

Abstract
Sorting-related receptor with A-type repeats (SORLA) is a sorting receptor for the amyloid precursor protein (APP) that prevents breakdown of APP into Aβ peptides, a hallmark of Alzheimer's disease (AD). Several cytosolic adaptors have been shown to interact with the cytoplasmic domain of SORLA, thereby controlling intracellular routing of SORLA/APP complexes in cell lines. However, the relevance of adaptor-mediated sorting of SORLA for amyloidogenic processes in vivo remained unexplored. We focused on the interaction of SORLA with phosphofurin acidic cluster sorting protein 1 (PACS1), an adaptor that shuttles proteins between the trans-Golgi network (TGN) and endosomes. By studying PACS1 knockdown in neuronal cell lines and investigating transgenic mice expressing a PACS1-binding-defective mutant form of SORLA, we found that disruption of SORLA and PACS1 interaction results in the inability of SORLA/APP complexes to sort to the TGN in neurons and in increased APP processing in the brain. Loss of PACS1 also impairs the proper expression of the cation-independent mannose 6-phosphate receptor and its target cathepsin B, a protease that breaks down Aβ. Thus, our data identified the importance of PACS1-dependent protein sorting for amyloidogenic-burden control via both SORLA-dependent and SORLA-independent mechanisms.
INTRODUCTION
Sorting-related receptor with A-type repeats (SORLA) is a member of the VPS10P domain receptor gene family, a group of sorting and signaling receptors expressed in neurons of the central and peripheral nervous systems (1, 2). Typically, newly synthesized VPS10P domain receptors, including SORLA, traffic through the secretory pathway to the cell surface once. Following internalization from the plasma membrane, the receptors shuttle between early endosomes and the trans-Golgi network (TGN), directing target proteins between secretory and endocytic compartments of the cell (reviewed in reference 3).
SORLA is best known for its role in intracellular sorting of the amyloid precursor protein (APP), an etiologic agent in Alzheimer's disease (AD) (4). APP follows a complex trafficking path through intracellular compartments of neurons that determines alternative processing into amyloidogenic and nonamyloidogenic products. Although the details still warrant clarification, it is believed that en route to the cell surface, most nascent APP molecules are cleaved by α-secretase at the plasma membrane to produce soluble APPα (sAPPα) (nonamyloidogenic pathway). However, some APP molecules at the cell surface remain intact and internalize into endosomes, where sequential processing by β- and γ-secretases produces sAPPβ and neurotoxic amyloid-β (Aβ) peptide, the constituent of senile plaques in the brains of AD patients (amyloidogenic pathway) (5). According to current models, SORLA retrieves APP molecules from early endosomes back to the TGN and delays Golgi network exit to the cell surface, blocking amyloidogenic and nonamyloidogenic pathways alike (6–8). Consequently, overexpression of the receptor impairs proteolytic breakdown of APP (6–8), while loss of receptor activity in gene-targeted mice enhances amyloidogenic processing and senile plaque deposition (9, 10). The significance of SORLA as an AD risk factor is underscored by epidemiological studies documenting the association of variants of SORL1 (the gene encoding SORLA) with the risk of sporadic AD on a genome-wide level (11–13).
Since altered APP transport is considered an important pathological mechanism contributing to enhanced Aβ production in AD, much attention has been focused on elucidating the cellular mechanisms that direct SORLA (and its target APP) between secretory and endocytic compartments. Conceptually, these studies should not only shed light on basic principles of neuronal protein sorting but also identify novel disease genes underlying aberrant transport processes in neurodegeneration. One factor implicated in protein sorting between endosomes and the TGN is phosphofurin acidic cluster sorting protein 1 (PACS1), a cytosolic adaptor that directs the movement of furin and cation-independent mannose 6-phosphate receptors (CI-MPR) between endosomes and the TGN (14, 15). Interestingly, PACS1 also binds to an acidic cluster motif in the cytoplasmic tail of SORLA and deletion of this motif impairs the ability of SORLA mutants to sort properly in Chinese hamster ovary (CHO) cells (6) and in human embryonic kidney (HEK293) cells (16). While these data suggested PACS1 as a candidate for SORLA-guided APP transport in nonneuronal cell lines, the significance of PACS1 for amyloidogenic processes in neurons in vivo remained unclear. Here, we performed PACS1 knockdown studies with neuronal cell lines and investigations with mice expressing a PACS1-binding-defective mutant form of SORLA to test the relevance of PACS1 for AD-related processes. Our studies not only confirm the importance of PACS1 for SORLA-dependent APP transport and amyloidogenic processing but also suggest an independent role for PACS1 in the biosynthesis of cathepsin B (CatB), an Aβ-degrading enzyme, in the brain.
MATERIALS AND METHODS
Reagents.
PACS1 (Accell small interfering RNAs [siRNAs] A006697-13 and A006697-16) and nontargeting (Accell siRNA D001910-01) siRNAs were purchased from Thermo Scientific. Commercially available antibodies to the following were used in the various immunodetection experiments: PACS1 (sc-136344; Santa Cruz Biotechnology), NeuN (MAB377; Millipore), Vti1b (BD611405; BD Transduction Laboratories), EEA1 (BD610457; BD Transduction Laboratories), γ-adaptin (BD610385; BD Transduction Laboratories), Rab5 (Synaptic Systems), CI-MPR (5230; Epitomics), CatB (C6243; Sigma-Aldrich), Na/K-ATPase (05-369; Millipore), AP2M1 (ab106542; Abcam), Lamp1 (BD553792; BD Pharmingen), and furin (ab3467; Abcam). Polyclonal antibodies directed against SORLA were kindly provided by Claus M. Petersen (Aarhus University). Antiserum directed against APP (1227) was produced in house.
Interaction of SORLA with PACS1 variants.
Vectors encoding the PACS1 domain were constructed by PCR-based cloning strategies with human PACS1 cDNA as the template (kindly provided by Gary Thomas, University of Pittsburgh). PCR products encompassing the ARR domain (residues 1 to 117, GenBank accession no. BC010096), the FBR domain (residues 117 to 266), or the MR domain (residues 267 to 541) were introduced into expression vector pcDNA3.1zeo (Invitrogen). For the FBR deletion mutant, the sequence corresponding to the FBR domain (residues 117 to 266) was deleted from the PACS1 cDNA by PCR cloning and introduced into the expression vector pcDNA3.1zeo as well. CHO cells stably expressing human SORLA (6) were transiently transfected with PACS1 expression constructs. After 48 h, cells were washed and lysed in Triton X-100–Nonidet P-40 buffer on ice. Immunoprecipitations from cell (or brain tissue) extracts were performed with anti-SORLA or anti-PACS1 antiserum and protein G-coupled Sepharose beads (Pierce) according to standard protocols.
Cell culture and PACS1 knockdown experiments.
SH-SY5Y cells were stably transfected with constructs encoding APP695 and either SORLAWT or SORLAΔCD as described elsewhere for CHO cells (6). For knockdown, the cells were treated with 1 μM siRNA for 72 h, after which the medium was replaced and conditioned for 24 h. The cell supernatant was collected, and the cells were lysed in lysis buffer containing 1% (wt/vol) NP-40, 1% (wt/vol) Triton X-100, 300 mM NaCl, 5 mM EDTA, and 50 mM Tris-HCl (pH 7.4). Cell media and lysates were stored at −80°C until further analysis by Western blotting or enzyme-linked immunosorbent assay (ELISA). All knockdown data were generated by siRNA A006697-13 targeting PACS1 expression (Thermo Scientific). Where indicated in Results (see Fig. 4B and 6D), these data were reproduced with a second siRNA, A006697-16 (Thermo Scientific). Primary hippocampal neurons from newborn mice were prepared and cultured for 5 days in Neurobasal-A medium (Life Technologies) supplemented with B27 and GlutaMAX (Life Technologies).
Fig 4.
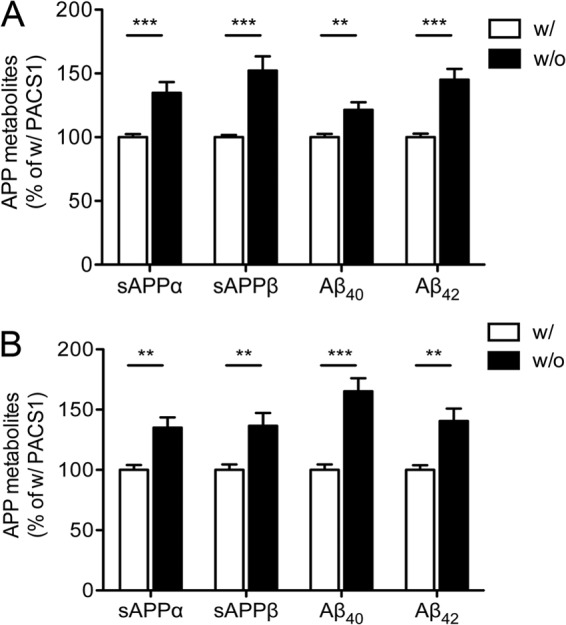
Knockdown of PACS1 expression enhances APP processing in SY5Y cells. (A) Levels of soluble APPα (sAPPα), sAPPβ, Aβ40, and Aβ42 were determined by ELISA in SY5Y-A/S cells treated with a siRNA directed against PACS1 (without [w/o] PACS1) or with a scrambled control siRNA (with [w/] PACS1). Data are the means ± the standard errors of the means of triplicate measurements of five independent experiments. Values were calculated as percentages of the scrambled siRNA control (with PACS1, set to 100%). (B) Same as panel A but with an alternative siRNA directed against PACS1 as described in Materials and Methods. Data represent the means ± the standard errors of the means of triplicate measurements of four independent experiments (**, P < 0.01; ***, P < 0.001).
Fig 6.
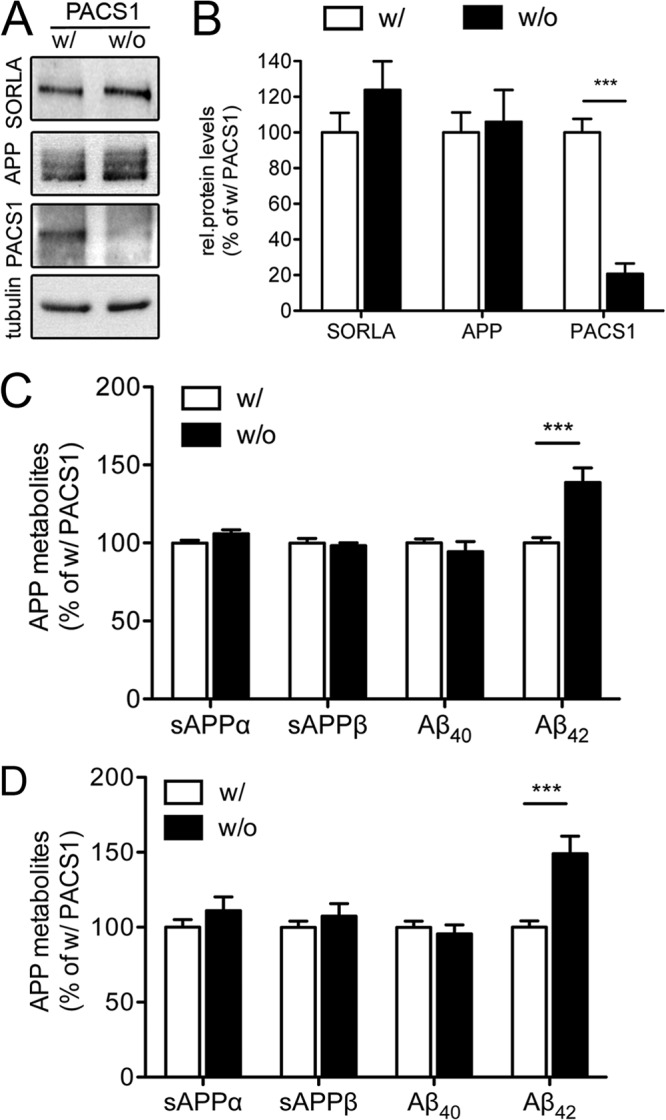
Effect of PACS1 knockdown on APP processing in SY5Y cells expressing a trafficking-defective mutant form of SORLA. (A and B) SY5Y-A/SΔCD mutant cells were treated with a siRNA directed against endogenous PACS1 (without [w/o] PACS1) or with a scrambled control siRNA (with [w/] PACS1). Western blot analysis of representative cell lysates (A) and densitometric scanning of replicate blots (B) document significant reduction of PACS1 levels in siRNA-treated SY5Y-A/SΔCD cells (number of samples per condition, 7). Levels of SORLA, APP, and tubulin (loading control) are not affected by PACS1 knockdown. rel., relative. (C) Levels of soluble APPα (sAPPα), sAPPβ, Aβ40, and Aβ42 were determined by ELISA in SY5Y-A/SΔCD cells treated with a siRNA directed against PACS1 (without PACS1 [w/o]) or with a scrambled control siRNA (with PACS1 [w/]). Data are the means ± the standard errors of the means of triplicate measurements in five independent experiments. Values were calculated as percentage of the scrambled siRNA control (with PACS1, set to 100%). (D) Same as panel C but with an alternative siRNA directed against PACS1 as described in Materials and Methods. Data represent the means ± the standard errors of the means of triplicate measurements of four independent experiments (***, P < 0.001).
CatB activity assay.
CatB activity in cell lysates was assayed by adding a 50 μM concentration of the fluorogenic CatB substrate z-RR-AMC (219392; EMD Millipore) in a buffer containing 50 mM sodium acetate, 4 mM EDTA, 8 mM dithiothreitol, and 1 mM phenylmethylsulfonyl fluoride (pH 6) as previously described (17). Fluorescence intensity was measured (excitation wavelength, 360 nm; emission wavelength, 480 nm) with a fluorescence reader.
Immunocytochemistry and image analysis.
Confocal immunofluorescence microscopy of primary hippocampal neurons or siRNA-treated SY5Y-A/S cells was performed according to standard protocols. For image analysis, stacks of z-sections (0.17 μm) were sequentially acquired with an SP5 laser scanning microscope. Images (1,024 by 1,024 pixels) were acquired with a 63× oil objective (numerical aperture 1.4) with the following settings: airy = 1, zoom 4. Image analysis was carried out with Fiji/ImageJ software. In detail, in each image, a region of interest encompassing the cell of interest was defined. Thresholded Manders (tM) values and Pearson's correlation coefficient (r) were measured. Pearson's correlation coefficient adopts values from +1 to −1 and represents a measurement of the pixel's covariance of two channels in an image. Whereas +1 indicates a positive relationship between the fluorescence intensities, −1 describes a negative relationship. tM values range from 0 (no colocalization) to 1 (perfect colocalization) and denote the degrees of overlap between the signals in the two channels. Statistically significant differences between two groups were evaluated with Student's t test.
Generation and analysis of mouse models.
Mice carrying an inducible human cDNA encoding SORLAWT or SORLAacidic (6) were generated by targeting the Rosa26 locus in murine embryonic stem cells with a vector described in reference 18. The vector includes a neomycin resistance cassette and a transcription stop site preceding the cDNA constructs (see Fig. 8A for details). Mice carrying the transgenes through their germ line were first crossed with the cre deleter strain of mice (Taconics) to remove the transcription stop site and to enable cDNA expression driven by the endogenous Rosa26 promoter. Next, these mice were crossed with the SORLA-deficient line (Sorl1−/−) (7) to remove the expression of endogenous murine SORLA and to derive lines expressing human SORLAWT or SORLAacidic variants only. For analysis of human APP processing, SORLAWT or SORLAacidic mice were crossed with the 5XFAD line, an established model of AD (19).
Fig 8.

Generation of transgenic mice expressing human SORLA variants. (A) Strategy for targeting of cDNAs encoding wild-type SORLA (SORLAWT) or a receptor variant lacking the PACS1 binding motif (SORLAacidic) to the endogenous Rosa26 gene locus in mice. The nucleotide sequences of the cytoplasmic domains of SORLAWT and SORLAacidic are shown at the top. The PACS1 binding motif is highlighted by a horizontal line. Nucleotides modified by site-directed mutagenesis in SORLAacidic are in bold. The organization of the targeting vector and the wild-type Rosa26 locus is indicated. Exons 1 and 2 of Rosa26 are shown as black bars. Triangles indicate loxP recombination sites. Removal of the loxP-flanked neomycin resistance cassette (neoR) by Cre recombinase enables transcription of the SORLA cDNA from the endogenous Rosa26 promoter (activated Rosa26 locus). s.a., splice acceptor site; polyA, polyadenylation site. (B) Western blot analysis of SORLA expression in cortex and hippocampus extracts from mice that lack endogenous SORLA but are homozygous for the SORLAWT and SORLAacidic cDNAs inserted into Rosa26. Levels of expression of SORLAWT and SORLAacidic are comparable to the endogenous receptor level in wild-type mice (Sorl1+/+). Extracts from animals genetically deficient in SORLA (Sorl1−/−) were used as a negative control. Detection of Na/K-ATPase and PSD95 served as loading controls. The arrowhead indicates the protein band representing SORLA in hippocampal extracts. The asterisk denotes a background band seen in all genotypes. (C and D) Immunohistological detection of SORLA (green) in the cortex and hippocampus of mice of the indicated genotypes. In panel C, neurons were costained for the marker NeuN (blue). In panel D, astroglia were costained for GFAP (red) and nuclei were counterstained with DAPI (gray). Expression of SORLAWT and SORLAacidic is restricted largely to neurons, comparable to the pattern of the endogenous receptor in Sorl1+/+ animals. No immunoreactivity for SORLA is seen in Sorl1−/− tissue. The insets highlight localization of SORLAWT to the perinuclear region (arrowheads) but a disperse subcellular pattern for SORLAacidic. Scale bars, 10 μm.
For analysis of protein expression, brain tissue was homogenized in ice-cold homogenization buffer (0.25 mM sucrose, 2 mM MgCl2, 20 mM Tris-HCl, pH 7.5) supplemented with protease (05 892 791 001; Roche) and phosphatase inhibitors (78440; Thermo Scientific). Following incubation for 20 min on ice, nuclei and cellular debris were removed by a centrifugation step (1,000 × g, 10 min). The membrane fraction was pelleted (100,000 × g, 1 h) and resuspended in lysis buffer for analysis of membrane-associated proteins. The supernatant was used to detect APP metabolites and all cytoplasmic proteins. For immunohistochemistry, 40-μm free-floating sagittal sections of adult mouse brains were produced and processed for confocal immunofluorescence microscopy by standard protocols.
Measurement of APP metabolites.
Human APP processing products were determined in multiplex biological assays (K15120W, K151BUE, and K15141; Meso Scale Discovery) with the SECTOR Imager 2400 (Meso Scale Discovery) as a readout device. All measurements were carried out according to the manufacturer's protocol. Statistical significance of data was determined with Student's t test.
RESULTS
Using coimmunoprecipitation experiments, we previously documented the ability of PACS1 to bind to an acidic cluster motif, D2190DLGEDDED (UniProt accession no. Q92673), in the cytoplasmic domain of SORLA (6). To further refine the molecular determinants of SORLA and PACS1 interaction, we dissected the polypeptide sequence of PACS1 into its main structural domains, designated the atrophin-1-related region (ARR), the furin-binding region (FBR), and the middle region (MR) (Fig. 1A) (20). When tested individually, the FBR, but not the ARR or the MR, was able to coimmunoprecipitate with SORLA from transfected CHO cells (Fig. 1B). To substantiate the FBR as the main interaction site for SORLA, we generated a PACS1 deletion mutant specifically lacking this domain (ΔFBR, Fig. 1A). Despite much stronger expression of ΔFBR than full-length PACS1 in CHO transfectants (Fig. 1C, input), only residual background binding to SORLA was observed for this deletion mutant (Fig. 1C; IP-SORLA). Interestingly, the FBR domain was identified as the binding region for the cytoplasmic domains of furin and CI-MPR before (15), suggesting the potential of various cargo molecules to compete for sorting by PACS1. Coimmunoprecipitation of SORLA and PACS1 was also shown for the endogenous proteins from brain extracts of mice (Fig. 1D).
Fig 1.

SORLA interacts with the furin-binding region in PACS1. (A) PACS1 constructs used for binding domain mapping. ARR, atrophin-1-related region; FBR, furin-binding region; MR, middle region; CTR, carboxyl-terminal region. (B) CHO cells stably expressing human SORLA were transiently transfected with constructs encoding hemagglutinin (HA)-tagged versions of ARR (ARR-HA; lane 1), FBR (FBR-HA; lane 2), and MR (MR-HA; lane 3) of PACS1. Cells transfected with the empty vector (lane 4) or with a vector encoding full-length, HA-tagged PACS1 (PACS1-HA; lane 5) were used as negative and positive controls, respectively. The input part of the panel represents Western blot analyses for SORLA (α-SORLA) and PACS1 variants (α-HA) in cell extracts prior to immunoprecipitation. The IP-SORLA part of the panel shows Western blot analyses for SORLA (α-SORLA) and PACS1 variants (α-HA) in samples immunoprecipitated with anti-SORLA IgG. Full-length PACS1 (lane 5) and the FBR domain (lane 2) are coprecipitated with SORLA. No coimmunoprecipitation is seen for ARR (lane 1) or MR (lane 3) or for the FBR when the anti-SORLA IgG was omitted (lane 6). The migration of marker proteins of the indicated molecular masses is shown. w/o, without. (C) CHO cells expressing human SORLA were transiently transfected with constructs encoding wild-type PACS1 (PACS1-HA, lane 1) or a deletion mutant form lacking the FBR (ΔFBR-HA, lane 2). The input part of the panel represents Western blot analyses for SORLA (α-SORLA) and PACS1 variants (α-HA) in cell extracts prior to immunoprecipitation. The IP-SORLA part of the panel shows Western blot analyses for SORLA (α-SORLA) and PACS1 variants (α-HA) in samples immunoprecipitated with anti-SORLA IgG. Efficient coimmunoprecipitation for full-length PACS1-HA (lane 1) but very little for ΔFBR-HA (lane 2) was seen. (D) Endogenous PACS1 and SORLA coimmunoprecipitate from brain lysate. The IP-PACS1 part of the panel shows that immunoprecipitation of PACS1 (α-PACS1) coprecipitates SORLA (α-SORLA) from wild-type (Sorl1+/+; lane 1) but not from SORLA-deficient (Sorl1−/−; lane 2) mouse brains. Also, no coimmunoprecipitation of SORLA from Sorl1+/+ brains was seen in the absence of anti-PACS1 antiserum (lane 3). The input part of the panel represents Western blot analyses for SORLA and PACS1 in tissue samples prior to immunoprecipitation.
When alanine mutations were introduced into the acidic cluster motif in SORLA (A2190ALGAAAA), the respective receptor variant SORLAacidic failed to properly localize to the TGN when expressed in CHO cells (6). Because this acidic cluster motif overlaps the binding sites for adaptor complex 1 (AP1) (antero/retrograde sorting) and AP2 (endocytosis), the proposed involvement of PACS1 in SORLA sorting was met with skepticism. To more rigorously test the relevance of PACS1 for SORLA transport in neurons, we established a novel cell model by using the neuroblastoma cell line SH-SY5Y, which stably overexpresses the human SORLA and APP695 transgenes (SY5Y-A/S). SH-SY5Y cells express low levels of endogenous SORLA, but these levels are negligible when SORLA transgenes are overexpressed (21). When transiently transfected with a siRNA directed against endogenous PACS1 (without PACS1), expression of the adaptor was strongly reduced compared to that in SY5Y-A/S cells transfected with a scrambled control siRNA (with PACS1) (Fig. 2A and B).
Fig 2.
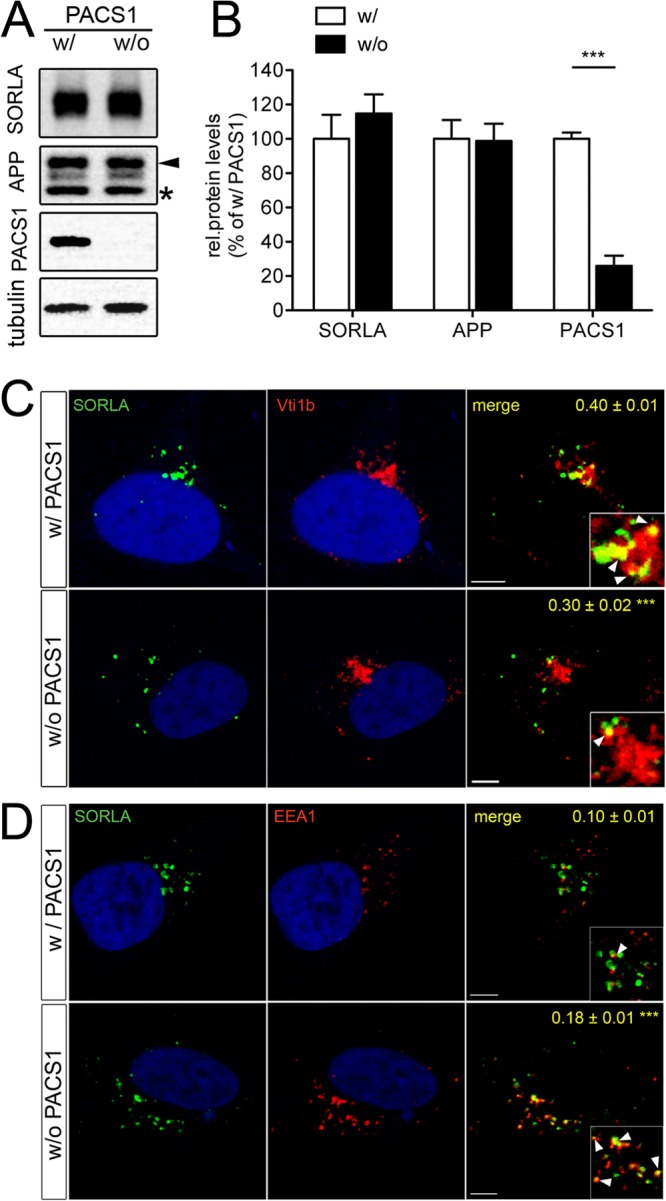
Loss of PACS1 alters trafficking of SORLA in SY5Y cells. (A and B) SH-SY5Y cells stably overexpressing human APP695 and human wild-type SORLA (SY5Y-A/S) were treated with a siRNA directed against endogenous PACS1 (without PACS1 [w/o PACS1]) or with a scrambled control siRNA (with PACS1 [w/PACS1]). Western blot analysis of representative cell lysates (A) and densitometric scanning of replicate blots (B; number of samples per condition, 12) show a significant reduction of PACS1 expression in siRNA-treated cells (***, P < 0.001). Levels of SORLA, APP, and tubulin (loading control) are not affected by PACS1 knockdown. In panel A, the protein bands representing the precursor form (asterisk) and the fully glycosylated mature form of APP (arrowhead) are indicated. (C and D) SY5Y-A/S cells were treated with a siRNA directed against PACS1 (without PACS1) or with a scrambled control siRNA (with PACS1). Subsequently, colocalization of SORLA (green) with markers (red) of the trans-Golgi network (C, Vti1b) or of early endosomes (D, EEA1) was tested by confocal immunofluorescence microscopy. Nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI). Pearson's correlation coefficient (r) for estimation of colocalization of the markers with SORLA is given in the respective merged images. Twenty to 22 cells per condition were scored (***, P < 0.001). The arrowheads in the inset indicate colocalization of SORLA with the marker proteins. Scale bars, 5 μm.
The overall levels of SORLA and APP were not affected by PACS1 knockdown (Fig. 2B). However, loss of PACS1 resulted in a distinct change in the subcellular localization of SORLA, as shown by confocal immunofluorescence microscopy. In the presence of PACS1 (with PACS1), the receptor showed the typical perinuclear pattern, significantly overlapping the TGN marker Vti1b (Pearson's coefficient r = 0.40 ± 0.01) (Fig. 2C). In contrast, following knockdown of PACS1 (without PACS1), receptor signals were dispersed to more distal vesicular structures and showed significantly less colocalization with Vti1b (r = 0.30 ± 0.02; P < 0.001) (Fig. 2C). Instead, in the absence of PACS1, SORLA exhibited greater colocalization with early endosome antigen 1 (EEA1)-positive early endosomes (r = 0.18 ± 0.01) than with PACS1-expressing control cells (r = 0.10 ± 0.01; P < 0.001) (Fig. 2D). This altered SORLA pattern in the absence of PACS1 was also confirmed by determination of the tM values for both markers (Table 1, top). Collectively, these findings suggested the inability of SORLA to properly localize to the TGN in the absence of PACS1.
Table 1.
Colocalization of SORLA and APP with marker proteins in SY5Y-A/S cellsa
| Colocalization | Mean tM value ± SEM |
P value | |
|---|---|---|---|
| With PACS1 | Without PACS1 | ||
| SORLA and Vti1b | |||
| tM1 (Vti1b-SORLA overlap) | 0.254 ± 0.017 | 0.186 ± 0.015 | <0.01 |
| tM2 (SORLA-Vti1b overlap) | 0.586 ± 0.023 | 0.430 ± 0.014 | <0.001 |
| SORLA and EEA1 | |||
| tM1 (EEA1-SORLA overlap) | 0.072 ± 0.004 | 0.117 ± 0.009 | <0.01 |
| tM2 (SORLA-EEA1 overlap) | 0.152 ± 0.017 | 0.220 ± 0.017 | <0.001 |
| APP and SORLA | |||
| tM1 (SORLA-APP overlap) | 0.504 ± 0.038 | 0.457 ± 0.039 | >0.05 |
| tM2 (APP-SORLA overlap) | 0.233 ± 0.018 | 0.251 ± 0.002 | >0.05 |
| APP and Vti1b | |||
| tM1 (Vti1b-APP overlap) | 0.134 ± 0.01 | 0.118 ± 0.007 | >0.05 |
| tM2 (APP-Vti1b overlap) | 0.84 ± 0.016 | 0.611 ± 0.028 | <0.001 |
| APP and EEA1 | |||
| tM1 (EEA1-APP overlap) | 0.059 ± 0.004 | 0.081 ± 0.004 | <0.001 |
| tM2 (APP-EEA1 overlap) | 0.448 ± 0.026 | 0.559 ± 0.027 | <0.01 |
Colocalization of SORLA and APP with the indicated proteins in SY5Y-A/S cells treated with a siRNA directed against endogenous PACS1 (without PACS1) or with a scrambled control siRNA (with PACS1) was tested by confocal immunofluorescence microscopy. The extent of colocalization was scored by determination of thresholded Manders (tM) values as detailed in Materials and Methods.
To explore the consequences of altered SORLA transport for APP routing, we repeated the above-described knockdown studies by testing the colocalization of APP with SORLA and with cellular markers. As shown in Fig. 3A, APP and SORLA showed similar degrees of colocalization in the presence and absence of PACS1 (r = 0.37 ± 0.02 versus 0.38 ± 0.03; P > 0.05), indicating that APP parallels SORLA routing under both conditions. This assumption was confirmed by loss of APP from TGN compartments marked by Vti1b (r = 0.27 ± 0.02 versus 0.35 ± 0.02 in the presence of PACS1; P < 0.001) (Fig. 3B) and by a concomitant accumulation in EEA1-positive early endosomes (r = 0.19 ± 0.01 versus 0.15 ± 0.01 in the presence of PACS1; P < 0.01) (Fig. 3C). Altered sorting of APP due to loss of PACS1 was confirmed by determination of tM values (Table 1, bottom). Defective routing of APP coincided with a significant increase in amyloidogenic and nonamyloidogenic processing as documented by comparing levels of sAPPα, sAPPβ, as well as Aβ40 and Aβ42, in supernatants of SY5Y-A/S cells with or without PACS1 (Fig. 4A). These findings were reproduced with an alternative siRNA targeting PACS1 (Fig. 4B).
Fig 3.

Loss of PACS1 alters trafficking of APP in SY5Y cells. SY5Y-A/S cells were treated with a siRNA directed against PACS1 (without PACS1 [w/o PACS1]) or with a scrambled control siRNA (with PACS1 [w/PACS1]). Subsequently, colocalization of APP (green) with SORLA (red) (A) or with markers (red) of the trans-Golgi network (B; Vti1b) or early endosomes (C; EEA1) was tested by confocal immunofluorescence microscopy. Pearson's correlation coefficient (r) for estimation of colocalization of APP with the three proteins is given in the merged images. Nineteen to 29 cells per condition were scored (**, P < 0.01; ***, P < 0.001). The arrowheads in the inset indicate colocalization of APP with the respective proteins. Scale bars, 5 μm.
Since PACS1 knockdown likely also affects the transport of cargo other than SORLA, the increase in APP processing products seen in the absence of PACS1 may be caused by SORLA-dependent or SORLA-independent mechanisms. To dissect these possibilities, we performed studies with SH-SY5Y cells expressing the APP695 transgene together with a SORLA variant that lacks the cytoplasmic domain (SORLAΔCD) and that is therefore not responsive to adaptor-mediated sorting (6). In line with previous findings with CHO cells (6), faulty trafficking of SORLAΔCD in the neuroblastoma cell line SY5Y-A/SΔCD resulted in enhanced APP processing, as shown by significantly elevated levels of processing products sAPPα, sAPPβ, Aβ40, and Aβ42 compared with SY5Y-A/S cells (Fig. 5). Knockdown of PACS1 in SY5Y-A/SΔCD worked efficiently (Fig. 6A and B). However, loss of PACS1 failed to affect levels of SORLA and APP (Fig. 6B) or of the APP processing products sAPPα, sAPPβ, and Aβ40 compared to SY5Y-A/SΔCD treated with scrambled control siRNA (Fig. 6C). Thus, the effect of PACS1 on these APP processing products required wild-type SORLA activity. In contrast to the levels of sAPPα, sAPPβ, and Aβ40, those of Aβ42 were still increased in SY5Y-A/SΔCD in response to PACS1 knockdown (Fig. 6C). This finding was reproduced with an alternative siRNA (Fig. 6D). This observation suggested a specific effect of PACS1 activity on Aβ42 metabolism that worked independently of SORLA and independently of APP processing (as sAPPα and sAPPβ levels were unchanged).
Fig 5.
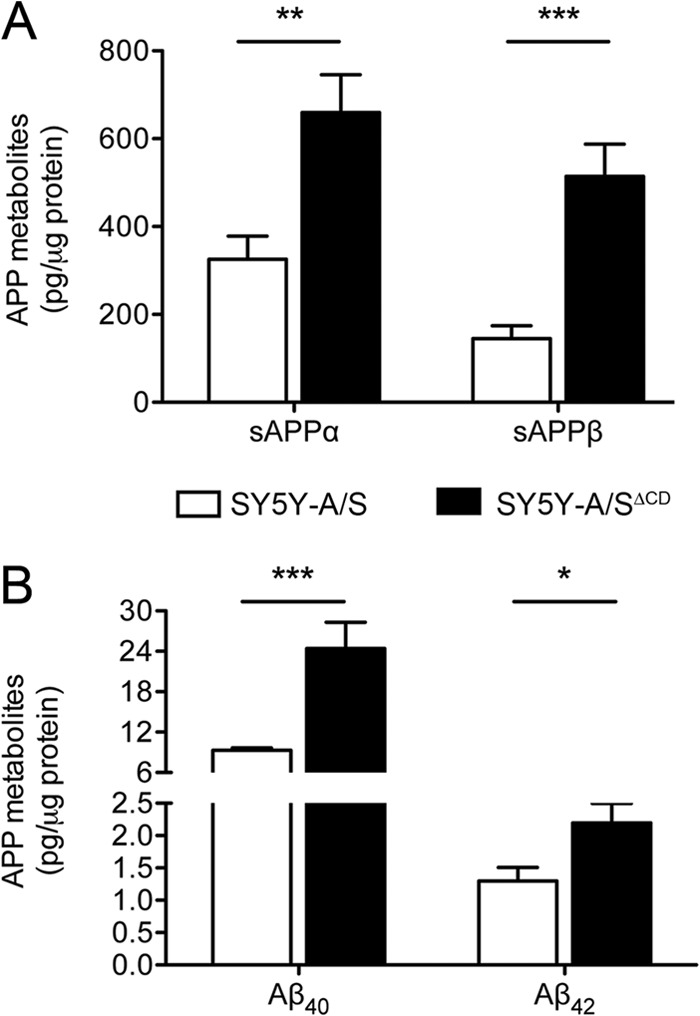
Defect in SORLA trafficking increases APP processing rates in SY5Y cells. A comparative analysis of APP processing products in SH-SY5Y cells stably overexpressing human APP695 and either wild-type SORLA (SY5Y-A/S) or a receptor variant lacking the cytoplasmic tail (SY5Y-A/SΔCD) is shown. Levels of soluble APPα (sAPPα) and sAPPβ (A), as well as of Aβ40 and Aβ42 (B), in cell supernatants are higher in cells expressing SORLAΔCD than in those expressing SY5Y-A/S, as tested by ELISA. Data are the means ± the standard errors of the means of triplicate measurements in four independent experiments (*, P < 0.05; **, P < 0.01; ***, P < 0.001).
One cargo trafficked by PACS1 is the CI-MPR, a sorting receptor that directs lysosomal enzymes from the TGN to endosomes and that requires PACS1 for its retrograde TGN retrieval (14). Among the enzymes sorted by CI-MPR is CatB, a cysteine protease that degrades peptides in the endosomal-lysosomal system but also in the extracellular space following secretion (22). Because CatB specifically degrades Aβ42 (23–25), we reasoned that the SORLA-independent effect of PACS1 knockdown on Aβ42 levels might work by impacting the CI-MPR–CatB system. This hypothesis was supported when we detected reduced levels of CI-MPR in cell lysates (Fig. 7A and B) and of mature CatB in the lysate and supernatant (Fig. 7C and D) of SY5Y-A/S cells treated with PACS1 siRNA. To confirm lower levels of CatB activity in cells lacking PACS1, we performed an activity assay of cell lysates with the fluorogenic CatB substrate z-RR-AMC (17). As shown in Fig. 7E, reduced levels of mature CatB protein in PACS1 knockdown cells resulted in significantly reduced levels of protease activity. Thus, impaired maturation and activity of CatB likely caused the SORLA-independent increase in Aβ42 levels in PACS1-deficient cells.
Fig 7.

Loss of PACS1 impairs expression of the cation-independent mannose 6-phosphate receptor and CatB in SY5Y cells. SY5Y-A/S cells were treated with a siRNA directed against PACS1 (without [w/o] PACS1) or with a scrambled control siRNA (with [w/] PACS1). Subsequently, levels of PACS1 and cation-independent mannose 6-phosphate receptor (CI-MPR) in cell lysate (A and B) and of mature CatB in cell lysate and medium (C and D) were determined by Western blotting and densitometric scanning of replicate blots (number of samples per condition, 9 to 13) Detection of tubulin (tub.) served as a loading control for cell lysates in panels A and C. (E) CatB activity was determined in lysates of SY5Y-A/S cells with fluorogenic CatB substrate z-RR-AMC (as detailed in Materials and Methods). Activity levels are significantly lower in cells treated with a siRNA directed against PACS1 (without PACS1) than in cells receiving a scrambled control siRNA (with PACS1). Data represent the means of duplicate measurements of four independent experiments (*, P < 0.05; ***, P < 0.001).
Taken together, our studies with cultured cells uncovered both SORLA-dependent and -independent PACS1 mechanisms in amyloidogenic processes in neuronal cells. To further test the relevance of PACS1-dependent sorting of SORLA for AD-related processes in vivo, we next generated a novel mouse model that expresses SORLAacidic, the receptor variant lacking the PACS1 binding motif in its cytoplasmic tail (6). In detail, we used homologous recombination in embryonic stem cells to introduce a human SORLAacidic cDNA construct into the murine Rosa26 gene locus (Fig. 8A). This strategy had been used before to achieve the expression of transgenes from the endogenous Rosa26 promoter (18). Mice carrying the activated SORLAacidic cDNA insertion were crossed with animals with targeted disruption of the endogenous SORLA gene locus (Sorl1−/−) (7) to eliminate the expression of the endogenous wild-type receptor. The resulting mouse strain is referred to as SORLAacidic. To control for possible differences in the levels of expression from the endogenous Sorl1 and Rosa26 loci, the same strategy was used to generate a control line carrying a wild-type human SORLA cDNA insertion in Rosa26 but lacking the endogenous gene locus (Sorl1−/−). This strain is referred to as SORLAWT. Western blot analyses confirmed equal levels of SORLA expression in the hippocampus and cortex in SORLAWT and SORLAacidic mice that were slightly higher than the levels seen for the endogenous murine receptor in wild-type control (Sorl1+/+) mice (Fig. 8B). Like the expression of the murine receptor, that of the two human receptor variants in the brain was restricted mainly to neurons, as shown by immunohistology (Fig. 8C). No significant coexpression of endogenous or transgenic SORLA variants with glial fibrillary acidic protein (GFAP), a marker of astroglia, was observed in the cortex or hippocampus in these animals (Fig. 8D). Curiously, restriction of SORLA transgene expression to cell types that normally express this receptor was also seen in other tissues. For example, the SORLA transgenes are strongly expressed in the kidney but overexpression is seen exclusively in cells of the distal convoluted tubule, the cell type expressing the endogenous receptor (data not shown). Although these findings need further rigorous testing, they suggest the existence of a posttranscriptional mechanism(s) required to sustain SORLA expression in distinct cell types.
In the brain, expression of SORLAacidic did not affect the levels of expression of PACS1 targets CI-MPR, CatB, and furin (Fig. 9A and B) or of various adaptor proteins, including PACS1, AP1 (γ-adaptin), and AP2 (μ subunit) (Fig. 9C and D). Thus, SORLAacidic was deemed a suitable model for specific testing of the consequences of loss of PACS1 binding for SORLA-dependent processes in the brain.
Fig 9.
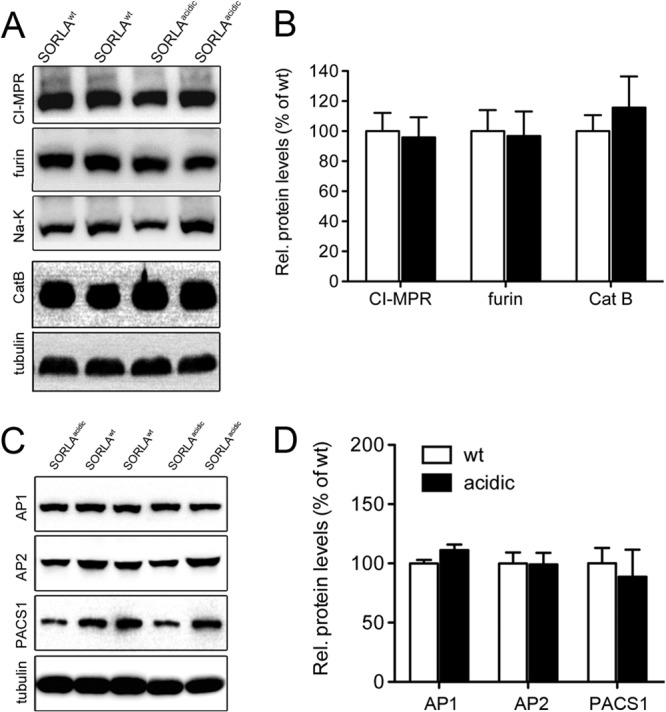
Expression of proteins in the brains of SORLAWT and SORLAacidic mice. Levels of expression of PACS1 targets CI-MPR, furin, and CatB (A and B) and of SORLA adaptors AP1 (γ-adaptin), AP2 (μ subunit), and PACS1 (C and D) were determined in cortical brain extracts by Western blotting and subsequent densitometric scanning of replicate blots (number of samples, 4 to 6). No statistically significant differences in any of the proteins were seen when comparing SORLAWT and SORLAacidic brain tissues. Detection of tubulin and Na-K-ATPase (Na-K) served as loading controls. Rel., relative; wt, wild type.
While the levels of expression of SORLAacidic in neurons were comparable to those of SORLAWT (Fig. 8B), we observed a distinct difference in the subcellular localization of the two receptor variants by immunohistology. Whereas the wild-type protein showed the typical perinuclear (Golgi-like) pattern, SORLAacidic immunoreactivity was more dispersed throughout the neuronal soma (insets in Fig. 8C). This notion was confirmed by immunocytochemical analysis of primary neurons derived from the two mouse lines. In line with the proposed role for PACS1 in TGN sorting of SORLA, the SORLAacidic variant showed a distinct loss of immunoreactivity from TGN compartments marked by Vti1b (Fig. 10A) and γ-adaptin (Fig. 10B) and a concomitant shift into vesicles positive for the early endosome marker Rab5 (Fig. 10C). Altered colocalization of SORLAacidic compared to SORLAWT was substantiated by determination of Pearson's coefficient (values in panels of Fig. 10) and of tM values (Table 2). Lack of the PACS1 binding motif did not result in an aberrant shift of SORLAacidic into the lysosomal compartment, as no apparent difference in the colocalization of SORLAacidic and SORLAWT with the lysosomal marker Lamp1 was observed (Fig. 10D; Table 2).
Fig 10.

Subcellular localization of SORLAWT and SORLAacidic in primary neurons. Primary hippocampal neurons from postnatal day 1 mice expressing human SORLAWT or SORLAacidic were analyzed by confocal immunofluorescence microscopy for localization of SORLA (green) and markers (red) of the trans-Golgi network (A, Vti1b; B, γ-adaptin), early endosomes (C, Rab5), and lysosomes (D, Lamp1). Pearson's correlation coefficient (r) for estimation of colocalization of the markers with SORLA is shown in the respective merged images. Twenty-one to 23 cells per condition were scored (**, P < 0.01; ***, P < 0.001). The arrowheads in the insets indicate colocalization of SORLA variants with the respective marker proteins. Scale bars, 5 μm.
Table 2.
Colocalization of SORLAWT and SORLAacidic with marker proteins in primary neuronsa
| Colocalization | Mean tM value ± SEM |
P value | |
|---|---|---|---|
| SORLAWT | SORLAacidic | ||
| SORLA and Vti1b | |||
| tM1 (Vti1b-SORLA overlap) | 0.279 ± 0,020 | 0.174 ± 0.011 | <0.001 |
| tM2 (SORLA-Vti1b overlap) | 0.685 ± 0.021 | 0.388 ± 0.025 | <0.001 |
| SORLA and γ-adaptin | |||
| tM1 (γ-adaptin–SORLA overlap) | 0.376 ± 0.033 | 0.163 ± 0.016 | <0.001 |
| tM2 (SORLA–γ-adaptin overlap) | 0.405 ± 0.027 | 0.215 ± 0.017 | <0.001 |
| SORLA and Rab5 | |||
| tM1 (Rab5-SORLA overlap) | 0.099 ± 0.007 | 0.180 ± 0.006 | <0.001 |
| tM2 (SORLA-Rab5 overlap) | 0.444 ± 0.029 | 0.521 ± 0.033 | >0.05 |
| SORLA and Lamp1 | |||
| tM1 (Lamp1-SORLA overlap) | 0.146 ± 0.015 | 0.141 ± 0.012 | >0.05 |
| tM2 (SORLA-Lamp1 overlap) | 0.416 ± 0.022 | 0.377 ± 0.028 | >0.05 |
Colocalization of SORLA with the indicated marker proteins in primary neurons from SORLAWT and SORLAacidic mice was tested by confocal immunofluorescence microscopy. The extent of colocalization was scored by determination of thresholded Manders (tM) values as detailed in Materials and Methods.
To study the consequence of aberrant SORLA trafficking for APP processing, we crossed the SORLAWT and SORLAacidic lines with the 5XFAD line, an established model of AD (19). Loss of the ability of SORLAacidic to engage PACS1 or other adaptors that bind to the acidic cluster motif did not affect the total levels of APP (Fig. 11A and B) but resulted in significantly higher levels of APP processing products, including Aβ40 and Aβ42, in the brains of SORLAacidic/5XFAD mice than in those of SORLAWT/5XFAD control animals (Fig. 11C and D).
Fig 11.

APP processing in the brains of SORLAWT/5XFAD and SORLAacidic/5XFAD mice. (A and B) Western blot analysis of levels of SORLA, APP, and tubulin (loading control) in the membrane fraction of cortical brain extracts of 5XFAD mice expressing either SORLAWT (lane 2) or SORLAacidic (lane 3). Brain tissue from a SORLA-deficient 5XFAD mouse (Sorl1−/−, lane 1) served as a negative control. Panel B depicts quantification of APP levels by densitometric scanning of replicate blots exemplified in panel A (number of samples, 14). (C and D) Levels of soluble APPα (sAPPα), sAPPβ, Aβ40, and Aβ42 were determined by ELISA in the cytoplasmic fraction of cortical brain extracts of mice of the indicated genotypes. Values are means ± the standard errors of the means (duplicate measurements of 14 or 15 animals per genotype; **, P < 0.01; ***, P < 0.001). wt, wild type.
In conclusion, our studies with the neuroblastoma cell line SH-SY5Y, as well as the brains of mice and primary neurons derived therefrom, all support a model whereby interaction with PACS1 (or other adaptors targeting the acidic cluster motif) is essential to enable retrograde sorting of SORLA and its target APP from the early endocytic pathway back to the TGN. TGN retrieval of SORLA/APP complexes reduces nonamyloidogenic and amyloidogenic processing at the plasma membrane and in endosomes, respectively (Fig. 12A). Loss of PACS1 expression or deletion of the PACS1 binding site in SORLA results in aberrant accumulation of APP molecules in the early endocytic compartment and coincides with enhanced proteolytic breakdown of APP (Fig. 12B).
Fig 12.
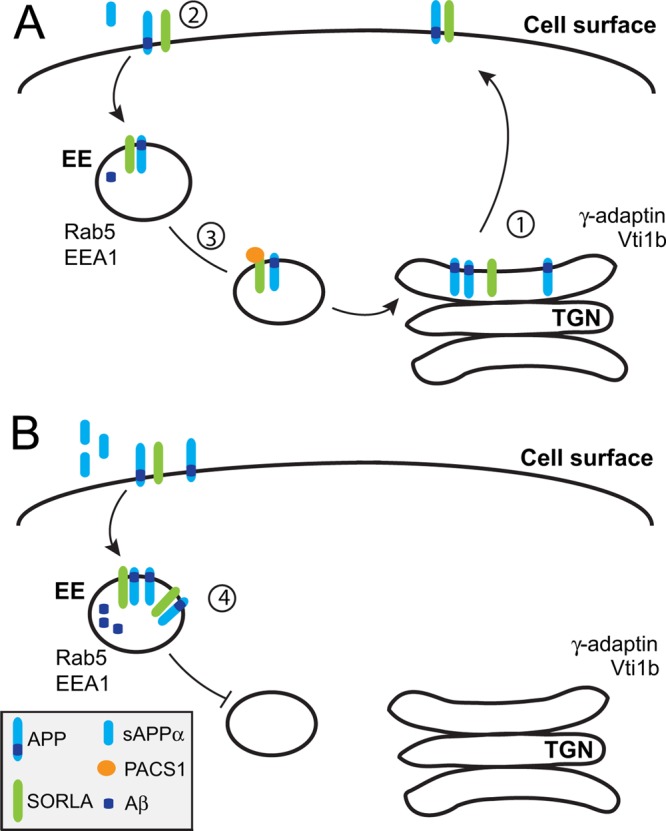
Model of PACS1 function in SORLA transport and APP processing.(A) In wild-type neurons, SORLA interacts with APP in the TGN to impair its export to the cell surface (step 1), thereby decreasing the extent of nonamyloidogenic processing by α-secretases at the plasma membrane (step 2). APP molecules that are not cleaved at the cell surface internalize through clathrin-mediated endocytosis and move to early endosomes (EE). From EE, APP molecules are sorted retrogradely by SORLA back to the TGN to reduce amyloidogenic processing in endosomal compartments (step 3). Retrograde sorting of APP requires interaction of SORLA with PACS1. (B) Retrograde sorting of APP/SORLA complexes is impaired by knockdown of PACS1 in SH-SY5Y cells or by expression of a SORLA variant lacking the PACS1 binding motif in neurons. As a consequence of blocked retrograde sorting, SORLA is depleted from the TGN but accumulates in early endosomes. Because of aberrant shuttling of SORLA/APP complexes between the cell surface and early endosomal compartments, both nonamyloidogenic (to sAPPα) and amyloidogenic processing (to sAPPβ and Aβ) pathways are accelerated (step 4).
DISCUSSION
Our investigations of the functional interaction of SORLA with PACS1 have uncovered three important concepts concerning neuronal transport and processing of APP. First, and foremost, we have substantiated the relevance of neuronal sorting of SORLA by an acidic cluster motif for amyloidogenic processing in vivo. Second, we have refined an underlying molecular mechanism by demonstrating the requirement of PACS1 for retrograde sorting of SORLA (and APP) in neuronal cell lines. Third, we have identified the relevance of PACS1 for the functional expression of mature CatB, a major Aβ42-catabolizing enzyme, in the central nervous system.
SORLA is a negative regulator of APP processing and is encoded by a gene that is a major risk factor for sporadic AD (reviewed in reference 26). On the basis of studies with established cell lines such as CHO and HEK293, SORLA is proposed to target APP to the TGN, thereby protecting the precursor protein from proteolytic breakdown at the cell surface and in endosomes (Fig. 12A) (6, 7). Although substantial evidence from studies with nonneuronal cell lines supports such a role for SORLA in the intracellular trafficking of APP, unequivocal evidence implicating SORLA in neuronal APP transport in vivo is lacking. For instance, complete loss of SORLA in gene-targeted mice results in enhanced Aβ production and plaque deposition, documenting an inverse correlation between receptor activity and amyloidogenic processing (9, 10). However, whether aggravated amyloidogenic processes in SORLA null mice are caused by abnormal APP trafficking or by loss of unrelated functions of SORLA remained difficult to assess. To rigorously test a role for SORLA in neuronal APP transport in vivo, we generated a novel mouse model that expresses a trafficking mutant form of SORLA lacking the acidic cluster motif. The inability of SORLAacidic to properly localize to the TGN has been documented in CHO cells before (6). In line with these earlier observations, SORLAacidic failed to correctly reside in the TGN in primary neurons (Fig. 10; Table 2) and caused enhanced production of amyloidogenic and nonamyloidogenic products in the brains of 5XFAD mice (Fig. 11C and D). The increase in amyloid-β peptide levels in the SORLAacidic/5XFAD line over those in the SORLAWT/5XFAD line was substantial and equaled the increases in amyloid-β peptides seen in SORLA-deficient mice (9).
As well as confirming the importance of sorting of SORLA for APP processing in vivo, our studies have substantiated the functional significance of an acidic cluster motif in the receptor tail and its interaction with PACS1 in these processes. The cytoplasmic domain of SORLA harbors a number of functional elements implicated in the control of protein trafficking in cultured cells. A binding site, D2207DVPMVIA, for the monomeric clathrin adaptors GGA (Golgi-localizing, γ-adaptin ear homology domain, ARF-interacting proteins) enables anterograde sorting of SORLA from the TGN to early endosomes (6, 27, 28). The VPS26 subunit of the retromer complex, which is part of the vesicle coat, targets the motif F2172ANSHY for retrograde endosome-to-TGN routing of the receptor (29–31). Finally, an acidic cluster motif, D2190DLGEDDED, in the tail of SORLA interacts with AP1, a tetrameric complex that links cargo to the clathrin coat of endosomal and TGN vesicles but also with PACS1 (6, 16). Since both AP1 and PACS1 bind to an overlapping site in SORLA and since both have been proposed to mediate anterograde, as well as retrograde, sorting, the functional relevance of this acidic cluster motif for TGN-endosome shuttling (and the adaptor involved) remained unresolved (6, 16).
To dissect the importance of PACS1 for SORLA trafficking, we performed knockdown studies with SY5Y-A/S cells, documenting defects in TGN targeting of SORLA and of APP in the absence of this adaptor (Fig. 2C and D and 3; Table 1). Faulty SORLA/APP transport resulted in increased processing of APP (Fig. 4), an effect that was not seen when SY5Y-A/SΔCD cells expressed a SORLA variant unable to bind trafficking adaptors (Fig. 6C and D). These changes in SORLA and APP transport and amyloidogenic processing seen with PACS1 knockdown were comparable to the alterations seen in primary neurons from SORLAacidic/5XFAD mice (Fig. 11C and D) and in CHO cells in an earlier study (6), strongly arguing for the involvement of PACS1 in SORLA transport. As the binding sites for AP1 and PACS1 overlap, we cannot exclude the possibility that changes in SORLA transport and APP processing seen in SORLAacidic mice also reflect defects in the binding of AP1 or other adaptors to this site. However, the similarity in the trafficking behavior of SORLAacidic in neurons to that of SORLA in SY5Y-A/S cells with PACS1 knockdown strongly argues for the functional relevance of PACS1 binding to this receptor. Concerning the exact trafficking route controlled by PACS1, the shift of SORLA (and APP) immunoreactivity from the TGN to early endosomes in SY5Y cells lacking PACS1 and in primary neurons from SORLAacidic mice suggests a function for the adaptor in TGN retrieval of SORLA. In fact, PACS1-mediated retrograde transport has also been shown for the CI-MPR, another adaptor ligand (14). In retrograde transport of SORLA, the adaptors PACS1 and retromer likely serve a redundant function (31, 32).
Studies of PACS1 knockdown in SY5Y-A/SΔCD cells expressing a tailless SORLA variant were intended to provide a negative control for off-target effects of the loss of this adaptor on APP processing. While these studies confirmed that the effects of PACS1 knockdown on sAPPα, sAPPβ, and Aβ40 production were dependent on SORLA activity, they also revealed a surprising SORLA-independent action of PACS1 in Aβ42 catabolism (Fig. 6C and D). Although the details still warrant clarification, we propose a mechanism whereby PACS1 deficiency impairs functional expression of the CI-MPR, a sorting receptor for CatB. CatB is produced as a precursor polypeptide that requires TGN-to-endosome sorting by CI-MPR to be proteolytically activated in the endosomal/lysosomal compartment. Mature CatB, in turn, catabolizes numerous polypeptides in lysosomes or in the extracellular space following secretion (33). Among the substrates for CatB is Aβ42, which is cleaved in preference to other amyloid-β peptide variants, such as Aβ40 (23). In line with this activity, loss of CatB in gene-targeted mice results in a specific increase in Aβ42 levels (23) while enhanced CatB activity (caused by gene inactivation of the protease inhibitor cystatin C) lowers Aβ42 concentrations in the brain (24, 25). Impaired functional expression of CI-MPR and its cargo CatB in SY5Y-A/S cells lacking PACS1 (Fig. 7) coincides with a specific increase in Aβ42, further supporting a role for CatB as an Aβ42-degrading enzyme, a function that likely requires proper PACS1-dependent sorting of CI-MPR–CatB complexes. Why PACS1 knockdown impacts levels of CI-MPR but not of SORLA is unclear. As SORLA interacts with multiple adaptors for retrograde sorting, including retromer, SNX1, and AP1 (reviewed in reference 3), the impact of PACS1 knockdown on SORLA routing may be partially compensated for by other adaptor pathways. Levels of expression of PACS1 and CI-MPR are normal in SORLAacidic mice (Fig. 9). Thus, the defects in APP transport and processing in this model can be attributed to abnormal trafficking of a SORLA variant unable to interact with PACS1. Still, our findings strongly suggest both SORLA-dependent and SORLA-independent functions for the sorting adaptor PACS1, both of which are important for reduction of the amyloidogenic burden in the brain.
ACKNOWLEDGMENTS
We are indebted to Z. Cseresnyes for help with confocal microscopy and to T. Pantzlaff, C. Kruse, and M. Schmeisser for expert technical assistance.
This study was funded by grants from the Helmholtz Association (HelMA and ICEMED) and the European Commission (Memories).
Footnotes
Published ahead of print 3 September 2013
REFERENCES
- 1.Jacobsen L, Madsen P, Moestrup SK, Lund AH, Tommerup N, Nykjaer A, Sottrup-Jensen L, Gliemann J, Petersen CM. 1996. Molecular characterization of a novel human hybrid-type receptor that binds the alpha2-macroglobulin receptor-associated protein. J. Biol. Chem. 271:31379–31383 [DOI] [PubMed] [Google Scholar]
- 2.Yamazaki H, Bujo H, Kusunoki J, Seimiya K, Kanaki T, Morisaki N, Schneider WJ, Saito Y. 1996. Elements of neural adhesion molecules and a yeast vacuolar protein sorting receptor are present in a novel mammalian low density lipoprotein receptor family member. J. Biol. Chem. 271:24761–24768 [DOI] [PubMed] [Google Scholar]
- 3.Willnow TE, Petersen CM, Nykjaer A. 2008. VPS10P-domain receptors—regulators of neuronal viability and function. Nat. Rev. Neurosci. 9:899–909 [DOI] [PubMed] [Google Scholar]
- 4.Selkoe DJ. 2001. Alzheimer's disease: genes, proteins, and therapy. Physiol. Rev. 81:741–766 [DOI] [PubMed] [Google Scholar]
- 5.Haass C, Kaether C, Thinakaran G, Sisodia S. 2012. Trafficking and proteolytic processing of APP. Cold Spring Harb. Perspect. Med. 2:a006270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schmidt V, Sporbert A, Rohe M, Reimer T, Rehm A, Andersen OM, Willnow TE. 2007. SorLA/LR11 regulates processing of amyloid precursor protein via interaction with adaptors GGA and PACS-1. J. Biol. Chem. 282:32956–32964 [DOI] [PubMed] [Google Scholar]
- 7.Andersen OM, Reiche J, Schmidt V, Gotthardt M, Spoelgen R, Behlke J, von Arnim CA, Breiderhoff T, Jansen P, Wu X, Bales KR, Cappai R, Masters CL, Gliemann J, Mufson EJ, Hyman BT, Paul SM, Nykjaer A, Willnow TE. 2005. Neuronal sorting protein-related receptor sorLA/LR11 regulates processing of the amyloid precursor protein. Proc. Natl. Acad. Sci. U. S. A. 102:13461–13466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Offe K, Dodson SE, Shoemaker JT, Fritz JJ, Gearing M, Levey AI, Lah JJ. 2006. The lipoprotein receptor LR11 regulates amyloid beta production and amyloid precursor protein traffic in endosomal compartments. J. Neurosci. 26:1596–1603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rohe M, Carlo AS, Breyhan H, Sporbert A, Militz D, Schmidt V, Wozny C, Harmeier A, Erdmann B, Bales KR, Wolf S, Kempermann G, Paul SM, Schmitz D, Bayer TA, Willnow TE, Andersen OM. 2008. Sortilin-related receptor with A-type repeats (SORLA) affects the amyloid precursor protein-dependent stimulation of ERK signaling and adult neurogenesis. J. Biol. Chem. 283:14826–14834 [DOI] [PubMed] [Google Scholar]
- 10.Dodson SE, Andersen OM, Karmali V, Fritz JJ, Cheng D, Peng J, Levey AI, Willnow TE, Lah JJ. 2008. Loss of LR11/SORLA enhances early pathology in a mouse model of amyloidosis: evidence for a proximal role in Alzheimer's disease. J. Neurosci. 28:12877–12886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rogaeva E, Meng Y, Lee JH, Gu Y, Kawarai T, Zou F, Katayama T, Baldwin CT, Cheng R, Hasegawa H, Chen F, Shibata N, Lunetta KL, Pardossi-Piquard R, Bohm C, Wakutani Y, Cupples LA, Cuenco KT, Green RC, Pinessi L, Rainero I, Sorbi S, Bruni A, Duara R, Friedland RP, Inzelberg R, Hampe W, Bujo H, Song YQ, Andersen OM, Willnow TE, Graff-Radford N, Petersen RC, Dickson D, Der SD, Fraser PE, Schmitt-Ulms G, Younkin S, Mayeux R, Farrer LA, St George-Hyslop P. 2007. The neuronal sortilin-related receptor SORL1 is genetically associated with Alzheimer disease. Nat. Genet. 39:168–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reitz C, Cheng R, Rogaeva E, Lee JH, Tokuhiro S, Zou F, Bettens K, Sleegers K, Tan EK, Kimura R, Shibata N, Arai H, Kamboh MI, Prince JA, Maier W, Riemenschneider M, Owen M, Harold D, Hollingworth P, Cellini E, Sorbi S, Nacmias B, Takeda M, Pericak-Vance MA, Haines JL, Younkin S, Williams J, van Broeckhoven C, Farrer LA, St George-Hyslop PH, Mayeux R. 2011. Meta-analysis of the association between variants in SORL1 and Alzheimer disease. Arch. Neurol. 68:99–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Naj AC, Jun G, Beecham GW, Wang LS, Vardarajan BN, Buros J, Gallins PJ, Buxbaum JD, Jarvik GP, Crane PK, Larson EB, Bird TD, Boeve BF, Graff-Radford NR, De Jager PL, Evans D, Schneider JA, Carrasquillo MM, Ertekin-Taner N, Younkin SG, Cruchaga C, Kauwe JS, Nowotny P, Kramer P, Hardy J, Huentelman MJ, Myers AJ, Barmada MM, Demirci FY, Baldwin CT, Green RC, Rogaeva E, St George-Hyslop P, Arnold SE, Barber R, Beach T, Bigio EH, Bowen JD, Boxer A, Burke JR, Cairns NJ, Carlson CS, Carney RM, Carroll SL, Chui HC, Clark DG, Corneveaux J, Cotman CW, Cummings JL, DeCarli C, DeKosky ST, Diaz-Arrastia R, Dick M, Dickson DW, Ellis WG, Faber KM, Fallon KB, Farlow MR, Ferris S, Frosch MP, Galasko DR, Ganguli M, Gearing M, Geschwind DH, Ghetti B, Gilbert JR, Gilman S, Giordani B, Glass JD, Growdon JH, Hamilton RL, Harrell LE, Head E, Honig LS, Hulette CM, Hyman BT, Jicha GA, Jin LW, Johnson N, Karlawish J, Karydas A, Kaye JA, Kim R, Koo EH, Kowall NW, Lah JJ, Levey AI, Lieberman AP, Lopez OL, Mack WJ, Marson DC, Martiniuk F, Mash DC, Masliah E, McCormick WC, McCurry SM, McDavid AN, McKee AC, Mesulam M, Miller BL, Miller CA, Miller JW, Parisi JE, Perl DP, Peskind E, Petersen RC, Poon WW, Quinn JF, Rajbhandary RA, Raskind M, Reisberg B, Ringman JM, Roberson ED, Rosenberg RN, Sano M, Schneider LS, Seeley W, Shelanski ML, Slifer MA, Smith CD, Sonnen JA, Spina S, Stern RA, Tanzi RE, Trojanowski JQ, Troncoso JC, Van Deerlin VM, Vinters HV, Vonsattel JP, Weintraub S, Welsh-Bohmer KA, Williamson J, Woltjer RL, Cantwell LB, Dombroski BA, Beekly D, Lunetta KL, Martin ER, Kamboh MI, Saykin AJ, Reiman EM, Bennett DA, Morris JC, Montine TJ, Goate AM, Blacker D, Tsuang DW, Hakonarson H, Kukull WA, Foroud TM, Haines JL, Mayeux R, Pericak-Vance MA, Farrer LA, Schellenberg GD. 2011. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer's disease. Nat. Genet. 43:436–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Scott GK, Fei H, Thomas L, Medigeshi GR, Thomas G. 2006. A PACS-1, GGA3 and CK2 complex regulates CI-MPR trafficking. EMBO J. 25:4423–4435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wan L, Molloy SS, Thomas L, Liu G, Xiang Y, Rybak SL, Thomas G. 1998. PACS-1 defines a novel gene family of cytosolic sorting proteins required for trans-Golgi network localization. Cell 94:205–216 [DOI] [PubMed] [Google Scholar]
- 16.Nielsen MS, Gustafsen C, Madsen P, Nyengaard JR, Hermey G, Bakke O, Mari M, Schu P, Pohlmann R, Dennes A, Petersen CM. 2007. Sorting by the cytoplasmic domain of the amyloid precursor protein binding receptor SorLA. Mol. Cell. Biol. 27:6842–6851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kapuria V, Peterson LF, Fang D, Bornmann WG, Talpaz M, Donato NJ. 2010. Deubiquitinase inhibition by small-molecule WP1130 triggers aggresome formation and tumor cell apoptosis. Cancer Res. 70:9265–9276 [DOI] [PubMed] [Google Scholar]
- 18.Hohenstein P, Slight J, Ozdemir DD, Burn SF, Berry R, Hastie ND. 2008. High-efficiency Rosa26 knock-in vector construction for Cre-regulated overexpression and RNAi. Pathogenetics 1:3. 10.1186/1755-8417-1-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, Guillozet-Bongaarts A, Ohno M, Disterhoft J, Van Eldik L, Berry R, Vassar R. 2006. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer's disease mutations: potential factors in amyloid plaque formation. J. Neurosci. 26:10129–10140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Crump CM, Xiang Y, Thomas L, Gu F, Austin C, Tooze SA, Thomas G. 2001. PACS-1 binding to adaptors is required for acidic cluster motif-mediated protein traffic. EMBO J. 20:2191–2201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schmidt V, Baum K, Lao A, Rateitschak K, Schmitz Y, Teichmann A, Wiesner B, Petersen CM, Nykjaer A, Wolf J, Wolkenhauer O, Willnow TE. 2012. Quantitative modelling of amyloidogenic processing and its influence by SORLA in Alzheimer's disease. EMBO J. 31:187–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roshy S, Sloane BF, Moin K. 2003. Pericellular cathepsin B and malignant progression. Cancer Metastasis Rev. 22:271–286 [DOI] [PubMed] [Google Scholar]
- 23.Mueller-Steiner S, Zhou Y, Arai H, Roberson ED, Sun B, Chen J, Wang X, Yu G, Esposito L, Mucke L, Gan L. 2006. Antiamyloidogenic and neuroprotective functions of cathepsin B: implications for Alzheimer's disease. Neuron 51:703–714 [DOI] [PubMed] [Google Scholar]
- 24.Sun B, Zhou Y, Halabisky B, Lo I, Cho SH, Mueller-Steiner S, Devidze N, Wang X, Grubb A, Gan L. 2008. Cystatin C-cathepsin B axis regulates amyloid beta levels and associated neuronal deficits in an animal model of Alzheimer's disease. Neuron 60:247–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang C, Sun B, Zhou Y, Grubb A, Gan L. 2012. Cathepsin B degrades amyloid-beta in mice expressing wild-type human amyloid precursor protein. J. Biol. Chem. 287:39834–39841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Willnow TE, Carlo AS, Rohe M, Schmidt V. 2010. SORLA/SORL1, a neuronal sorting receptor implicated in Alzheimer's disease. Rev. Neurosci. 21:315–329 [DOI] [PubMed] [Google Scholar]
- 27.Jacobsen L, Madsen P, Nielsen MS, Geraerts WP, Gliemann J, Smit AB, Petersen CM. 2002. The sorLA cytoplasmic domain interacts with GGA1 and -2 and defines minimum requirements for GGA binding. FEBS Lett. 511:155–158 [DOI] [PubMed] [Google Scholar]
- 28.Herskowitz JH, Offe K, Deshpande A, Kahn RA, Levey AI, Lah JJ. 2012. GGA1-mediated endocytic traffic of LR11/SorLA alters APP intracellular distribution and amyloid-beta production. Mol. Biol. Cell 23:2645–2657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Seaman MN. 2007. Identification of a novel conserved sorting motif required for retromer-mediated endosome-to-TGN retrieval. J. Cell Sci. 120:2378–2389 [DOI] [PubMed] [Google Scholar]
- 30.Seaman MN. 2004. Cargo-selective endosomal sorting for retrieval to the Golgi requires retromer. J. Cell Biol. 165:111–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fjorback AW, Seaman M, Gustafsen C, Mehmedbasic A, Gokool S, Wu C, Militz D, Schmidt V, Madsen P, Nyengaard JR, Willnow TE, Christensen EI, Mobley WB, Nykjaer A, Andersen OM. 2012. Retromer binds the FANSHY sorting motif in SorLA to regulate amyloid precursor protein sorting and processing. J. Neurosci. 32:1467–1480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Muhammad A, Flores I, Zhang H, Yu R, Staniszewski A, Planel E, Herman M, Ho L, Kreber R, Honig LS, Ganetzky B, Duff K, Arancio O, Small SA. 2008. Retromer deficiency observed in Alzheimer's disease causes hippocampal dysfunction, neurodegeneration, and Abeta accumulation. Proc. Natl. Acad. Sci. U. S. A. 105:7327–7332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lorenzo K, Ton P, Clark JL, Coulibaly S, Mach L. 2000. Invasive properties of murine squamous carcinoma cells: secretion of matrix-degrading cathepsins is attributable to a deficiency in the mannose 6-phosphate/insulin-like growth factor II receptor. Cancer Res. 60:4070–4076 [PubMed] [Google Scholar]