

Abstract
Cdc42 is a Ras-related GTPase that plays an important role in the regulation of a range of cellular functions, including cell migration, proliferation, and survival. Consistent with its critical functions in vitro, the inactivation of Cdc42 in mice has been shown to result in embryonic lethality at embryonic day 6.5 (E6.5) before blood vessel formation. To determine the role of Cdc42 in new blood vessel formation, we have generated vascular endothelial cell (EC)-specific Cdc42 knockout mice by crossing Cdc42flox/flox mice with Tie2-Cre mice. The deletion of Cdc42 in ECs caused embryonic lethality with vasculogenesis and angiogenesis defects. We observed that Cdc42 is critical for EC migration and survival but not for cell cycle progression. Moreover, we found that the inactivation of Cdc42 in ECs decreased the level of vascular endothelial growth factor receptor 2 (VEGFR2) protein on the EC surface and promoted the production of a 75-kDa membrane-associated C-terminal VEGFR2 fragment. Using cultured primary mouse ECs and human umbilical vein ECs, we have demonstrated that the deletion of Cdc42 increased ADAM17-mediated VEGFR2 shedding. Notably, inhibition of ADAM17 or overexpression of VEGFR2 can partially reverse Cdc42 deletion-induced EC apoptosis. These data indicate that Cdc42 is essential for VEGFR2-mediated signal transduction in blood vessel formation.
INTRODUCTION
Aberrant blood vessel formation induces embryonic lethality as well as contributing to the pathogenesis of many human diseases, including coronary heart disease, diabetes, and cancer (1–3). Blood vessels can form by vasculogenesis, in which a primitive vascular network is formed by pluripotent mesenchymal progenitor differentiation and proliferation in situ in avascular tissue, or by angiogenesis, which refers to the growth of new blood vessels from preexisting vessels (4–6). Vascular endothelial cells (ECs) line the whole vascular system and play central roles in angiogenesis and vasculogenesis. Many EC surface receptors and their ligands have been found to play essential roles in the formation of new blood vessels; these receptors include the vascular endothelial growth factor receptor (VEGFR), Tie1 (tyrosine kinase with immunoglobulin-like and EGF-like domains 1) and Tie2 receptors, Eph receptors, integrins, and Notch (7–9). However, there is still much to learn regarding the intracellular signal transduction mechanisms involved in angiogenesis and vasculogenesis.
VEGF plays a key role in regulating angiogenesis and vasculogenesis in both embryogenesis and pathogenesis in human diseases, such as cancer metastasis (2, 6). After binding to its major receptor, VEGFR2, in ECs, VEGF induces the dimerization of VEGFR2 and activates many different signal transduction pathways. Rho GTPase-mediated signal transduction is one of the pathways activated by VEGFR2. Cdc42 is a Rho GTPase family member that cycles between an inactive GDP-bound state and an active GTP-bound state in response to extracellular stimuli (10, 11). VEGF stimulation induces time-dependent activation of Cdc42 in human umbilical vein endothelial cells (HUVECs) (4, 12, 13). EC morphogenesis, including vacuole and lumen formation, is important for angiogenesis. A series of seminal in vitro studies has demonstrated that Cdc42 and its downstream effectors, including p21-activated kinase 2 (PAK2), PAK4, partitioning-defective 3 homolog (Par3), and Par6, are crucial for EC morphogenesis (14, 15). Overexpression of either constitutively active Cdc42 or dominant negative Cdc42 by use of a recombinant adenovirus (Ad) has been shown to inhibit EC vacuole formation in experiments utilizing a 3-dimensional extracellular matrix, suggesting that proper cycling of Cdc42 between its GDP- and GTP-bound states is required for EC morphogenesis and angiogenesis (16). A recent study using cultured mouse embryonic stem cells also demonstrated the importance of Cdc42 for vasculogenesis through its downstream effectors protein kinase C and glycogen synthase kinase-3β (17).
Accumulating evidence indicates that Cdc42 plays an important role in EC function and vascular development (13, 18–22); however, far less is known about the in vivo functions of Cdc42 in blood vessel formation during embryonic development. Mice with a total knockout of Cdc42 die before embryonic day 6.5 (E6.5) (23), which limits the usefulness of this mouse model in studying the role of Cdc42 in the later stages of embryonic development and in adulthood. In this study, we used a conditional Cdc42 knockout mouse model to examine these critical issues. The mouse mutant, in which the Cdc42 locus was modified by adding 2 flanking loxP sites (24), was crossed with Tie2-Cre transgenic mice that expressed Cre recombinase in their ECs (25, 26). Our results revealed that Cdc42 is essential for vasculogenesis during embryonic development. Cdc42 deletion reduced the survival and migration of ECs, leading to defects in blood vessel formation. The upregulation of disintegrin and metalloprotease 17 (ADAM17)-mediated VEGFR2 shedding, reducing the density of VEGFR2 on the cell surface, is an underlying molecular mechanism for the vascular defects in Cdc42 knockout embryos.
MATERIALS AND METHODS
Generation of Cdc42 EC-specific knockout mice.
Cdc42flox/flox mice were generated by inserting two LoxP sites to flank exon 2 of the Cdc42 gene (24). Cdc42 EC-specific knockout mice were created by crossing Cdc42flox/flox mice with Tie2-Cre mice (mixed C57BL/6 × S129/S4 background) (24–26). The deletion of exon 2 upon Cre-mediated recombination results in a truncated small peptide that lacks the majority of the Cdc42 amino acid residues. All study protocols were approved by the Institutional Animal Care and Use Committee of the Texas A&M Health Science Center and conform to the NIH Guide for the Care and Use of Laboratory Animals (27).
siRNA transfection.
HUVECs (2 × 105/well) were plated in 6-well plates and were incubated with various small interfering RNAs (siRNAs) (20 nM) and HiPerFect transfection reagent (Qiagen) for 72 h, according to the manufacturer's instructions. Subsequently, HUVECs were used for tube formation, bromodeoxyuridine (BrdU) incorporation, or biotinylation assays, and aliquots of cell lysates were blotted to confirm the efficiency of RNA interference (RNAi).
Generation of a VEGFR2-expressing adenovirus.
Hemagglutinin (HA)-tagged wild-type VEGFR2 was released from the pKH3 vector by XhoI and SalI restriction enzymes and was then subcloned into the pAdTrack-CMV vector. After homologous recombination, the AdEasy-1 vector (Stratagene), which contains HA-tagged wild-type VEGFR2, was transfected into Ad-293 cells for virus packaging. Ten days later, adenovirus was harvested from cell lysates and was stored in a −80°C freezer for future use (28, 29).
Tube formation assay.
HUVECs transfected with siRNA or infected with adenoviruses were plated on 24-well plates coated with a thin layer of Matrigel (BD Biosciences) at 5 × 104 cells/well in EBM-2 medium (Lonza). HUVECs were then cultured for 18 h in a 37°C humidified CO2 incubator to form tube structures. The tube structures were photographed under a microscope with a charge-coupled device (CCD) camera, and the lengths of the tubes were determined as described previously (30).
Isolation and culture of primary ECs of mice.
ECs were isolated from neonatal Cdc42flox/flox mice by using magnetic beads (Invitrogen) coupled with rat anti-mouse platelet endothelial cell adhesion molecule 1 (anti-PECAM-1) (BD Biosciences), as described previously (31, 32). ECs were cultured in EBM-2 endothelial cell medium. The purity of the ECs was confirmed by identifying EC markers (vascular endothelial cadherin [VE-cadherin]) using flow cytometry.
Flow cytometric analysis.
Confluent cultured ECs were detached using 0.05% EDTA and were washed with phosphate-buffered saline (PBS). Subsequently, ECs were fixed in 4% paraformaldehyde (PFA) and were incubated with normal rabbit IgG or an antibody against VE-cadherin (Enzo). Cells were washed, stained with Alexa Fluor 488-conjugated goat anti-rabbit IgG (Invitrogen), and analyzed using a BD Biosciences FACSCalibur flow cytometer. For analysis of apoptosis, HUVECs were treated with scrambled siRNA, Cdc42 siRNA, or Cdc42 siRNA plus ADAM17 siRNA. The siRNA-treated HUVECs were stained with annexin V by using a fluorescein isothiocyanate (FITC)-annexin V staining kit (Invitrogen) and were then analyzed with a BD Biosciences FACSCalibur flow cytometer.
X-Gal and PECAM-1 whole-mount staining.
Isolated embryos were fixed in 4% PFA in PBS and were then stained with 1 mg/ml 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal) overnight at room temperature as described previously (28). For PECAM-1 whole-mount staining, the 4% PFA-fixed embryos and yolk sacs were dehydrated by methanol, and the endogenous peroxidases were quenched with 5% H2O2 in methanol for 4 to 5 h. After rehydration in methanol and PBS, samples were blocked in 3% nonfat dry milk with 0.1% Triton X-100 in PBS. The embryos were then incubated with anti-PECAM-1 (1:100) antibodies (BD Biosciences) at 4°C overnight, followed by staining with horseradish peroxidase (HRP)-conjugated secondary antibodies.
TUNEL assay.
Embryo sections or isolated ECs infected with Ad-LacZ, or with Ad-Cre with or without TAPI-1 treatment, were examined for apoptosis by a terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end-labeling (TUNEL) assay by using the In Situ cell death detection kit (Roche) according to the product instructions, as described previously (33).
Real-time PCR.
Cultured mouse primary ECs were infected with Ad-LacZ or Ad-Cre. Three days later, total RNA was isolated by using an RNeasy kit (Qiagen) according to the manufacturer's instructions. The samples were then reverse transcribed to cDNA, which was assayed by real-time PCR experiments using SYBR green master mix (Applied Biosystems), and the results were analyzed using the 7900HT Fast real-time PCR system (Applied Biosystems).
Cell surface biotinylation and internalization assay.
Cell surface protein biotinylation and internalization assays were performed essentially as described previously (34). In brief, isolated mouse ECs or HUVECs were cultured in 6-well plates and were incubated with 0.5 mg/ml of EZ-Link NHS-SS-biotin (Pierce) in PBS for 60 min at 4°C. Whole-cell lysates were precipitated with streptavidin-conjugated agarose beads, and the level of VEGFR2 on the cell surface was assessed by Western blotting with an anti-VEGFR2 antibody. For the VEGFR2 internalization assay, biotin-labeled cells were incubated at 37°C with 50 ng/ml VEGF in Dulbecco's modified Eagle's medium (DMEM) for 5, 10, or 20 min. The biotin tags from proteins remaining on the cell surface were removed by washing twice with glutathione. The whole-cell lysates were precipitated with streptavidin-agarose beads and were analyzed by Western blotting with an anti-VEGFR2 antibody.
Boyden chamber cell migration assay.
Cell migration assays were performed in Boyden chambers (Neuro Probe), as described previously (25). A total of 1 × 104 cells in 200 μl serum-free DMEM were added to each upper well, and either 1 ng/ml VEGF in DMEM or DMEM alone as a control was added to the bottom wells. Cells were incubated for 6 h in a 37°C humidified CO2 incubator. The cells remaining in the upper wells were removed by gently rubbing the upper membrane surface. Cells migrating to the lower surface of the membrane were fixed with methanol and were stained with modified Giemsa stain (Sigma-Aldrich).
Membrane fractionation assay.
The membrane and cytosol fractions of HUVECs were separated as described previously (35). Briefly, HUVECs (∼2 × 105/well) were seeded in a 6-well plate and were treated with various siRNAs for 72 h. HUVECs were permeabilized with digitonin (20 μg/ml) in 200 μl HEPES buffer and were kept at 25°C for 5 min and on ice for an additional 30 min (releasing cytosolic proteins without solubilizing membrane proteins). The supernatant was collected as the cytosolic fraction. Subsequently, the pellet of HUVECs was lysed using radioimmunoprecipitation assay (RIPA) lysis buffer (Millipore) and was centrifuged at 13,000 rpm for 15 min at 4°C to remove the nuclear fraction. The supernatant contained the membrane fraction.
Statistical analysis.
Data are presented as means ± standard errors (SE). Means were compared by a 2-tailed Student t test, or by 1-way analysis of variance (ANOVA) for the comparison of multiple groups. A P value of ≤0.05 was considered statistically significant.
RESULTS
Specific deletion of Cdc42 in vascular endothelial cells causes embryonic lethality.
To explore the role of Cdc42 in angiogenesis and vasculogenesis in vivo, we generated vascular EC-specific Cdc42 knockout mice by crossing Cdc42flox/flox mice with Tie2-Cre mice, in which the expression of Cre recombinase was driven by the EC-specific Tie2 promoter and enhancer (25, 26, 33). Of the 148 viable offspring, we obtained 50 Cdc42flox/flox pups (designated controls), 51 Tie2-Cre+; Cdc42flox/+ pups, and 47 Cdc42flox/+ pups. The lack of viable Tie2-Cre+; Cdc42flox/flox Cdc42 EC knockout (CEKO) mice strongly suggests that the inactivation of Cdc42 in ECs results in a recessive lethal phenotype. By immunofluorescence staining, we verified that Cdc42 was specifically deleted in the ECs of E9.5 embryos (Fig. 1). Specifically, CEKO (Fig. 1B, D, and F) and control (Fig. 1A, C, and E) embryos were sectioned and double-stained with antibodies against Cdc42 and platelet endothelial cell adhesion molecule 1 (PECAM-1), a marker for vascular ECs. PECAM-1-positive cells were detected in control (Fig. 1C and E) and CEKO (Fig. 1D and F) embryos. However, Cdc42 was not expressed in the PECAM-1-positive cells of CEKO embryos (Fig. 1B and F), in contrast to the controls (Fig. 1A and E).
Fig 1.
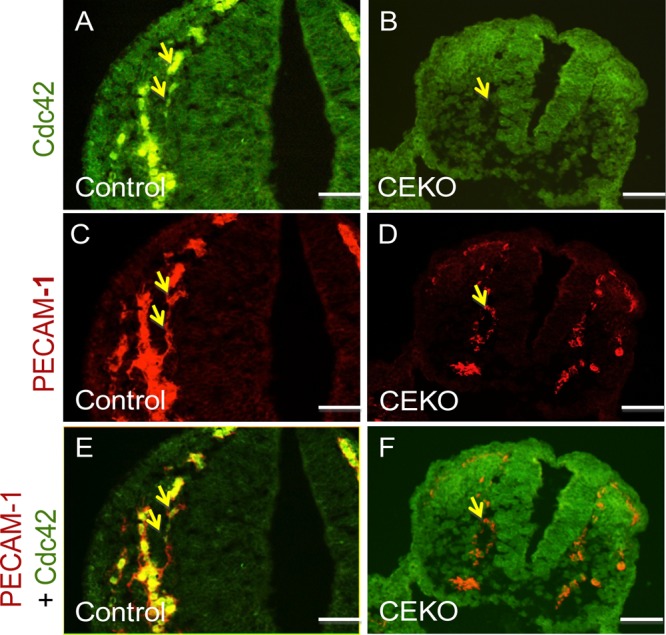
Specific deletion of Cdc42 in vascular ECs of CEKO embryos. Sections from E9.5 control (A, C, and E) and CEKO (B, D, and F) embryos were stained with antibodies against Cdc42 (A and B) and PECAM-1 (C and D). (A and B) Cdc42 expression was detected in the ECs of control embryos (A) but not in those of CEKO embryos (B). (C and D) PECAM-1-positive ECs were present in both control (C) and CEKO (D) embryos. (E and F) Merged images. Cdc42 was colocalized with PECAM-1 in control (E) but not in CEKO (F) embryo sections. Arrows indicate blood vessels. Bars, 25 μm.
To determine the time at which the effects of EC Cdc42 deficiency occurred, we examined embryos from Cdc42flox/flox female mice with timed pregnancies who had been mated with Tie2-Cre+; Cdc42flox/+ male mice. In contrast to the Cdc42 total-knockout embryos, which died before E6.5, no overtly abnormal phenotype (embryo size and stage) was seen in CEKO embryos at E8.5, compared with their littermate controls (compare Fig. 2A and B). However, by E9.5, more than half of the CEKO embryos showed progressively retarded growth (Fig. 2D) and had smaller bodies than their littermate controls (Fig. 2C). In addition, heart looping was delayed in the CEKO embryos, and the ventricular wall was transparent (Fig. 2D). The CEKO embryos failed to survive beyond E10.5.
Fig 2.

Deletion of Cdc42 in vascular ECs caused vasculogenesis defects in the yolk sac. (A through D) Gross examination of E8.5 control (A) and CEKO (B) embryos showed similar morphologies. Side views of E9.5 control (C) and CEKO (D) embryos revealed that CEKO embryos had smaller bodies. (E and F) The control E9.5 yolk sac presented highly organized vasculature (arrows) (E), but only the blood islands were evident in the yolk sacs of CEKO embryos (F). (G and H) Whole-mount staining with PECAM-1 performed on the yolk sacs of control (G) and CEKO (H) embryos. (I and J) Hematoxylin- and eosin-stained sections of yolk sacs from control (I) and CEKO (J) embryos. Arrows indicate blood vessels in the yolk sacs. Bars, 25 μm.
Vasculogenesis defects in the CEKO yolk sac and embryo.
The yolk sac is the first extraembryonic vasculogenesis site and undergoes extensive angiogenesis during E8.5 to E10.5 (36). The vessels of control E9.5 yolk sacs were highly organized, with branching vitelline vessels (Fig. 2E). In CEKO yolk sacs, the vessels were nearly completely absent; only the blood islands were evident (Fig. 2F). To visualize the vascular ECs and confirm the vasculogenesis defects in CEKO yolk sacs, whole-mount PECAM-1 staining was performed. PECAM-1 staining of E9.5 control yolk sacs showed an extensive honeycomb-like network of capillaries (Fig. 2G), while no blood vessel formation was evident in CEKO yolk sacs (Fig. 2H). In agreement with the whole-mount staining observation, histological analysis revealed that the control E9.5 yolk sac contained a series of blood vessels that attached to the undersurface of the mesodermal layer of the yolk sac (Fig. 2I). In contrast, there was a lack of obvious blood vessels in the CEKO yolk sac (Fig. 2J). These data suggest that Cdc42 plays an indispensable role in vasculogenesis in the yolk sac.
To define the role of Cdc42 in embryonic blood vessel formation, we performed whole-mount PECAM-1 and LacZ staining to visualize the morphology of the blood vessels. PECAM-1 immunofluorescence staining experiments showed well-formed intersomitic blood vessels in control embryos (Fig. 3A) but not in CEKO embryos (Fig. 3B). In addition, newly formed blood vessels were detected in the hearts of control embryos (Fig. 3C) but not in those of Tie2-Cre+; Cdc42flox/+ embryos (Fig. 3D). Moreover, PECAM-1 whole-mount staining revealed normal vascular networks in the brains of E9.5 control embryos (Fig. 4A and C). However, the brain vascular network was disrupted in CEKO embryos (Fig. 4B and D). To exclude the possibility that the inactivation of Cdc42 impairs EC differentiation and affects the expression of PECAM-1, we performed an alternative experiment using LacZ staining to visualize blood vessels. We introduced a floxed LacZ transgene into CEKO embryos by crossing Tie2-Cre+; Cdc42flox/+ mice with Rosa26 mice. X-Gal staining showed ECs lining the blood vessels in control embryos (Fig. 4E), while blood vessel formation in the CEKO mutant embryos was totally disrupted (Fig. 4F). Further histological analysis of controls revealed that a flat, elongated layer of ECs formed a patent vascular lumen (Fig. 4G). In contrast, the ECs in CEKO embryos exhibited a cuboidal epithelial morphology and mirrored the phenotype of integrin β1 EC knockout mice (Fig. 4H) (37). Moreover, the ECs in CEKO embryos failed to connect to each other, indicating that Cdc42 deletion impaired vascular integrity (Fig. 4I).
Fig 3.

Inactivation of Cdc42 impaired blood vessel formation in the trunk region and the heart. Whole-mount staining with PECAM-1 was performed on E9.5 control (A and C) and CEKO (B and D) embryos. Arrows indicate blood vessels in the trunks (A) and hearts (C) of control embryos. No obvious blood vessels were detected in the trunks (B) and hearts (D) of CEKO embryos. Bars, 25 μm.
Fig 4.

Blood vessel formation defects in CEKO embryos. (A through D) PECAM-1 whole-mount staining of E9.5 control (A) and CEKO (B) embryos. The head region in the control (C) and CEKO (D) embryos was examined at a higher magnification. Red arrows indicate the organized intersomitic blood vessels (A), and white arrows indicate the branched vessels in the brain (C). (E and F) X-Gal staining showed well-organized vessels in control embryos (arrow) (E), whereas blood vessels were absent from CEKO embryos (F). (G through I) PECAM-1 immunofluorescence staining of cross sections of control (G) and CEKO (H and I) embryos. A flat and elongated layer of ECs formed a patent vascular lumen in control embryos (white arrows) (G), but the ECs in CEKO embryos showed a cuboidal epithelial morphology (yellow arrow) (H) and failed to connect to each other (white arrows) (I). Bars, 25 μm.
Inhibition of Cdc42 in HUVECs disrupted tube formation.
Blood flow-induced shear stress has emerged as one of the most critical factors for vascular formation in chicken and mouse yolk sacs (38, 39). To exclude the influence of shear stress and determine the physiological functions of Cdc42 in capillary morphogenesis, we performed an assay of tube formation, a process mimicking sprouting during angiogenesis (30). HUVECs infected with a control adenovirus differentiated into well-defined tube-like structures (Fig. 5A), whereas the overexpression of a HA-tagged Cdc42-T17N mutant (Fig. 5F), which serves as a dominant negative mutant by preventing Cdc42 from binding to GTP, disrupted EC tube formation (Fig. 5B). To explore the endogenous physiological functions of Cdc42 in tube formation, we performed an RNAi experiment with cultured HUVECs. We have screened three siRNA fragments targeting Cdc42 and found fragment 15 to be an efficient inhibitor of Cdc42 expression, so this fragment was used for the following experiments (Fig. 5H). HUVECs transfected with scrambled siRNA differentiated into tube-like structures (Fig. 5C); however, tube formation was impaired when Cdc42 expression was knocked down by siRNA treatment (Fig. 5D). Quantitative analysis confirmed that the average lengths of the tubes in HUVECs overexpressing Cdc42-T17N were significantly decreased from those in control cells (Fig. 5E), and in HUVECs where Cdc42 was knocked down, the tube length was reduced by ∼40% from that in cells transfected with scrambled siRNA (Fig. 5G). These data suggest that Cdc42 is necessary for tube formation.
Fig 5.

Interference with Cdc42 function in HUVECs disrupted tube formation. (A through D) Representative images showing tube formation in HUVECs infected/transfected with a control green fluorescent protein (GFP)-tagged adenovirus (A), an adenovirus expressing HA-tagged dominant negative Cdc42-T17N (B), scrambled siRNA (C), or Cdc42-specific siRNA (D). (E) Quantitative analysis of tube length in HUVECs infected with a control GFP-tagged adenovirus or the HA-tagged Cdc42-T17N mutant adenovirus. *, P < 0.05. (F) Western blotting with anti-HA antibodies was carried out to confirm the overexpression of exogenous Cdc42. (G) Quantitative analysis of tube length in HUVECs transfected with scrambled or Cdc42-specific siRNA. (H) Western blotting showed that Cdc42 expression was lower in cells treated with Cdc42 siRNA than in control cells. Vinculin was utilized as a loading control.
Cdc42 is critical for EC survival and migration.
The coordinated regulation of EC proliferation, migration, and apoptosis is critical for the formation of functional blood vessels. To better understand the underlying mechanism by which Cdc42 regulates new blood vessel formation, we performed histological analysis of E9.5 CEKO and control embryos. Hematoxylin and eosin staining revealed dramatic increases in the numbers of pyknotic and fragmented nuclei, both of which are signs of apoptosis, as an outcome of Cdc42 deletion (Fig. 6B and D). No apoptotic cells were evident in control embryos (Fig. 6A and C). It is not clear whether the apoptotic cells were ECs (due to the inactivation of Cdc42) or non-ECs. To examine the role of Cdc42 in controlling EC survival, we performed TUNEL assays and PECAM-1 double staining on CEKO (Fig. 7B, D, and F) and control (Fig. 7A, C, and E) embryos. In agreement with the histology results, Cdc42 inactivation induced massive apoptosis, with increased numbers of apoptotic ECs in CEKO embryos (Fig. 7B). Interestingly, many of the apoptotic cells were negative for PECAM-1 staining (Fig. 7F). It has been reported that Cre recombinase, the expression of which was driven by the Tie2 promoter and enhancer, was expressed not only in vascular ECs but also in non-ECs, such as hematopoietic cells (26). It is possible that the PECAM-1-negative apoptotic cells resulted from the deletion of Cdc42 in hematopoietic cells. Another possibility is that the apoptotic ECs and non-ECs we detected in CEKO embryos were caused by a secondary effect due to dysfunctional blood vessels. To address this question, we isolated ECs from the hearts of neonatal Cdc42flox/flox mice. Flow cytometric analysis showed that the percentage of cells positive for VE-cadherin (a marker of EC) was more than 80% (data not shown). The cultured Cdc42flox/flox ECs were infected with recombinant adenoviruses encoding Cre recombinase or LacZ as a control. The Cdc42 expression level was significantly decreased after 48 h in ECs infected with Ad-Cre, and this effect was maintained as long as 96 h after virus inoculation (data not shown). In agreement with the in vivo results, levels of cleaved caspase 3, a marker for apoptosis, were increased in Cdc42-null ECs, and this effect was reversed by the reexpression of wild-type Cdc42 (Fig. 7G, H, and I). These results suggest that Cdc42 is critical for EC survival during blood vessel formation.
Fig 6.

Deletion of Cdc42 induced massive apoptosis during embryogenesis. (A and B) Hematoxylin and eosin staining analyses were performed on E9.5 control (A) and CEKO (B) embryos. Bars, 200 μm. (C and D) Higher-magnification microphotographs of control (C) and CEKO (D) embryos. Arrowheads (D) indicate massive pyknotic and fragmented nuclei. Bars, 25 μm.
Fig 7.

Inactivation of Cdc42 caused EC apoptosis. (A through F) TUNEL assays (A and B) and PECAM-1 staining (C and D) were performed on E9.5 control (A, C, and E) and CEKO (B, D, and F) embryos. (E and F) Merged images. Bars, 25 μm. (G) Cdc42flox/flox ECs were infected by Ad-LacZ or by Ad-Cre with or without HA-tagged wild-type Cdc42. Cell lysates were analyzed by Western blotting using various antibodies, as indicated. (H and I) The relative intensities of cleaved caspase 3 (H) and Cdc42 (I) were analyzed quantitatively for three independent experiments. *, P < 0.05.
EC proliferation and migration are essential steps for vasculogenesis and angiogenesis. To determine the role of Cdc42 in EC proliferation, we stained embryos with phosphorylated histone H3 (pH3), a marker for proliferation. We did not detect any significant differences between the levels of pH3-positive cells in CEKO and control embryos (Fig. 8). In addition, we also performed BrdU incorporation experiments on cultured Cdc42 knockdown (Fig. 9D, E, and F) and control (Fig. 9A, B, and C) HUVECs. Consistently, we observed that the inactivation of Cdc42 had no significant effect on cell cycle progression induced by fetal bovine serum (Fig. 9G), suggesting that Cdc42 does not play a role in EC proliferation in angiogenesis during embryo development.
Fig 8.

Deletion of Cdc42 has no effect on EC proliferation in vivo. (A through F) Sections from E9.5 control (A, C, and E) and CEKO (B, D, and F) embryos were stained with antibodies against pH3 (A and B) and PECAM-1 (C and D). The numbers of pH3 and PECAM-1 double-positive cells in control (E) and CEKO (F) embryos are comparable. Bars, 25 μm.
Fig 9.

Inactivation of Cdc42 did not influence the cell cycle progression of HUVECs but impaired EC migration. (A through F) Cultured HUVECs were transfected with scrambled siRNA (A, B, and C) or Cdc42 siRNA (D, E, and F). Three days later, BrdU was added and was incubated for 18 h. BrdU (A and D) and VE-cadherin (B and E) staining was then performed. (C and F) Merged images. Bars, 50 μm. (G) Quantitative analysis of BrdU incorporation shows no significant difference between control and Cdc42 knockdown cells with or without fetal bovine serum (FBS) stimulation. (H) Cultured Cdc42flox/flox ECs were infected with Ad-LacZ, Ad-Cre, or Ad-Cre plus Ad-Cdc42 (wild type). The infected ECs were subjected to Boyden chamber analysis in response to VEGF (1 ng/ml). *, P < 0.05.
To determine the function of Cdc42 in EC migration, we performed Boyden chamber analysis. Figure 9H shows that Cdc42 deletion significantly decreased the level of EC migration from that for the control. To further confirm the importance of Cdc42 in EC migration, we performed a rescue experiment by overexpressing wild-type Cdc42. As expected, overexpression of the wild-type Cdc42 rescued EC migration defects.
Deletion of Cdc42 in ECs decreased the level of VEGFR2 protein on the plasma membrane.
Cdc42 plays critical roles in regulating protein hemostasis, including gene transcription, protein trafficking, endocytosis, and degradation (10, 40, 41). To determine the molecular mechanisms of Cdc42 in regulating blood vessel formation, we first isolated ECs from Cdc42flox/flox mouse hearts and then infected them with Ad-Cre or Ad-LacZ. We performed Western blotting to examine the effect of deletion of Cdc42 on plasma membrane receptors that are important for new blood vessel formation. We observed that the deletion of Cdc42 had no significant influence on PECAM-1, VE-cadherin, and VEGFR1 protein levels but significantly decreased the VEGFR2 level (Fig. 10A and B). To confirm the Western blotting result, we performed VEGFR2 and PECAM-1 double staining in CEKO (Fig. 10E and F) and control (Fig. 10C and D) E9.5 embryos. In agreement with data presented above (Fig. 4H and I), Cdc42 inactivation had no effect on EC PECAM-1 staining in CEKO embryos (Fig. 10F) compared to the results from the controls (Fig. 10D). However, we found that the level of VEGFR2 staining in the CEKO embryos was significantly decreased (Fig. 10E) from that in control embryos (Fig. 10C). Due to the importance of VEGFR2 in blood vessel formation, these data strongly suggest that impaired VEGFR2-mediated signal transduction is likely one of the underlying reasons for EC apoptosis and vasculogenesis defects in CEKO embryos.
Fig 10.

Deletion of Cdc42 in ECs decreased the VEGFR2 protein level. (A) Primary ECs isolated from Cdc42flox/flox mouse hearts were infected by Ad-Cre or by Ad-LacZ as a control. Whole-cell lysates were prepared and were blotted with antibodies against VEGFR1, VEGFR2, VE-cadherin, PECAM-1, Cdc42, and vinculin, as indicated. (B) Quantitative analysis shows that the VEGFR2 level was significantly decreased in Cdc42-null ECs. (C through F) Sections of E9.5 control (C and D) and CEKO (E and F) embryos were stained by antibodies against VEGFR2 (green) and PECAM-1 (red). Blood vessels are indicated by arrows, and dashed lines indicate the boundary of the neural tube (NT). Bars, 25 μm. (G) The membrane surface proteins of primary mouse ECs were labeled with biotin and were pulled down using streptavidin-conjugated beads. The precipitated complex was then analyzed by Western blotting using anti-VEGFR2 antibodies. (H) The intensities of VEGFR2 75-kDa fragment bands from three independent experiments were quantitatively analyzed. *, P < 0.05.
In order to determine how Cdc42 influences VEGFR2 levels, we performed real-time PCR experiments to assess the effect of Cdc42 on VEGFR2 transcription. Our results showed that the mRNA levels of VEGFR2 in Cdc42-null ECs were comparable to those for the control (data not shown), suggesting that the regulation of VEGFR2 protein levels by Cdc42 is mediated through posttranscriptional mechanisms. Using a plasma membrane-impermeant biotin (EZ-Link NHS-SS-biotin; Pierce) probe, which can bind to lysine residues on cell surface proteins, we found that VEGFR2 levels on the plasma membrane surface were significantly lower in Cdc42-null mouse ECs (Ad-Cre) than in control cells (Ad-LacZ) (Fig. 10G and H).
To further determine how Cdc42 influences VEGFR2 levels, we knocked down Cdc42 in cultured HUVECs using siRNA against Cdc42 and performed a VEGFR2 internalization analysis. For this experiment, we first labeled cell surface proteins with biotin and then stimulated HUVECs with VEGF for the times indicated in Fig. 11A. Subsequently, the biotin tags from proteins remaining on the cell surface were removed by washing twice with glutathione. The internalized VEGFR2 fragments were precipitated by streptavidin-agarose beads and were detected by Western blotting with an anti-VEGFR2 antibody. We detected a significant increase in the amount of a 75-kDa fragment in Cdc42-knockdown HUVECs (Fig. 11A, right) over that in the controls (Fig. 11A, left). Because this 75-kDa fragment could be precipitated by streptavidin and recognized by an antibody against the C-terminal domain of VEGFR2, it likely contains the full transmembrane domain, the cytosolic domain, and a portion of the extracellular domain of VEGFR2. We also detected a 130-kDa band in HUVECs, recognized by the anti-VEGFR2 antibody; its expression was independent of the presence of Cdc42. However, this band was not present in mouse ECs (Fig. 10G). The inactivation of Cdc42 resulted in decreased expression of VEGFR2 on the cell surface and increased expression of a 75-kDa VEGFR2 fragment in the cytosol (Fig. 10G and 11A and B), suggesting that Cdc42 deletion might increase VEGFR2 shedding. To confirm that the 75-kDa fragment came from VEGFR2 shedding, we infected cultured HUVECs with an adenovirus that encodes C-terminally HA-tagged wild-type VEGFR2. Western blotting showed that the 75-kDa VEGFR2 fragment can be recognized by both anti-HA and anti-VEGFR2 antibodies (data not shown), suggesting that the 75-kDa fragment is a part of VEGFR2. Moreover, we overexpressed C-terminally HA- and His-tagged wild-type VEGFR2 in HEK 293 cells. We found that nickel beads or anti-HA antibody-conjugated agarose beads pulled down the 75-kDa VEGFR2 fragment (data not shown). These results further confirmed that the 75-kDa fragment is a part of VEGFR2. The level of cytosolic biotinylated full-length VEGFR2 in unstimulated Cdc42 knockdown HUVECs (incubated at 37°C for 20 min without VEGF stimulation) was higher than that in the controls (Fig. 11A), suggesting that the deletion of Cdc42 might enhance VEGFR2 autointernalization. Furthermore, we examined the effect of knocking down Cdc42 on VEGF-stimulated signal transduction. We found that Cdc42 inactivation can inhibit VEGF-induced Erk phosphorylation (Fig. 11C and D), supporting our hypothesis that the inactivation of Cdc42 interferes with VEGF signal transduction.
Fig 11.

Inactivation of Cdc42 promoted VEGFR2 shedding and impaired VEGF-induced Erk phosphorylation. (A) HUVECs were transfected with either scrambled siRNA (left) or Cdc42-targeted siRNA (right) and were then labeled with biotin. After treatment with 50 ng/ml VEGF from 0 to 20 min, the biotin tags on the cell surface were removed by l-glutathione (reduced), and the internalized biotin-labeled proteins were analyzed by Western blotting with anti-VEGFR2 antibodies. Full-length VEGFR2 (230 kDa) and the smaller VEGFR2 fragments detected (130 kDa and 75 kDa) are indicated by arrows. Cdc42 expression was assessed to confirm the efficiency of siRNA treatment against Cdc42, and vinculin was utilized as a loading control. (B) Quantitative analysis shows that Cdc42 knockdown significantly increased the production of the 75-kDa VEGFR2 fragment. *, P < 0.05. (C) HUVECs were transfected with scrambled siRNA or Cdc42 siRNA and were challenged with VEGF at the indicated time points. Whole-cell lysates were analyzed by Western blotting with specific antibodies against phosphorylated Erk (P-Erk) and total Erk (T-Erk). (D) Quantitative data show that Cdc42 knockdown significantly decreased the P-Erk level in response to VEGF stimulation at 15 min.
Cdc42 deletion increased ADAM17-mediated VEGFR2 shedding.
The ADAM family of proteases cleaves the ectodomains of growth factors and their receptors (42, 43). To determine whether ADAMs are involved in VEGFR2 shedding in Cdc42-depleted ECs, we added TAPI-1, an inhibitor of ADAMs (44), to HUVECs in which Cdc42 had been knocked down. We found that TAPI-1 decreased the level of production of the 75-kDa fragment with or without VEGF stimulation (Fig. 12A and B), suggesting that one or several ADAM family members are responsible for the production of this fragment. Consistently, we observed that TAPI-1 can reverse cell apoptosis induced by the deletion of Cdc42 in cultured primary mouse ECs (Fig. 12C to N). Among 27 ADAM family members, ADAM17 and ADAM10 have been reported to function as “sheddases” for VEGFR2 (45, 46). Using a more specific ADAM inhibitor (INCB3619) that attenuates the activity of ADAM10, ADAM17, and metalloproteases (MMPs) (47), we observed results similar to those obtained with TAPI-1 (data not shown). Furthermore, we examined the effects of TAPI-1 and INCB3619 on the production of cleaved caspase 3. We discovered that deletion of Cdc42 increased the production of cleaved caspase 3, suggesting that the level of EC apoptosis was increased. Notably, we found that both TAPI-1 and INCB3619 can inhibit the production of cleaved caspase 3 induced by the deletion of Cdc42 in ECs (Fig. 13A and B), indicating that ADAM10, ADAM17, and/or MMPs are very likely the candidate enzymes for VEGFR2 shedding.
Fig 12.

TAPI inhibited ADAM17-mediated VEGFR2 shedding and EC apoptosis induced by the inactivation of Cdc42. (A) HUVECs transfected with Cdc42 siRNA were either left untreated or treated with TAPI-1 (50 μM) for 4 h, and internalized VEGFR2 was then analyzed by Western blotting. Samples were also blotted with antibodies against Cdc42 and vinculin. (B) Quantitative analysis results show that TAPI treatment significantly decreased the level of production of 75-kDa VEGFR2 fragments. (C through N) Cultured Cdc42flox/flox ECs were infected either with Ad-LacZ (C through F), with Ad-Cre without further treatment (G through J), or with Ad-Cre plus TAPI-1 (50 μM) (K through N). TUNEL assays, PECAM-1 staining, and Hoechst staining for nuclei were then performed. Bars, 25 μm.
Fig 13.

Inhibition of ADAM17 decreased the production of the 75-kDa VEGFR2 fragment. (A) Cultured mouse Cdc42flox/flox ECs were infected with Ad-Cre or Ad-LacZ and were then incubated with or without TAPI-1 (50 μM) or INCB3619 (2 μM) for 4 h. Cell lysates were reacted with antibodies for cleaved caspase 3 (top), Cdc42 (center), or vinculin (bottom). (B) The intensities of bands from three independent experiments were quantitatively analyzed. *, P < 0.05. (C) HUVECs were either left untreated or treated with PMA (25 ng/ml) for 1 h and were then analyzed by Western blotting using antibodies against VEGFR2 and vinculin. (D) The intensities of bands from three independent experiments were quantitatively analyzed. (E) HUVECs transfected with Cdc42 siRNA, alone or together with ADAM17 siRNA, were incubated with or without VEGF (50 ng/ml) for 30 min and were then subjected to an internalization assay (top). The cell lysates from each sample were analyzed by Western blotting with antibodies against Cdc42, ADAM17, and vinculin, as indicated. (F) The relative intensities of 75-kDa VEGFR2 fragments were quantitatively analyzed. (G) HUVECs transfected with scrambled siRNA, Cdc42 siRNA alone, or Cdc42 siRNA together with ADAM17 siRNA. Cellular membrane and cytosol fractions were separated and were then blotted with various antibodies as indicated.
To identify which ADAM family member(s) is involved in the production of the 75-kDa VEGFR2 fragment, we stimulated HUVECs with phorbol-12-myristate-13-acetate (PMA) or ionomycin (IM), which have been reported to activate ADAM17 or ADAM10, respectively (45, 46, 48). We found that treatment with PMA, but not IM (data not shown), increased the production of the 75-kDa VEGFR2 fragment, indicating that ADAM17 triggers VEGFR2 shedding in HUVECs (Fig. 13C and D). To further test this hypothesis, we knocked down ADAM17 from Cdc42-depleted HUVECs and assessed the level of the 75-kDa VEGFR2 fragment in these cells. Consistent with the TAPI-1 result, knocking down ADAM17 significantly inhibited the production of the 75-kDa VEGFR2 fragment (Fig. 13E and F). These data strongly indicate that ADAM17 is the sheddase that mediates VEGFR2 cleavage in Cdc42-depleted HUVECs. Moreover, we performed membrane fractionation assays and examined the role of ADAM17 in VEGFR2 shedding. Our data showed that inactivation of Cdc42 decreased the amount of full-length VEGFR2, and that deletion of ADAM17 prevented this decrease, on the membrane (Fig. 13G). At the same time, inactivation of Cdc42 enhanced the production of the 75-kDa VEGFR2 fragment, and inhibition of ADAM17 attenuated the amount of 75-kDa VEGFR2, on the membrane (Fig. 13G). PECAM-1 and α-tubulin were blotted as the markers for the membrane and cytosol fractions, respectively. Cdc42 cycles between the plasma membrane and the cytosol, so we detected Cdc42 expression in both the membrane and cytosol fractions. VEGFR2 and ADAM17 contain transmembrane domains, and we detected the signals of VEGFR2 and ADAM17 only in the membrane fraction, because the method we utilized for fractionation cannot separate the plasma membrane from the cytosolic vesicles.
Next, we examined the effect of ADAM17 on Cdc42 deletion-induced EC apoptosis by determining the levels of the proapoptotic protein Bax and the antiapoptotic protein Bcl-2. In line with our previous results, we found that inhibition of Cdc42 increased Bax and decreased Bcl-2 protein levels (Fig. 14A and B), results consistent with our finding that Cdc42 plays an indispensable role in EC survival. Importantly, the inactivation of ADAM17 can reverse the Cdc42 deletion-induced Bax increase and Bcl-2 decrease, suggesting that ADAM17 is an important effector of Cdc42 in maintaining EC survival. Consistently, the results of flow cytometric analysis experiments showed that the inactivation of Cdc42 increased HUVEC apoptosis, by comparison of Cdc42 knockdown cells (Fig. 14D) to control cells (Fig. 14C). Cdc42 deletion-induced apoptosis could be reversed by knocking down ADAM17 (Fig. 14E and F). To further confirm the importance of ADAM17 in Cdc42 deletion-induced HUVEC apoptosis, we performed TUNEL analysis. Our data showed that knocking down Cdc42 increased HUVEC apoptosis (Fig. 14I and J) over that for the control (Fig. 14G and H) and that inactivation of ADAM17 significantly decreased Cdc42 deletion-induced HUVEC apoptosis (Fig. 14K, L, and M).
Fig 14.

Inactivation of ADAM17 reversed Cdc42 deletion-induced EC apoptosis. (A) Cell lysates of HUVECs transfected with scrambled siRNA, Cdc42 siRNA, or Cdc42 siRNA plus ADAM17 siRNA were analyzed by Western blotting using specific antibodies against Bax or Bcl-2. (B) The intensities of bands from three different experiments were quantitatively analyzed. (C through F) HUVECs were transfected with scrambled siRNA (C), Cdc42 siRNA (D), or Cdc42 siRNA plus Adam17 siRNA (E) and were cultured for 72 h. HUVEC nuclei were then labeled by propidium iodide (PI), and apoptotic cells were labeled by annexin V. (F) Quantitative analysis data show that ADAM17 knockdown significantly decreased Cdc42 deletion-induced apoptosis. (G through M) TUNEL staining revealed increased HUVEC apoptosis in Cdc42 knockdown cells (I and J) relative to that in control cells (G and H). The silencing of ADAM17 significantly reduced HUVEC apoptosis induced by the deletion of Cdc42 (K and L). The percentage of apoptotic HUVECs was quantitatively analyzed (M).
To determine whether ADAM17-mediated VEGFR2 shedding is the cause of EC survival and migration defects induced by Cdc42 deletion, we isolated primary ECs from homozygous Cdc42flox/flox mice. The cultured Cdc42flox/flox ECs were infected with Ad-LacZ, Ad-Cre, or Ad-Cre plus Ad-HA-VEGFR2. Cell lysates were then analyzed by a Western blotting assay using cleaved caspase 3 antibodies. We discovered that deletion of Cdc42 increased the production of cleaved caspase 3 and that overexpression of VEGFR2 hampered the production of cleaved caspase 3, indicating that VEGFR2 can reverse Cdc42 deletion-induced EC apoptosis (Fig. 15A and B). However, we noted that overexpression of VEGFR2 cannot reverse Cdc42 deletion-induced EC migration defects, indicating that VEGFR2 shedding is not responsible for the migration defects in Cdc42-null ECs (Fig. 15C). All these data strongly suggest that ADAM17-mediated VEGFR2 shedding is a major cause of induction of EC apoptosis in blood vessel formation defects in CEKO embryos.
Fig 15.

Overexpression of VEGFR2 reversed Cdc42 deletion-induced EC apoptosis but not migration defects. (A) Lysates from primary cultured mouse ECs infected with Ad-LacZ, Ad-Cre, or Ad-Cre plus Ad-VEGFR2 were analyzed by Western blotting using different antibodies as indicated. (B) The intensities of bands for cleaved caspase 3 and VEGFR2 were quantitatively analyzed. *, P < 0.05. (C) Cultured primary mouse ECs were infected with Ad-LacZ, Ad-Cre, or Ad-Cre plus Ad-VEGFR2 and were subjected to Boyden chamber analysis following the response to VEGF (1 ng/ml). *, P < 0.05.
DISCUSSION
As an essential molecular switch in signal transduction, Cdc42 has been shown to play a critical role in regulating cell proliferation, migration, differentiation, and apoptosis (10). However, the putative function and molecular mechanisms of Cdc42 in new blood vessel formation in vivo remain unclear due to the early embryonic lethality (E6.5) exhibited by Cdc42 total-knockout embryos. Using the Cre/LoxP system, we found that the inactivation of Cdc42 in ECs induced embryonic lethality (E10.5) and blood vessel formation defects. Moreover, we demonstrated that Cdc42 is a pivotal regulator of EC migration and survival but does not play a role in regulating EC proliferation. Furthermore, we observed that Cdc42 deletion decreased the levels of VEGFR2 in ECs through ADAM17-mediated VEGFR2 shedding. Inhibition of ADAM17 activity or overexpression of VEGFR2 can partially reverse EC apoptosis induced by Cdc42 deletion. Taking our findings together, we conclude that Cdc42 plays a crucial role in EC survival and migration and that deletion of Cdc42 impairs blood vessel formation during embryogenesis (Fig. 16).
Fig 16.
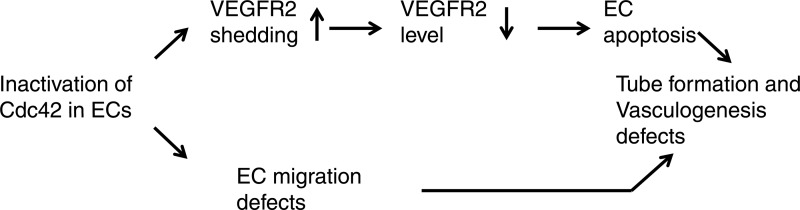
Working model of Cdc42 in tube formation and vasculogenesis. Inactivation of Cdc42 induced EC survival and migration defects. The increased ADAM17-mediated VEGFR2 shedding contributed to the enhancement of EC apoptosis.
Vascular endothelial cells line the entire circulatory system and are the sensors and responders for blood vessel formation (3, 49). Previous studies have shown the importance of Cdc42 in regulating EC cytoskeleton organization and adherens junction assembly (50, 51). Here, we showed that Cdc42 deletion in ECs reduced the levels of VEGFR2 on the cell surface and increased the production of a 75-kDa membrane-associated C-terminal VEGFR2 fragment through ADAM17. Combining these findings with previous studies, we would propose that Cdc42 functions, at least in two ways, to control proangiogenic signals. First, Cdc42 could mediate important signal transduction in ECs during vascular development. Following the stimulation of ECs by growth factors or the extracellular matrix (e.g., fibronectin), activated Cdc42 can bind to its downstream effectors, stimulate EC migration, and maintain the viability of ECs, which are critical events for angiogenesis and vasculogenesis. Inactivation of Cdc42 may interfere with the ability of growth factors and/or integrins to trigger signaling events, thus leading to the disruption of blood vessel formation. Second, Cdc42 is also actively involved in regulating the levels of VEGFR2 on the cell surface. Cdc42 deletion in ECs increased the production of a 75-kDa VEGFR2 fragment and caused a reduction in the level of full-length VEGFR2 on the membrane. This function is important for controlling the magnitude and duration of VEGF-initiated proangiogenic signals and for preventing the premature termination of VEGF signals. Interestingly, previous studies have shown that the secreted VEGFR2 (sVEGFR2) extracellular domain competes with VEGFR2 by binding to VEGF (52). The apparent size of the VEGFR2 extracellular domain (around 150 kDa) generated by ADAM17-mediated VEGFR2 shedding is very close to the size of sVEGFR2. It is possible that this putative VEGFR2 extracellular domain competes for VEGF and inhibits VEGF-stimulated signaling.
ECs need to coordinate and properly respond to multiple extracellular stimuli, including growth factors, the extracellular matrix, and cell-cell adhesion, to maintain cell survival (1, 53, 54). We show that Cdc42 deletion in ECs dramatically increased EC apoptosis both in vivo and in vitro and that inactivation of ADAM17 or overexpression of VEGFR2 reversed Cdc42 deletion-induced EC apoptosis. VEGF plays an essential role in regulating EC survival, and Cdc42 deletion compromised VEGFR2 homeostasis; thus, aberrant VEGFR2-mediated signal transduction is likely one of the causes of EC apoptosis. Cdc42 is located at the convergence of many signal transduction pathways, including integrins and receptor tyrosine kinases (11). Because overexpression of VEGFR2 or knockdown of ADAM17 cannot fully reverse EC apoptosis, it is possible that other Cdc42-regulated signal transduction pathways are also involved in controlling EC survival. In addition, we showed that the decrease in VEGFR2 had no influence on EC proliferation. It is possible that when VEGFR2 expression is decreased, ECs rely on other growth factors to stimulate proliferation. We noticed significant increases in the levels of the 75-kDa VEGFR2 fragment in Cdc42-depleted HUVECs. The function of this 75-kDa VEGFR2 fragment remains unclear. The 75-kDa fragment may bind to its downstream effectors to elicit specific physiological functions. It would be interesting to determine the relationship between the 75-kDa fragment and EC migration defects.
In our studies we found that three fragments of VEGFR2 (230 kDa, 130 kDa, and 75 kDa) were pulled down by streptavidin beads. Previous studies have shown that PMA stimulation can increase ADAM17-mediated VEGFR2 shedding and generate a 150-kDa N-terminal VEGFR2 fragment (48). We found that the deletion of Cdc42 enhanced ADAM17-mediated VEGFR2 shedding and increased the production of a C-terminal 75-kDa fragment. However, the production of the 130-kDa VEGFR2 fragment is not affected by Cdc42 deletion and is inhibited by TAPI-1. Interestingly, knocking down ADAM17 does not influence the production of the 130-kDa fragment. All of these data strongly suggest that the 130-kDa VEGFR2 fragment is generated through another ADAM family enzyme and that Cdc42 does not play a direct role in its regulation.
The underlying molecular mechanisms by which Cdc42 regulates ADAM17-mediated VEGFR2 shedding remain to be determined. Previous studies have reported that the mature form of ADAM17 is enriched in lipid (membrane) rafts and that depletion of cholesterol, which disrupts the structure of lipid rafts, increases ADAM-17-mediated substrate shedding (55). Interestingly, the maintenance of lipid rafts relies on polarized actin, which is directed by Cdc42 (10, 56). Therefore, Cdc42 may play a positive role in limiting the amount of ADAM17 in lipid rafts and preventing its interaction with VEGFR2. Deletion of Cdc42 may disrupt the lipid raft structure, due to the loss of support from actin, and release the mature ADAM17 to induce abnormal VEGFR2 shedding. It is well documented that Cdc42 plays an essential role in regulating receptor endocytosis, recycling, and trafficking (40, 57). Thus, it is also possible that Cdc42 deletion interferes with VEGFR2 internalization and trafficking. Consequently, VEGFR2 may be sorted to the proteasome for degradation, reducing VEGFR2 levels in Cdc42-deficient ECs.
In addition to VEGF signaling stimulation, extracellular matrix and integrin receptors are also important for blood vessel formation (53, 54). Deletion of fibronectin or integrins in ECs induces angiogenesis and vasculogenesis defects (53, 54). Since Cdc42 is an important signaling mediator of integrins, its inactivation may also disrupt integrin-mediated signal transduction. A recent report showed that the deletion of β1 integrin in ECs interfered with EC polarity via a Par3-dependent mechanism (37). Our data show that Cdc42 deletion caused EC remodeling defects, and Cdc42-null ECs showed a cuboidal epithelial morphology instead of the long, thin morphology characteristic of ECs. Cdc42 can form a complex with Par6, Par3, and atypical protein kinase C (aPKC), and this complex is critical for cell polarity (10, 58). The loss of Cdc42 in ECs disrupts complex formation between these proteins and interferes with cell polarity. N-WASP and Arp2/3 are other important effectors of Cdc42 that are involved in cell polarity. Cdc42-null ECs may not properly regulate actin polarization, and this is another possible mechanism for the observed defects in EC remodeling.
In conclusion, we have demonstrated that Cdc42 plays a pivotal role in ECs during blood vessel formation. The results generated from the in vitro and in vivo studies suggest that Cdc42 functions as a signal transducer and an active regulator of VEGFR2 to properly guide the EC response to VEGF stimulation.
ACKNOWLEDGMENTS
This research was supported by an American Heart Association Scientist Development Grant (0735543T), a Beginning-Grant-in-Aid (13BGIA14470002), a Texas A&M Health Science Center startup grant to X. Peng, and a National Institutes of Health (HL068838) grant to X. Peng and D. E. Dostal.
We are grateful to Peggy Scherle (Incyte Corporation) for providing INCB03619 and to Lena Claesson-Welsh (Uppsala University) for sharing the wild-type VEGFR2 plasmid. We appreciate the efforts of Richard Tobin and M. Karen Newell in helping us perform the flow cytometric analysis experiments. We also appreciate Carl Blobel, Harris Granger, and Lih Kuo for scientific discussions and Enoch Kuo for manuscript proofreading.
Footnotes
Published ahead of print 26 August 2013
REFERENCES
- 1.Carmeliet P. 2005. Angiogenesis in life, disease and medicine. Nature 438:932–936 [DOI] [PubMed] [Google Scholar]
- 2.Carmeliet P, Jain RK. 2011. Molecular mechanisms and clinical applications of angiogenesis. Nature 473:298–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Eilken HM, Adams RH. 2010. Dynamics of endothelial cell behavior in sprouting angiogenesis. Curr. Opin. Cell Biol. 22:617–625 [DOI] [PubMed] [Google Scholar]
- 4.Coultas L, Chawengsaksophak K, Rossant J. 2005. Endothelial cells and VEGF in vascular development. Nature 438:937–945 [DOI] [PubMed] [Google Scholar]
- 5.Hanahan D. 1997. Signaling vascular morphogenesis and maintenance. Science 277:48–50 [DOI] [PubMed] [Google Scholar]
- 6.Red-Horse K, Crawford Y, Shojaei F, Ferrara N. 2007. Endothelium-microenvironment interactions in the developing embryo and in the adult. Dev. Cell 12:181–194 [DOI] [PubMed] [Google Scholar]
- 7.Eliceiri BP, Cheresh DA. 2001. Adhesion events in angiogenesis. Curr. Opin. Cell Biol. 13:563–568 [DOI] [PubMed] [Google Scholar]
- 8.Rossant J, Howard L. 2002. Signaling pathways in vascular development. Annu. Rev. Cell Dev. Biol. 18:541–573 [DOI] [PubMed] [Google Scholar]
- 9.Yancopoulos GD, Davis S, Gale NW, Rudge JS, Wiegand SJ, Holash J. 2000. Vascular-specific growth factors and blood vessel formation. Nature 407:242–248 [DOI] [PubMed] [Google Scholar]
- 10.Etienne-Manneville S, Hall A. 2002. Rho GTPases in cell biology. Nature 420:629–635 [DOI] [PubMed] [Google Scholar]
- 11.Cerione RA. 2004. Cdc42: new roads to travel. Trends Cell Biol. 14:127–132 [DOI] [PubMed] [Google Scholar]
- 12.Olsson AK, Dimberg A, Kreuger J, Claesson-Welsh L. 2006. VEGF receptor signalling—in control of vascular function. Nat. Rev. Mol. Cell Biol. 7:359–371 [DOI] [PubMed] [Google Scholar]
- 13.Lamalice L, Houle F, Jourdan G, Huot J. 2004. Phosphorylation of tyrosine 1214 on VEGFR2 is required for VEGF-induced activation of Cdc42 upstream of SAPK2/p38. Oncogene 23:434–445 [DOI] [PubMed] [Google Scholar]
- 14.Koh W, Sachidanandam K, Stratman AN, Sacharidou A, Mayo AM, Murphy EA, Cheresh DA, Davis GE. 2009. Formation of endothelial lumens requires a coordinated PKCε-, Src-, Pak- and Raf-kinase-dependent signaling cascade downstream of Cdc42 activation. J. Cell Sci. 122:1812–1822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koh W, Mahan RD, Davis GE. 2008. Cdc42- and Rac1-mediated endothelial lumen formation requires Pak2, Pak4 and Par3, and PKC-dependent signaling. J. Cell Sci. 121:989–1001 [DOI] [PubMed] [Google Scholar]
- 16.Bayless KJ, Davis GE. 2002. The Cdc42 and Rac1 GTPases are required for capillary lumen formation in three-dimensional extracellular matrices. J. Cell Sci. 115:1123–1136 [DOI] [PubMed] [Google Scholar]
- 17.Qi Y, Liu J, Wu X, Brakebusch C, Leitges M, Han Y, Corbett SA, Lowry SF, Graham AM, Li S. 2011. Cdc42 controls vascular network assembly through protein kinase Cι during embryonic vasculogenesis. Arterioscler. Thromb. Vasc. Biol. 31:1861–1870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moreau V, Tatin F, Varon C, Anies G, Savona-Baron C, Genot E. 2006. Cdc42-driven podosome formation in endothelial cells. Eur. J. Cell Biol. 85:319–325 [DOI] [PubMed] [Google Scholar]
- 19.Tatin F, Varon C, Genot E, Moreau V. 2006. A signalling cascade involving PKC, Src and Cdc42 regulates podosome assembly in cultured endothelial cells in response to phorbol ester. J. Cell Sci. 119:769–781 [DOI] [PubMed] [Google Scholar]
- 20.Tzima E, Kiosses WB, del Pozo MA, Schwartz MA. 2003. Localized cdc42 activation, detected using a novel assay, mediates microtubule organizing center positioning in endothelial cells in response to fluid shear stress. J. Biol. Chem. 278:31020–31023 [DOI] [PubMed] [Google Scholar]
- 21.Wojciak-Stothard B, Ridley AJ. 2003. Shear stress-induced endothelial cell polarization is mediated by Rho and Rac but not Cdc42 or PI 3-kinases. J. Cell Biol. 161:429–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hu GD, Chen YH, Zhang L, Tong WC, Cheng YX, Luo YL, Cai SX, Zhang L. 2011. The generation of the endothelial specific cdc42-deficient mice and the effect of cdc42 deletion on the angiogenesis and embryonic development. Chin. Med. J. (Engl.) 124:4155–4159 [PubMed] [Google Scholar]
- 23.Chen F, Ma L, Parrini MC, Mao X, Lopez M, Wu C, Marks PW, Davidson L, Kwiatkowski DJ, Kirchhausen T, Orkin SH, Rosen FS, Mayer BJ, Kirschner MW, Alt FW. 2000. Cdc42 is required for PIP(2)-induced actin polymerization and early development but not for cell viability. Curr. Biol. 10:758–765 [DOI] [PubMed] [Google Scholar]
- 24.Peng X, Lin Q, Liu Y, Jin Y, Druso JE, Antonyak MA, Guan JL, Cerione RA. 2013. Inactivation of Cdc42 in embryonic brain results in hydrocephalus with ependymal cell defects in mice. Protein Cell 4:231–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shen TL, Park AY, Alcaraz A, Peng X, Jang I, Koni P, Flavell RA, Gu H, Guan JL. 2005. Conditional knockout of focal adhesion kinase in endothelial cells reveals its role in angiogenesis and vascular development in late embryogenesis. J. Cell Biol. 169:941–952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Koni PA, Joshi SK, Temann UA, Olson D, Burkly L, Flavell RA. 2001. Conditional vascular cell adhesion molecule 1 deletion in mice: impaired lymphocyte migration to bone marrow. J. Exp. Med. 193:741–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Institute of Laboratory Animal Resources 1996. Guide for the care and use of laboratory animals, 7th ed. NIH publication no. 85-23 National Academy Press, Washington, DC [Google Scholar]
- 28.Peng X, Wu X, Druso JE, Wei H, Park AY, Kraus MS, Alcaraz A, Chen J, Chien S, Cerione RA, Guan JL. 2008. Cardiac developmental defects and eccentric right ventricular hypertrophy in cardiomyocyte focal adhesion kinase (FAK) conditional knockout mice. Proc. Natl. Acad. Sci. U. S. A. 105:6638–6643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Luo J, Deng ZL, Luo X, Tang N, Song WX, Chen J, Sharff KA, Luu HH, Haydon RC, Kinzler KW, Vogelstein B, He TC. 2007. A protocol for rapid generation of recombinant adenoviruses using the AdEasy system. Nat. Protoc. 2:1236–1247 [DOI] [PubMed] [Google Scholar]
- 30.Kubota Y, Kleinman HK, Martin GR, Lawley TJ. 1988. Role of laminin and basement membrane in the morphological differentiation of human endothelial cells into capillary-like structures. J. Cell Biol. 107:1589–1598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Peng X, Ueda H, Zhou H, Stokol T, Shen TL, Alcaraz A, Nagy T, Vassalli JD, Guan JL. 2004. Overexpression of focal adhesion kinase in vascular endothelial cells promotes angiogenesis in transgenic mice. Cardiovasc. Res. 64:421–430 [DOI] [PubMed] [Google Scholar]
- 32.Jin Y, Liu Y, Antonyak M, Peng X. 2012. Isolation and characterization of vascular endothelial cells from murine heart and lung. Methods Mol. Biol. 843:147–154 [DOI] [PubMed] [Google Scholar]
- 33.Zhao X, Peng X, Sun S, Park AY, Guan JL. 2010. Role of kinase-independent and -dependent functions of FAK in endothelial cell survival and barrier function during embryonic development. J. Cell Biol. 189:955–965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu X, Gan B, Yoo Y, Guan JL. 2005. FAK-mediated src phosphorylation of endophilin A2 inhibits endocytosis of MT1-MMP and promotes ECM degradation. Dev. Cell 9:185–196 [DOI] [PubMed] [Google Scholar]
- 35.Zhen Y, Sorensen V, Skjerpen CS, Haugsten EM, Jin Y, Walchli S, Olsnes S, Wiedlocha A. 2012. Nuclear import of exogenous FGF1 requires the ER-protein LRRC59 and the importins Kpnα1 and Kpnβ1. Traffic 13:650–664 [DOI] [PubMed] [Google Scholar]
- 36.Auerbach R, Gilligan B, Lu LS, Wang SJ. 1997. Cell interactions in the mouse yolk sac: vasculogenesis and hematopoiesis. J. Cell. Physiol. 173:202–205 [DOI] [PubMed] [Google Scholar]
- 37.Zovein AC, Luque A, Turlo KA, Hofmann JJ, Yee KM, Becker MS, Fassler R, Mellman I, Lane TF, Iruela-Arispe ML. 2010. β1 integrin establishes endothelial cell polarity and arteriolar lumen formation via a Par3-dependent mechanism. Dev. Cell 18:39–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lucitti JL, Jones EA, Huang C, Chen J, Fraser SE, Dickinson ME. 2007. Vascular remodeling of the mouse yolk sac requires hemodynamic force. Development 134:3317–3326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.le Noble F, Moyon D, Pardanaud L, Yuan L, Djonov V, Matthijsen R, Breant C, Fleury V, Eichmann A. 2004. Flow regulates arterial-venous differentiation in the chick embryo yolk sac. Development 131:361–375 [DOI] [PubMed] [Google Scholar]
- 40.Harris KP, Tepass U. 2010. Cdc42 and vesicle trafficking in polarized cells. Traffic 11:1272–1279 [DOI] [PubMed] [Google Scholar]
- 41.Stengel K, Zheng Y. 2011. Cdc42 in oncogenic transformation, invasion, and tumorigenesis. Cell. Signal. 23:1415–1423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Blobel CP. 2005. ADAMs: key components in EGFR signalling and development. Nat. Rev. Mol. Cell Biol. 6:32–43 [DOI] [PubMed] [Google Scholar]
- 43.Murphy G. 2008. The ADAMs: signalling scissors in the tumour microenvironment. Nat. Rev. Cancer 8:929–941 [DOI] [PubMed] [Google Scholar]
- 44.Black RA, Rauch CT, Kozlosky CJ, Peschon JJ, Slack JL, Wolfson MF, Castner BJ, Stocking KL, Reddy P, Srinivasan S, Nelson N, Boiani N, Schooley KA, Gerhart M, Davis R, Fitzner JN, Johnson RS, Paxton RJ, March CJ, Cerretti DP. 1997. A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature 385:729–733 [DOI] [PubMed] [Google Scholar]
- 45.Sahin U, Weskamp G, Kelly K, Zhou HM, Higashiyama S, Peschon J, Hartmann D, Saftig P, Blobel CP. 2004. Distinct roles for ADAM10 and ADAM17 in ectodomain shedding of six EGFR ligands. J. Cell Biol. 164:769–779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Donners MM, Wolfs IM, Olieslagers S, Mohammadi-Motahhari Z, Tchaikovski V, Heeneman S, van Buul JD, Caolo V, Molin DG, Post MJ, Waltenberger J. 2010. A disintegrin and metalloprotease 10 is a novel mediator of vascular endothelial growth factor-induced endothelial cell function in angiogenesis and is associated with atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 30:2188–2195 [DOI] [PubMed] [Google Scholar]
- 47.Zhou BB, Peyton M, He B, Liu C, Girard L, Caudler E, Lo Y, Baribaud F, Mikami I, Reguart N, Yang G, Li Y, Yao W, Vaddi K, Gazdar AF, Friedman SM, Jablons DM, Newton RC, Fridman JS, Minna JD, Scherle PA. 2006. Targeting ADAM-mediated ligand cleavage to inhibit HER3 and EGFR pathways in non-small cell lung cancer. Cancer Cell 10:39–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Swendeman S, Mendelson K, Weskamp G, Horiuchi K, Deutsch U, Scherle P, Hooper A, Rafii S, Blobel CP. 2008. VEGF-A stimulates ADAM17-dependent shedding of VEGFR2 and crosstalk between VEGFR2 and ERK signaling. Circ. Res. 103:916–918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.De Val S, Black BL. 2009. Transcriptional control of endothelial cell development. Dev. Cell 16:180–195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tzima E. 2006. Role of small GTPases in endothelial cytoskeletal dynamics and the shear stress response. Circ. Res. 98:176–185 [DOI] [PubMed] [Google Scholar]
- 51.Broman MT, Kouklis P, Gao X, Ramchandran R, Neamu RF, Minshall RD, Malik AB. 2006. Cdc42 regulates adherens junction stability and endothelial permeability by inducing alpha-catenin interaction with the vascular endothelial cadherin complex. Circ. Res. 98:73–80 [DOI] [PubMed] [Google Scholar]
- 52.Albuquerque RJ, Hayashi T, Cho WG, Kleinman ME, Dridi S, Takeda A, Baffi JZ, Yamada K, Kaneko H, Green MG, Chappell J, Wilting J, Weich HA, Yamagami S, Amano S, Mizuki N, Alexander JS, Peterson ML, Brekken RA, Hirashima M, Capoor S, Usui T, Ambati BK, Ambati J. 2009. Alternatively spliced vascular endothelial growth factor receptor-2 is an essential endogenous inhibitor of lymphatic vessel growth. Nat. Med. 15:1023–1030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Avraamides CJ, Garmy-Susini B, Varner JA. 2008. Integrins in angiogenesis and lymphangiogenesis. Nat. Rev. Cancer 8:604–617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rupp PA, Little CD. 2001. Integrins in vascular development. Circ. Res. 89:566–572 [DOI] [PubMed] [Google Scholar]
- 55.Tellier E, Canault M, Rebsomen L, Bonardo B, Juhan-Vague I, Nalbone G, Peiretti F. 2006. The shedding activity of ADAM17 is sequestered in lipid rafts. Exp. Cell Res. 312:3969–3980 [DOI] [PubMed] [Google Scholar]
- 56.Chichili GR, Rodgers W. 2009. Cytoskeleton-membrane interactions in membrane raft structure. Cell. Mol. Life Sci. 66:2319–2328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.McCormack J, Welsh NJ, Braga VM. 2013. Cycling around cell-cell adhesion with Rho GTPase regulators. J. Cell Sci. 126:379–391 [DOI] [PubMed] [Google Scholar]
- 58.Heasman SJ, Ridley AJ. 2008. Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat. Rev. Mol. Cell Biol. 9:690–701 [DOI] [PubMed] [Google Scholar]