurkat T cells (J14) expressing wild-type SLP-76-mYFP and wild-type ADAP-mCerulean proteins were plated onto coverslips coated with stimulatory antibodies and imaged with a spinning-disk confocal system. Live-cell imaging began shortly after the cell contacted the coverslip, and the first image was defined as t = 0. The images are maximum-intensity projections of z-stacks (four sections 0.5 μm apart) of selected time points. For each time point, the ADAP-mCerulean fluorescence signal is shown at the top and the SLP-76-mYFP fluorescence signal is shown in the center. In the merged images shown at the bottom, comigration (shown in yellow) of SLP-76 (shown in green) and ADAP (shown in red) is apparent.
SLP-76 microclusters require multivalent interactions with ADAP for assembly and persistence.
Our biophysical data demonstrated direct binding of SLP-76 SH2 to three phosphorylated ADAP sites and showed that binding to multiple ADAP sites oligomerizes SLP-76 in vitro. In order to extend these observations and ask whether the potential for oligomerization translates to microcluster stability, we used the SLP-76-deficient Jurkat T cell line J14 to generate nine cell lines stably expressing wild-type or mutated SLP-76-mYFP and ADAP-mCerulean chimeric proteins. We generated all possible combinations of Y-F mutations at the three ADAP sites: Y595, Y651, and Y771. The cells were sorted for matched expression of fluorescent proteins, which was further confirmed by Western blot analysis (data not shown). Prior to experiments, siRNAs targeting the noncoding region of ADAP were used to reduce the endogenous ADAP levels. Despite a substantial reduction, endogenous ADAP protein was still expressed in excess of the exogenous Cerulean-tagged, chimeric ADAP proteins.
To examine whether the three binding sites of ADAP contribute to the assembly of SLP-76-mYFP-positive microclusters, we fixed the cell lines on anti-CD3-coated coverslips after 3 min of stimulation and quantified the area of SLP-76 microclusters in single cells. As reported previously, the R448K mutation of the SLP-76 SH2 domain nearly abolished all SLP-76 microclusters (Fig. 4A, SLP-76-SH2*) (8). Interestingly, expression of the 3YF mutation of ADAP resulted in a loss of SLP-76 microclusters that was statistically indistinguishable from that of SLP-76-SH2*. This indicates that the assembly of SLP-76 microclusters requires one or more of the three ADAP binding sites. Adding back any single binding site was not sufficient to increase clustering of SLP-76, as all three 2YF ADAP mutations were similar to the 3YF mutation. Therefore, the assembly of SLP-76 microclusters requires multiple binding sites. Interestingly, adding back any two of the three binding sites resulted in a significant increase in SLP-76 microclusters. However, the levels were significantly reduced compared to those of cells expressing wild-type ADAP-mCerulean. Note that all of the effects of the 2YF and 3YF ADAP mutants take place in the presence of higher levels of endogenous ADAP. This fact suggests a dominant-negative effect (see Discussion). Regardless of this effect, our data support a model in which SLP-76 microclusters are stabilized by multipoint binding of the SLP-76 SH2 domain to the three ADAP binding sites, pY595, pY651, and pY771.
Fig 4.

Assembly and persistence of SLP-76 microclusters requires multiple ADAP binding sites. (A) Area of TCR-induced SLP-76 microclusters in stable cell lines. The indicated cell lines were plated on stimulatory coverslips, fixed after incubation at 37°C for 3 min, and imaged with a spinning-disk confocal system. Single z-slices with the highest intensity punctae at the cell surface were chosen, and the total area of SLP-76 microclusters in each cell was calculated using Imaris software and plotted. Means are presented along with error bars representing ±SEM. Significant deviations from the values of cells expressing wild-type proteins or SLP-76-SH2* are indicated, respectively, as follows: * or #, P = 0.01 to 0.05; ** or ##, P = 0.001 to 0.01; *** or ###, P = 0.0001 to 0.001; **** or ####, P < 0.0001. The number of cells analyzed for each sample is shown above the symbols. (B and C) Analysis of SLP-76 microclusters from live-cell imaging. SLP-76-deficient Jurkat T cells (J14) expressing wild-type SLP-76-mYFP and either wild-type ADAP-mCerulean or ADAP-Y595F-Y651F-mCerulean proteins were imaged with a spinning-disk confocal system. Live-cell imaging began shortly after the cell contacted the coverslip. Movies were prepared with Slidebook software from z-stacks by making a maximum-intensity projection of each time point and assembling a sequence of all projections. Microcluster lifetimes were determined using particle tracking from Slidebook software. Mutations to Y595 and Y651 reduce the number (B) and lifetime (C) of SLP-76 microclusters in live cells.
As the microcluster areas were quantified at a fixed time point, we reasoned that the loss of microclusters in cells expressing ADAP mutations could result from either reduced assembly or reduced persistence. If the ADAP binding sites control SLP-76 microcluster assembly, there should be fewer microclusters at all time points in cells expressing mutated ADAP. However, if the binding sites stabilize microclusters, ADAP mutations should decrease microcluster persistence. To address these issues, we performed live imaging studies of stimulated cells expressing either the wild type or one of the 2YF ADAP-mCerulean constructs (Y595F/Y651F) and quantified the movement of SLP-76 microclusters by single-particle tracking. Our results demonstrate that both the number and persistence of SLP-76 microclusters are reduced by mutations to ADAP (Fig. 4B and C). These results are consistent with a role for ADAP in both microcluster assembly and stabilization.
SLP-76 and ADAP association and recruitment into microclusters depend on multipoint binding.
Although mutations to either the SLP-76 SH2 domain or the ADAP binding sites reduce SLP-76 microclusters, even in the most severe cases (SLP-76-SH2* or ADAP-3YF), SLP-76 microclusters are not completely absent (Fig. 4A). In cells expressing SLP-76-SH2*-mYFP, the remaining SLP-76 clusters might result from indirect interactions with clustered LAT. There is also evidence for SH2-independent interactions between SLP-76 and ADAP (36). In cells expressing exogenous wild-type SLP-76-mYFP, the clusters could also result from interactions with endogenous ADAP. These possibilities raised the question of whether SLP-76 microclusters from cells expressing mutated forms of SLP-76-mYFP or ADAP-mCerulean are quantitatively different from those of cells expressing wild-type proteins. To address this, we compared the colocalization of SLP-76-mYFP and ADAP-mCerulean in SLP-76 microclusters by calculating Pearson's correlation coefficients. Association of wild-type SLP-76-mYFP with endogenous ADAP is expected to reduce the Pearson's coefficient between SLP-76-mYFP and ADAP-mCerulean proteins; however, that association is expected to occur similarly in all of the stable cell lines. For cells expressing wild-type proteins, there is a high Pearson's coefficient, indicating an association in microclusters (Fig. 5A). Either the SLP-76 SH2 domain mutation or the 3YF mutation to ADAP significantly lowered the Pearson's coefficients, indicating a reduced association. Interestingly, adding back any single ADAP binding site did not increase colocalization, indicating yet again that a single binding site is not sufficient to increase the association between SLP-76 and ADAP (Fig. 5A, ADAP 2YF mutations). Adding back a combination of Y595 and either Y651 or Y771 increased the Pearson's coefficients; however, they were reduced compared to those of cells expressing wild-type ADAP-mCerulean. These data demonstrate that mutations to the SLP-76 SH2 domain or the ADAP binding sites impair interactions between the two proteins in microclusters. Moreover, the data indicate that two SLP-76 binding sites in ADAP suffice for binding, but all three binding sites are required for optimal interactions.
Fig 5.

Altered molecular composition of SLP-76 microclusters with a SLP-76 SH2 mutation or ADAP Y-F mutations. The single z-slices selected to determine the area of SLP-76 microclusters shown in Fig. 4A were further analyzed. (A) Colocalization between SLP-76 and ADAP was determined by Imaris software as described in Materials and Methods. Wild-type SLP-76 and ADAP proteins colocalize in the SLP-76 punctae, but mutations to either SLP-76 or ADAP reduce the Pearson's coefficient between the two proteins. Means are presented along with error bars representing ±SEM. Significant deviations from the values of cells expressing wild-type proteins or SLP-76-SH2* are indicated, respectively, as follows: * or #, P = 0.01 to 0.05; ** or ##, P = 0.001 to 0.01; *** or ###, P = 0.0001 to 0.001; **** or ####, P < 0.0001. The number of cells analyzed for each sample is shown above the symbols. (B and C) Recruitment of SLP-76-mYFP and ADAP-mCerulean into SLP-76 punctae. The average fluorescence of wild-type or mutated SLP-76-mYFP and ADAP-mCerulean in punctae was calculated and normalized to the average fluorescence in a cytoplasmic region of the same z-slice. Mutations to either SLP-76 or ADAP reduce the amount of SLP-76 (B) and ADAP (C) recruited into punctae.
We also observed that SLP-76 microclusters from cells with mutations to SLP-76 or ADAP had reduced fluorescence intensities compared to those of cells expressing wild-type proteins. The fluorescence intensity is proportional to protein concentration, which suggests a reduced recruitment of SLP-76 and ADAP to these microclusters. To investigate this, we calculated the average fluorescence intensity for both SLP-76 and ADAP within SLP-76 microclusters and normalized these values to the corresponding average fluorescence intensity in a cytoplasmic region not containing microclusters from the same z-slice. Upon comparing the average normalized fluorescence intensities, we found that either the SH2 domain mutation to SLP-76 or the 3YF mutation to ADAP caused a reduction in average recruitment of both proteins to microclusters (Fig. 5B and C).
Therefore, both the SLP-76 SH2 domain and the three ADAP binding sites are required for associations between the two proteins and their recruitment into microclusters. Collectively, these data provide quantitative evidence that the molecular composition of SLP-76 microclusters, which do assemble in cells expressing mutated SLP-76 or ADAP proteins, are different from those assembled from wild-type proteins. We anticipated that these differences are also associated with signaling defects.
Impaired calcium signaling and adhesion results from mutations to the SLP-76 SH2 domain or the SLP-76 binding sites of ADAP.
Mutations to the ADAP residues Y595 and Y651 have been shown to reduce NFAT activity, cytokine production, and calcium signaling in transfected T cells (21, 37, 38). Also, defects in TCR-induced adhesion to integrin ligands are associated with mutations to ADAP, including the 1YF mutation Y771F and the 2YF mutation Y595F/Y651F, as well as a mutation to the SLP-76 SH2 domain (24, 38, 39). In order to compare the functional consequences of mutations to the three ADAP binding sites in our stable cell lines, we evaluated calcium signaling and adhesion. TCR-induced calcium flux is an early signaling event which coincides with both the assembly of SLP-76 microclusters and the time scale of our imaging experiments. We observed that, relative to cells expressing wild-type SLP-76 and ADAP, cells with changes to either protein had reduced TCR-induced calcium flux (Fig. 6A). TCR-induced calcium fluxes of cells expressing the Y595F mutation were not consistent among four experiments, varying between the levels of wild-type ADAP and the Y651F mutation (data not shown). Cells expressing the Y651F mutation had higher TCR-induced calcium flux than cells expressing Y771F or any 2YF mutation. While cells expressing 2YF mutations had detectable TCR-induced calcium flux under these conditions, cells expressing either the 3YF ADAP mutation or the SLP-76 SH2 mutation had no detectable TCR-induced calcium flux (Fig. 6A). Similarly, we observed adhesion defects resulting from mutations to the three ADAP sites or the SLP-76 SH2 domain (Fig. 6B). Of the 1YF ADAP mutations, only Y771F resulted in reduced TCR-induced cell adhesion to VCAM-1. Reduced adhesion was also observed from cells expressing the 2YF and 3YF ADAP mutations. A mutation to the SLP-76 SH2 domain caused the most severe adhesion phenotype. These functional data are comparable to our imaging results and are consistent with a model in which multipoint binding of SLP-76 promotes the assembly of SLP-76 microclusters to drive signaling.
Fig 6.

Mutations to the SLP-76 SH2 domain or the three ADAP binding sites cause defects in calcium signaling and adhesion. (A) Calcium signaling is reduced in cell lines expressing SLP-76 or ADAP mutations. SLP-76-deficient Jurkat T cell lines (J14) stably expressing wild-type or mutated versions of SLP-76 and ADAP (as indicated) were stimulated with anti-CD3 (OKT3), and the Ca2+ influx was measured as described in Materials and Methods. (B) The percentage of cells retained in the wells of 96-well plates coated with anti-CD3 (OKT3) and recombinant human VCAM-1 is shown. The percentage of cells remaining was determined after high shear stress was applied. Averages from 8 replicates were calculated from each experiment, and the means ± SEM from two independent experiments are shown.
DISCUSSION
Multipoint binding to ADAP oligomerizes SLP-76.
Using biophysical methods, we have verified that SLP-76 associates directly with three ADAP binding sites: pY595, pY651, and pY771. Due to the presence of multiple binding sites for SLP-76, we hypothesized that ADAP oligomerizes SLP-76 by multipoint binding. As predicted, in examining the doubly phosphorylated ADAP peptide, our AUC data are consistent with a trimolecular complex containing two SLP-76 SH2 domains and the peptide (Fig. 1, red line). A global analysis of AUC data indicates two binding sites, one with strong affinity and one with moderate affinity, which is consistent with the affinities determined for the singly phosphorylated peptides (Table 2 and Fig. 2D). The results of these studies provide direct evidence for oligomerization of SLP-76 by ADAP in vitro.
Our studies with the ADAP-70-pY595 and ADAP-70-pY651 peptides indicated that the pY595 site binds with weaker affinity to SLP-76 SH2 than the pY651 site (Table 2). This is consistent with previous coimmunoprecipitation data, which showed that the Y651F mutation resulted in a greater loss of SLP-76 association than did the Y595F mutation (21). However, it was surprising that the affinity of the SLP-76 SH2 domain to the pY595 site was substantially different between the large and small peptides, whereas the affinity to the pY651 site was similar (Tables 1 and 2). This discrepancy in the pY595 site might result from adding back more of the native ADAP sequence to the larger peptide.
The affinity of the SLP-76 SH2 domain to ADAP-14-pY771 was weaker than that to ADAP-14-pY595 and ADAP-14-pY651. We were unable to obtain a large peptide also including the pY771 site. Therefore, we cannot address whether the affinity of pY771 changes as more of the native ADAP sequence is restored. Nevertheless, our imaging and functional data clearly indicate an important role for pY771 in microcluster assembly, calcium signaling, and adhesion.
Our AUC data allowed us to exclude the presence of strong negative cooperative interactions in binding of SLP-76 SH2 to pY595 and pY651 on the large ADAP peptides. Other regions of the peptide sequence might modulate binding of SLP-76 to this site, although circular dichroism experiments did not suggest any secondary structural changes to the ADAP-70-pY595-pY651 peptide upon binding SLP-76 SH2 (data not shown). Interestingly, there are two isoforms of ADAP (ADAP-120 and ADAP-130) that differ only by the addition of a 46-amino-acid sequence insertion positioned directly between Y595 and Y651 (40). The insertion in the larger isoform might serve to alter binding of SLP-76 SH2 to these sites.
Unexpectedly, our AUC results indicated an interaction of SLP-76 SH2 with the nonphosphorylated ADAP-70 peptide (Table 2 and Fig. 2A), and these data fit well to a model of this peptide having two weak-affinity sites. Binding of SLP-76 SH2 to the singly phosphorylated peptides ADAP-70-pY595 and ADAP-70-pY651 is described best by a two-site model, with each weak-affinity site having an affinity similar to that of the nonphosphorylated peptide. Weak-affinity binding of the SLP-76 SH2 domain to ADAP-14-Y595 and ADAP-14-Y651 was observed by ITC (Table 1), which strongly suggests that the SLP-76 SH2 domain binds the nonphosphorylated ADAP sites of the 70-amino-acid ADAP peptides. Such binding is not unprecedented, as the Src SH2 domain was shown to bind a nonphosphorylated peptide with an affinity four orders of magnitude weaker than that of the corresponding phosphorylated peptide (41). Also, dissociation rates in the micromolar range were observed for the Grb2 and Grb7 SH2 domains binding nonphosphorylated peptides. Such binding is attributed to highly polar residues at the −1 or −2 position relative to tyrosine, which partially compensate for the nonphosphorylated tyrosine (42–45). The Y595 and Y651 sites of ADAP share the same motifs, EVYDDV, with glutamate at the −2 position. Therefore, a similar association mechanism might underlie the weak binding events observed in our studies.
Multipoint binding of SLP-76 to ADAP mediates the assembly and persistence of SLP-76 microclusters.
Our in vitro biophysical results, indicating a role for ADAP binding sites in oligomerizing SLP-76, were supported and extended by our imaging studies in live and fixed cells. Importantly, either a 3YF ADAP mutation or a mutation to the SLP-76 SH2 domain resulted in a similar loss of SLP-76 microclusters (Fig. 4A). This indicates a functional link between the SH2 domain and the ADAP binding sites in microcluster assembly. The presence of only a single SH2 binding site on ADAP did not increase SLP-76 clustering; however, microclusters were significantly increased with any combination of two SLP-76 binding sites. Of the 1YF mutations, Y771F appeared most severe, although the difference was not statistically significant. Together, our results indicate that all three sites are critical for SLP-76 microcluster assembly. Moreover, the colocalization data indicate that the three sites are required for optimal interactions between SLP-76 and ADAP.
We further demonstrated that the integrity of the three ADAP binding sites has functional consequences that correlate with the extent of SLP-76 clustering. We observed reduced TCR-induced calcium flux and adhesion to VCAM-1 from cells expressing either mutated SLP-76 or ADAP proteins (Fig. 6). Interestingly, the reduction in TCR-induced calcium flux for a given cell line correlates, to an extent, with the amount of SLP-76 microclusters observed. 1YF ADAP mutations resulted in both reduced SLP-76 clustering and calcium flux, while the 2YF and 3YF mutations resulted in substantial reductions of TCR-stimulated SLP-76 microcluster assembly and calcium flux. In contrast to calcium signaling, not all ADAP mutations resulted in apparent adhesion defects. The only 1YF ADAP mutation associated with a reduction in adhesion was Y771F, which is consistent with previous studies of the three 1YF mutations (24). The 2YF and 3YF mutations also reduce adhesion, comparable to reported studies of the 2YF mutation Y595F-Y651F (24, 38). Finally, our results are consistent with studies showing adhesion defects due to the SLP-76 SH2 domain mutation (39). Unexpectedly, the SLP-76 SH2 domain mutation caused a more severe adhesion phenotype than the ADAP 3YF mutation. We attribute this difference to the presence of wild-type endogenous ADAP in the stable cell lines used for this study.
Despite our best efforts to eliminate endogenous ADAP protein from the stable cell lines, Western blot analysis indicated that the exogenous ADAP-mCerulean proteins are expressed at only 5 to 10% of the levels of endogenous ADAP (data not shown). Nevertheless, we still observed significant effects on SLP-76 microcluster assembly, calcium signaling, and adhesion. Due to the strong phenotypes we observed, the mutated ADAP constructs described in this study appear to function as dominant-negative proteins with very disruptive effects on cluster assembly and persistence. Photobleaching studies have shown that SLP-76 in microclusters remains in rapid exchange with cytoplasmic pools (46). Rapid exchange among signaling proteins could allow low levels of mutant ADAP proteins to significantly disrupt SLP-76 microclusters. Therefore, our data might underestimate the effect of mutating the SLP-76 binding sites of ADAP on SLP-76 microcluster assembly, calcium signaling, and adhesion.
Multivalent interactions with ADAP might stabilize SLP-76 microclusters by multiple mechanisms.
Both SLP-76 and LAT are phosphorylated in microclusters, indicating that they are in an activated state and able to interact with other signaling proteins (46). Consistent with this, microclusters have been shown to contain numerous adapter and effector molecules, including SLP-76, LAT, Grb2, Gads, NCK, WASP, ADAP, Zap-70, Sos, PLC-γ1, Vav, and c-Cbl (6, 7, 9, 36, 47). If SLP-76 in microclusters is in rapid exchange with cytoplasmic pools, it is possible that multipoint binding to ADAP acts to shift the equilibrium of SLP-76 toward the bound and phosphorylated state in microclusters to promote signaling. Without multipoint binding, SLP-76 would have a higher dissociation rate and would be more accessible to phosphatases. The result would be reductions in both the number and persistence of microclusters, which is consistent with our analyses of microclusters in live cells (Fig. 4B and C). Moreover, we observed reduced recruitment of SLP-76 and ADAP into microclusters of cells expressing either the SLP-76-SH2* or ADAP-3YF proteins (Fig. 5B and C). Although our biophysical results suggest different binding affinities for the three ADAP sites to the SLP-76 SH2 domain, our imaging results indicate that all three binding sites contribute similarly to microcluster assembly. During the early stages of TCR signaling, it is possible that high local concentrations of SLP-76 recruited to the plasma membrane drive binding and minimize any differences in affinity among the ADAP binding sites. The presence of weaker affinity binding sites may be more critical to microcluster disassembly.
The SLP-76 SH2 domain can also bind to the serine threonine kinase HPK1 (48). Upon association, it was shown that HPK1 phosphorylates SLP-76, leading to a reduction of SLP-76 microclusters (49). Occupancy of the SLP-76 SH2 domain by ADAP would block interactions with HPK1 and prevent microcluster disassembly. According to this model, HPK1 and ADAP compete for interactions with SLP-76. Our ITC titrations with 14-amino-acid phosphopeptides indicate that SLP-76 binds the HPK1 site with an affinity (Kd) of 8 nM (range, 3 to 17 nM) (data not shown), which is 5 to 30 times stronger than the affinities to the 14-amino-acid ADAP phosphopeptides. HPK1 could be at an advantage to compete with ADAP for SLP-76, depending on the cellular concentrations of ADAP and HPK1. However, HPK1 only contains a single binding site for SLP-76, so multipoint binding of the SH2 domain to HPK1 is not possible (48). Therefore, ADAP might promote SLP-76 microclusters by oligomerizing SLP-76 as well as preventing HPK1-mediated microcluster disassembly. These mechanisms are not mutually exclusive, and it is possible that both contribute to the stability of microclusters (Fig. 7A).
Fig 7.

(A) Multipoint binding of SLP-76 to ADAP induces oligomerization of SLP-76 upon TCR stimulation. The binding of ADAP favors SLP-76 clustering and interferes with the binding of HPK1. (B) Separate models for oligomerization of LAT and SLP-76/LAT complexes. (1) Interactions between bivalent LAT and Grb2/Sos complexes can induce oligomerization of LAT into chains. However, if LAT binds 3 Grb2/Sos complexes, the oligomerization potential increases. (Adapted from reference 52 with permission.) (2) Association of the Gads/SLP-76 complex with LAT reduces the number of Grb2 binding sites to two, allowing only the assembly of chains. Multipoint binding of SLP-76 to ADAP oligomerizes SLP-76 and increases the oligomerization potential of LAT complexes.
Oligomerization of SLP-76 has implications for LAT clustering.
The roles of three ADAP binding sites in facilitating SLP-76 clustering and calcium signaling is reminiscent of two other examples of multipoint binding required in TCR-mediated signaling. First, studies from our laboratory showed that three LAT phosphotyrosines (pY171, pY191, and pY226) contribute to LAT signaling and clustering. While 1YF mutations to LAT were tolerated, 2YF and 3YF mutations resulted in altered TCR-induced Erk and NFAT activation (50), similar to the effects of the 2YF and 3YF ADAP mutations on TCR-induced calcium flux. To bind LAT, Grb2 requires multiple binding sites and did not associate with mutated LAT molecules containing individual tyrosine motifs. At a minimum, two of three binding sites were required for Grb2 to associate with LAT (51). Similarly, colocalization between SLP-76 and ADAP required two of the three ADAP binding sites (Fig. 5A).
Second, in vitro studies by Houtman and colleagues extended the results described above and demonstrated that Grb2 can bind all three LAT phosphotyrosines in vitro (9, 32). Mixtures of Grb2, Sos, and LAT resulted in 1:2:1 complexes of LAT-Grb2-Sos along with larger complexes, thereby demonstrating evidence supporting the potential for the Grb2/Sos complex to cross-link LAT. Functional imaging data also supported this model. Nag and colleagues have described a model for LAT oligomerization which showed that the number of available binding sites for Grb2 could have a dramatic effect on the extent of oligomerization. If LAT contains two available Grb2 binding sites, association of a Grb2/Sos complex can induce linear chains. However, the model predicted a dramatic rise in oligomerization when the number of sites was increased from two to three (52). According to this model, if a Gads/SLP-76 complex competed for Grb2 and associated with one of the three Grb2 binding sites of LAT, oligomerization by the Grb2/Sos complex would be limited to the assembly of linear chains. However, the association of ADAP would add two additional SLP-76 binding sites and dramatically increase the oligomerization potential of LAT and SLP-76 (Fig. 7B). Our imaging data show that reducing the number of ADAP sites from three to two substantially reduces the extent of SLP-76 oligomerization.
Our studies indicate that multipoint binding of SLP-76 to ADAP is essential for the assembly and persistence of SLP-76 microclusters initiated by the TCR. The theme for all of these studies, i.e., the requirement for multipoint binding of LAT, oligomerization mediated by Grb2-Sos and by SLP-76-ADAP interactions, is that the multiple levels of cooperative interactions are required for optimal TCR-mediated signaling. Ultimately, this allows for a highly ordered, organized, and stable association of kinases, effectors, and adapters, which are necessary for appropriate T cell responses to different types of stimuli.
ACKNOWLEDGMENTS
We thank Connie L. Sommers, Robert L. Kortum, Lisa M. Miller Jenkins, Huaying Zhao, Mira Barda-Saad, and Zhuyin Li for helpful discussions. Thomas Burlin performed amino acid analysis of peptides. We thank Barbara J. Taylor, Robert P. Wersto, and William G. Telford for performing cell sorting.
This research was supported by the Intramural Research Programs of the Center for Cancer Research, NCI, and NIBIB, National Institutes of Health.
Footnotes
Published ahead of print 26 August 2013
REFERENCES
- 1.Balagopalan L, Coussens NP, Sherman E, Samelson LE, Sommers CL. 2010. The LAT story: a tale of cooperativity, coordination, and choreography. Cold Spring Harb. Perspect. Biol. 2:a005512. 10.1101/cshperspect.a005512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bartelt RR, Houtman JC. 2013. The adaptor protein LAT serves as an integration node for signaling pathways that drive T cell activation. Wiley Interdiscip. Rev. Syst. Biol. Med. 5:101–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barda-Saad M, Shirasu N, Pauker MH, Hassan N, Perl O, Balbo A, Yamaguchi H, Houtman JC, Appella E, Schuck P, Samelson LE. 2010. Cooperative interactions at the SLP-76 complex are critical for actin polymerization. EMBO J. 29:2315–2328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sangani D, Venien-Bryan C, Harder T. 2009. Phosphotyrosine-dependent in vitro reconstitution of recombinant LAT-nucleated multiprotein signalling complexes on liposomes. Mol. Membr. Biol. 26:159–170 [DOI] [PubMed] [Google Scholar]
- 5.Houtman JC, Higashimoto Y, Dimasi N, Cho S, Yamaguchi H, Bowden B, Regan C, Malchiodi EL, Mariuzza R, Schuck P, Appella E, Samelson LE. 2004. Binding specificity of multiprotein signaling complexes is determined by both cooperative interactions and affinity preferences. Biochemistry 43:4170–4178 [DOI] [PubMed] [Google Scholar]
- 6.Bunnell SC, Hong DI, Kardon JR, Yamazaki T, McGlade CJ, Barr VA, Samelson LE. 2002. T cell receptor ligation induces the formation of dynamically regulated signaling assemblies. J. Cell Biol. 158:1263–1275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barda-Saad M, Braiman A, Titerence R, Bunnell SC, Barr VA, Samelson LE. 2005. Dynamic molecular interactions linking the T cell antigen receptor to the actin cytoskeleton. Nat. Immunol. 6:80–89 [DOI] [PubMed] [Google Scholar]
- 8.Bunnell SC, Singer AL, Hong DI, Jacque BH, Jordan MS, Seminario MC, Barr VA, Koretzky GA, Samelson LE. 2006. Persistence of cooperatively stabilized signaling clusters drives T-cell activation. Mol. Cell. Biol. 26:7155–7166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Houtman JC, Yamaguchi H, Barda-Saad M, Braiman A, Bowden B, Appella E, Schuck P, Samelson LE. 2006. Oligomerization of signaling complexes by the multipoint binding of GRB2 to both LAT and SOS1. Nat. Struct. Mol. Biol. 13:798–805 [DOI] [PubMed] [Google Scholar]
- 10.Nguyen K, Sylvain NR, Bunnell SC. 2008. T cell costimulation via the integrin VLA-4 inhibits the actin-dependent centralization of signaling microclusters containing the adaptor SLP-76. Immunity 28:810–821 [DOI] [PubMed] [Google Scholar]
- 11.Sylvain NR, Nguyen K, Bunnell SC. 2011. Vav1-mediated scaffolding interactions stabilize SLP-76 microclusters and contribute to antigen-dependent T cell responses. Sci. Signal. 4:ra14. 10.1126/scisignal.2001178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yokosuka T, Sakata-Sogawa K, Kobayashi W, Hiroshima M, Hashimoto-Tane A, Tokunaga M, Dustin ML, Saito T. 2005. Newly generated T cell receptor microclusters initiate and sustain T cell activation by recruitment of Zap70 and SLP-76. Nat. Immunol. 6:1253–1262 [DOI] [PubMed] [Google Scholar]
- 13.Campi G, Varma R, Dustin ML. 2005. Actin and agonist MHC-peptide complex-dependent T cell receptor microclusters as scaffolds for signaling. J. Exp. Med. 202:1031–1036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hashimoto-Tane A, Yokosuka T, Ishihara C, Sakuma M, Kobayashi W, Saito T. 2010. T-cell receptor microclusters critical for T-cell activation are formed independently of lipid raft clustering. Mol. Cell. Biol. 30:3421–3429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.da Silva AJ, Li Z, de Vera C, Canto E, Findell P, Rudd CE. 1997. Cloning of a novel T-cell protein FYB that binds FYN and SH2-domain-containing leukocyte protein 76 and modulates interleukin 2 production. Proc. Natl. Acad. Sci. U. S. A. 94:7493–7498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Musci MA, Hendricks-Taylor LR, Motto DG, Paskind M, Kamens J, Turck CW, Koretzky GA. 1997. Molecular cloning of SLAP-130, an SLP-76-associated substrate of the T cell antigen receptor-stimulated protein tyrosine kinases. J. Biol. Chem. 272:11674–11677 [DOI] [PubMed] [Google Scholar]
- 17.Yablonski D, Kadlecek T, Weiss A. 2001. Identification of a phospholipase C-gamma1 (PLC-gamma1) SH3 domain-binding site in SLP-76 required for T-cell receptor-mediated activation of PLC-gamma1 and NFAT. Mol. Cell. Biol. 21:4208–4218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Burns JC, Corbo E, Degen J, Gohil M, Anterasian C, Schraven B, Koretzky GA, Kliche S, Jordan MS. 2011. The SLP-76 Src homology 2 domain is required for T cell development and activation. J. Immunol. 187:4459–4466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Peterson EJ, Woods ML, Dmowski SA, Derimanov G, Jordan MS, Wu JN, Myung PS, Liu QH, Pribila JT, Freedman BD, Shimizu Y, Koretzky GA. 2001. Coupling of the TCR to integrin activation by Slap-130/Fyb. Science 293:2263–2265 [DOI] [PubMed] [Google Scholar]
- 20.Griffiths EK, Krawczyk C, Kong YY, Raab M, Hyduk SJ, Bouchard D, Chan VS, Kozieradzki I, Oliveira-Dos-Santos AJ, Wakeham A, Ohashi PS, Cybulsky MI, Rudd CE, Penninger JM. 2001. Positive regulation of T cell activation and integrin adhesion by the adapter Fyb/Slap. Science 293:2260–2263 [DOI] [PubMed] [Google Scholar]
- 21.Geng L, Raab M, Rudd CE. 1999. Cutting edge: SLP-76 cooperativity with FYB/FYN-T in the up-regulation of TCR-driven IL-2 transcription requires SLP-76 binding to FYB at Tyr595 and Tyr651. J. Immunol. 163:5753–5757 [PubMed] [Google Scholar]
- 22.Boerth NJ, Judd BA, Koretzky GA. 2000. Functional association between SLAP-130 and SLP-76 in Jurkat T cells. J. Biol. Chem. 275:5143–5152 [DOI] [PubMed] [Google Scholar]
- 23.Lange S, Sylvester M, Schumann M, Freund C, Krause E. 2010. Identification of phosphorylation-dependent interaction partners of the adapter protein ADAP using quantitative mass spectrometry: SILAC vs (18)O-labeling. J. Proteome Res. 9:4113–4122 [DOI] [PubMed] [Google Scholar]
- 24.Sylvester M, Kliche S, Lange S, Geithner S, Klemm C, Schlosser A, Grossmann A, Stelzl U, Schraven B, Krause E, Freund C. 2010. Adhesion and degranulation promoting adapter protein (ADAP) is a central hub for phosphotyrosine-mediated interactions in T cells. PLoS One 5:e11708. 10.1371/journal.pone.0011708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brown PH, Balbo A, Schuck P. 2008. Characterizing protein-protein interactions by sedimentation velocity analytical ultracentrifugation. Curr. Protoc. Immunol. Chapter 18:Unit 18.15. 10.1002/0471142735.im1815s81 [DOI] [PubMed] [Google Scholar]
- 26.Schuck P. 2000. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys. J. 78:1606–1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhao H, Brown PH, Schuck P. 2011. On the distribution of protein refractive index increments. Biophys. J. 100:2309–2317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cohn EJ, Edsall JT. 1943. Density and apparent specific volume of proteins. Van Nostrand-Reinhold, Princeton, NJ [Google Scholar]
- 29.Brautigam CA. 2011. Using Lamm-equation modeling of sedimentation velocity data to determine the kinetic and thermodynamic properties of macromolecular interactions. Methods 54:4–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dam J, Velikovsky CA, Mariuzza RA, Urbanke C, Schuck P. 2005. Sedimentation velocity analysis of heterogeneous protein-protein interactions: Lamm equation modeling and sedimentation coefficient distributions c(s). Biophys. J. 89:619–634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Keller S, Vargas C, Zhao H, Piszczek G, Brautigam CA, Schuck P. 2012. High-precision isothermal titration calorimetry with automated peak-shape analysis. Anal. Chem. 84:5066–5073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Houtman JC, Brown PH, Bowden B, Yamaguchi H, Appella E, Samelson LE, Schuck P. 2007. Studying multisite binary and ternary protein interactions by global analysis of isothermal titration calorimetry data in SEDPHAT: application to adaptor protein complexes in cell signaling. Protein Sci. 16:30–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zacharias DA, Violin JD, Newton AC, Tsien RY. 2002. Partitioning of lipid-modified monomeric GFPs into membrane microdomains of live cells. Science 296:913–916 [DOI] [PubMed] [Google Scholar]
- 34.Yablonski D, Kuhne MR, Kadlecek T, Weiss A. 1998. Uncoupling of nonreceptor tyrosine kinases from PLC-gamma1 in an SLP-76-deficient T cell. Science 281:413–416 [DOI] [PubMed] [Google Scholar]
- 35.Bunnell SC, Barr VA, Fuller CL, Samelson LE. 2003. High-resolution multicolor imaging of dynamic signaling complexes in T cells stimulated by planar substrates. Sci. STKE 2003:PL8. 10.1126/stke.2003.177.pl8 [DOI] [PubMed] [Google Scholar]
- 36.Pauker MH, Reicher B, Fried S, Perl O, Barda-Saad M. 2011. Functional cooperation between the proteins Nck and ADAP is fundamental for actin reorganization. Mol. Cell. Biol. 31:2653–2666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang H, Wei B, Bismuth G, Rudd CE. 2009. SLP-76-ADAP adaptor module regulates LFA-1 mediated costimulation and T cell motility. Proc. Natl. Acad. Sci. U. S. A. 106:12436–12441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang H, McCann FE, Gordan JD, Wu X, Raab M, Malik TH, Davis DM, Rudd CE. 2004. ADAP-SLP-76 binding differentially regulates supramolecular activation cluster (SMAC) formation relative to T cell-APC conjugation. J. Exp. Med. 200:1063–1074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baker RG, Hsu CJ, Lee D, Jordan MS, Maltzman JS, Hammer DA, Baumgart T, Koretzky GA. 2009. The adapter protein SLP-76 mediates “outside-in” integrin signaling and function in T cells. Mol. Cell. Biol. 29:5578–5589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang H, Rudd CE. 2008. SKAP-55, SKAP-55-related and ADAP adaptors modulate integrin-mediated immune-cell adhesion. Trends Cell Biol. 18:486–493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bradshaw JM, Mitaxov V, Waksman G. 1999. Investigation of phosphotyrosine recognition by the SH2 domain of the Src kinase. J. Mol. Biol. 293:971–985 [DOI] [PubMed] [Google Scholar]
- 42.Spuches AM, Argiros HJ, Lee KH, Haas LL, Pero SC, Krag DN, Roller PP, Wilcox DE, Lyons BA. 2007. Calorimetric investigation of phosphorylated and non-phosphorylated peptide ligand binding to the human Grb7-SH2 domain. J. Mol. Recognit. 20:245–252 [DOI] [PubMed] [Google Scholar]
- 43.Long YQ, Voigt JH, Lung FD, King CR, Roller PP. 1999. Significant compensatory role of position Y-2 conferring high affinity to non-phosphorylated inhibitors of Grb2-SH2 domain. Bioorg. Med. Chem. Lett. 9:2267–2272 [DOI] [PubMed] [Google Scholar]
- 44.Li P, Zhang M, Peach ML, Zhang X, Liu H, Nicklaus M, Yang D, Roller PP. 2003. Structural basis for a non-phosphorus-containing cyclic peptide binding to Grb2-SH2 domain with high affinity. Biochem. Biophys. Res. Commun. 307:1038–1044 [DOI] [PubMed] [Google Scholar]
- 45.Lung FD, Chang CW, Chong MC, Liou CC, Li P, Peach ML, Nicklaus MC, Lou BS, Roller PP. 2005. Small nonphosphorylated Grb2-SH2 domain antagonists evaluated by surface plasmon resonance technology. Biopolymers 80:628–635 [DOI] [PubMed] [Google Scholar]
- 46.Barr VA, Balagopalan L, Barda-Saad M, Polishchuk R, Boukari H, Bunnell SC, Bernot KM, Toda Y, Nossal R, Samelson LE. 2006. T-cell antigen receptor-induced signaling complexes: internalization via a cholesterol-dependent endocytic pathway. Traffic 7:1143–1162 [DOI] [PubMed] [Google Scholar]
- 47.Braiman A, Barda-Saad M, Sommers CL, Samelson LE. 2006. Recruitment and activation of PLCgamma1 in T cells: a new insight into old domains. EMBO J. 25:774–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sauer K, Liou J, Singh SB, Yablonski D, Weiss A, Perlmutter RM. 2001. Hematopoietic progenitor kinase 1 associates physically and functionally with the adaptor proteins B cell linker protein and SLP-76 in lymphocytes. J. Biol. Chem. 276:45207–45216 [DOI] [PubMed] [Google Scholar]
- 49.Lasserre R, Cuche C, Blecher-Gonen R, Libman E, Biquand E, Danckaert A, Yablonski D, Alcover A, Di Bartolo V. 2011. Release of serine/threonine-phosphorylated adaptors from signaling microclusters down-regulates T cell activation. J. Cell Biol. 195:839–853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang W, Trible RP, Zhu M, Liu SK, McGlade CJ, Samelson LE. 2000. Association of Grb2, Gads, and phospholipase C-gamma 1 with phosphorylated LAT tyrosine residues. Effect of LAT tyrosine mutations on T cell antigen receptor-mediated signaling. J. Biol. Chem. 275:23355–23361 [DOI] [PubMed] [Google Scholar]
- 51.Zhu M, Janssen E, Zhang W. 2003. Minimal requirement of tyrosine residues of linker for activation of T cells in TCR signaling and thymocyte development. J. Immunol. 170:325–333 [DOI] [PubMed] [Google Scholar]
- 52.Nag A, Monine MI, Faeder JR, Goldstein B. 2009. Aggregation of membrane proteins by cytosolic cross-linkers: theory and simulation of the LAT-Grb2-SOS1 system. Biophys. J. 96:2604–2623 [DOI] [PMC free article] [PubMed] [Google Scholar]