

Abstract
The stimulation of Toll-like receptors (TLRs) on macrophages by pathogen-associated molecular patterns (PAMPs) results in the activation of intracellular signaling pathways that are required for initiating a host immune response. Both phosphatidylinositol 3-kinase (PI3K)–Akt and p38 mitogen-activated protein kinase (MAPK) signaling pathways are activated rapidly in response to TLR activation and are required to coordinate effective host responses to pathogen invasion. In this study, we analyzed the role of the p38-dependent kinases MK2/3 in the activation of Akt and show that lipopolysaccharide (LPS)-induced phosphorylation of Akt on Thr308 and Ser473 requires p38α and MK2/3. In cells treated with p38 inhibitors or an MK2/3 inhibitor, phosphorylation of Akt on Ser473 and Thr308 is reduced and Akt activity is inhibited. Furthermore, BMDMs deficient in MK2/3 display greatly reduced phosphorylation of Ser473 and Thr308 following TLR stimulation. However, MK2/3 do not directly phosphorylate Akt in macrophages but act upstream of PDK1 and mTORC2 to regulate Akt phosphorylation. Akt is recruited to phosphatidylinositol 3,4,5-trisphosphate (PIP3) in the membrane, where it is activated by PDK1 and mTORC2. Analysis of lipid levels in MK2/3-deficient bone marrow-derived macrophages (BMDMs) revealed a role for MK2/3 in regulating Akt activity by affecting availability of PIP3 at the membrane. These data describe a novel role for p38α-MK2/3 in regulating TLR-induced Akt activation in macrophages.
INTRODUCTION
The phosphatidylinositol 3-kinase (PI3K) signaling system is highly conserved across eukaryotic species and plays critical roles in transducing a wide range of signals into appropriate cellular responses. Class I PI3Ks phosphorylate phosphatidylinositol 4,5-bisphosphate (PIP2) to generate phosphatidylinositol 3,4,5-trisphosphate (PIP3), and this results in the activation of several downstream pathways. One important, and extensively studied, enzyme activated downstream of PI3K is the Ser/Thr kinase Akt (also known as protein kinase B [PKB]), of which 3 distinct isoforms have been identified in mammalian cells (1–3). Akt has attracted considerable interest due to its roles in cell survival and insulin signaling, although PI3K-Akt signaling also has roles in immunity. PI3K signaling is involved in the development and proliferation of T and B cells (4). The PI3K-Akt signaling pathway has also been suggested to regulate the responses of macrophages to inflammatory stimuli (5, 6). Studies using early PI3K inhibitors such as wortmannin or LY-294002 have produced conflicting reports with both positive and negative roles in regulating the production of inflammatory cytokines being described (5, 7). Some of these differences could be explained by off-target activities of these inhibitors. Genetic studies have also provided evidence for a role for PI3K in innate immunity, as dendritic cells (DCs) lacking the p85 subunit of PI3K express higher levels of interleukin 12 (IL-12) (8) and transgenic mice expressing constitutively active Akt (Myr-Akt) express higher levels of IL-10 following lipopolysaccharide (LPS) challenge (9), lending support for the notion that PI3K-Akt activation downstream of Toll-like receptor (TLR) signaling may act as a safety mechanism to limit the response to pathogens. Furthermore, PI3K is involved in macrophage migration and phagocytosis (10, 11), with p85 alpha and beta double-knockout cells demonstrating a key role for PI3K in phagosomal cup formation (12).
The generation of PIP3 by PI3K induces Akt activation by recruiting it to the membrane via interaction of its pleckstrin homology (PH) domain with PIP3. Once at the membrane, Akt is then activated by phosphorylation on two sites: Thr308 in the T-loop of the kinase domain and Ser473 in the C-terminal hydrophobic motif. Thr308 phosphorylation is critical for Akt activation, and Thr308 is phosphorylated in vivo by PDK1 (3-phosphoinositide-dependent protein kinase 1), which is also recruited by PIP3 to the membrane via its own PH domain (13, 14). Embryonic stem (ES) cells from mice lacking PDK1 are unable to phosphorylate Akt on Thr308, while ES cells with a mutation of the PH domain in PDK1 that prevents its recruitment to the membrane show significantly reduced activation of Akt in response to insulin-like growth factor (IGF) (15–17).
While PDK1 has been clearly established as the kinase responsible for Thr308 phosphorylation, the kinase responsible for phosphorylation of the hydrophobic motif has been more elusive. Several kinases have been proposed as Ser473 kinases for Akt, including MK2 (mitogen-activated protein kinase [MAPK]-activated protein kinase 2), integrin-linked kinase (ILK), double-stranded DNA-dependent protein kinase (DNA-PK), protein kinase Cα, and ataxia telangiectasia mutated gene product (ATM) (18–22). However, the relevance of these kinases to in vivo phosphorylation has been controversial (23). It has since been established that mTORC2 (mammalian target of rapamycin complex 2), consisting of mTOR, Rictor, Sin1, mLST8, and Protor, controls the phosphorylation of Ser473 (24, 25). Studies using Rictor knockout cells have confirmed these results and shown greatly reduced Ser473 phosphorylation following IGF, platelet-derived growth factor (PDGF), or insulin stimulation in Rictor knockouts compared to that in wild-type cells (1, 26–28).
MK2 is activated downstream of the p38α MAPK cascade (29) and was one of the first kinases shown to phosphorylate Akt on Ser473 in vitro (18). A role for MK2 in regulating Ser473 phosphorylation in vivo had initially been discounted since under some circumstances where there is strong activation of Akt, for instance, stimulation of HEK293 cells with IGF, there is little or no activation of p38 or MK2 (18). Furthermore, p38 inhibitors do not block IGF- or H2O2-stimulated Akt activation (30), which is consistent with a role for mTORC2 in Akt Ser473 phosphorylation in these pathways. However, there are some circumstances under which p38 inhibitors have been shown to block Akt activation. For example, the p38 inhibitor SB203580 blocks Ser473 phosphorylation in human neutrophils, while angiotensin II-stimulated Ser473 phosphorylation is blocked in vascular smooth muscle cells at concentrations similar to those required for inhibition of p38α (31, 32). It has also been suggested that MK2 and Akt exist in a complex mediated by Hsp27 in polymorphonuclear leukocytes (33). However, genetic proof that MK2 can act as an in vivo Ser473 kinase is lacking.
MK2 has important roles in macrophages and DCs, including the regulation of pinocytosis and the production of tumor necrosis factor alpha (TNF-α) downstream of TLR signaling (29, 34). It has also been shown that MK2 and the related kinase MK3 play a role in the activation of ribosomal S6 kinase (RSK) in DCs in response to LPS stimulation (34). RSK possesses two kinase domains, of which the N-terminal domain is related to the kinase domain of Akt. The C-terminal kinase domain normally phosphorylates the hydrophobic motif in the N-terminal kinase domain of RSK; however, following stimulation of DCs with LPS, MK2 and MK3 were able to phosphorylate this residue (34). Interestingly, the ability of MK2/3 to phosphorylate RSK seems to be restricted to TLR signaling, as in most cells RSK activation is completely independent of p38α and MK2/3. However, as the hydrophobic domain site in RSK that is phosphorylated by MK2/3 is analogous to Ser473 in Akt, it raises the possibility that MK2 and/or MK3 also regulates Akt phosphorylation downstream of TLR signaling. We therefore assessed the requirement for MK2 and -3 in the activation of Akt in macrophages.
MATERIALS AND METHODS
Mice.
Mice with a T106M knock-in mutation in p38α or both p38α and p38β and MK2/MK3 knockouts have been described previously (34, 35). To generate mice with a conditional loss of function for PDK1 or mTORC2 in hematopoietic cells, mice with either PDK1 (36) or Rictor (37) floxed alleles were crossed with mice expressing a Vav-Cre transgene (38). Mice with floxed hspB1 (Hsp25) were crossed with CMV-Cre mice to generate the hspB1+/del strain (J. Crowe, A. Aubareda, K. McNamee, P. M. Przybycien, X. Lu, R. O. Williams, G. Bou-Gharios, J. Saklatvala, and J. L. Dean, submitted for publication). Heterozygotes were then intercrossed to generate homozygous hspB1del/del and hspB1+/+ mice. Knockout mice were backcrossed onto C57BL/6J mice for at least 6 generations before use.
Cell culture.
RAW264.7 cells were maintained in Dulbecco modified Eagle medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS), 2 mM l-glutamine, 100 U/ml of penicillin G, and 100 μg/ml of streptomycin. HeLa cells were maintained in DMEM supplemented with 10% FBS, 2 mM l-glutamine, 100 U/ml of penicillin G, and 100 μg/ml of streptomycin. BMDMs were isolated as described previously (39). Freshly isolated bone marrow cells were differentiated in bacterial-grade plastic dishes in BMDM medium (DMEM supplemented with 10% heat-inactivated fetal bovine serum, 2 mM l-glutamine, 100 U/ml of penicillin G, 100 μg/ml of streptomycin, 0.25 μg/ml of amphotericin [Invitrogen], and 5 ng/ml of macrophage colony-stimulating factor [M-CSF; R&D Systems]). After 7 days, macrophages were passaged onto tissue culture plastic and used the following day. For Rictor knockout BMDMs, frozen bone marrow cells were thawed and cultured in bacterial-grade plastic dishes in complete RPMI medium (RPMI medium supplemented with 10% heat-inactivated FBS, 2 mM l-glutamine, 100 U/ml of penicillin G, 100 μg/ml of streptomycin, 0.1 mM nonessential amino acids, 1 mM sodium pyruvate, 10 mM HEPES buffer, and 100 μM β-mercaptoethanol) combined with 30% L929-conditioned medium. Cells were fed after 3 to 4 days by the addition of 50% more medium and passaged onto tissue culture plastic in complete RPMI medium without L929-conditioned medium after 7 days. Splenic macrophages were cultured from PDK1 Vav-Cre mice as described above for BMDMs.
To generate sufficient numbers of primary macrophages for the time-resolved fluorescence resonance energy transfer (TRFRET) assay, bone marrow cells were isolated and expanded in culture using a method adapted from reference 40. Cells were expanded in bacterial-grade plastic dishes in BMDM medium without recombinant colony-stimulating factor (rCSF) but with the addition of recombinant murine IL-3 (10 ng/ml), recombinant murine IL-6 (10 ng/ml), and stem cell factor (SCF) (50 ng/ml). Both adherent and nonadherent cells were passaged every 2 to 3 days, and after 13 days, cells were plated in medium without IL-3, IL-6, or SCF but with rCSF for a further 3 days to allow differentiation into macrophages. On day 16, adherent cells were passaged onto tissue culture plastic for experimentation the following day.
Stimulation of cells.
Cells were stimulated with 100 ng/ml of LPS, 10 μg/ml of anisomycin, 1 μg/ml of Pam3CSK4, 10 μg/ml of poly(I·C), 1 μg/ml of CL097, 2 μM CpG-ODN1826, 200 μg/ml of zymosan, 10 μg/ml of curdlan, 50 ng/ml of granulocyte-macrophage colony-stimulating factor (GM-CSF), 10 nM C5a, or 0.5 to 5 mM H2O2 for various times. Where indicated, cells were preincubated for 1 h with 5 μM SB203580, 0.1 μM BIRB0796, 1 μM VX-745, 1 μM PI-103, 1 μM GDC0941, 1 μM MK2206, 1 μM Ku-0063794, 0.5 μM PIK75, 1 μM TGX221, 1 μM IC87114, 2 μM PD184352, or 3 μM BI-D1870 or PF3644022 (concentrations indicated in figure legends).
Immunoblotting.
For immunoblotting, cells were lysed in 50 mM Tris-HCl (pH 7.5), 1 mM EGTA, 1 mM EDTA, 1 mM sodium orthovanadate, 50 mM sodium fluoride, 1 mM sodium pyrophosphate, 0.27 M sucrose, 1% (vol/vol) Triton X-100, 0.1% (vol/vol) 2-mercaptoethanol, 1 μM microcystin-LR, and complete proteinase inhibitor cocktail (Roche). Lysates were clarified by centrifugation (13,000 rpm for 10 min at 4°C) and supernatants snap-frozen and stored at −80°C. The protein concentration was determined with Coomassie protein assay reagent (Thermo Scientific). Proteins were separated on 10% polyacrylamide gels, and immunoblotting was carried out using standard techniques. Antibodies against phospho-Thr308 Akt, phospho-Ser473 Akt, phospho-Thr334 MK2, phospho-Thr180/Tyr182 p38, total MK2, total p38, phospho-Ser82 Hsp27, total Hsp27, phospho-Thr573 p90RSK, phospho-Thr24/Thr32 FOXO1/FOXO3a, phospho-Thr389 p70 S6 kinase, phospho-Ser21/9 glycogen synthase kinase 3α/β (GSK3α/β), phospho-Thr202/Tyr204 extracellular signal-regulated kinase 1/2 (ERK1/2), total ERK1/2, and total Jun N-terminal protein kinase (JNK) were from Cell Signaling. The antibodies against total RSK and total GSK3β were from Transduction Laboratories, the antibody against total FOXO3a was from Biolegend, the antibody against phospho-Thr183/Tyr185 JNK was from Invitrogen, the antibodies against total PTEN and SHIP1 were from Santa Cruz, and the antibody against total PDK1 was from BD Biosciences. The antibodies against total Akt (41), total SHIP2 (42), and total p70 S6 kinase (43) were raised by the Division of Signal Transduction Therapy (DSTT) and have been described previously. Equal protein concentrations in the lysates were confirmed by blotting for total ERK1/2 in addition to the total antibodies to the relevant phospho-specific antibodies. Most membranes were probed with a single antibody; however, where indicated, membranes were stripped using Re-Blot Plus buffer (Millipore) before reprobing with a second antibody.
For Fig. 3B, membranes were cut after transfer; the upper part was blotted for phospho-Ser473 Akt and the lower part for total ERK1/2. Membranes were imaged using a charge-coupled-device (CCD) camera and quantified using Aida software. A standard curve for the antibody response was obtained using serial dilutions of the sample stimulated with 5 mM H2O2 that were run on the same gel by curve fitting to a 3-parameter logistic function in SigmaPlot. This standard curve was used to determine the amount of Ser473 phosphorylation present in the LPS-stimulated samples relative to the sample stimulated with 5 mM H2O2. Values were then corrected for loading using the quantification of the ERK1/2 signal.
Fig 3.

MK2 and -3 regulate Akt phosphorylation in response to multiple TLR agonists. (A) BMDMs were cultured from wild-type or MK2/3 knockouts and stimulated with 1 μg/ml of Pam3CSK4, 10 μg/ml of poly(I·C), 100 ng/ml of LPS, 1 μg/ml of CL097, 2 μM CpG-ODN1826, 200 μg/ml of zymosan, 10 μg/ml of curdlan, 50 ng/ml of GM-CSF, or 10 nM C5a for 30 min. Cells were then lysed and the levels of the indicated phosphorylated and total proteins determined by immunoblotting. Gray arrowheads indicate MK2 bands, while the open arrowhead indicates a nonspecific band. Blots are representative of a minimum of two independent experiments. (B) BMDMs were cultured from wild-type mice and stimulated for 15 min with LPS or H2O2 as indicated. Cells were then lysed and the levels of phospho-Ser473 Akt and total ERK1/2 measured by immunoblotting on the same membrane. Akt Ser473 phosphorylation was quantified as described in Materials and Methods. Results of the quantification are shown on the left, and a representative membrane is shown on the right. Error bars represent the standard deviations of 3 independent experiments.
Akt kinase assay.
Assay of Akt kinase activity was carried out as described previously (15). Briefly, cells were lysed in 50 mM Tris-HCl (pH 7.5), 1 mM EDTA, 1 mM EGTA, 1% (wt/vol) Triton X-100, 1 mM sodium orthovanadate, 50 mM sodium fluoride, 5 mM sodium pyrophosphate, 10 mM sodium glycerophosphate, 0.27 M sucrose, and 0.1% (vol/vol) 2-mercaptoethanol (lysis buffer). Akt was immunoprecipitated from 500 μg of lysate using a sheep anti-Akt antibody coupled to protein G-Sepharose. Immunoprecipitates were washed once in lysis buffer with 0.5 M NaCl, once in 50 mM Tris-HCl (pH 7.5), 0.1 mM EGTA, 0.5 M NaCl, and 0.1% (vol/vol) 2-mercaptoethanol, and once in 50 mM Tris-HCl (pH 7.5), 0.1 mM EGTA, and 0.1% (vol/vol) 2-mercaptoethanol. Kinase activity was measured against 30 μM Crosstide peptide (GRPRTSSFAEG) in 50 mM Tris-HCl (pH 7.5), 0.1 mM EGTA, 2.5 μM PKI (TTYADFIASGRTGRRNAIHD, a peptide inhibitor of cyclic AMP [cAMP]-dependent protein kinase), 10 mM magnesium acetate, 0.1 mM [γ-32P]ATP (∼500 cpm/pmol), and 0.1% (vol/vol) 2-mercaptoethanol. Reactions were terminated by transfer onto P81 paper and washing in 100 mM orthophosphoric acid, and 32P radioactivity was measured by Cerenkov counting in a liquid scintillation counter. One unit of protein kinase activity was the amount that catalyzed the incorporation of 1 nmol of phosphate into the substrate in 1 min at 30°C.
Measurement of lipid levels.
PIP3 levels were measured using a TRFRET displacement assay as described previously (44). Briefly, following treatment of cells (∼2×107 cells per data point), medium was aspirated and cells were precipitated with 0.5 M trichloroacetic acid (TCA) on ice. Neutral lipids were extracted from the pelleted precipitate with methanol-chloroform (2:1) by vortexing 3 or 4 times in 10 min at room temperature, and the solvent was discarded. Acidic lipids were extracted from the pellet with chloroform-methanol-12 M HCl (40:80:1) with occasional vortexing for 15 min at room temperature. The organic phase was separated by the addition of chloroform and 0.1 M HCl, followed by vortexing and centrifugation. The organic phase was collected into a clean tube and dried in a SpeedVac centrifuge. Lipids were resuspended by sonication in 60 μl of assay buffer (50 mM Tris-HCl [pH 7], 150 mM NaCl, 1 mM dithiothreitol [DTT], 0.5 mM EGTA, 1.2% Na cholate). The mass of inositol lipid was estimated by adding 25 μl of the resuspended lipid to 25 μl of sensor complex [50 mM Tris (pH 7.5), 150 mM NaCl, 2 mM DTT, 0.02% Na cholate containing streptavidin-allophycocyanin at 3.3 μg/ml, glutathione S-transferase (GST)–GRP1 PH domain at 3.5 μg/ml, Eu chelate-labeled anti-GST antibody at 2.56 μg/ml, and 120 nM biotinylated di-C6 PI(3,4,5)P3]. Comparison to a standard curve from 5,000 pmol to 0.064 pmol/well gives an estimate of PIP3 in the sample. Plates were read using an LJL Analyst.
RESULTS
LPS-induced phosphorylation of Akt on Thr308 and Ser473 requires p38.
To examine the activation of Akt in macrophages, RAW264.7 cells were stimulated with the TLR4 agonist LPS and phosphorylation of Akt was analyzed by immunoblotting. LPS induced the phosphorylation of Akt on both Thr308 and Ser473, which was maximal at 15 to 30 min (Fig. 1A). The activation of Akt has been shown to be dependent on PI3K signaling in other cell types (45), and in line with this, the PI3K inhibitors PI-103 (46) and GDC-0941 (47) were able to block Akt phosphorylation in response to LPS (Fig. 1B). Furthermore, the allosteric Akt inhibitor MK-2206, which inhibits phosphorylation at both Thr308 and Ser473 (48), was also able to block phosphorylation of both these sites in response to LPS (Fig. 1B).
Fig 1.

LPS-induced phosphorylation of Akt on Thr308 and Ser473 requires p38. (A) RAW264.7 cells were stimulated for the indicated times with 100 ng/ml of LPS. Cells were lysed and the levels of phospho-Thr308 Akt, phospho-Ser473 Akt, total Akt, phospho-p38 and total p38, phospho-MK2 and total MK2, and total ERK determined by immunoblotting. (B) Same as panel A except that cells were preincubated for 1 h with 1 μM PI-103 (PI), 1 μM GDC-0941 (GDC), or 1 μM MK-2206 (MK) where indicated. (C) Same as panel A except that cells were preincubated for 1 h with 1 μM GDC-0941 (GDC), 0.5 μM PIK75 (PIK), 1 μM TGX221 (TGX), or 1 μM IC87114 (IC8) where indicated. (D) Same as panel A except that cells were preincubated for 1 h with 5 μM SB203580 (SB), 1 μM VX-745 (VX), or 0.1 μM BIRB-0796 (BI) where indicated. (E) RAW264.7 cells were preincubated for 1 h with 5 μM SB203580 (SB), 0.1 μM BIRB0796 (BI), or 1 μM PI-103 (PI) where indicated. Cells were then stimulated with 100 ng/ml of LPS for 30 min and lysed, and Akt activity was determined as described in Materials and Methods. Error bars represent the standard deviations for 5 independent stimulations. A P value of less than 0.01 relative to the LPS-stimulated sample is indicated by double asterisks. Each panel is representative of at least 2 or 3 independent experiments.
To assess whether there was PI3K isoform specificity in the regulation of Akt phosphorylation in macrophages stimulated with LPS, we examined the phosphorylation of Ser473 and Thr308 in the presence of isoform-specific PI3K inhibitors (49). Phosphorylation of both sites was blocked in the presence of the PI3K p110α inhibitor PIK75 but not with the PI3K p110β and p110δ inhibitors TGX221 and IC87114, respectively (Fig. 1C).
In RAW264.7 cells, LPS also induced phosphorylation of p38α and its downstream kinase MK2. Like Akt phosphorylation, this could be detected by 15 min of stimulation; however, p38α activation was more transient than Akt phosphorylation (Fig. 1A). The phosphorylation of p38α and MK2 was unaffected by the inhibition of PI3K (Fig. 1C). To examine the potential role for the p38 pathway in Akt activation in macrophages, three structurally unrelated p38α/β inhibitors, SB203580, VX-745, and BIRB0796, were used. LPS-induced Akt phosphorylation on both Thr308 and Ser473 was greatly reduced by the p38 inhibitors SB203580, VX-745, and BIRB0796 (Fig. 1D). The phosphorylation of Akt on both Thr308 and Ser473 is required for full activation of Akt. In line with the ability of LPS to promote Akt phosphorylation, it was also able to activate Akt as judged by immunoprecipitation kinase assays (Fig. 1E). Preincubation of the cells with the PI3K inhibitor PI-103 or the p38α/β inhibitors SB203580 and BIRB0796 was able to inhibit the ability of LPS to induce Akt kinase activity (Fig. 1E).
LPS-induced Akt phosphorylation requires p38α and MK2/3.
While the above-described experiments with p38α/β inhibitors are consistent with a role for p38α or -β in regulating Akt activity in macrophages, a drawback with the use of kinase inhibitors is that they can, in some cases, have off-target effects. Although SB203580, BIRB0796, and VX-745 have a good selectivity for p38α and p38β (Table 1) (46), it is possible that their effect on Akt phosphorylation is due to an off-target activity. To confirm that SB203580 was acting via p38 and to examine if p38α or p38β was the critical isoform, we utilized cells from mice in which endogenous p38α is mutated so that it is no longer inhibited by SB203580 (35). SB203580 inhibits p38α by binding to the ATP-binding pocket, and much of the specificity SB203580 has for p38α/β is due to the presence of a Thr at position 106 in p38α and the equivalent residue in p38β. In most kinases, this position is occupied by an amino acid with a larger side chain, and this prevents the binding of SB203580 to these kinases. Mutation of Thr106 to Met in the ATP binding pocket of p38α is sufficient to reduce the sensitivity to inhibition by SB203580, but it does not affect the ability of the kinase to bind ATP or phosphorylate its normal substrates (35). LPS was able to stimulate Ser473 phosphorylation in bone marrow-derived macrophages (BMDMs), and this was inhibited by SB203580 in wild-type cells (Fig. 2A). In wild-type cells, SB203580 also inhibited the phosphorylation of the p38α substrate MK2. However, in BMDMs from p38α Thr106Met knock-in mice, SB203580 was unable to block the phosphorylation of MK2, confirming that the Thr106Met mutation had generated a p38α that is resistant to SB203580 (Fig. 2A). SB203580 was also unable to inhibit Ser473 Akt phosphorylation in the Thr106Met knock-in cells, confirming that the action of SB203580 in wild-type cells was via p38α and not an off-target effect of the inhibitor (Fig. 2A). Similar results were also obtained for mice carrying the Thr106Met knock-in in both p38α and p38β (Fig. 2A).
Table 1.
Specificity of VX-745 as a p38 inhibitora
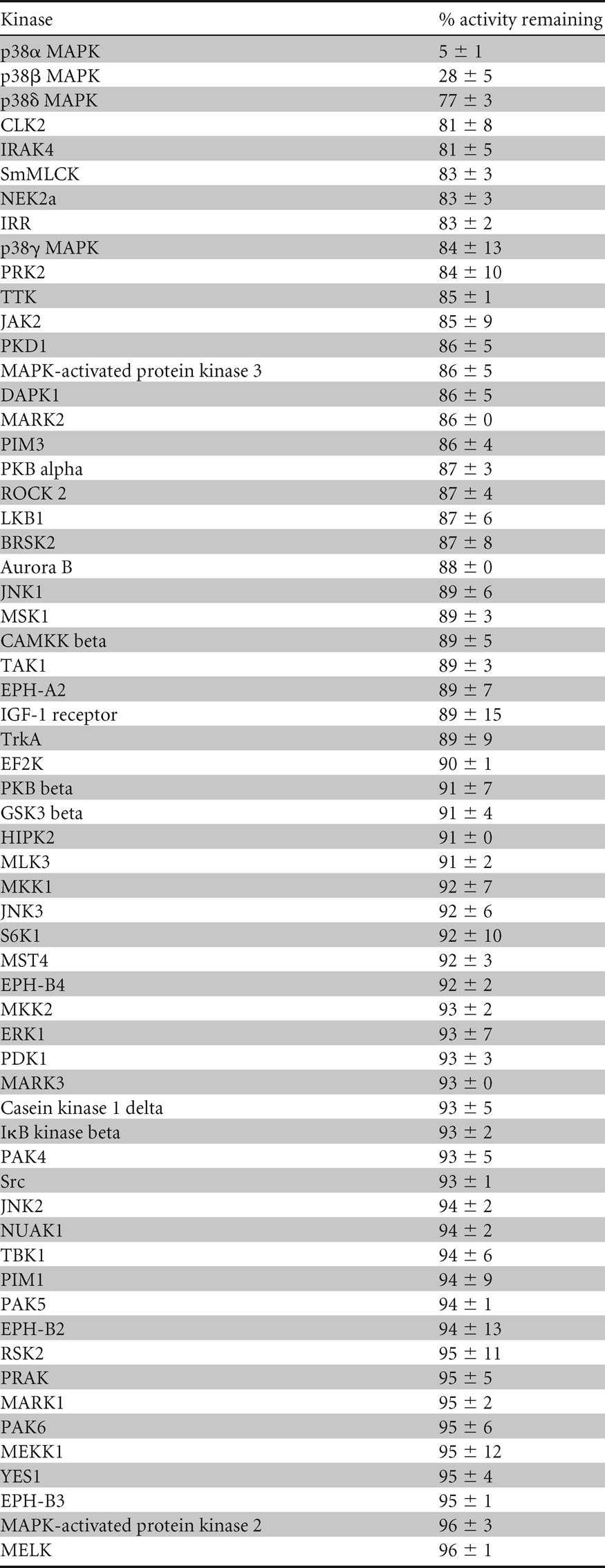
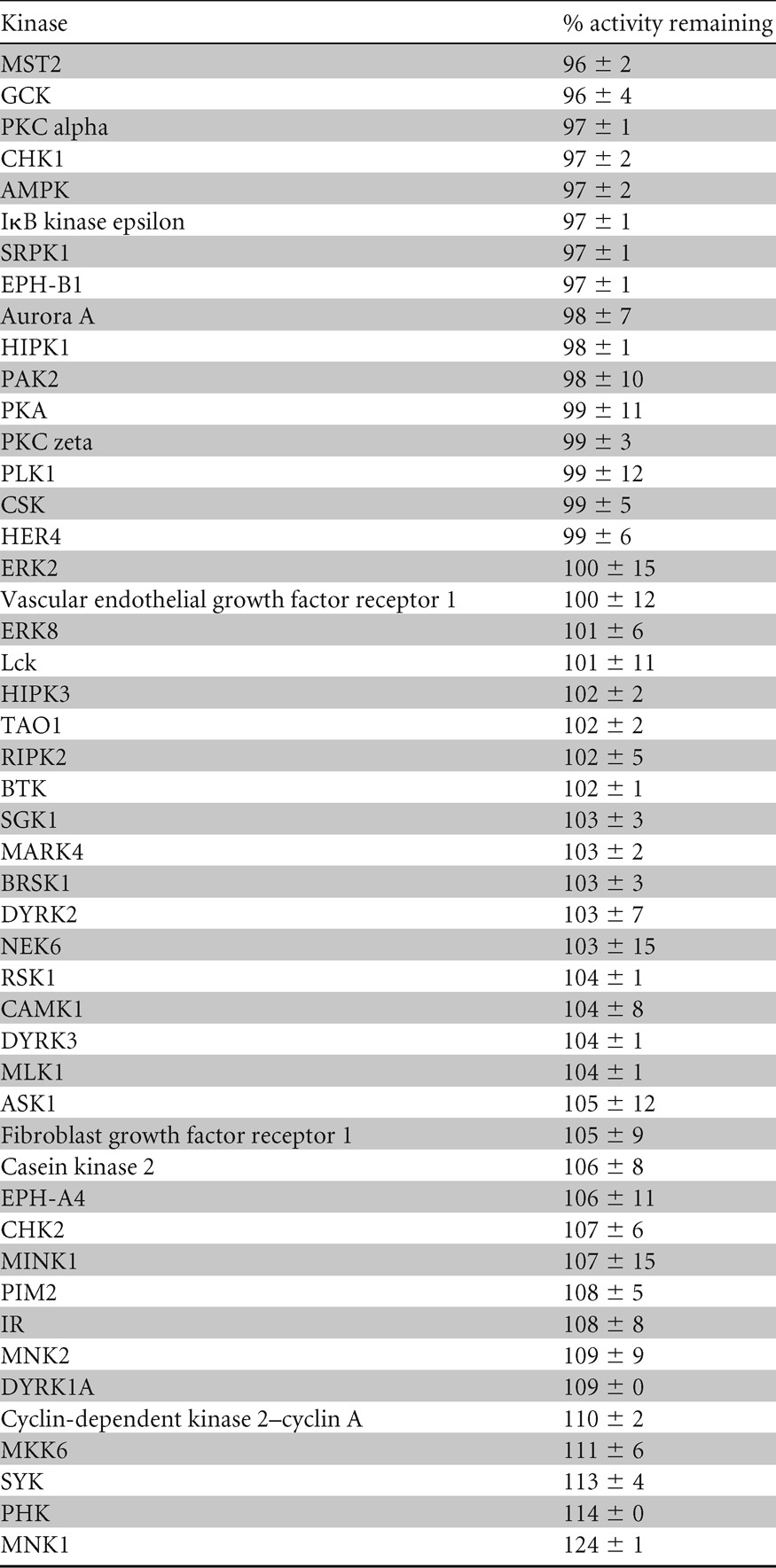
Kinase selectivity profiling for VX-745 was carried out in vitro as described previously (http://www.kinase-screen.mrc.ac.uk/) (46). Kinases were ranked in order of the percent activity remaining at 0.1 μM VX-745. Values represent the averages ± the variances of duplicate assays.
Fig 2.

LPS-induced Akt phosphorylation requires p38α and MK2/3. (A) BMDMs were cultured from wild-type, T106M p38α knock-in (p38α KI), or T106M p38α/T106M p38β double-knock-in (p38αβ KI) mice. Where indicated, cells were preincubated for 1 h with 5 μM SB203580 (SB) and then stimulated with 100 ng/ml of LPS for 30 min. Cells were lysed and levels of the indicated phosphorylated and total proteins determined by immunoblotting. (B) BMDMs were cultured from wild-type, MK2, MK3, or MK2/MK3 knockouts. Cells were stimulated for the indicated times with 100 ng/ml of LPS and lysed, and the levels of the indicated phosphorylated and total proteins were determined by immunoblotting. Gray arrowheads indicate MK2 bands, while the open arrowhead indicates a nonspecific band. (C) HeLa cells were preincubated for 1 h with PF3644022 (PF) and then stimulated for 30 min with 10 μg/ml of anisomycin. Cells were lysed and the levels of phospho-Hsp27 and total Hsp27, phospho-MK2 and total MK2, phospho-p38 and total p38, and total ERK measured by immunoblotting. (D) RAW264.7 cells were preincubated for 1 h with PF3644022 (PF) and then stimulated for 30 min with 100 ng/ml of LPS. Cells were lysed and the levels of the indicated phosphorylated and total proteins determined by immunoblotting. (E) BMDMs were cultured from wild-type mice and stimulated with 100 ng/ml of LPS for 15 min. Where indicated, cells were pretreated with 1 μM MK-2206, 2 μM PD184352, or 3 μM BI-D1870 for 1 h. Following stimulation, cells were lysed and the levels of the indicated phosphorylated and total proteins determined by immunoblotting. Blots are representative of a minimum of two independent experiments.
p38α is a proline-directed kinase and therefore unlikely to directly phosphorylate either Thr308 or Ser473 in Akt, as these residues are not followed by a proline. However, Ser473 does lie in a consensus sequence for MK2 phosphorylation. Consistent with this, it is known that MK2 is an efficient in vitro kinase for Ser473 in Akt (18), although the direct phosphorylation of Akt by MK2 has not been shown in cells (30). MK2 and the closely related kinase MK3 are activated by p38α in response to LPS stimulation in macrophages (Fig. 1 and 2) (50). To examine the role of MK2/3 in regulating LPS-induced phosphorylation of Akt, we isolated BMDMs from MK2, MK3, and MK2/3 double-knockout mice. Knockout of either MK2 or MK3 alone had little effect on Ser473 and Thr308 phosphorylation of Akt, although BMDMs with a knockout of both MK2 and MK3 displayed greatly reduced phosphorylation of Ser473 and Thr308 (Fig. 2B). It should be noted that in MK2 or MK2/3 knockouts, p38α is expressed at lower levels than in wild-type cells. MK2 has been shown to be involved in the stabilization of p38α, and a reduction in p38 levels in MK2 or MK2/3 knockout mice has been previously reported (50). It could be argued that the observed effects on Akt phosphorylation may be due to a reduction of p38α levels in the MK2/3 knockouts rather than a direct role for MK2 and -3. To examine this, we used the recently described MK2/3 inhibitor PF3644022 (51). To confirm that the inhibitor was effective at blocking MK2 activity in cells, we analyzed the phosphorylation of the MK2 substrate Hsp27 in HeLa cells in response to anisomycin stimulation (Fig. 2C). This demonstrated that 5 μM PF3644022 was able to completely inhibit MK2/3 activity in cells. We were unable to examine the phosphorylation of Hsp25, the murine homolog of Hsp27, in macrophages, as we were unable to detect expression in these cells by immunoblotting, which is consistent with previous studies (52). In macrophages, 5 μM PF3644022 was able to block the phosphorylation of Akt on both Thr308 and Ser473 (Fig. 2D).
GSK3 and FOXO1/3 have previously been shown to be direct substrates for Akt (1). We therefore tested the effect of MK2/3 knockout on the ability of LPS to induce GSK3 and FOXO phosphorylation. LPS was found to induce FOXO phosphorylation using an antibody that recognizes FOXO1 or -3 phosphorylated on the N-terminal Akt sites. In line with the reduced activation of Akt, the phosphorylation of FOXO was reduced in MK2/3 double-knockout cells (Fig. 2B). LPS also induced GSK3 phosphorylation; however, this was not affected by loss of MK2 and 3 (Fig. 2B). In addition to being phosphorylated by Akt, GSK3 can also be phosphorylated on the same sites by the ERK1/2-activated kinase RSK (53). To examine this possibility, BMDMs were treated with kinase inhibitors against either the Akt or ERK1/2 pathway before LPS stimulation. While the Akt inhibitor MK2206 blocked FOXO phosphorylation, it did not have a major effect on GSK phosphorylation (Fig. 2E). Treatment of the cells with the RSK inhibitor BI-D1870 or the MEK1/2 inhibitor PD184352 (which blocks ERK1/2 and therefore RSK activation) did modestly reduce GSK3 phosphorylation, although a combination of MEK1/2 and Akt inhibitors had a greater effect. This suggests that Akt is not the only GSK3 kinase downstream of LPS and that GSK3 is targeted by multiple pathways in response to LPS stimulation of macrophages (Fig. 2E). Akt can also phosphorylate TSC2 and thereby promote mTORC1 activation (1), which leads to p70S6 kinase phosphorylation on Thr389. LPS stimulated Thr389 phosphorylation of p70S6 kinase, and this was reduced by double knockout of MK2 and -3 (Fig. 2B).
MK2 and -3 are required for Akt phosphorylation downstream of multiple TLR agonists.
LPS acts via TLR4, which can utilize both the MyD88 and Trif adapter proteins to couple to downstream signaling pathways. With the exception of TLR3, which uses only Trif, all other TLRs use MyD88 as an adapter. We therefore tested the role of MK2/3 in Akt activation downstream of other TLRs. Stimulation of BMDMs using poly(I·C) as an agonist for TLR3 also promoted Akt phosphorylation on Thr308 and Ser473, and consistent with our observations with LPS, TLR3-induced Akt phosphorylation was also inhibited by the knockout of MK2/3 (Fig. 3A). Stimulation of BMDMs with any of the MyD88-dependent agonists Pam3CSK4 (TLR1/2), CpG (TLR9), and CL097 (TLR7/8) was also able to induce Akt phosphorylation, and again, this was reduced in MK2/3 knockout cells (Fig. 3A). Thus, MK2/3 knockout reduces Akt activation in BMDMS in response to both MyD88-dependent (TLR1/2, TLR4, TLR7/8, and TLR9) and Trif-dependent (TLR3) stimuli. As TLRs are not the only pattern recognition receptors (PRRs) in macrophages, we also assessed the activation of Akt with agonists of dectin-1, a receptor involved in the recognition of fungal ligands. Zymosan is an extract of yeast cell wall that stimulates macrophages via a combination of dectin-1 and TLR2 (54). In response to zymosan, the phosphorylation of Akt was reduced in MK2/3 knockout cells relative to that in wild-type BMDMs, although the effect was less pronounced than that observed for TLR agonists (Fig. 3A; see Fig. 5C). Curdlan is a selective dectin-1 agonist in BMDMs (55). Curdlan was a much weaker stimulus than zymosan for both Akt and p38; however, the phosphorylation of Akt in response to curdlan was still slightly reduced by MK2/3 knockout (Fig. 3A). We also analyzed the activation of Akt by non-PRR stimuli. C5a and GM-CSF have been reported to activate Akt in macrophages (56, 57). In agreement with this, both of these stimuli induced the phosphorylation of Akt, and the level of phosphorylation was comparable to that seen with LPS. Both C5a and GM-CSF were, however, very poor activators of the p38 pathway, and in line with this, knockout of MK2 and 3 did not have any major effects on Akt Thr308 or Ser473 phosphorylation in C5a- or GM-CSF-stimulated cells (Fig. 3A). To further assess the degree to which Akt was phosphorylated in BMDMs in response to LPS, wild-type cells were stimulated with increasing concentrations of either LPS or H2O2, a stimulus known to be a strong activator of Akt (30). In BMDMs, H2O2 gave a maximal phosphorylation of Akt on Ser473 at 5 mM (Fig. 3B), and at concentrations above 5 mM H2O2, the phosphorylation of Akt was diminished (data not shown). Stimulation with 100 ng/ml of LPS gave approximately 20% of the total phosphorylation achievable with 5 mM H2O2 (Fig. 3B). Increasing the concentration of LPS did not further increase Akt phosphorylation.
Fig 5.

Phosphorylation of Akt downstream of MK2/3 is indirect. (A) Splenic macrophages were cultured from wild-type or conditional PDK1 knockouts. Cells were stimulated for 30 min with 100 ng/ml of LPS and lysed, and the levels of phospho-Thr308 Akt, total Akt, and total PDK1 were measured by immunoblotting. (B) BMDMs were cultured from wild-type or MK2/3 knockouts. Cells were pretreated with 5 μM SB203580 (SB) or 1 μM Ku-0063794 for 1 h and then stimulated for the indicated times with 100 ng/ml of LPS. Cells were lysed and the levels of phospho-Thr308 Akt, phospho-Ser473 Akt, total Akt, phospho- and total p38, phospho- and total MK2, and total ERK measured by immunoblotting. For total Akt and total ERK, membranes were stripped and reprobed from the phospho-Thr308 Akt and total p38, respectively. (C) BMDMs were cultured from wild-type or MK2/3 knockouts. Cells were pretreated with 5 mM SB203580 (SB) or 1 mM KU-0063794 for 1 h and then stimulated for the indicated times with 200 mg/ml of zymosan. Cells were lysed and the levels of phospho-Thr308 Akt, phospho-Ser473 Akt, total Akt, phospho-p38 and total p38, phospho-MK2 and total MK2, and total ERK measured by immunoblotting. For total Akt and total ERK, membranes were stripped and reprobed from the phospho-Thr308 Akt and total p38, respectively. (D) BMDMs were cultured from wild-type or conditional Rictor knockouts. Cells were preincubated for 1 h with 5 μM SB203580 (SB) or 1 μM Ku-0063794 (KU) and then stimulated for the indicated times with 100 ng/ml of LPS. Cells were lysed and the levels of phospho-Thr308 Akt, phospho-Ser473 Akt, total Akt, phospho-p38 and total p38, phospho-MK2 and total MK2, and total ERK measured by immunoblotting. For total Akt and total ERK, membranes were stripped and reprobed from the phospho-Thr308 Akt and total p38, respectively.
Regulation of Akt by MK2/3 is independent of Hsp25.
It has been proposed that Hsp27, the human homolog of Hsp25, acts as a scaffold to regulate Akt phosphorylation downstream of p38/MK2 signaling in human polymorphonuclear leukocytes and transfected L929 cells (33, 58). We therefore sought to determine whether this mechanism also functioned in primary murine macrophages. Since we were unable to detect Hsp25 in BMDMs by immunoblotting (data not shown), we isolated BMDMs from HspB1 (Hsp25) knockout animals (Crowe et al., submitted) and assessed the phosphorylation of Akt in response to LPS (Fig. 4). In both wild-type and Hsp25 knockout cells, LPS was able to induce phosphorylation of both Thr308 and Ser473 on Akt, which was prevented by pretreatment of cells with SB203580. These data demonstrate that the genetic deletion of Hsp25 does not prevent activation of Akt downstream of p38-MK2 signaling in macrophages, suggesting that Hsp25 is dispensable for the regulation of Akt activity in response to TLR4 signaling.
Fig 4.

Hsp25 (HspB1) is dispensable for regulating Akt activity in BMDMs. BMDMs were cultured from wild-type or HspB1 knockouts. Cells were preincubated for 1 h with 5 mM SB203580 and then stimulated for the indicated times with 100 ng/ml of LPS. Cells were lysed and the levels of phospho-Thr308 Akt, phospho-Ser473 Akt, total Akt, phospho-MK2 and total MK2, and total ERK measured by immunoblotting. Data are shown from two mice per genotype.
PDK1 and mTORC2 are required for Akt phosphorylation in macrophages.
The data shown in Fig. 1 to 3 demonstrate that MK2 and -3 are required for the phosphorylation of Akt in response to TLR stimulation. This could be consistent with either a direct phosphorylation of Akt by MK2/3 or a role for MK2 and -3 in controlling the activity of an upstream component of the Akt pathway. However, while MK2 is an efficient in vitro kinase for Ser473 on Akt (18), Thr308 does not lie in an MK2 consensus sequence, making a role for direct phosphorylation of Akt by MK2/3 less likely. Importantly, knockout of MK2 and -3 did not result in a global inhibition of TLR signaling, as both the ERK1/2 and JNK MAPK pathways were activated normally in response to TLR agonists in MK2/3 knockout BMDMs (Fig. 3A). We therefore investigated if MK2/3 regulated Akt phosphorylation via a direct or indirect mechanism. It is well established that PDK1 is required for Thr308 phosphorylation of Akt in response to IGF stimulation (15). To determine if PDK1 played a role in Thr308 phosphorylation downstream of TLR signaling, we isolated spleen-derived macrophages from mice with a conditional knockout of PDK1 in hematopoietic cells. These mice are viable, and although T and B cell development is compromised, macrophages are present in the spleens of these mice (59). In LPS-stimulated PDK1 null splenic macrophages, there was no detectable Thr308 phosphorylation, confirming that PDK1 is indeed the Thr-308 kinase in macrophages (Fig. 5A). Splenic macrophages were used for this, as the knockout of PDK1 using Vav-Cre blocks the differentiation of BMDMs in vitro (unpublished data).
As the mTORC2 complex (consisting of mTOR, Rictor, mLST8, sin1, and Protor) has been shown to be required for phosphorylation of Ser473 in response to multiple stimuli that activate Akt (1), we examined the role of mTORC2 in regulating Ser473 phosphorylation in macrophages using the mTOR inhibitor Ku-0063794 (60). In wild-type BMDMs, LPS-induced Akt phosphorylation was significantly reduced when cells were pretreated with Ku-0063794 (Fig. 5B). Interestingly, however, in BMDMs lacking MK2/3, there was no induction of either Ser473 or Thr308 phosphorylation in response to LPS, and the basal phosphorylation of Ser473 was further reduced in the presence of Ku-0063794 (Fig. 5B). The induction of both Ser473 and Thr308 phosphorylation in response to zymosan stimulation was also reduced in MK2/3 knockout BMDMs, and phosphorylation on both sites was further reduced in the presence of Ku-0063794 (Fig. 5C). The decrease in Thr308 phosphorylation in the presence of Ku-0063794 is consistent with the original report describing the action of this inhibitor, which was interpreted as Ser473 phosphorylation inducing a conformational change that protects Thr308 from dephosphorylation (60).
To examine this further, we analyzed Akt phosphorylation in BMDMs derived from conditional Rictor knockout mice (37). We did not observe any Ser473 phosphorylation in Rictor knockout cells, either basally or in response to LPS stimulation (Fig. 5D), demonstrating that Rictor/mTORC2 is absolutely required for phosphorylation of Akt on Ser473 in macrophages. Thr308 phosphorylation was still inducible in Rictor knockouts in response to LPS, albeit at a lower level than in wild-type cells. This may be expected in the absence of Ser473 phosphorylation, since this site has been proposed to protect Thr308 from dephosphorylation (60). These data demonstrate that PDK1 and mTORC2 are required for Akt phosphorylation in macrophages and indicate that MK2 and -3 do not directly phosphorylate Akt in macrophages. Instead, MK2 and -3 regulate the ability of PDK1 and mTORC2 to phosphorylate Akt. As Akt activation is dependent on its recruitment to the membrane and subsequent phosphorylation by both PDK1 and mTORC2, this could suggest that MK2/3 regulate the levels of PIP3 in TLR-stimulated macrophages.
Accumulation of PIP3 following LPS stimulation of macrophages requires MK2/3.
A key step in the activation of Akt is its recruitment via its PH domain to PIP3 in the membrane. Without this, PDK1 and mTORC2 are unable to gain access to the Thr308 and Ser473 sites in order to phosphorylate them. We therefore examined if p38α and MK2/3 affected PIP3 production in response to LPS by directly analyzing the levels of PIP3 in macrophages. In RAW264.7 cells, the LPS-induced production of PIP3 was inhibited by the p38 inhibitors SB203580, VX-745, and BIRB0796 (Fig. 6A). PIP3 production was also blocked by the MK2 inhibitor PF3644022 (Fig. 6B). To confirm a role for MK2/3 in regulating PIP3 production, we analyzed lipid levels in BMDMs isolated from wild-type and MK2/3 double-knockout mice. In wild-type BMDMs, LPS stimulation resulted in the generation of PIP3, but this did not occur in BMDMs derived from MK2/3 knockout mice (Fig. 6C). This suggests that MK2/3 regulate Akt activity by affecting the levels of PIP3 in macrophages.
Fig 6.

Accumulation of PIP3 following LPS stimulation requires MK2/3. (A) RAW264.7 cells were preincubated for 1 h with 5 μM SB203580, 1 μM VX-745, or 0.1 μM BIRB0796 and then stimulated for 30 min with 100 ng/ml of LPS. Lipids were isolated and relative levels of PIP3 were measured using a TRFRET displacement assay as described in Materials and Methods. Error bars represent the SEMs of 12 replicates per condition. (B) RAW264.7 cells were preincubated for 1 h with 5 μM SB203580 or 10 μM PF-3644022 and then stimulated for 30 min with 100 ng/ml of LPS. PIP3 levels were determined as for panel A. Error bars represent the SEMs of 5 to 8 replicates per condition. (C) BMDMs were cultured from wild-type or MK2/3 knockouts and expanded as described in Materials and Methods. Cells were stimulated for 30 min with 100 ng/ml of LPS and PIP3 levels determined as for panel A. Error bars represent the SEMs of 4 replicates from 2 mice per genotype. In all panels, a P value (Student's t test) of less than 0.05 is indicated by a single asterisk, a P value of less than 0.01 is indicated by double asterisks, and a P value of less than 0.001 is indicated by triple asterisks.
DISCUSSION
The data presented in this paper demonstrate that the kinases MK2 and MK3, which are activated downstream of p38, are important for regulating the activation of Akt in response to TLR signaling. We have utilized macrophages deficient in key components of the p38 and Akt signaling pathways together with kinase inhibitors to show that MK2/3 regulate Akt by affecting the availability of PIP3 at the membrane. In the absence of MK2/3 activity, there is a marked reduction in the production of PIP3 in response to LPS, which equates to a lower level of both Ser473 and Thr308 Akt phosphorylation and, hence, Akt activity. This requirement for MK2/3 in Akt activation was found for a variety of TLR ligands in macrophages. It should be noted, however, that this is not a universal effect for all Akt-activating stimuli, as C5a and GM-CSF could induce Akt activation independently of the p38-MK2/3 axis, while p38 has been shown to be dispensable for Akt activation in other pathways such as those downstream of heat shock and H2O2 in fibroblasts (30). Interestingly, however, a recent report has demonstrated a deficiency in Akt activation in p38α−/− T cells following TCR stimulation, suggesting that the effect of the p38-MK2 pathway on Akt activation may not be completely restricted to TLR signaling (61).
A requirement for MK2/3 activity to elevate PIP3 levels in response to LPS suggests that MK2/3 could function either by stimulating PI3K activity or by downregulating the activity of a PIP3 phosphatase(s). Class IA PI3Ks are comprised of a p110 catalytic subunit (p110α/β/δ) and a p85 regulatory subunit (p85α/β). p85α and p85β contain two Src homology 2 (SH2) domains that recruit the p110 subunit to tyrosine-phosphorylated proteins via interaction with a phosphorylated YXXM motif, bringing it into proximity of its lipid substrate (11). The exact mechanism for PI3K activation in response to TLR engagement is not fully understood. p85 may bind the TLR receptor directly, or PI3K may be recruited to the receptor via YXXM-containing adapter proteins such as MyD88 (62, 63). TLR2, -3, and -5 contain YXXM motifs, which, in the case of TLR2 and TLR3, have been shown to be required for downstream signaling (64, 65). However, a YXXM motif has not been identified in TLR4, suggesting that a YXXM-containing adapter protein may facilitate the activation of PI3K in response to LPS. Consistent with this, we have not been able to observe recruitment of PI3K to TLR4 in coimmunoprecipitation experiments (data not shown), although it is possible that a weak or transient recruitment of PI3K would have been missed.
MyD88 is proposed to associate with p85 in response to TLR4 engagement (62), and although the interaction between MyD88 and p85 in response to TLR9 stimulation with CpG is abrogated when MyD88 carries a point mutation in the YXXM motif (Y257F) (66), this has not been demonstrated in the case of TLR4 activation. The interaction between MyD88 and TLR4 is mediated by TIRAP (Toll-interleukin 1 receptor [TIR] domain containing adapter protein), which itself is recruited to the membrane by PIP2 (67). As the accumulation of PIP3 is dependent on MK2/3 in response to LPS (Fig. 6), MK2/3 might function to enhance the association of PI3K with the receptor complex to facilitate activation of its lipid kinase activity.
Our data obtained through the use of selective PI3K inhibitors suggest that p110α may be the major isoform required for Akt activation by TLR4 (Fig. 1C). It should be noted that our data do not exclude a role for p110γ, since PIK75 can block p110γ activity in addition to p110α (49). It is likely, however, that some degree of compensation occurs between p110 isoforms and that different isoforms may be utilized depending on the stimuli and cell type. Based on short hairpin RNA (shRNA) knockdown, a role for p110α has also been found in the RAW264.7 macrophage cell line downstream of the Fcγ receptor (68). Despite this, it has been reported by the same group that knockdown of p110β reduces LPS-induced Akt phosphorylation, while knockdown of p110α or knockout of p110γ has no effect (56), suggesting that isoform specificity varies with respect to the receptor that is activated. Furthermore, a recent study with dendritic cells has shown that a knock-in mutation rendering p110δ catalytically inactive reduced LPS-induced Akt activation (69). Further work will be required to resolve these conflicting results.
It is possible that the p38-MK2/3 pathway regulates PIP3 levels via controlling PIP3-specific phosphatases rather than PI3K itself. SHIP (Src homology 2-containing inositol 5-phosphate) and PTEN (phosphatase and tensin homolog deleted on chromosome 10) hydrolyze PIP3 to generate PI(3,4)P2 and PI(4,5)P2, respectively, and therefore, both phosphatases can function to antagonize PI3K pathway activation. Consistent with this, macrophages deficient in either SHIP or PTEN have elevated levels of Akt phosphorylation (70, 71). Thus, it is tempting to speculate that MK2/3 may affect PIP3 production by modulating the activity of one of these phosphatases, although this does not appear to occur via affecting protein levels of the phosphatases, as these remain unchanged in MK2/3 knockout BMDMs (Fig. 3A). It has been proposed, based on a comparison of knockout models, that PTEN acts constitutively to maintain low levels of PIP3 while SHIP is recruited to the plasma membrane in response to PI3K activation (72), so one possibility is that MK2/3 can interfere with SHIP recruitment to the membrane in response to LPS-induced signaling. The activation of TLRs is established to promote the induction of reactive oxygen species (ROS) in macrophages, and oxidative stress is known to activate Akt. Interestingly, the reversible oxidation of PTEN′s active-site cysteine has been proposed to regulate its activity (73). In RAW264.7 macrophages, blocking the generation of ROS has been shown to reduce the activation of Akt and its phosphorylation on Ser473 in response to LPS (73). In line with this, LPS promoted the oxidation and inhibition of PTEN (73). This suggests that one mechanism for the activation of Akt in response to TLR signaling is indirect and may involve MK2/3-dependent ROS production, which, in turn, inhibits PTEN to result in elevated PIP3 levels and Akt activation. This could also suggest that the generation of PIP3 in this system can occur in part via basal PI3K activity, which might explain the conflicting results (discussed above) on the p110 isoform involved. Further work will, however, be required in order to determine how p38-MK2/3 signaling fits into such a model, particularly as MK2 has recently been reported to either promote or inhibit ROS production depending on the cell type and stimulus (74).
It is interesting that the Cot/tpl2-ERK1/2 pathway has also been reported to cross talk with the Akt pathway in TLR-activated macrophages (75, 76). The regulation of Akt by Cot/tpl2 is distinct from that by MK2/3, as it is restricted to phosphorylation of Ser473. In Cot/tlp2 knockout BMDMs, there is no defect in Thr308 phosphorylation in response to LPS, but Ser473 is diminished along with Ser422 of SGK1, suggesting that Cot/tpl2 regulates Akt phosphorylation through mTORC2 activity (76).
In summary, we show here that MK2 and MK3 are required for Akt activation following TLR stimulation of macrophages. While MK2/3 do not phosphorylate Akt directly on Ser473 or Thr308, these kinases are required for the accumulation of PIP3 in response to LPS and therefore regulate Akt activity by affecting the PI3K pathway. The mechanism for exactly how this happens is currently unclear, but it may involve regulating the activity of a PIP3 phosphatase or modulating PI3K activation. The data we present demonstrate cross talk between the p38α-MK2/3 and Akt pathways in macrophages and describe novel regulation of Akt activity in TLR-mediated signaling.
ACKNOWLEDGMENTS
This work was supported by the Medical Research Council (United Kingdom) and Arthritis Research UK as well as by the DSTT in Dundee, United Kingdom, which is supported by AstraZeneca, Boehringer-Ingelheim, GlaxoSmithKline, Merck KgaA, Janssen Pharmaceutica, and Pfizer. M.G. is supported by the Deutsche Forschungsgemeinschaft.
We thank Dario Alessi for providing PDK1 floxed mice and the National Centre for Protein Kinase Profiling (http://www.kinase-screen.mrc.ac.uk) for analyzing the in vitro specificity of VX-745.
Footnotes
Published ahead of print 26 August 2013
REFERENCES
- 1.Manning BD, Cantley LC. 2007. AKT/PKB signaling: navigating downstream. Cell 129:1261–1274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Leevers SJ, Vanhaesebroeck B, Waterfield MD. 1999. Signalling through phosphoinositide 3-kinases: the lipids take centre stage. Curr. Opin. Cell Biol. 11:219–225 [DOI] [PubMed] [Google Scholar]
- 3.Franke TF. 2008. PI3K/Akt: getting it right matters. Oncogene 27:6473–6488 [DOI] [PubMed] [Google Scholar]
- 4.So L, Fruman DA. 2012. PI3K signalling in B- and T-lymphocytes: new developments and therapeutic advances. Biochem. J. 442:465–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hazeki K, Nigorikawa K, Hazeki O. 2007. Role of phosphoinositide 3-kinase in innate immunity. Biol. Pharm. Bull. 30:1617–1623 [DOI] [PubMed] [Google Scholar]
- 6.Lee YG, Lee J, Byeon SE, Yoo DS, Kim MH, Lee SY, Cho JY. 2011. Functional role of Akt in macrophage-mediated innate immunity. Front. Biosci. 16:517–530 [DOI] [PubMed] [Google Scholar]
- 7.Fukao T, Koyasu S. 2003. PI3K and negative regulation of TLR signaling. Trends Immunol. 24:358–363 [DOI] [PubMed] [Google Scholar]
- 8.Fukao T, Tanabe M, Terauchi Y, Ota T, Matsuda S, Asano T, Kadowaki T, Takeuchi T, Koyasu S. 2002. PI3K-mediated negative feedback regulation of IL-12 production in DCs. Nat. Immunol. 3:875–881 [DOI] [PubMed] [Google Scholar]
- 9.Pengal RA, Ganesan LP, Wei G, Fang H, Ostrowski MC, Tridandapani S. 2006. Lipopolysaccharide-induced production of interleukin-10 is promoted by the serine/threonine kinase Akt. Mol. Immunol. 43:1557–1564 [DOI] [PubMed] [Google Scholar]
- 10.Koyasu S. 2003. The role of PI3K in immune cells. Nat. Immunol. 4:313–319 [DOI] [PubMed] [Google Scholar]
- 11.Katso R, Okkenhaug K, Ahmadi K, White S, Timms J, Waterfield MD. 2001. Cellular function of phosphoinositide 3-kinases: implications for development, homeostasis, and cancer. Annu. Rev. Cell Dev. Biol. 17:615–675 [DOI] [PubMed] [Google Scholar]
- 12.Vieira OV, Botelho RJ, Rameh L, Brachmann SM, Matsuo T, Davidson HW, Schreiber A, Backer JM, Cantley LC, Grinstein S. 2001. Distinct roles of class I and class III phosphatidylinositol 3-kinases in phagosome formation and maturation. J. Cell Biol. 155:19–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Downward J. 1998. Mechanisms and consequences of activation of protein kinase B/Akt. Curr. Opin. Cell Biol. 10:262–267 [DOI] [PubMed] [Google Scholar]
- 14.Chan TO, Rittenhouse SE, Tsichlis PN. 1999. AKT/PKB and other D3 phosphoinositide-regulated kinases: kinase activation by phosphoinositide-dependent phosphorylation. Annu. Rev. Biochem. 68:965–1014 [DOI] [PubMed] [Google Scholar]
- 15.Williams MR, Arthur JS, Balendran A, van der Kaay J, Poli V, Cohen P, Alessi DR. 2000. The role of 3-phosphoinositide-dependent protein kinase 1 in activating AGC kinases defined in embryonic stem cells. Curr. Biol. 10:439–448 [DOI] [PubMed] [Google Scholar]
- 16.McManus EJ, Collins BJ, Ashby PR, Prescott AR, Murray-Tait V, Armit LJ, Arthur JS, Alessi DR. 2004. The in vivo role of PtdIns(3,4,5)P3 binding to PDK1 PH domain defined by knockin mutation. EMBO J. 23:2071–2082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bayascas JR, Wullschleger S, Sakamoto K, Garcia-Martinez JM, Clacher C, Komander D, van Aalten DM, Boini KM, Lang F, Lipina C, Logie L, Sutherland C, Chudek JA, van Diepen JA, Voshol PJ, Lucocq JM, Alessi DR. 2008. Mutation of the PDK1 PH domain inhibits protein kinase B/Akt, leading to small size and insulin resistance. Mol. Cell. Biol. 28:3258–3272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alessi DR, Andjelkovic M, Caudwell B, Cron P, Morrice N, Cohen P, Hemmings BA. 1996. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 15:6541–6551 [PMC free article] [PubMed] [Google Scholar]
- 19.Persad S, Attwell S, Gray V, Mawji N, Deng JT, Leung D, Yan J, Sanghera J, Walsh MP, Dedhar S. 2001. Regulation of protein kinase B/Akt-serine 473 phosphorylation by integrin-linked kinase: critical roles for kinase activity and amino acids arginine 211 and serine 343. J. Biol. Chem. 276:27462–27469 [DOI] [PubMed] [Google Scholar]
- 20.Feng J, Park J, Cron P, Hess D, Hemmings BA. 2004. Identification of a PKB/Akt hydrophobic motif Ser-473 kinase as DNA-dependent protein kinase. J. Biol. Chem. 279:41189–41196 [DOI] [PubMed] [Google Scholar]
- 21.Partovian C, Simons M. 2004. Regulation of protein kinase B/Akt activity and Ser473 phosphorylation by protein kinase Calpha in endothelial cells. Cell. Signal. 16:951–957 [DOI] [PubMed] [Google Scholar]
- 22.Viniegra JG, Martinez N, Modirassari P, Hernandez Losa J, Parada Cobo C, Sanchez-Arevalo Lobo VJ, Aceves Luquero CI, Alvarez-Vallina L, Ramon y Cajal S, Rojas JM, Sanchez-Prieto R. 2005. Full activation of PKB/Akt in response to insulin or ionizing radiation is mediated through ATM. J. Biol. Chem. 280:4029–4036 [DOI] [PubMed] [Google Scholar]
- 23.Dong LQ, Liu F. 2005. PDK2: the missing piece in the receptor tyrosine kinase signaling pathway puzzle. Am. J. Physiol. Endocrinol. Metab. 289:E187–E196. 10.1152/ajpendo.00011.2005 [DOI] [PubMed] [Google Scholar]
- 24.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. 2005. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307:1098–1101 [DOI] [PubMed] [Google Scholar]
- 25.Hresko RC, Mueckler M. 2005. mTOR.RICTOR is the Ser473 kinase for Akt/protein kinase B in 3T3-L1 adipocytes. J. Biol. Chem. 280:40406–40416 [DOI] [PubMed] [Google Scholar]
- 26.Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, Markhard AL, Sabatini DM. 2006. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol. Cell 22:159–168 [DOI] [PubMed] [Google Scholar]
- 27.Shiota C, Woo JT, Lindner J, Shelton KD, Magnuson MA. 2006. Multiallelic disruption of the rictor gene in mice reveals that mTOR complex 2 is essential for fetal growth and viability. Dev. Cell 11:583–589 [DOI] [PubMed] [Google Scholar]
- 28.Kumar A, Harris TE, Keller SR, Choi KM, Magnuson MA, Lawrence JC., Jr 2008. Muscle-specific deletion of rictor impairs insulin-stimulated glucose transport and enhances basal glycogen synthase activity. Mol. Cell. Biol. 28:61–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gaestel M. 2006. MAPKAP kinases—MKs—two's company, three's a crowd. Nat. Rev. Mol. Cell Biol. 7:120–130 [DOI] [PubMed] [Google Scholar]
- 30.Shaw M, Cohen P, Alessi DR. 1998. The activation of protein kinase B by H2O2 or heat shock is mediated by phosphoinositide 3-kinase and not by mitogen-activated protein kinase-activated protein kinase-2. Biochem. J. 336(Part 1):241–246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rane MJ, Coxon PY, Powell DW, Webster R, Klein JB, Pierce W, Ping P, McLeish KR. 2001. p38 kinase-dependent MAPKAPK-2 activation functions as 3-phosphoinositide-dependent kinase-2 for Akt in human neutrophils. J. Biol. Chem. 276:3517–3523 [DOI] [PubMed] [Google Scholar]
- 32.Taniyama Y, Ushio-Fukai M, Hitomi H, Rocic P, Kingsley MJ, Pfahnl C, Weber DS, Alexander RW, Griendling KK. 2004. Role of p38 MAPK and MAPKAPK-2 in angiotensin II-induced Akt activation in vascular smooth muscle cells. Am. J. Physiol. Cell Physiol. 287:C494–C499. 10.1152/ajpcell.00439.2003 [DOI] [PubMed] [Google Scholar]
- 33.Wu R, Kausar H, Johnson P, Montoya-Durango DE, Merchant M, Rane MJ. 2007. Hsp27 regulates Akt activation and polymorphonuclear leukocyte apoptosis by scaffolding MK2 to Akt signal complex. J. Biol. Chem. 282:21598–21608 [DOI] [PubMed] [Google Scholar]
- 34.Zaru R, Ronkina N, Gaestel M, Arthur JS, Watts C. 2007. The MAPK-activated kinase Rsk controls an acute Toll-like receptor signaling response in dendritic cells and is activated through two distinct pathways. Nat. Immunol. 8:1227–1235 [DOI] [PubMed] [Google Scholar]
- 35.O'Keefe SJ, Mudgett JS, Cupo S, Parsons JN, Chartrain NA, Fitzgerald C, Chen SL, Lowitz K, Rasa C, Visco D, Luell S, Carballo-Jane E, Owens K, Zaller DM. 2007. Chemical genetics define the roles of p38alpha and p38beta in acute and chronic inflammation. J. Biol. Chem. 282:34663–34671 [DOI] [PubMed] [Google Scholar]
- 36.Lawlor MA, Mora A, Ashby PR, Williams MR, Murray-Tait V, Malone L, Prescott AR, Lucocq JM, Alessi DR. 2002. Essential role of PDK1 in regulating cell size and development in mice. EMBO J. 21:3728–3738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee K, Nam KT, Cho SH, Gudapati P, Hwang Y, Park DS, Potter R, Chen J, Volanakis E, Boothby M. 2012. Vital roles of mTOR complex 2 in Notch-driven thymocyte differentiation and leukemia. J. Exp. Med. 209:713–728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.de Boer J, Williams A, Skavdis G, Harker N, Coles M, Tolaini M, Norton T, Williams K, Roderick K, Potocnik AJ, Kioussis D. 2003. Transgenic mice with hematopoietic and lymphoid specific expression of Cre. Eur. J. Immunol. 33:314–325 [DOI] [PubMed] [Google Scholar]
- 39.Beardmore VA, Hinton HJ, Eftychi C, Apostolaki M, Armaka M, Darragh J, McIlrath J, Carr JM, Armit LJ, Clacher C, Malone L, Kollias G, Arthur JS. 2005. Generation and characterization of p38beta (MAPK11) gene-targeted mice. Mol. Cell. Biol. 25:10454–10464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Weintz G, Olsen JV, Fruhauf K, Niedzielska M, Amit I, Jantsch J, Mages J, Frech C, Dolken L, Mann M, Lang R. 2010. The phosphoproteome of toll-like receptor-activated macrophages. Mol. Syst. Biol. 6:371. 10.1038/msb.2010.29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Walker KS, Deak M, Paterson A, Hudson K, Cohen P, Alessi DR. 1998. Activation of protein kinase B beta and gamma isoforms by insulin in vivo and by 3-phosphoinositide-dependent protein kinase-1 in vitro: comparison with protein kinase B alpha. Biochem. J. 331(Part 1):299–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Batty IH, van der Kaay J, Gray A, Telfer JF, Dixon MJ, Downes CP. 2007. The control of phosphatidylinositol 3,4-bisphosphate concentrations by activation of the Src homology 2 domain containing inositol polyphosphate 5-phosphatase 2, SHIP2. Biochem. J. 407:255–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vitari AC, Deak M, Collins BJ, Morrice N, Prescott AR, Phelan A, Humphreys S, Alessi DR. 2004. WNK1, the kinase mutated in an inherited high-blood-pressure syndrome, is a novel PKB (protein kinase B)/Akt substrate. Biochem. J. 378:257–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gray A, Olsson H, Batty IH, Priganica L, Downes CP. 2003. Nonradioactive methods for the assay of phosphoinositide 3-kinases and phosphoinositide phosphatases and selective detection of signaling lipids in cell and tissue extracts. Anal. Biochem. 313:234–245 [DOI] [PubMed] [Google Scholar]
- 45.Hemmings BA. 1997. Akt signaling: linking membrane events to life and death decisions. Science 275:628–630 [DOI] [PubMed] [Google Scholar]
- 46.Bain J, Plater L, Elliott M, Shpiro N, Hastie CJ, McLauchlan H, Klevernic I, Arthur JS, Alessi DR, Cohen P. 2007. The selectivity of protein kinase inhibitors: a further update. Biochem. J. 408:297–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Folkes AJ, Ahmadi K, Alderton WK, Alix S, Baker SJ, Box G, Chuckowree IS, Clarke PA, Depledge P, Eccles SA, Friedman LS, Hayes A, Hancox TC, Kugendradas A, Lensun L, Moore P, Olivero AG, Pang J, Patel S, Pergl-Wilson GH, Raynaud FI, Robson A, Saghir N, Salphati L, Sohal S, Ultsch MH, Valenti M, Wallweber HJ, Wan NC, Wiesmann C, Workman P, Zhyvoloup A, Zvelebil MJ, Shuttleworth SJ. 2008. The identification of 2-(1H-indazol-4-yl)-6-(4-methanesulfonyl-piperazin-1-ylmethyl)-4-morpholin-4-yl-t hieno[3,2-d]pyrimidine (GDC-0941) as a potent, selective, orally bioavailable inhibitor of class I PI3 kinase for the treatment of cancer. J. Med. Chem. 51:5522–5532 [DOI] [PubMed] [Google Scholar]
- 48.Yap TA, Yan L, Patnaik A, Fearen I, Olmos D, Papadopoulos K, Baird RD, Delgado L, Taylor A, Lupinacci L, Riisnaes R, Pope LL, Heaton SP, Thomas G, Garrett MD, Sullivan DM, de Bono JS, Tolcher AW. 2011. First-in-man clinical trial of the oral pan-AKT inhibitor MK-2206 in patients with advanced solid tumors. J. Clin. Oncol. 29:4688–4695 [DOI] [PubMed] [Google Scholar]
- 49.Knight ZA, Gonzalez B, Feldman ME, Zunder ER, Goldenberg DD, Williams O, Loewith R, Stokoe D, Balla A, Toth B, Balla T, Weiss WA, Williams RL, Shokat KM. 2006. A pharmacological map of the PI3-K family defines a role for p110alpha in insulin signaling. Cell 125:733–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ronkina N, Kotlyarov A, Dittrich-Breiholz O, Kracht M, Hitti E, Milarski K, Askew R, Marusic S, Lin LL, Gaestel M, Telliez JB. 2007. The mitogen-activated protein kinase (MAPK)-activated protein kinases MK2 and MK3 cooperate in stimulation of tumor necrosis factor biosynthesis and stabilization of p38 MAPK. Mol. Cell. Biol. 27:170–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mourey RJ, Burnette BL, Brustkern SJ, Daniels JS, Hirsch JL, Hood WF, Meyers MJ, Mnich SJ, Pierce BS, Saabye MJ, Schindler JF, South SA, Webb EG, Zhang J, Anderson DR. 2010. A benzothiophene inhibitor of mitogen-activated protein kinase-activated protein kinase 2 inhibits tumor necrosis factor alpha production and has oral anti-inflammatory efficacy in acute and chronic models of inflammation. J. Pharmacol. Exp. Ther. 333:797–807 [DOI] [PubMed] [Google Scholar]
- 52.Alford KA, Glennie S, Turrell BR, Rawlinson L, Saklatvala J, Dean JL. 2007. Heat shock protein 27 functions in inflammatory gene expression and transforming growth factor-beta-activated kinase-1 (TAK1)-mediated signaling. J. Biol. Chem. 282:6232–6241 [DOI] [PubMed] [Google Scholar]
- 53.Sapkota GP, Cummings L, Newell FS, Armstrong C, Bain J, Frodin M, Grauert M, Hoffmann M, Schnapp G, Steegmaier M, Cohen P, Alessi DR. 2007. BI-D1870 is a specific inhibitor of the p90 RSK (ribosomal S6 kinase) isoforms in vitro and in vivo. Biochem. J. 401:29–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gantner BN, Simmons RM, Canavera SJ, Akira S, Underhill DM. 2003. Collaborative induction of inflammatory responses by dectin-1 and Toll-like receptor 2. J. Exp. Med. 197:1107–1117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Elcombe SE, Naqvi S, Van Den Bosch MW, MacKenzie KF, Cianfanelli F, Brown GD, Arthur JS. 2013. Dectin-1 regulates IL-10 production via a MSK1/2 and CREB dependent pathway and promotes the induction of regulatory macrophage markers. PLoS One 8:e60086. 10.1371/journal.pone.0060086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tsukamoto K, Hazeki K, Hoshi M, Nigorikawa K, Inoue N, Sasaki T, Hazeki O. 2008. Critical roles of the p110 beta subtype of phosphoinositide 3-kinase in lipopolysaccharide-induced Akt activation and negative regulation of nitrite production in RAW 264.7 cells. J. Immunol. 180:2054–2061 [DOI] [PubMed] [Google Scholar]
- 57.Hirsch E, Katanaev VL, Garlanda C, Azzolino O, Pirola L, Silengo L, Sozzani S, Mantovani A, Altruda F, Wymann MP. 2000. Central role for G protein-coupled phosphoinositide 3-kinase gamma in inflammation. Science 287:1049–1053 [DOI] [PubMed] [Google Scholar]
- 58.Zheng C, Lin Z, Zhao ZJ, Yang Y, Niu H, Shen X. 2006. MAPK-activated protein kinase-2 (MK2)-mediated formation and phosphorylation-regulated dissociation of the signal complex consisting of p38, MK2, Akt, and Hsp27. J. Biol. Chem. 281:37215–37226 [DOI] [PubMed] [Google Scholar]
- 59.Venigalla RK, McGuire VA, Clarke R, Patterson-Kane JC, Najafov A, Toth R, McCarthy PC, Simeons F, Stojanovski L, Arthur JS. 2013. PDK1 regulates VDJ recombination, cell-cycle exit and survival during B-cell development. EMBO J. 32:1008–1022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.García-Martínez JM, Moran J, Clarke RG, Gray A, Cosulich SC, Chresta CM, Alessi DR. 2009. Ku-0063794 is a specific inhibitor of the mammalian target of rapamycin (mTOR). Biochem. J. 421:29–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hu P, Nebreda AR, Liu Y, Carlesso N, Kaplan M, Kapur R. 2012. P38alpha negatively regulates T helper type 2 responses by orchestrating multiple TCR-associated signals. J. Biol. Chem. 10.1074/jbc.M112.355594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ojaniemi M, Glumoff V, Harju K, Liljeroos M, Vuori K, Hallman M. 2003. Phosphatidylinositol 3-kinase is involved in Toll-like receptor 4-mediated cytokine expression in mouse macrophages. Eur. J. Immunol. 33:597–605 [DOI] [PubMed] [Google Scholar]
- 63.Laird MH, Rhee SH, Perkins DJ, Medvedev AE, Piao W, Fenton MJ, Vogel SN. 2009. TLR4/MyD88/PI3K interactions regulate TLR4 signaling. J. Leukoc. Biol. 85:966–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Arbibe L, Mira JP, Teusch N, Kline L, Guha M, Mackman N, Godowski PJ, Ulevitch RJ, Knaus UG. 2000. Toll-like receptor 2-mediated NF-kappa B activation requires a Rac1-dependent pathway. Nat. Immunol. 1:533–540 [DOI] [PubMed] [Google Scholar]
- 65.Sarkar SN, Peters KL, Elco CP, Sakamoto S, Pal S, Sen GC. 2004. Novel roles of TLR3 tyrosine phosphorylation and PI3 kinase in double-stranded RNA signaling. Nat. Struct. Mol. Biol. 11:1060–1067 [DOI] [PubMed] [Google Scholar]
- 66.Gelman AE, LaRosa DF, Zhang J, Walsh PT, Choi Y, Sunyer JO, Turka LA. 2006. The adaptor molecule MyD88 activates PI-3 kinase signaling in CD4+ T cells and enables CpG oligodeoxynucleotide-mediated costimulation. Immunity 25:783–793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kagan JC, Medzhitov R. 2006. Phosphoinositide-mediated adaptor recruitment controls Toll-like receptor signaling. Cell 125:943–955 [DOI] [PubMed] [Google Scholar]
- 68.Tamura N, Hazeki K, Okazaki N, Kametani Y, Murakami H, Takaba Y, Ishikawa Y, Nigorikawa K, Hazeki O. 2009. Specific role of phosphoinositide 3-kinase p110alpha in the regulation of phagocytosis and pinocytosis in macrophages. Biochem. J. 423:99–108 [DOI] [PubMed] [Google Scholar]
- 69.Aksoy E, Taboubi S, Torres D, Delbauve S, Hachani A, Whitehead MA, Pearce WP, Berenjeno-Martin I, Nock G, Filloux A, Beyaert R, Flamand V, Vanhaesebroeck B. 2012. The p110delta isoform of the kinase PI(3)K controls the subcellular compartmentalization of TLR4 signaling and protects from endotoxic shock. Nat. Immunol. 13:1045–1054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fang H, Pengal RA, Cao X, Ganesan LP, Wewers MD, Marsh CB, Tridandapani S. 2004. Lipopolysaccharide-induced macrophage inflammatory response is regulated by SHIP. J. Immunol. 173:360–366 [DOI] [PubMed] [Google Scholar]
- 71.Cao X, Wei G, Fang H, Guo J, Weinstein M, Marsh CB, Ostrowski MC, Tridandapani S. 2004. The inositol 3-phosphatase PTEN negatively regulates Fc gamma receptor signaling, but supports Toll-like receptor 4 signaling in murine peritoneal macrophages. J. Immunol. 172:4851–4857 [DOI] [PubMed] [Google Scholar]
- 72.Sly LM, Rauh MJ, Kalesnikoff J, Buchse T, Krystal G. 2003. SHIP, SHIP2, and PTEN activities are regulated in vivo by modulation of their protein levels: SHIP is up-regulated in macrophages and mast cells by lipopolysaccharide. Exp. Hematol. 31:1170–1181 [DOI] [PubMed] [Google Scholar]
- 73.Leslie NR, Bennett D, Lindsay YE, Stewart H, Gray A, Downes CP. 2003. Redox regulation of PI 3-kinase signalling via inactivation of PTEN. EMBO J. 22:5501–5510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kobayashi Y, Qi X, Chen G. 2012. MK2 regulates Ras oncogenesis through stimulating ROS production. Genes Cancer 3:521–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.López-Pelaez M, Soria-Castro I, Bosca L, Fernandez M, Alemany S. 2011. Cot/tpl2 activity is required for TLR-induced activation of the Akt p70 S6k pathway in macrophages: implications for NO synthase 2 expression. Eur. J. Immunol. 41:1733–1741 [DOI] [PubMed] [Google Scholar]
- 76.López-Pelaez M, Fumagalli S, Sanz C, Herrero C, Guerra S, Fernandez M, Alemany S. 2012. Cot/tpl2-MKK1/2-Erk1/2 controls mTORC1-mediated mRNA translation in Toll-like receptor-activated macrophages. Mol. Biol. Cell 23:2982–2992 [DOI] [PMC free article] [PubMed] [Google Scholar]