

Abstract
Cell adhesion-mediated drug resistance contributes to minimal residual disease and relapse in hematological malignancies. Here, we show that adhesion of Jurkat T-acute lymphoblastic leukemia cells to substrates engaging α4β1-integrin or α5β1-integrin promotes chemoresistance to doxorubicin-induced apoptosis. Reconstituted expression of α4δ, a truncated α4-integrin with KXGFFKR as the cytoplasmic motif, in α4-deficient cells promoted chemoresistance to doxorubicin in a manner independent of α4-mediated adhesion. The adhesion-independent chemoresistance did not require β1-integrin as the heterodimeric pair, since expression of Tacδ, a monomeric nonintegrin transmembrane protein fused to the juxtamembrane KXGFFKR, was sufficient to reproduce the phenomenon. The requirement for integrin-mediated adhesion in stimulation of Akt phosphorylation and activation was bypassed for cells expressing α4δ and Tacδ. Cells expressing α4δ and Tacδ exhibited a high influx of extracellular Ca2+, and inhibition of Ca2+ channels with verapamil attenuated the adhesion-independent chemoresistance. Tacδ cells also exhibited greater rates of drug efflux. α4δ and Tacδ interacted with the Ca2+-binding protein calreticulin, in a manner dependent on the KXGFFKR motif. Adhesion-mediated engagement of α4-integrins promoted an increased calreticulin-α4 association and greater influx of extracellular Ca2+ than in nonadherent cells. The α-integrin KXGFFKR motif is involved in adhesion-mediated control of chemoresistance in T cells.
INTRODUCTION
Acquired chemoresistance is a significant contributor to minimal residual disease and treatment relapse in hematological malignancies (1, 2). Multiple studies have implicated the role of an integrin-substratum ligand interaction in promotion of tumor cell prosurvival signaling and chemoresistance, a process termed cell adhesion-mediated drug resistance (CAM-DR) (3–9). These processes are deemed to occur in hematopoietic niches, such as the bone marrow stroma, where tumor cell interactions with microenvironmental factors, including adhesion, promote their survival and potentiate minimal residual disease following chemotherapy (10).
Integrins are heterodimeric cell adhesion receptors that consist of α- and β-subunits; their extracellular domains mediate cell attachment to extracellular matrix proteins or cell adhesion molecules, and their cytoplasmic domains couple signaling and linkage with the cytoskeleton (11, 12). The α4-integrins are highly expressed in leukocytes and play critical roles in their recruitment and trafficking to hematopoietic niches (13). Cell adhesion mediated via α4-integrins also contributes to chemoresistance (3, 4, 9), which can be overcome by neutralization of the extracellular α4-integrin–substrate interactions (5, 14–16). However, adhesion via integrins other than α4 that are expressed by lymphocytes also contributes to chemoresistance (6–8, 17), suggesting a common regulatory mechanism governed by integrin-mediated adhesion as the chemoprotective switch. The adhesion-mediated chemoresistance is often attributed to β1-integrin-mediated stimulation of Akt activity and subsequent regulation of prosurvival signaling (3, 18, 19).
By comparison, the contribution of α-integrins in chemoresistance and prosurvival signaling remains little characterized. The cytoplasmic domains of α-integrins share few sequence similarities, with the exception of the highly conserved membrane-proximal KXGFFKR motif (11). This motif is required to maintain the α-β-integrin heterodimer by forming a salt bridge with its β-cytoplasmic domain counterpart (11, 20). The KXGFFKR motif also mediates interactions with proteins that regulate various aspects of integrin function, including sharpin (21), MDGI (22), Mss4 (23), CIB (24), and calreticulin (25). The role for these interactions in regulating CAM-DR remains to be characterized, but their likely role is implicated since they modulate aspects of integrin-mediated adhesion.
The α4-cytoplasmic domain interacts with several proteins, including paxillin (26), type I protein kinase A (PKA) (27), and nonmuscle myosin IIA (28), to regulate cell spreading and migration. These interactions are specific to α4-integrin, as supported by mutational analyses that implicated sequences C-terminal of the KXGFFKR motif that are unique to α4-integrin. Given that these interactions modulate α4-dependent adhesion, we undertook this study to investigate the requirement of the α4-integrin cytoplasmic domain in regulation of α4-dependent CAM-DR in a T cell model for acute lymphoblastic leukemia (ALL). We found that engagement of different integrins in Jurkat T-ALL cells equally promoted CAM-DR. Expression of a truncated α4-integrin with only KXGFFKR as the cytoplasmic motif resulted in a chemoresistant cell line that bypassed the requirement for cell adhesion. Further characterization revealed that several signaling events normally requiring adhesion as the trigger are constitutively activated by cells expressing the juxtamembrane KXGFFKR. Thus, α-integrin KXGFFKR-mediated interactions constitute a common regulatory mechanism with the potential to impact prosurvival signaling and tumor cell chemoresistance.
MATERIALS AND METHODS
Cells.
Jurkat T cells were obtained from the American Type Culture Collection. JB4 is a Jurkat derivative lacking α4-integrin expression and has been described previously (29). Cells were cultured at 37°C, 5% CO2 in complete RPMI (RPMI 1640 supplemented with 10% fetal bovine serum [FBS; Sigma-Aldrich], l-glutamine, penicillin-streptomycin, and nonessential amino acids [Invitrogen]). Cell transfections were performed using an Amaxa nucleofection kit V (Lonza) and selected accordingly for hygromycin or G418 (Invitrogen) resistance. Cells stably expressing the desired receptor levels were sorted to homogeneity following surface immunolabeling with antibodies.
Plasmids.
The truncated α4δ-integrin corresponding to human α4 amino acids 1 to 1007 was amplified by PCR using primers, adding a stop codon following the KAGFFKR sequence, and subcloning into pcDNA3.1 (Invitrogen). Tacδ was cloned as a fusion of Tac (human CD25 amino acids 1 to 263) to KLGFFKR encoded by double-stranded oligonucleotides. For Tacδscr, the oligonucleotides encoded KLRFGFK. Expression plasmids for full-length α4-integrin and the Tac epitope were gifts from Mark Ginsberg (UCSD).
Recombinant proteins and fibronectin.
Glutathione S-transferase (GST)-tagged proteins were purified from BL21 Escherichia coli lysates by affinity chromatography through glutathione-Sepharose (GE Healthcare) according to the manufacturer's instructions. GST-CS1 and GST-Fn9.11 are GST fusions to the fibronectin CS1 region (30) and repeats 9 to 11 (31), respectively. Expression vectors for GST-KLGFFKR and GST-KLRFGFK proteins were made as double-stranded oligonucleotides encoding the peptides fused C-terminal to GST and encoded by pGEX-4T (GE Healthcare). Fibronectin from human plasma was purified by affinity chromatography through gelatin-Sepharose (GE Healthcare).
Antibodies.
Antibodies used for flow cytometry labeling of cell surface proteins were the following: β1-integrin (sc-53711; Santa Cruz Biotechnology) and Tac (BC96) and α4-integrin (9F10) from BioLegend. Antibodies used for immunoblotting were Akt (40D4), α4-integrin (4600), and phospho-Akt substrate (23C8D2) from Cell Signaling Technology; α4-integrin (sc-365209) and Tac (sc-665) from Santa Cruz Biotechnology; GAPDH (FF26A/F9; BioLegend), phospho-Thr308 Akt (EP2107Y; Epitomics), phospho-Ser473 Akt (EP2109Y; Epitomics), and calreticulin (PA3-900; Thermo Scientific). Antibodies used for immunoprecipitation were α4-integrin (sc-365209 [Santa Cruz Biotechnology] or HP2/1 [Beckman-Coulter]) and Tac (BC96; BioLegend).
Flow cytometry.
Flow cytometry work was conducted at the Child & Family Research Institute (CFRI) Flow Core facility. Analytical work was conducted on FACSCalibur and FACSCanto instruments, while fluorescence-activated cell sorting was conducted on a FACSAria apparatus (BD Biosciences). Postacquisition analysis was done using FlowJo (Tree Star).
Preparation of adhesion substrates.
Tissue culture dishes (Corning Costar) were incubated with 40 μg/ml of GST-CS1, GST-Fn9.11, GST, or 1% bovine serum albumin (BSA) in phosphate-buffered saline (PBS; 137 mM NaCl, 2.7 mM KCl, 1.5 mM KH2PO4, 8 mM Na2HPO4 [pH 7.4]; Sigma) overnight at 4°C and then blocked with 1% BSA–PBS for 1 h. Following 2 washes with PBS, the coated dishes were ready for seeding with cells.
Cell apoptosis.
Twelve-well substrate-coated plates were seeded with cells at 2 × 105 cells/well in 1.0 ml complete RPMI for 4 h before addition of doxorubicin to the desired concentration. The half-maximal effective concentrations (EC50s) of doxorubicin for Jurkat and JB4 cells were determined to be 0.05 and 0.03 μg/ml, respectively. After 48 h of incubation, cells were resuspended and incubated with annexin V-Cy5 (BD Biosciences) according to the manufacturer's instructions prior to flow cytometry analysis. In some experiments, cells were treated with (i) Akt inhibitor IV (Millipore) at the indicated concentrations or (ii) 0.6 mM EGTA (Sigma) or 60 μM verapamil (Enzo), either alone or with doxorubicin for 48 h before determination of apoptosis.
Cell adhesion assay.
Cells were plated on substrate-coated 6-well dishes for 30 min before imaging. For each condition, 12 different fields of view (FOVs) were acquired before physical agitation of the dishes, followed by reacquisition of the same FOVs. Alignment markers imprinted on the dish were used for registration of images. Imaging was performed with an Olympus IX81 microscope equipped with a 20× phase objective, CoolSnap HQ2 camera (Photometrics), H-117 linear-encoded stage (Prior Scientific), and control with MetaMorph (Molecular Devices). Postacquisition image processing was performed with ImageJ (http://rsb.info.nih.gov/ij/) before and after images were pseudocolored, aligned, and overlaid. Nontranslocated cells were scored as adhered, and displaced cells were scored as nonadhered. The percent cell adhesion was calculated for each FOV (number of adhered cells/total number cells), with means and standard deviations computed for the 12 FOVs. On average, each FOV had ∼150 cells, with ∼1,800 cells scored per cell-substrate combination.
Affinity chromatography, immunoprecipitation, and Western blot analysis.
Cell lysates were routinely prepared in PN buffer [10 mM piperazine-N,N′-bis(2-ethanesulfonic acid), 50 mM NaCl, 150 mM sucrose, 50 mM NaF, 40 mM Na4P2O7 · 10H2O, 1 mM CaCl2, 1 mM MgCl2, 1% Triton X-100, Complete protease inhibitors (Roche)]. For phospho-Akt analysis, cells were serum starved in 0.5% FBS–RPMI for 48 h before plating on 100-mm-diameter substrate-coated dishes. For immunoprecipitations, 1 mg cell lysate was incubated with 2 μg antibodies for 14 h. Then, 10 μl bed-volume protein A/G-Sepharose (Pierce) was added, and the mixture was incubated a further 2 h before washes and elution in SDS-PAGE sample buffer. For recombinant GST-protein pulldown of calreticulin, 1 mg Jurkat lysate was incubated with 12 μg GST-KLGFFKR or GST-KLRFGFK immobilized on GSH-Sepharose for 14 h. Loading of GST-protein fusions was done by Coomassie blue staining of an excised portion of the SDS-PAGE gel. For Western blot analyses, SDS-PAGE-separated proteins transferred onto nitrocellulose membranes (Bio-Rad) were incubated with primary and infrared dye-conjugated secondary antibodies (Pierce and Rockland). Blots were imaged on an Odyssey imaging system (LI-COR). The Akt activation index (in relative units) was calculated as the fluorescence intensity of Akt-phosphorylated substrates divided by that of GAPDH, the loading reference.
Intracellular calcium.
Cells resuspended in Ca2+-free PBS were incubated with 1 μM Fluo-4-AM, 0.02% (wt/vol) Pluronic F-127 (Invitrogen), 0.1% dimethyl sulfoxide (Sigma-Aldrich) for 30 min at 22°C. Following washes, cells were resuspended in PBS, 1 mM CaCl2–PBS, or 1 mM CaCl2–5 mM EGTA–PBS for 10 min at 22°C and then kept chilled on ice preceding flow cytometry measurements. The median fluorescent intensity (MFI) values were used to compare intracellular Ca2+ levels within one experiment. Intracellular calcium levels were identical for cells incubated with PBS alone or with 1 mM CaCl2–5 mM EGTA–PBS, indicating insignificant changes resulting from Ca2+ efflux for the assay duration. Ca2+ influx was calculated as the intracellular Fluo-4-AM fluorescence levels in 1 mM CaCl2–PBS subtracted from measurements in PBS alone. To monitor adhesion-mediated changes in intracellular Ca2+, aliquots containing 1 × 105 Fluo-4-AM-labeled cells resuspended in complete RPMI or complete RPMI–0.6 mM EGTA were seeded onto BSA-, CS1-, or fibronectin-coated wells at 37°C, and microplate fluorescence readings were taken at intervals following cell plating (Enspire; Perkin-Elmer).
Calcein-AM efflux assay.
Harvested cells were resuspended into prewarmed (37°C) complete RPMI and seeded as 100-μl aliquots containing 2 × 105 cells per well in 96-well plates. Plates were incubated for 15 min at 37°C, 5% CO2 prior to addition of an equal volume of calcein-AM (Invitrogen) dissolved in prewarmed complete RPMI at a final concentration of 250 nM. Cellular esterases convert the nonfluorescent calcein-AM to fluorescent calcein, which is retained in the cell. Calcein fluorescence measurements (excitation at 485 nm, emission at 520 nm) were collected immediately (t = 0) and thereafter at the indicated time points at room temperature. Between t = 15 and t = 30 min, the cells were reincubated at 37°C, 5% CO2. The data were plotted as the fluorescence values subtracted from values for cell-free wells (taken as blank measurements).
Doxorubicin efflux assay.
To measure doxorubicin effux, harvested cells were resuspended into complete RPMI and incubated with 2 μg/ml doxorubicin for 2 h at 37°C. Cells were then washed free of extracellular doxorubicin, resuspended in PBS or 1 mM CaCl2–PBS at 6 × 106 cells/ml, and incubated at 37°C, 5% CO2. At the indicated time points, aliquots of cell suspensions were removed and centrifuged to pellet cells, and 150 μl of the supernatant was recovered to assay for effluxed doxorubicin in a microplate format (excitation at 488 nm, emission at 578 nm). The data are plotted as the fluorescence values minus the solution-only blank measurements.
RESULTS
Jurkat T cells that have adhered to integrin α4β1-integrin or α5β1-integrin substrates exhibit a decreased apoptotic response to doxorubicin.
To determine if T cells gain chemoresistance upon integrin-mediated adhesion, we plated Jurkat cells on the recombinant protein substrates GST-CS1, which engages α4β1-integrins (30), or on GST-Fn9.11, which engages α5β1-integrins (31). For no integrin engagement, cells were plated on GST alone or on BSA. The plated cells were left untreated or treated with doxorubicin at the EC50 (which had been determined beforehand for Jurkat cells) and assayed by flow cytometry based on annexin V binding to cell surface phosphatidylserine as a marker for apoptotic cells. In the absence of drug treatment, the fractions of apoptotic Jurkat cells plated on CS1, Fn9.11, or control substrate were not significantly different (Fig. 1A). In contrast, the fractions of apoptotic Jurkat cells plated on CS1 or Fn9.11 and treated with doxorubicin were 50% less than those plated on control substrate, indicating that adhesion via α4β1- or α5β1-integrins confers enhanced chemoresistance to doxorubicin in Jurkat T cells.
Fig 1.
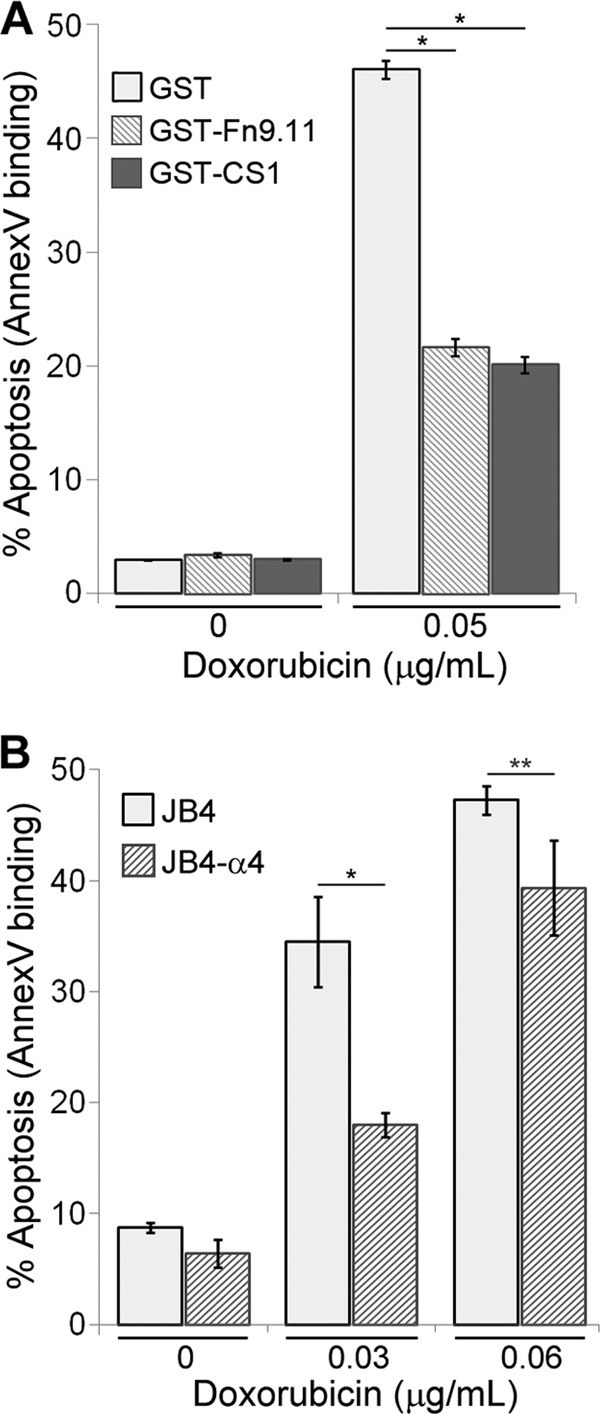
Adhesion on integrin substrates confers resistance to doxorubicin in Jurkat T cells. (A) Jurkat T cells were plated on dishes coated with GST-CS1, GST-Fn9.11, or GST and left untreated or treated with 0.05 μg/ml doxorubicin for 48 h. The data plotted are the percentage of cells that were apoptotic, based on flow cytometry determination of Cy5-annexin V binding. Data are means ± standard deviations (n = 3). *, P < 0.001. (B) JB4 (lacking α4) or JB4-α4 (reconstituted with α4 expression) cells were plated on GST-CS1-coated dishes and left untreated or treated with doxorubicin at 0.03 or 0.06 μg/ml for 48 h. The data plotted are the percentage of cells that were apoptotic, based on flow cytometry determination of Cy5-annexin V binding. Data are means ± standard deviations (n = 3). *, P < 0.02; **, P < 0.05.
To confirm the requirement of α4β1-integrin in CAM-DR, we made use of JB4 cells, a Jurkat-derivative cell line lacking α4-integrin expression (29), and JB4-α4 cells, which we reconstituted with α4β1-integrin expression. JB4 and JB4-α4 cells were plated on CS1 and left untreated or treated with doxorubicin. Treatment of JB4 cells at the EC50 level resulted in a 2-fold-greater fraction of apoptotic cells than with JB4-α4 cells (Fig. 1B). The enhanced chemoresistance exhibited by JB4-α4 cells could be directly attributed to α4β1-integrin function, since JB4 cells lack α4 expression and thus cannot adhere to the α4β1-specific CS1 ligand.
Cells expressing α4-integrin with a truncated cytoplasmic tail exhibited enhanced resistance to doxorubicin-induced apoptosis without adhesion.
The α4 cytoplasmic tail mediates interactions with several proteins, including paxillin (32), type I PKA (27), and nonmuscle myosin IIA (28), that are important in controlling α4-dependent cell migration. Much of the cytoplasmic tail is also required to support cell adhesion to α4-specific extracellular ligands (20). To assess the requirement of α4 tail sequences in supporting α4-mediated CAM-DR, we constructed the tail-truncated α4δ (Fig. 2A). The unique C-terminal portion of the α4 tail was deleted, retaining only the juxtamembrane KAGFFKR sequence, which is essential for heterodimerization with β1 and maintaining surface α4 expression (20). JB4-α4 and JB4-α4δ cells had comparable surface α4 expression levels, as determined by flow cytometry (Fig. 2A). In comparison to the parental JB4 cells, β1-integrin expression was correspondingly increased in JB4-α4 and JB4-α4δ cells (Fig. 2A), an indication that the expressed α4 or α4δ are heterodimerized with β1-integrins. We also assessed several morphological parameters for all the cell lines used in this study and cultured under standard conditions. No significant differences were observed for general morphology (size and shape [see Fig. S1 in the supplemental material]), F-actin distribution, or nuclei number (data not shown) compared to either Jurkat cells or the parental JB4 cells.
Fig 2.
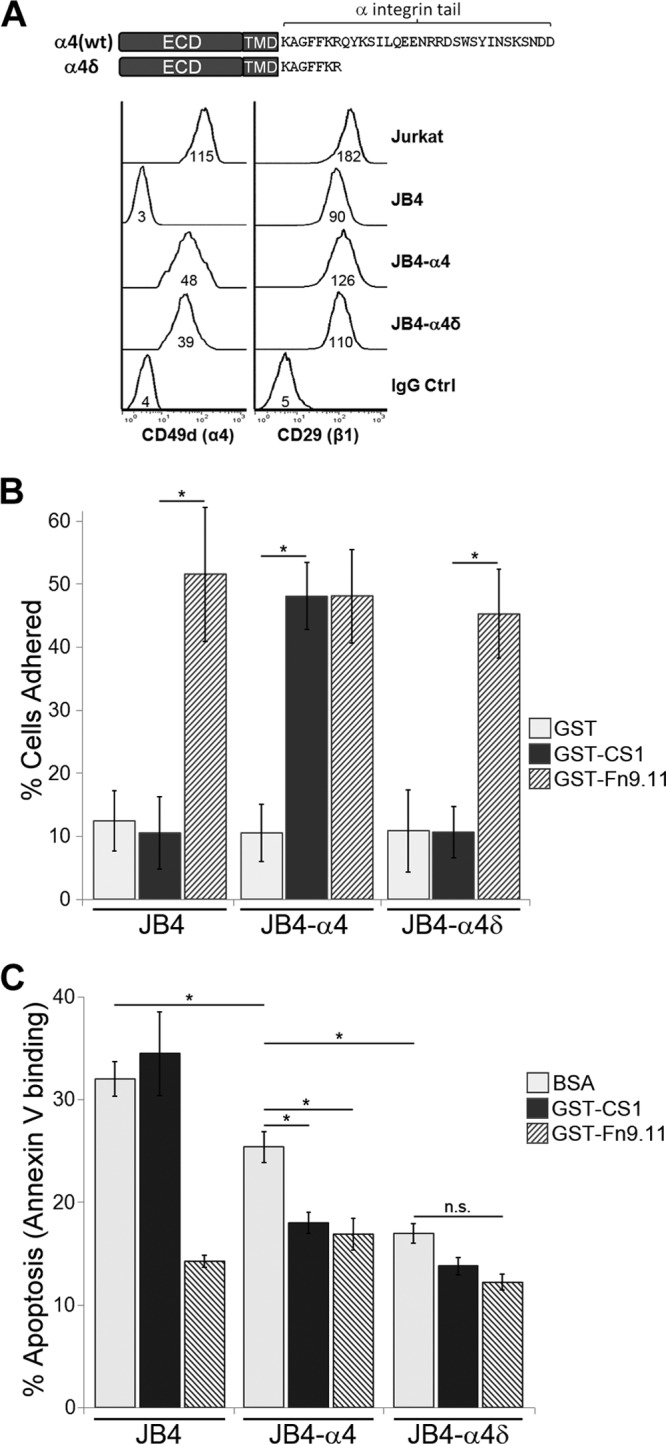
Expression of the α4 tail-truncated variant, α4δ, confers enhanced chemoresistance in an adhesion-independent manner. (A, top) Schematic of the α4-integrin constructs used, highlighting the cytoplasmic domain sequences. α4δ is truncated as indicated and retains the KAGFFKR portion of the cytoplasmic tail. ECD, extracellular domain; TMD, transmembrane domain. (Bottom) Flow cytometry determination of cell surface α4-integrin and β1-integrin expression in Jurkat, JB4, JB4-α4, and JB4-α4δ cells. Numbers under the histogram are the MFI (median fluorescence intensity). (B) JB4-α4, JB4-α4δ, and JB4 cells were plated on dishes coated with GST-CS1, GST-Fn9.11, or GST for 30 min and scored as described in Materials and Methods for adherent versus nonadherent cells. Data plotted are the mean percentage of adherent cells, calculated from 12 FOVs per cell type and treatment condition (means ± standard deviations; n = 12 FOVs). *, P < 0.0001. (C) JB4-α4, JB4-α4δ, and JB4 cells were plated on dishes coated with GST-CS1, GST-Fn9.11, or BSA and left untreated or treated with 0.03 μg/ml doxorubicin for 48 h. Data plotted are the percentage of cells that are apoptotic, based on flow cytometry determination of Cy5-annexin V binding (means ± standard deviations; n = 3). *, P < 0.02; n.s., not significant.
To determine their ligand-binding properties, a cell adhesion assay with CS1, Fn9.11, and control substrate was performed (Fig. 2B). As anticipated, JB4, JB4-α4, and JB4-α4δ cells adhered to GST-Fn9.11 and not to the control GST substrate. JB4 and JB4-α4δ cells failed to adhere to GST-CS1, confirming that the truncated α4δ cannot support adhesion. Furthermore, α4δ expression did not affect cell adhesion via other integrins, such as α5β1-integrin to Fn9.11 (Fig. 2B).
To determine if cells expressing α4δ still mediate chemoresistance, JB4-α4δ cells were plated on various integrin substrates and treated with doxorubicin, and the level of cell apoptosis was determined. Consistent with their adhesion properties, JB4 and JB4-α4 cells exhibited significantly reduced apoptosis when plated on the integrin substrates to which they are adherent, in this case, JB4 on Fn9.11 and JB4-α4 on CS1 or Fn9.11 (Fig. 2C). We noted that for cells plated on BSA, JB4-α4 cells exhibited less doxorubicin-induced apoptosis than JB4 cells, suggesting that α4 expression alone can mediate a low level of drug resistance without adhesion. JB4-α4δ cells plated on CS1 or BSA, to which they are not adherent, exhibited low rates of doxorubicin-induced apoptosis that were comparable to integrin-adhered cells (Fig. 2C). Thus, JB4-α4δ cells exhibit a form of chemoresistance that is independent of integrin-mediated adhesion.
Expression of a carrier epitope bearing the membrane-proximal KXGFFKR motif of α-integrin is sufficient to confer adhesion-independent chemoresistance.
The adhesion-independent chemoresistance observed with JB4-α4δ cells could be attributed to the gain of β1-subunit-mediated signals, as α4δβ1, or to the truncated α4δ tail that encodes the juxtamembrane KAGFFKR. To rule out the involvement of β1-integrin in the observed chemoresistance, we created JB4-Tacδ and JB4-Tacδscr cells (Fig. 3A). Tac encodes the extracellular epitope and transmembrane domains of CD25 with no cytoplasmic domain (33). Tacδ is a fusion of the C-terminal KLGFFKR peptide to Tac, while the control Tacδscr contains a scrambled KLRFGFK version of the tail. Importantly, Tacδ is a monomer that won't heterodimerize with β1-integrin. In the absence of integrin-mediated adhesion, JB4-Tacδ cells, but not JB4-Tacδscr cells, exhibited low levels of doxorubicin-induced apoptosis comparable to those for JB4-α4δ cells (Fig. 3B), indicating that the juxtamembrane KXGFFKR motif is sufficient to promote an adhesion-independent form of chemoresistance.
Fig 3.
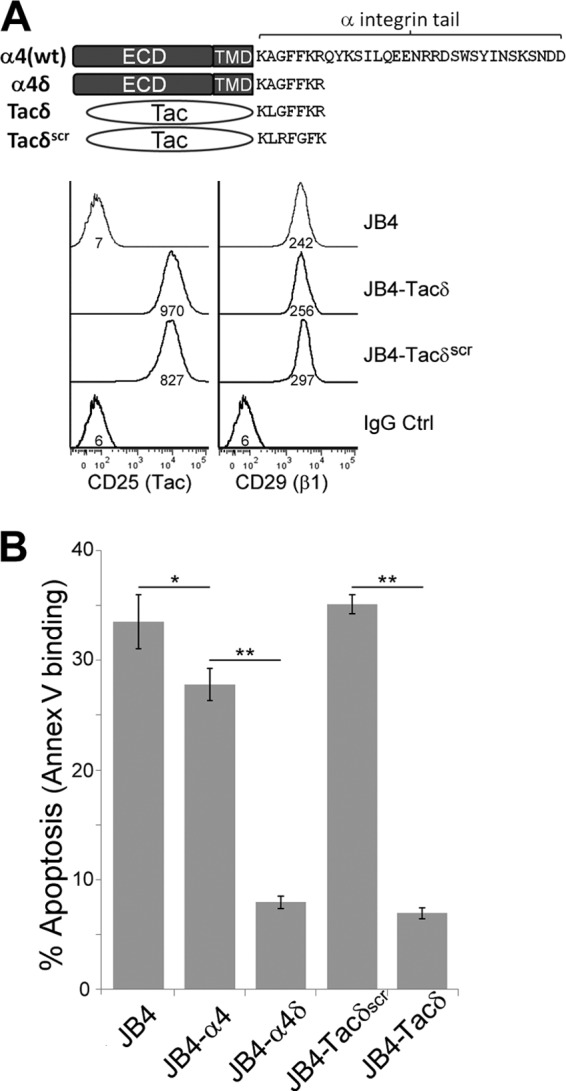
Expression of the membrane-proximal GFFKR motif of the α-integrin tail confers enhanced chemoresistance in an adhesion-independent manner. (A, top) Schematic of additional fusion constructs that express the extracellular and transmembrane domains of the carrier epitope Tac, in comparison with α4 and α4δ. Tacδ and Tacδscr are fusions of Tac with the cytoplasmic peptide, KLGFFKR, and the scrambled version, KLRFGFK, respectively. (Bottom) Flow cytometry determination of cell surface Tac and integrin-β1 expression in JB4, JB4-Tacδ, and JB4-Tacδscr cells. Numbers under the histogram are the MFI (scale, 0.1×). (B) JB4, JB4-α4, JB4-α4δ, JB4-Tacδ, and JB4-Tacδscr cells were plated on GST-coated dishes and treated with 0.03 μg/ml doxorubicin for 48 h. Data plotted are the percentage of cells that were apoptotic, based on flow cytometry determination of Cy5-annexin V binding (means ± standard deviations; n = 3). *, P < 0.03; **, P < 0.01.
α4δ and Tacδ expression circumvents the requirement for adhesion-mediated stimulation of Akt phosphorylation and activation.
Cell adhesion to integrin substrates is known to stimulate activation of Akt (18), which is often implicated in promotion of cell survival (34). To determine if Akt is involved in T cell CAM-DR, we plated cells on integrin substrates and immunoblotted lysates to detect Thr308-phosphorylated Akt. JB4-α4 cells plated on GST-CS1, to engage α4β1, exhibited increased levels of phospho(T308)-Akt at 40 and 60 min poststimulation (Fig. 4A). In contrast, phospho(T308)-Akt levels were unchanged for JB4-α4 cells plated on the control GST substrate, or for JB4 cells plated on GST-CS1, indicating the requirement for integrin ligation to stimulate Akt phosphorylation.
Fig 4.

α4δ and Tacδ expression circumvents the requirement for cell adhesion-mediated stimulation of Akt phosphorylation and activation. (A) JB4 or JB4-α4 cells were plated on dishes coated with GST-CS1 or GST for the indicated times, and cell lysates were immunoblotted to detect T308-phosphorylated Akt (pAkt) and total Akt levels. (B) JB4-Tacδ, JB4-Tacδscr, JB4, JB4-α4δ, and JB4-α4 cells were plated on dishes coated with GST-CS1, GST-Fn9.11, or BSA for 45 min, and cell lysates were immunoblotted to detect pAkt and total Akt levels. (C) JB4-Tacδ and JB4-Tacδscr cells were plated on dishes coated with GST-Fn9.11 (adhesion +ve) or GST (adhesion -ve) for 45 min, and cell lysates were immunoblotted to detect T308- or S473-phosphorylated Akt (pAkt) and total Akt levels. (D) Lysates of JB4-Tacδ and JB4-Tacδscr cells were blotted to detect Akt-phosphorylated substrates and GAPDH. The Akt activation index was calculated as the total fluorescence of Akt-phosphorylated substrates divided by GAPDH activity (means ± standard deviations; 3 independent experiments). *, P < 0.05. (E) Lysates of Jurkat cells plated on dishes coated with BSA or fibronectin for 30 min were blotted to detect Akt-phosphorylated substrates and GAPDH as described for panel D. Data are plotted as the Akt activation index (means ± standard deviations; 3 independent experiments). *, P < 0.03. (F) JB4-Tacδ and JB4-Tacδscr cells were treated with Akt inhibitor IV at the indicated concentrations for 48 h. Data are plotted as the percentage of apoptotic cells, based on flow cytometry determination of Cy5-annexin V binding (means ± standard deviations; n = 3). *, P < 0.02; ns, not significant.
We then compared phospho(T308)-Akt levels for the cell lines plated on CS1, Fn9.11, or BSA. Consistent with the results obtained for doxorubicin-induced apoptosis, cells adherent on ligand substrates corresponding to the expressed integrins had elevated phospho(T308)-Akt levels—in this case, JB4 and JB4-Tacδscr on Fn9.11 and JB4-α4 on CS1 or Fn9.11 (Fig. 4B). Furthermore, JB4-α4δ and JB4-Tacδ cells exhibited elevated levels of phospho(T308)-Akt regardless of substrate (Fig. 4B). Dual phosphorylation of Akt at the T308 and S473 residues is required for its full kinase activity. To determine if KXGFFKR expression differentially regulates Akt phosphorylation at these sites, we compared the lysates of nonadherent and adherent JB4-Tacδ and JB4-Tacδscr cells in immunoblot analyses (Fig. 4C). As before, JB4-Tacδ cells exhibited constitutively high phospho(T308)-Akt levels in an adhesion-independent manner. In contrast, levels of phospho(S473)-Akt were upregulated upon adhesion for both JB4-Tacδ and JB4-Tacδscr cells. Thus, expression of the juxtamembrane KXGFFKR motif led to constitutive phosphorylation of Akt at T308, while phosphorylation of S473 remained adhesion dependent.
To assess the activity of Akt, cell lysates were immunoblotted with an antibody that recognized phosphorylated Akt substrates. Nonadherent JB4-Tacδ cells exhibited higher levels of phosphorylated Akt substrates than did JB4-Tacδscr cells (Fig. 4D). This enhanced level of Akt activation was also observed for Jurkat cells adherent on fibronectin, compared to nonadherent conditions (Fig. 4E). Finally, to assess if the enhanced Akt activity mediated resistance to apoptosis, cells were treated with an inhibitor that blocks Akt activation. We found that a higher concentration of an Akt inhibitor was required to induce a comparable level of apoptosis of JB4-Tacδ cells as that in JB4-Tacδscr cells (Fig. 4F). A higher concentration of inhibitor was also required to reduce the Akt activity observed in lysates of JB4-Tacδ cells relative to JB4-Tacδscr cells (see Fig. S2 in the supplemental material). Taken together, these results indicate that adhesion via integrin ligation to substrate promotes Akt phosphorylation and activation, and also enhanced resistance to apoptosis. The requirement for integrin-mediated adhesion is bypassed for cells expressing the minimal KXGFFKR cytoplasmic tail.
Chemoresistance to doxorubicin is coupled to calcium influx via L-type Ca2+ channels.
We noted with interest in previous studies that T cell adhesion on various substrates promoted elevations of intracellular Ca2+ (35) and that Ca2+ channel-blocking agents can restore chemosensitivity (36). This led us to explore the possibility that integrin-mediated chemoresistance is coupled to regulation of Ca2+ flux in CAM-DR.
First, we measured intracellular Ca2+ levels by using the cell-permeant fluorescent Ca2+ indicator Fluo-4-AM in the various JB4 cells under nonadherent conditions. Cells were prelabeled with Fluo-4-AM and incubated in the presence or absence of available extracellular Ca2+. In the absence of extracellular Ca2+, the intracellular fluorescence of Fluo-4-AM was not significantly different between the cell lines tested (data not shown). In contrast, the presence of extracellular Ca2+ modulated the level of intracellular Ca2+ that is attributable to influx. The influx of extracellular Ca2+ was determined to be highest for JB4-α4δ and JB4-Tacδ cells, intermediate for JB4-α4, and lowest for JB4 and JB4-Tacδscr cells (Fig. 5A). Thus, cells exhibiting enhanced adhesion-independent chemoresistance (Fig. 3B) also exhibited increased Ca2+ influx (Fig. 5A).
Fig 5.

Blockade of L-type calcium channels attenuates chemoresistance to doxorubicin. (A) Intracellular Ca2+ measurements of JB4-derived cell lines via flow cytometry. The indicated cells were labeled with Fluo-4-AM as described in Materials and Methods, washed free of the dye, and resuspended in PBS with or without 1 mM CaCl2 at 22°C for 10 min prior to measurements. Data plotted are the intracellular calcium measurements obtained from 1 mM CaCl2–PBS subtracted from fluorescence with PBS alone (means ± standard deviations; n = 4). *, P < 0.01. (B) JB4-Tacδ and JB4-Tacδscr cells were treated with combinations of 0.04 μg/ml doxorubicin and/or 0.6 mM EGTA for 48 h. Data plotted are the percentage of apoptotic cells, based on flow cytometry determination of Cy5-annexin V binding (means ± standard deviations; n = 4). *, P < 0.001. (C) Fluo-4-AM fluorescence measurements of intracellular Ca2+ in JB4-Tacδ and JB4-Tacδscr cells incubated with combinations of extracellular 1 mM Ca2+ and/or 0.6 mM EGTA (means ± standard deviations; n = 3). *, P < 0.03. (D) JB4-Tacδ and JB4-Tacδscr cells were treated with combinations of 0.03 μg/ml doxorubicin and/or 60 μM verapamil for 48 h. Data plotted are the percentage of apoptotic cells, based on flow cytometry determination of Cy5-annexin V binding (means ± standard deviations; n = 3). *, P < 0.002; **, P < 0.0001. (E) Fluo-4-AM fluorescence measurements of intracellular Ca2+ in JB4-Tacδ and JB4-Tacδscr cells incubated with 1 mM Ca2+ and 0.6 mM EGTA or 60 μM verapamil (means ± standard deviations; n = 3). *, P < 0.001.
Next, we sought to inhibit the effects of KXGFFKR-mediated chemoresistance observed for JB4-Tacδ cells by blocking Ca2+ influx. We assessed the apoptotic indices of cells treated with a combination of doxorubicin and/or EGTA at a concentration sufficient to chelate all extracellular Ca2+ (Fig. 5B and C). Blockade of Ca2+ influx with EGTA increased the apoptotic index of doxorubicin-treated JB4-Tacδscr cells by 2-fold over that of doxorubicin treatment alone. In contrast, EGTA increased the apoptotic index of the chemoresistant JB4-Tacδ cells by 9-fold over that of doxorubicin treatment alone. We then performed the assay in the presence and absence of the Ca2+ channel inhibitor verapamil, to assess if the Ca2+ influx in T cells is also mediated via L-type channels (Fig. 5D and E). At a concentration that blocked ∼70% of extracellular Ca2+ influx, verapamil enhanced the chemosensitivity of JB4-Tacδ cells to doxorubicin by 21-fold over that of doxorubicin alone. By comparison, for the already-chemosensitive JB4-Tacδscr cells, verapamil exerted only a 1.5-fold increase. These results suggested that blockade of extracellular Ca2+ influx via an L-type channel potentiates the apoptotic effects of doxorubicin in an otherwise-chemoresistant cell line.
Finally, we determined if integrin engagement of T cells is sufficient to stimulate increases in intracellular Ca2+. Jurkat cells prelabeled with Fluo-4-AM were seeded onto substrate-coated dishes, and fluorescence was monitored over time (Fig. 6). As expected, all wells exhibited comparable fluorescence at t = 0, indicating that comparable quantities of cells were seeded before significant adhesion had occurred. Over the next 35 min, the intracellular Ca2+ levels of cells plated on CS1 or fibronectin were significantly higher than cells plated on BSA. No changes in intracellular Ca2+ were observed when EGTA was added to chelate extracellular Ca2+, indicating that integrin-mediated adhesion promoted influx of extracellular Ca2+ into cells.
Fig 6.
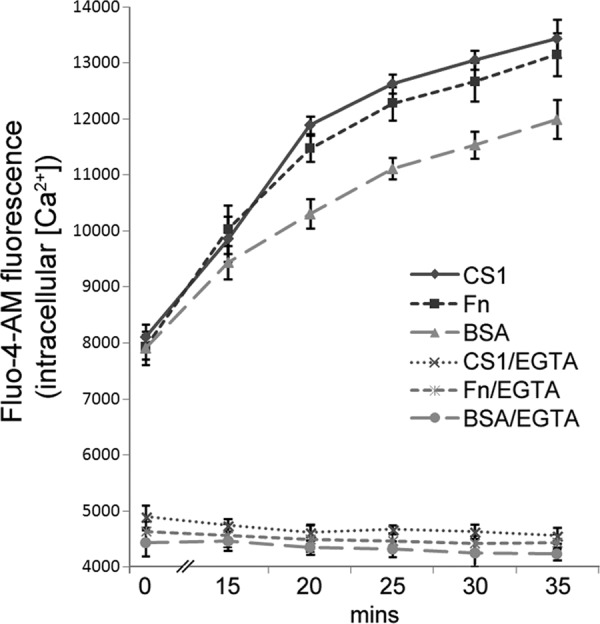
Cell adhesion promotes increases in intracellular Ca2+. Aliquots of Fluo-4-AM-labeled Jurkat cells were seeded onto dishes coated with CS1, fibronectin (Fn), or BSA with or without EGTA added to the extracellular medium. Fluo-4-AM fluorescence were determined at the indicated times following cell seeding. Data plotted are means ± standard deviations (n = 5 replicate wells). P < 0.01 for t = 20 to 35 min for Fn versus BSA; the difference was not significant for t = 0 to 15 min.
Expression of the membrane-proximal KXGFFKR motif leads to enhanced drug efflux.
A well-known cellular physiological adaptation contributing to chemoresistance in leukemia is enhanced expression and activity of drug efflux transporters (36). A recent CAM-DR study reported that integrin-β1 mediated adhesion of Jurkat cells stimulated the expression of the p-glycoprotein transporter MRP1 and decreased intracellular accumulation of doxorubicin (37).
To determine if the adhesion-independent chemoresistance observed for Tacδ expression was attributable to drug efflux activity, cells were incubated with calcein-AM, an indicator substrate used to assess the activity of certain p-glycoprotein-based transporters (38), including MRP1. As shown in Fig. 7A, JB4-Tacδ cells accumulated fluorescent calcein at a significantly lower rate than JB4-Tacδscr cells, indicating that Tacδ expression led to enhanced efflux of calcein-AM. To assess drug efflux, we took advantage of the inherent fluorescence of doxorubicin. Cells were incubated with, and then washed free of, extracellular doxorubicin before being incubated in fresh buffer with or without calcium. Release of cellular doxorubicin back to the buffer was assayed by measuring the fluorescence of the cell-free supernatant. When incubated in Ca2+-supplemented buffer, the supernatant of JB4-Tacδ cells accumulated a significantly higher level of doxorubicin than did that from JB4-Tacδscr cells (Fig. 7B). In contrast, incubation of JB4-Tacδ cells in a calcium-free buffer led to lower rates of doxorubicin efflux (Fig. 7B), indicating that available extracellular Ca2+ is an important modulator of drug transport. Taken together, these results indicate that KXGFFKR expression leads to chemoresistance to doxorubicin that is correlated with enhanced Ca2+ influx and enhanced drug efflux.
Fig 7.
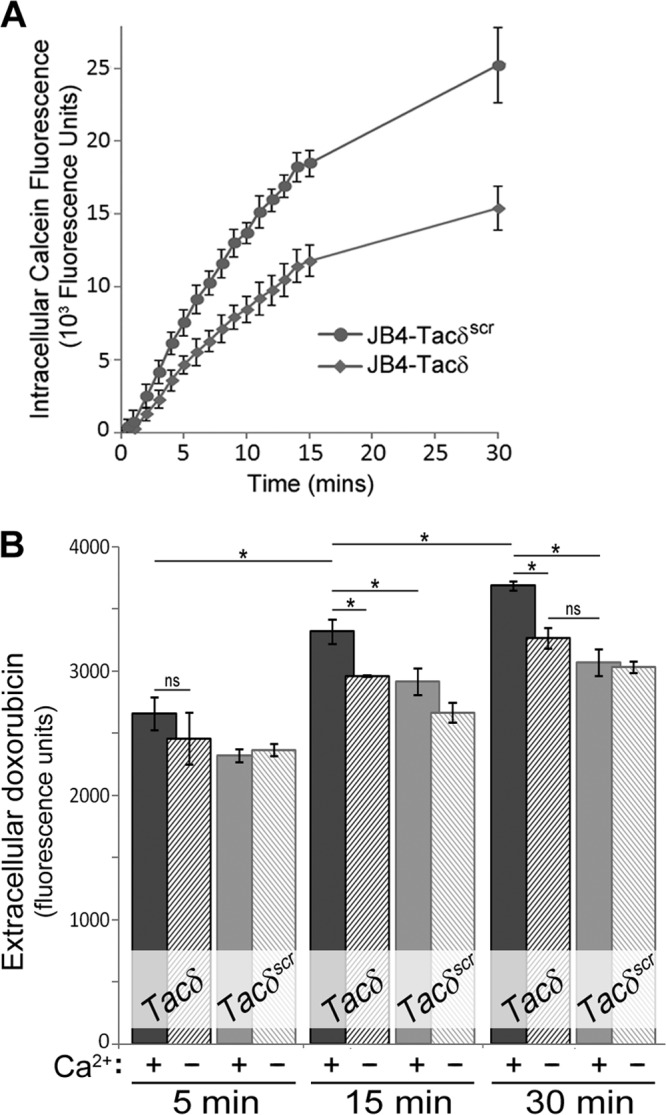
Effects of GFFKR expression on drug efflux. (A) JB4-Tacδ and JB4-Tacδscr cells were incubated with the cell-permeant calcein-AM substrate, and fluorescence measurements were made at the indicated times. Nonfluorescent calcein-AM is hydrolyzed to the highly fluorescent calcein by intracellular esterases; thus, the rate of calcein accumulation is an indirect and inverse measure of the cellular efflux rates of calcein-AM. Data plotted are the mean values ± standard deviations for 4 replicate wells. ns, not significant for t = 0 to 2 min; P < 0.002 for t > 5 min. (B) JB4-Tacδ and JB4-Tacδscr cells were incubated with high doses of doxorubicin, washed free, and resuspended in PBS with (+) or without (−) 1 mM Ca2+. At the indicated times, cell-free supernatants were assessed for fluorescence as an indication of doxorubicin efflux from cells. Sampling for the earliest time point was estimated at 5 min. Data plotted are the means ± standard deviations for 3 replicate cell aliquots. *, P < 0.009; ns, not significant.
Calreticulin interacts with α4, α4δ, and Tacδ via the KXGFFKR motif.
To infer the possible mechanisms responsible for the KXGFFKR-mediated chemoresistance, we evaluated the published literature for α-integrin KXGFFKR-interacting proteins. Calreticulin (CRT), a ubiquitous multifunctional Ca2+-binding protein, was a favored candidate, since CRT is implicated in regulation of apoptosis (39), integrin-mediated adhesion (25), focal adhesion assembly (40), and adhesion-mediated Ca2+ influx (25, 41), and it associates with several α-integrins (41–43).
We first determined if expression of the various constructs impacted total CRT levels, since cell sensitivity to chemotherapeutics is modulated by changes in total CRT (39). By Western blotting analyses, we found comparable total CRT levels in all of our JB4-based cell lines (Fig. 8A). To determine if CRT interacts with KXGFFKR, Jurkat cell lysates were incubated with matrix-immobilized GST-KLGFFKR or GST-KLRFGFK fusion proteins. CRT was detected at higher levels in a complex with GST-KLGFFKR, indicating the specificity of the interaction (Fig. 8B). To determine if CRT interacts with α4-integrin, we performed immunoprecipitation assays. Using lysates derived from nonadherent cells, we detected much higher levels of CRT immunoprecipitated with α4δ than with α4 (Fig. 8C). In a similar fashion, higher levels of CRT immunoprecipitated with Tacδ than with Tacδscr (Fig. 8D). Since cells expressing wild-type α4 exhibited enhanced Akt signaling and chemoresistance only in the adherent state, we determined if adhesion may also stimulate enhanced CRT association with α4. Adhesion of Jurkat cells on either CS1 or fibronectin stimulated increased levels of CRT associated with immunoprecipitated α4 (Fig. 8E). Thus, calreticulin associates with the truncated α4δ in a manner requiring the KXGFFKR peptide motif, and adhesion acts as a stimulus to enhance the interaction of calreticulin with wild-type α4.
Fig 8.
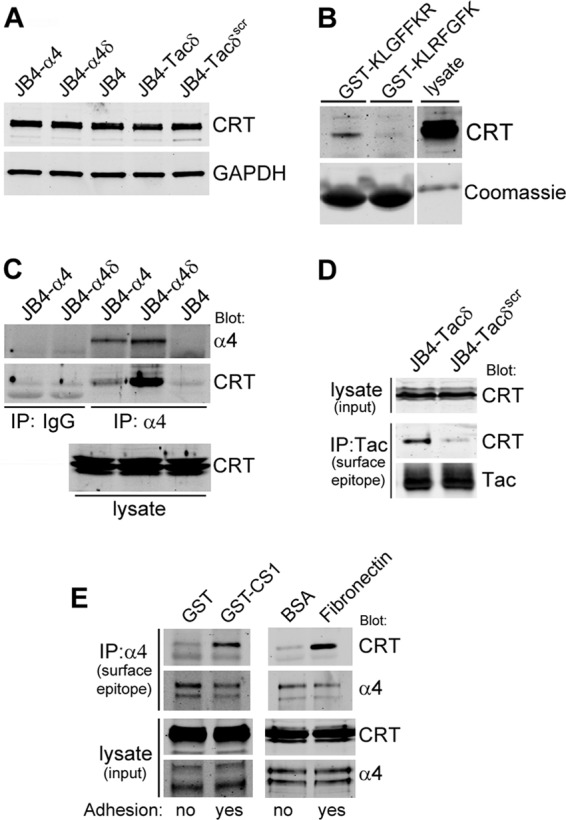
Calreticulin interacts with α4 and Tacδ via the GFFKR motif. (A) Comparison of total CRT expression in lysates of JB4-α4, JB4-α4δ, JB4, JB4-Tacδ, and JB4-Tacδscr cells by Western blotting. Equal protein loading was assessed by immunoblotting for GAPDH. (B) Affinity chromatography using matrix-immobilized GST-KLGFFKR and GST-KLRFGFK (scrambled) recombinant proteins incubated with Jurkat cell lysates. Binding of CRT was assessed by Western blotting. Loading of the GST recombinant proteins was visualized by Coomassie blue staining. (C) Lysates prepared from JB4-α4, JB4-α4δ, and JB4 cells were immunoprecipitated (IP) with an α4 surface epitope antibody or IgG control, and associated proteins were detected with antibodies against CRT and α4. (D) Lysates prepared from JB4-Tacδ and JB4-Tacδscr cells were immunoprecipitated with a Tac surface epitope antibody, and associated proteins were detected with antibodies against CRT and Tac. (E) Lysates prepared from Jurkat cells were plated on the indicated substrates for 45 min and then immunoprecipitated with an α4 surface epitope antibody; associated proteins were detected by blotting with antibodies against CRT and α4.
DISCUSSION
Our study describes the contribution of the juxtamembrane KXGFFKR cytoplasmic motif of α-integrins to chemoresistance in a T-lymphocyte model. Using cells reconstituted with wild-type α4 expression, we confirmed that T cell CAM-DR requires α4β1-mediated engagement with its substrate. CAM-DR in T cells may be supported by other β1-containing integrins as well, as cells lacking α4 expression exhibit chemoresistance when adhered to substrates for the corresponding expressed integrin. Reconstituted expression with the mutant α4δ, where the cytoplasmic domain is truncated to the minimal KXGFFKR motif, revealed a form of chemoresistance that was adhesion independent. Expression of a nonintegrin transmembrane fusion protein bearing KXGFFKR as the cytoplasmic domain also conferred an adhesion-independent chemoresistance phenotype. Thus, the KXGFFKR sequence conserved in α-integrins constitutes a common prosurvival regulatory motif.
As a major integrin expressed by hematopoietic cells, α4-integrin has been implicated in CAM-DR of various hematologic malignancies (3–5, 44, 45). However, chemoresistance upon adhesion to substrates engaging other integrins, including α5, α6, and α2 (6–8, 17), hints at the generality of this phenomenon. A common denominator for these α-integrins is their pairing with β1, without which the adhesion receptor is incomplete. Another denominator is the conserved KXGFFKR motif, which is found in 15 out of the 18 known human α-integrins (11), and of which the α-tail sequences C-terminal to KXGFFKR are largely divergent. Interactions involving the α4-tail have been characterized, with reported effects on cell adhesion and migration (27, 28, 32). We had anticipated abrogation of CAM-DR upon reconstituted expression of the tail-truncated α4δ. Although cell adhesion to the α4β1-specific substrate was predictably disrupted (20), α4δ-expressing cells exhibited chemoresistance in the absence of integrin-mediated adhesion. The contribution of β1 (as α4δβ1) in adhesion-independent chemoresistance may be negated upon expression of the monomeric fusion Tacδ construct, indicating that the KXGFFKR motif is sufficient to promote chemoresistance normally stimulated by integrin-mediated adhesion.
Cells expressing the Tacδ construct favored an opportunity to assess the role of a conserved motif found in α-integrins in regulatory roles and survival signaling typically stimulated by adhesion. We found that T308-Akt was constitutively highly phosphorylated in KXGFFKR-expressing cells exhibiting high levels of Ca2+ influx, while levels of S473-Akt phosphorylation remained adhesion regulated. Importantly, higher pT308-Akt levels correlated with higher levels of Akt activity detected in lysates of nonadherent Tacδ cells, suggesting that pT308-Akt represent an important prosurvival event that is regulated downstream of the α-integrin KXGFFKR motif. Our results are also consistent with the hypothesis postulated by other groups that the phosphorylation status of T308-Akt may be a more reliable indicator for Akt activity (46), and this measure may serve as a better prognosticator for certain tumor outcomes that include acute leukemias (47).
The increased Ca2+ influx may also explain the adhesion-independent high pT308-Akt and Akt activation levels that we observed in α4δ- and Tacδ-expressing cells, as well as for adhered cells expressing wild-type α4. As reported previously, the Ca2+/calmodulin-dependent protein kinase kinase can directly phosphorylate T308-Akt in a Ca2+-dependent manner (48). This raises the interesting possibility that high levels of intracellular Ca2+ sustain Akt activation following the initial activation via the classical phosphatidylinositol kinase-dependent processes (49).
Rises of intracellular Ca2+ levels stimulated by integrin-mediated adhesion have been reported in cell types that include myocytes (50) and T lymphocytes (35). The Ca2+ increases appear to involve influx of extracellular of Ca2+ via L-type channels in the plasma membrane, as well as Ca2+ release from intracellular stores (41). In our assays in which we used Fluo-4-AM to measure free cytosolic Ca2+, we observed that the availability of extracellular Ca2+ is an important determinant for obtaining the intracellular Ca2+ increases mediated by integrin signaling. Since extracellular Ca2+ is required to support integrin-mediated adhesion, we cannot conclude that adhesion-stimulated rises in intracellular Ca2+ can occur without influx of extracellular Ca2+. However, cells expressing α4δ or Tacδ did not require adhesion to promote the measured increases in intracellular Ca2+, as long as extracellular Ca2+ was available. Verapamil was able to block the Ca2+ influx associated with Tacδ expression; thus, our findings are consistent with the involvement of L-type channels in α-integrin KXGFFKR motif-mediated Ca2+ transport (41).
Ca2+ influx has been associated with the drug efflux function mediated by p-glycoprotein transporters and is thus a possible modulator of chemoresistance (51). This relationship is decidedly complex, as use of various Ca2+ indicators as well as Ca2+ channel inhibitors, such as verapamil, revealed interactions with the p-glycoprotein transporters themselves and a possible source of complication. We attempted to control for these effects in our assays, as follows. Drug efflux was assessed using both the calcein-AM assay as well as measuring release of doxorubicin from cells. In both cases, enhanced efflux was obtained for cells expressing Tacδ. Fluo-4-AM, which is used to assess intracellular Ca2+, may itself be a substrate for efflux by p-glycoproteins. However, our assay conditions revealed higher intracellular Fluo-4-AM fluorescence (and hence Ca2+) for α4δ and Tacδ cells; thus, any loss due to Fluo-4-AM efflux was minimal. We assessed the contribution of Ca2+ influx to chemoresistance either by chelating extracellular Ca2+ with EGTA or by inhibiting L-type Ca2+ channels with verapamil. At concentrations that appreciably reduced Ca2+ influx, we were able to show a synergistic effect on the apoptosis-inducing effects of doxorubicin. Thus, our results support a correlation between intracellular Ca2+ levels, drug efflux, and apoptosis.
The α-integrin KXGFFKR motif interacts with several proteins that have been described to regulate integrin function, including sharpin, MDGI, Mss4, CIB, and calreticulin (21–25). We analyzed the published data to identify the candidate effector that best described the data obtained in our studies. Both sharpin and MDGI act as inhibitors of integrin activation and cell adhesion (21, 22). If α4δ or Tacδ expression resulted in sharpin or MDGI binding to the exposed KXGFFKR, then the adhesion and chemoresistance observed via other integrins, such as α5β1 ligation to Fn9.11, would be expected to be increased in α4δ- or Tacδ-expressing cells, a phenomenon we did not observe. Mss4 is implicated in secretion of matrix metalloproteinases and fibronectin remodeling (23), neither of which is applicable with our assay system. CIB is a calcium- and integrin-binding protein; however, detailed interaction studies have indicated specificity for αIIb-integrin sequences N-terminal to and in addition to KXGFFKR (24), sequences not found in α4δ or Tacδ.
This leaves calreticulin, a ubiquitous calcium-binding and chaperone protein found predominantly within the lumen of the endoplasmic reticulum (ER) (52) and whose reported interactions with integrins α2 and α7 are associated with transient Ca2+ fluxes upon integrin-substrate ligation (25, 41). In immunoprecipitation studies, we showed an increased interaction of calreticulin with the truncated KXGFFKR motif of nonadherent cells and with α4-integrins from adhesion-stimulated cells. Thus, this study adds to the list of α-integrins that show this adhesion-stimulated phenomenon, which now includes integrins α2, α3, α4, α6, and α7 (41–43, 53, 54). It remains unclear how an ER-resident protein like calreticulin may bind to the plasma membrane-proximal cytoplasmic tail of α-integrins. The stoichiometry of the interaction that we observed was estimated to be very low. This is not surprising, considering the disparate locales of the interacting partners, with most of the calreticulin found within the ER lumen. Our attempts to visualize and quantitate their colocalization by immunofluorescence imaging in intact cells were inconclusive (data not shown), as we were hampered by the abundance of ER-resident calreticulin relative to levels found in the cytosol. Thus, the connection between the CRT–α-integrin interaction with that of Ca2+-mediated chemoresistance remains a correlative one.
Yet, it remains plausible that the hypothetical integrin-calreticulin interaction can occur, and this deserves discussion. An increasing body of work has highlighted the non-ER-resident localization and function of calreticulin, which includes cytosolic, cell surface, and secreted forms (55). Several elegant studies have highlighted mechanisms that can account for the minor cytosolic localization of calreticulin (56, 57). The demonstration that crt null cells have impaired integrin-mediated adhesion and adhesion-stimulated Ca2+ influx (25) suggests a more intimate role for calreticulin and integrin function. This was further supported by the finding that reconstituted expression of a cytosolic targeted form of CRT was able to rescue the adhesion defect exhibited by crt null fibroblasts (56).
It has now been established from extensive structure-function-based studies that activated integrins undergo conformational changes that include the physical separation of the cytoplasmic domains of α- and β-integrins (11, 58). Extrapolating from the available evidence, we postulate that for cells expressing the minimal KXGFFKR motif (α4δ and Tacδ), KXGFFKR is potentially accessible for binding to a prosurvival factor (such as CRT) in the absence of adhesion. For cells expressing full-length α4- or α5-integrins, structural changes within the integrin dimer accompanying integrin-mediated adhesion may facilitate the increased association. Influx of Ca2+ may be mediated via CRT's L-type channel regulatory function and/or the Ca2+ buffering capacity, triggering Akt-mediated prosurvival signaling and in the case for chemoresistance, increased drug efflux. Thus, cell adhesion via integrins forms a switch for activation of prosurvival signaling and chemoresistance.
Supplementary Material
ACKNOWLEDGMENTS
We extend our appreciation to Catherine Pallen and Chris Maxwell for providing critical input throughout the project. Qian Li provided initial technical support.
This work was supported by grants from the Leukemia and Lymphoma Society of Canada, Canadian Institutes of Health Research, and the Canada Foundation for Innovation to C.J.L. C.C.L. received a graduate award from the Michael Cuccione Foundation/Child and Family Research Institute. C.J.L. was supported by the Michael Cuccione Foundation and holds an investigatorship from the Child and Family Research Institute.
Footnotes
Published ahead of print 3 September 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.00580-13.
REFERENCES
- 1.Estey E, Dohner H. 2006. Acute myeloid leukaemia. Lancet 368:1894–1907 [DOI] [PubMed] [Google Scholar]
- 2.Pui CH, Evans WE. 2006. Treatment of acute lymphoblastic leukemia. N. Engl. J. Med. 354:166–178 [DOI] [PubMed] [Google Scholar]
- 3.Matsunaga T, Takemoto N, Sato T, Takimoto R, Tanaka I, Fujimi A, Akiyama T, Kuroda H, Kawano Y, Kobune M, Kato J, Hirayama Y, Sakamaki S, Kohda K, Miyake K, Niitsu Y. 2003. Interaction between leukemic-cell VLA-4 and stromal fibronectin is a decisive factor for minimal residual disease of acute myelogenous leukemia. Nat. Med. 9:1158–1165 [DOI] [PubMed] [Google Scholar]
- 4.Damiano JS, Cress AE, Hazlehurst LA, Shtil AA, Dalton WS. 1999. Cell adhesion mediated drug resistance (CAM-DR): role of integrins and resistance to apoptosis in human myeloma cell lines. Blood 93:1658–1667 [PMC free article] [PubMed] [Google Scholar]
- 5.Matsunaga T, Fukai F, Miura S, Nakane Y, Owaki T, Kodama H, Tanaka M, Nagaya T, Takimoto R, Takayama T, Niitsu Y. 2008. Combination therapy of an anticancer drug with the FNIII14 peptide of fibronectin effectively overcomes cell adhesion-mediated drug resistance of acute myelogenous leukemia. Leukemia 22:353–360 [DOI] [PubMed] [Google Scholar]
- 6.Damiano JS, Hazlehurst LA, Dalton WS. 2001. Cell adhesion-mediated drug resistance (CAM-DR) protects the K562 chronic myelogenous leukemia cell line from apoptosis induced by BCR/ABL inhibition, cytotoxic drugs, and gamma-irradiation. Leukemia 15:1232–1239 [DOI] [PubMed] [Google Scholar]
- 7.De Toni-Costes F, Despeaux M, Bertrand J, Bourogaa E, Ysebaert L, Payrastre B, Racaud-Sultan C. 2010. A new α5β1 integrin-dependent survival pathway through GSK3β activation in leukemic cells. PLoS One 5(3):e9807. 10.1371/journal.pone.0009807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yamakawa N, Kaneda K, Saito Y, Ichihara E, Morishita K. 2012. The increased expression of integrin α6 (ITGA6) enhances drug resistance in EVI1high leukemia. PLoS One 7(1):e30706. 10.1371/journal.pone.0030706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mudry RE, Fortney JE, York T, Hall BM, Gibson LF. 2000. Stromal cells regulate survival of B-lineage leukemic cells during chemotherapy. Blood 96:1926–1932 [PubMed] [Google Scholar]
- 10.Meads MB, Gatenby RA, Dalton WS. 2009. Environment-mediated drug resistance: a major contributor to minimal residual disease. Nat. Rev. Cancer 9:665–674 [DOI] [PubMed] [Google Scholar]
- 11.Abram CL, Lowell CA. 2009. The ins and outs of leukocyte integrin signaling. Annu. Rev. Immunol. 27:339–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hynes RO. 2002. Integrins: bidirectional, allosteric signaling machines. Cell 110:673–687 [DOI] [PubMed] [Google Scholar]
- 13.Rose DM, Han J, Ginsberg MH. 2002. α4 integrins and the immune response. Immunol. Rev. 186:118–124 [DOI] [PubMed] [Google Scholar]
- 14.Mraz M, Zent CS, Church AK, Jelinek DF, Wu X, Pospisilova S, Ansell SM, Novak AJ, Kay NE, Witzig TE, Nowakowski GS. 2011. Bone marrow stromal cells protect lymphoma B-cells from rituximab-induced apoptosis and targeting integrin alpha-4-beta-1 (VLA-4) with natalizumab can overcome this resistance. Br. J. Haematol. 155:53–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kurtova AV, Tamayo AT, Ford RJ, Burger JA. 2009. Mantle cell lymphoma cells express high levels of CXCR4, CXCR5, and VLA-4 (CD49d): importance for interactions with the stromal microenvironment and specific targeting. Blood 113:4604–4613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rettig MP, Ansstas G, DiPersio JF. 2012. Mobilization of hematopoietic stem and progenitor cells using inhibitors of CXCR4 and VLA-4. Leukemia 26:34–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Naci D, El Azreq MA, Chetoui N, Lauden L, Sigaux F, Charron D, Al-Daccak R, Aoudjit F. 2012. α2β1 integrin promotes chemoresistance against doxorubicin in cancer cells through extracellular signal-regulated kinase (ERK). J. Biol. Chem. 287:17065–17076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.King WG, Mattaliano MD, Chan TO, Tsichlis PN, Brugge JS. 1997. Phosphatidylinositol 3-kinase is required for integrin-stimulated AKT and Raf-1/mitogen-activated protein kinase pathway activation. Mol. Cell. Biol. 17:4406–4418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aoudjit F, Vuori K. 2012. Integrin signaling in cancer cell survival and chemoresistance. Chemother. Res. Pract. 2012:283181. 10.1155/2012/283181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kassner PD, Kawaguchi S, Hemler ME. 1994. Minimum alpha chain cytoplasmic tail sequence needed to support integrin-mediated adhesion. J. Biol. Chem. 269:19859–19867 [PubMed] [Google Scholar]
- 21.Rantala JK, Pouwels J, Pellinen T, Veltel S, Laasola P, Mattila E, Potter CS, Duffy T, Sundberg JP, Kallioniemi O, Askari JA, Humphries MJ, Parsons M, Salmi M, Ivaska J. 2011. SHARPIN is an endogenous inhibitor of β1-integrin activation. Nat. Cell Biol. 13:1315–1324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nevo J, Mai A, Tuomi S, Pellinen T, Pentikainen OT, Heikkila P, Lundin J, Joensuu H, Bono P, Ivaska J. 2010. Mammary-derived growth inhibitor (MDGI) interacts with integrin alpha-subunits and suppresses integrin activity and invasion. Oncogene 29:6452–6463 [DOI] [PubMed] [Google Scholar]
- 23.Knoblauch A, Will C, Goncharenko G, Ludwig S, Wixler V. 2007. The binding of Mss4 to alpha-integrin subunits regulates matrix metalloproteinase activation and fibronectin remodeling. FASEB J. 21:497–510 [DOI] [PubMed] [Google Scholar]
- 24.Barry WT, Boudignon-Proudhon C, Shock DD, McFadden A, Weiss JM, Sondek J, Parise LV. 2002. Molecular basis of CIB binding to the integrin alpha IIb cytoplasmic domain. J. Biol. Chem. 277:28877–28883 [DOI] [PubMed] [Google Scholar]
- 25.Coppolino MG, Woodside MJ, Demaurex N, Grinstein S, St-Arnaud R, Dedhar S. 1997. Calreticulin is essential for integrin-mediated calcium signalling and cell adhesion. Nature 386:843–847 [DOI] [PubMed] [Google Scholar]
- 26.Nishiya N, Kiosses WB, Han J, Ginsberg MH. 2005. An α4 integrin-paxillin-Arf-GAP complex restricts Rac activation to the leading edge of migrating cells. Nat. Cell Biol. 7:343–352 [DOI] [PubMed] [Google Scholar]
- 27.Lim CJ, Han J, Yousefi N, Ma Y, Amieux PS, McKnight GS, Taylor SS, Ginsberg MH. 2007. α4 integrins are type I cAMP-dependent protein kinase-anchoring proteins. Nat. Cell Biol. 9:415–421 [DOI] [PubMed] [Google Scholar]
- 28.Rivera Rosado LA, Horn TA, McGrath SC, Cotter RJ, Yang JT. 2011. Association between α4 integrin cytoplasmic tail and non-muscle myosin IIA regulates cell migration. J. Cell Sci. 124:483–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rose DM, Liu S, Woodside DG, Han J, Schlaepfer DD, Ginsberg MH. 2003. Paxillin binding to the alpha 4 integrin subunit stimulates LFA-1 (integrin alpha L beta 2)-dependent T cell migration by augmenting the activation of focal adhesion kinase/proline-rich tyrosine kinase-2. J. Immunol. 170:5912–5918 [DOI] [PubMed] [Google Scholar]
- 30.Jongewaard IN, Tsai PM, Smith JW. 1996. The type III connecting segment of fibronectin contains an aspartic acid residue that regulates the rate of binding to integrin alpha 4 beta 1. Cell Adhes. Commun. 3:487–495 [DOI] [PubMed] [Google Scholar]
- 31.Ramos JW, DeSimone DW. 1996. Xenopus embryonic cell adhesion to fibronectin: position-specific activation of RGD/synergy site-dependent migratory behavior at gastrulation. J. Cell Biol. 134:227–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu S, Thomas SM, Woodside DG, Rose DM, Kiosses WB, Pfaff M, Ginsberg MH. 1999. Binding of paxillin to α4 integrins modifies integrin-dependent biological responses. Nature 402:676–681 [DOI] [PubMed] [Google Scholar]
- 33.Bodeau AL, Berrier AL, Mastrangelo AM, Martinez R, LaFlamme SE. 2001. A functional comparison of mutations in integrin beta cytoplasmic domains: effects on the regulation of tyrosine phosphorylation, cell spreading, cell attachment and β1 integrin conformation. J. Cell Sci. 114:2795–2807 [DOI] [PubMed] [Google Scholar]
- 34.Song G, Ouyang G, Bao S. 2005. The activation of Akt/PKB signaling pathway and cell survival. J. Cell. Mol. Med. 9:59–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weismann M, Guse AH, Sorokin L, Broker B, Frieser M, Hallmann R, Mayr GW. 1997. Integrin-mediated intracellular Ca2+ signaling in Jurkat T lymphocytes. J. Immunol. 158:1618–1627 [PubMed] [Google Scholar]
- 36.Xia CQ, Smith PG. 2012. Drug efflux transporters and multidrug resistance in acute leukemia: therapeutic impact and novel approaches to mediation. Mol. Pharmacol. 82:1008–1021 [DOI] [PubMed] [Google Scholar]
- 37.El Azreq MA, Naci D, Aoudjit F. 2012. Collagen/β1 integrin signaling up-regulates the ABCC1/MRP-1 transporter in an ERK/MAPK-dependent manner. Mol. Biol. Cell 23:3473–3484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Homolya L, Hollo Z, Germann UA, Pastan I, Gottesman MM, Sarkadi B. 1993. Fluorescent cellular indicators are extruded by the multidrug resistance protein. J. Biol. Chem. 268:21493–21496 [PubMed] [Google Scholar]
- 39.Nakamura K, Bossy-Wetzel E, Burns K, Fadel MP, Lozyk M, Goping IS, Opas M, Bleackley RC, Green DR, Michalak M. 2000. Changes in endoplasmic reticulum luminal environment affect cell sensitivity to apoptosis. J. Cell Biol. 150:731–740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Goicoechea S, Orr AW, Pallero MA, Eggleton P, Murphy-Ullrich JE. 2000. Thrombospondin mediates focal adhesion disassembly through interactions with cell surface calreticulin. J. Biol. Chem. 275:36358–36368 [DOI] [PubMed] [Google Scholar]
- 41.Kwon MS, Park CS, Choi K, Ahnn J, Kim JI, Eom SH, Kaufman SJ, Song WK. 2000. Calreticulin couples calcium release and calcium influx in integrin-mediated calcium signaling. Mol. Biol. Cell 11:1433–1443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Coppolino M, Leung-Hagesteijn C, Dedhar S, Wilkins J. 1995. Inducible interaction of integrin alpha 2 beta 1 with calreticulin. Dependence on the activation state of the integrin. J. Biol. Chem. 270:23132–23138 [DOI] [PubMed] [Google Scholar]
- 43.Coppolino MG, Dedhar S. 1999. Ligand-specific, transient interaction between integrins and calreticulin during cell adhesion to extracellular matrix proteins is dependent upon phosphorylation/dephosphorylation events. Biochem. J. 340:41–50 [PMC free article] [PubMed] [Google Scholar]
- 44.de la Fuente MT, Casanova B, Moyano JV, Garcia-Gila M, Sanz L, Garcia-Marco J, Silva A, Garcia-Pardo A. 2002. Engagement of α4β1 integrin by fibronectin induces in vitro resistance of B chronic lymphocytic leukemia cells to fludarabine. J. Leukoc. Biol. 71:495–502 [PubMed] [Google Scholar]
- 45.Gattei V, Bulian P, Del Principe MI, Zucchetto A, Maurillo L, Buccisano F, Bomben R, Dal-Bo M, Luciano F, Rossi FM, Degan M, Amadori S, Del Poeta G. 2008. Relevance of CD49d protein expression as overall survival and progressive disease prognosticator in chronic lymphocytic leukemia. Blood 111:865–873 [DOI] [PubMed] [Google Scholar]
- 46.Vincent EE, Elder DJ, Thomas EC, Phillips L, Morgan C, Pawade J, Sohail M, May MT, Hetzel MR, Tavare JM. 2011. Akt phosphorylation on Thr308 but not on Ser473 correlates with Akt protein kinase activity in human non-small cell lung cancer. Br. J. Cancer 104:1755–1761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gallay N, Dos Santos C, Cuzin L, Bousquet M, Simmonet Gouy V, Chaussade C, Attal M, Payrastre B, Demur C, Recher C. 2009. The level of AKT phosphorylation on threonine 308 but not on serine 473 is associated with high-risk cytogenetics and predicts poor overall survival in acute myeloid leukaemia. Leukemia 23:1029–1038 [DOI] [PubMed] [Google Scholar]
- 48.Yano S, Tokumitsu H, Soderling TR. 1998. Calcium promotes cell survival through CaM-K kinase activation of the protein-kinase-B pathway. Nature 396:584–587 [DOI] [PubMed] [Google Scholar]
- 49.Vanhaesebroeck B, Alessi DR. 2000. The PI3K-PDK1 connection: more than just a road to PKB. Biochem. J. 346:561–576 [PMC free article] [PubMed] [Google Scholar]
- 50.Wu X, Davis GE, Meininger GA, Wilson E, Davis MJ. 2001. Regulation of the L-type calcium channel by alpha 5 beta 1 integrin requires signaling between focal adhesion proteins. J. Biol. Chem. 276:30285–30292 [DOI] [PubMed] [Google Scholar]
- 51.Sulova Z, Seres M, Barancik M, Gibalova L, Uhrik B, Polekova L, Breier A. 2009. Does any relationship exist between P-glycoprotein-mediated multidrug resistance and intracellular calcium homeostasis. Gen. Physiol. Biophys. 28(Focus Iss):F89–F95 [PubMed] [Google Scholar]
- 52.Michalak M, Groenendyk J, Szabo E, Gold LI, Opas M. 2009. Calreticulin, a multi-process calcium-buffering chaperone of the endoplasmic reticulum. Biochem. J. 417:651–666 [DOI] [PubMed] [Google Scholar]
- 53.Tran H, Pankov R, Tran SD, Hampton B, Burgess WH, Yamada KM. 2002. Integrin clustering induces kinectin accumulation. J. Cell Sci. 115:2031–2040 [DOI] [PubMed] [Google Scholar]
- 54.Zhu Q, Zelinka P, White T, Tanzer ML. 1997. Calreticulin-integrin bidirectional signaling complex. Biochem. Biophys. Res. Commun. 232:354–358 [DOI] [PubMed] [Google Scholar]
- 55.Gold LI, Eggleton P, Sweetwyne MT, Van Duyn LB, Greives MR, Naylor SM, Michalak M, Murphy-Ullrich JE. 2010. Calreticulin: non-endoplasmic reticulum functions in physiology and disease. FASEB J. 24:665–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Afshar N, Black BE, Paschal BM. 2005. Retrotranslocation of the chaperone calreticulin from the endoplasmic reticulum lumen to the cytosol. Mol. Cell. Biol. 25:8844–8853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shaffer KL, Sharma A, Snapp EL, Hegde RS. 2005. Regulation of protein compartmentalization expands the diversity of protein function. Dev. Cell 9:545–554 [DOI] [PubMed] [Google Scholar]
- 58.Kim C, Ye F, Ginsberg MH. 2011. Regulation of integrin activation. Annu. Rev. Cell Dev. Biol. 27:321–345 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.