

Abstract
Mutation in CYP1B1 has been reported for patients with congenital glaucoma. However, the underlying mechanisms remain unknown. Here we show increased diurnal intraocular pressure (IOP) in Cyp1b1-deficient (Cyp1b1−/−) mice. Cyp1b1−/− mice presented ultrastructural irregular collagen distribution in their trabecular meshwork (TM) tissue along with increased oxidative stress and decreased levels of periostin (Postn). Increased levels of oxidative stress and decreased levels of Postn were also detected in human glaucomatous TM tissues. Furthermore, Postn-deficient mice exhibited TM tissue ultrastructural abnormalities similar to those of Cyp1b1−/− mice. Administration of the antioxidant N-acetylcysteine (NAC) restored structural abnormality of TM tissue in Cyp1b1−/− mice. In addition, TM cells prepared from Cyp1b1−/− mice exhibited increased oxidative stress, altered adhesion, and decreased levels of Postn. These aberrant cellular responses were reversed in the presence of NAC or by restoration of Cyp1b1 expression. Cyp1b1 knockdown or inhibition of CYP1B1 activity in Cyp1b1+/+ TM cells resulted in a Cyp1b1−/− phenotype. Thus, metabolic activity of CYP1B1 contributes to oxidative homeostasis and ultrastructural organization and function of TM tissue through modulation of Postn expression.
INTRODUCTION
Glaucoma is the second leading cause of blindness worldwide. It is a type of optic neuropathy leading to progressive damage of the optic nerve that can further result in loss of vision if left untreated (1). The major risk factor and the main focus of treatment for glaucoma is an increased intraocular pressure (IOP) that is related mainly to defects in the drainage of the aqueous humor (2). The aqueous humor outflow system is dependent primarily on the trabecular meshwork (TM), a tissue composed of an irregular network of connective tissue beams lined by endothelial cells that is circumferentially present at the base of the iris and peripheral cornea (iridocorneal angle). The TM is responsible for draining the aqueous humor from the anterior chamber and maintaining homeostasis of IOP (3). Primary congenital glaucoma (PCG) is related mainly to developmental defects of the TM (3), but the underlying mechanisms remain elusive.
Cytochrome P450 family 1 subfamily B polypeptide 1 (CYP1B1) belongs to the CYP450 family that catalyzes NADPH-supported monooxygenation of diverse xenobiotics and endogenous molecules (4, 5). In these reactions, one atom of oxygen is inserted into an organic substrate, while the other oxygen is reduced to water (6). Specifically, CYP1B1 is involved in the metabolism of steroids (7), retinol, retinal (8, 9), arachidonate (9), and melatonin (10–12). CYP1B1 is expressed in extrahepatic epithelia, specifically mesenchymal cells, and exhibits a developmentally regulated pattern of expression (13). Its expression is conserved in early embryos across several species during the development of the ocular tissues, neural crest, and hindbrain. Strong expression of CYP1B1 in eyes suggests that CYP1B1 may play important roles in normal eye development and function (14–16).
Genetic linkage analyses and mutation studies have identified CYP1B1 as a causative gene in PCG (17–20). Cyp1b1−/− mice exhibit similar defects in the development of TM as those reported for PCG patients (21). The severity of these defects is enhanced in albino mice with a deficiency in tyrosinase, an enzyme important for production of melanin and l-dihydroxyphenylalanine (l-Dopa). In addition, the symptoms can be alleviated by the administration of l-Dopa (21), perhaps as a result of reduced oxidative stress. However, detailed mechanisms of how a lack of CYP1B1 expression and/or activity contributes to the pathogenesis of glaucoma remain unknown.
Oxidative stress is regarded as an imbalance between prooxidant and/or free radical production and opposing antioxidant defenses. It can be exacerbated when there is a deficiency of enzymes that detoxify free radicals (22). Oxidative stress appears to be a common disturbance in the pathogenesis of diseases related to collagen degeneration, including skeletal and dental dysgenesis. Glaucoma has also been linked to oxidative degenerative processes affecting the TM and its endothelial cells (22). Given the nature of CYP1B1-catalyzed reactions, we hypothesized that Cyp1b1 is closely related to the oxidative homeostasis of TM tissue.
Fibrillar collagen is a supporting framework of the TM tissue (23). TM tissue shares essential similarity with other collagen-rich tissues under constant mechanical stress, including bone, cartilage, and teeth. These tissues are all of neural crest origin (24) and express many common genes (25), including the periostin (Postn) gene. Postn is a secreted 90-kDa extracellular matrix (ECM) protein of the fasciclin family (26). It is strongly expressed in collagen-rich connective tissues, with an essential role in collagen fibrillogenesis (27). Postn interacts with ECM proteins such as collagen I and is involved in the remodeling of tissues during developmental stages (28–31). Its deficiency often leads to impaired collagen fibril formation and reduced mechanical strength of relevant tissues (27, 30). However, its potential function in TM tissue, and its contribution to the pathogenesis of glaucoma, has not been explored previously.
Here we tested the hypothesis that Cyp1b1 deficiency has a significant impact on TM cell function and that its metabolic activity contributes to oxidative homeostasis and development of TM tissue. We showed increased IOP in Cyp1b1−/− mice compared to Cyp1b1+/+ mice. Using transmission electron microscopy (TEM), we revealed abnormal collagen distribution in Cyp1b1−/− TM tissues. This was associated with increased oxidative stress in Cyp1b1−/− TM tissues and was reversed by administration of the antioxidant N-acetylcysteine (NAC). To delineate the Cyp1b1 cell-autonomous mechanisms involved, we isolated TM cells from Cyp1b1+/+ and Cyp1b1−/− transgenic mice. The lack of Cyp1b1 had a significant impact on the TM cell oxidative state and decreased Postn production. Cy1b1−/−TM cells exhibited reduced levels of Postn, which were restored to normal levels in the presence of NAC. The Postn level was also decreased in TM tissues from Cyp1b1−/− mice as well as glaucomatous human eyes. Furthermore, Postn-deficient (Postn−/−) mice exhibited TM tissue defects similar to those of Cyp1b1−/− mice. Thus, appropriate expression of Postn is essential for the organization and structural integrity of TM tissue, and its expression is impacted by Cyp1b1 expression and the cellular oxidative state.
MATERIALS AND METHODS
Animals and human specimens.
All experiments were carried out in accordance with the Association for Research in Vision and Ophthalmology Statement for the Use of Animals in Ophthalmic and Vision Research and were approved by the Institutional Animal Care and Use Committee of the University of Wisconsin School of Medicine and Public Health. For TM cell isolation, Immorto Cyp1b1−/− mice expressing a temperature-sensitive simian virus 40 (SV40) large T antigen (Charles River Laboratories, Wilmington, MA) in a C57BL/6 background were generated as previously described (13). Postn-deficient mice were generated in a C57BL/6 background as previously described (32). Normal and glaucomatous human TM tissues (see Table S1 in the supplemental material) were obtained from T. Michael Nork (University of Wisconsin—Madison) and processed in the laboratory of Donna Peters (University of Wisconsin—Madison) under institutional review board (IRB) approval.
Tonometer measurements of IOP in mice.
We used a commercial rebound tonometer for rodents (ICare Finland Ltd.) for noninvasive IOP measurements in mice. The calibration procedure for the tonometer was described previously (33). Mice were anesthetized by intraperitoneal injection of a ketamine-xylazine mixture (100 mg/kg of body weight ketamine hydrochloride and 5 mg/kg xylazine). All the IOPs were measured at a specific time, beginning between 10 a.m. and 1 p.m. during daylight. All measurements were taken during 4 to 7 min after anesthetization to minimize the effect of anesthesia on the IOP. The IOP was measured six times per eye at each time point, and the average value was calculated.
Transmission electron microscopy and TM morphometric analysis.
Murine eyes were enucleated and immersion fixed in 2% paraformaldehyde (PFA) and 2.5% glutaraldehyde in 0.1 M phosphate-buffered saline (PBS; pH 7.4) at 4°C overnight. We sampled the superior and inferior aspects in each animal. Eyes were sectioned on the midsagittal vertical plane and processed for routine TEM. Briefly, the globes were postfixed in 2% PFA and 2.5% glutaraldehyde at 4°C and treated with 1% OsO4 in PBS for 2 h at room temperature, followed by three 10-min washes with 0.1 M sodium acetate buffer. Tissues were then stained with 2% uranyl acetate in sodium acetate buffer for 1 h at room temperature, washed in buffer, dehydrated in a graded ethanol series (40% to 100%), and infiltrated with propylene oxide-812 resin (1005 Embed 812; EMS, Fort Washington, PA). The samples were embedded with fresh 100% 812 resin in molds and polymerized in a 60°C oven for 36 h. Ultrathin sections (90 nm) were analyzed by using a Philips 410 transmission electron microscope (Philips Medical System, Andover, MA).
In order to quantify the density of collagen fibrils in the TM, five nonconsecutive TEM images of the anterior, mid-, and posterior TM at a ×15,000 magnification were processed and analyzed by using FIJI software (http://fiji.sc/Fiji). Briefly, after the spatial calibration of the system using the image's scale bar, images were processed with the following algorithm: (i) background subtraction was performed on the image with the Process>Subtract Background option, (ii) segmentation of the collagen was performed with the Image>Adjust>Threshold tool option, (iii) the threshold was manually adjusted to select the collagen fibers, and (v) the percentage of collagen fibrils in the image and the area of collagen fibrils in μm2 were measured in the threshold collagen with the Analyze>Measure tool.
The ultrastructural morphology of the collagen and trabecular meshwork cells was also analyzed by a semiquantitative scoring system. Collagen morphology was evaluated with scores ranging from “0” to “4,” with 0 indicating no lesion, 1 indicating mild and multifocal areas of collagen fiber disarrangement, 2 indicating moderate and multifocal areas of collagen disarrangements, 3 indicating moderate and diffuse collagen disarrangements, and 4 indicating severe and diffuse collagen disarrangements. Trabecular meshwork cell morphology was analyzed by accessing the following ultrastructural cell lesion criteria: irregular cell surface, loss of contact with basement membrane, cytoplasmic vacuolization, cell swelling, and the presence of irregular cytoplasmic material. Samples were scored from “0” to “3,” with 0 indicating no lesion, 1 indicating lesions with 1 to 2 criteria present, 2 indicating those with 3 to 4 criteria present, and 3 indicating those with all 5 criteria present.
Immunohistochemistry.
Eyes were fixed and paraffin embedded by using standard procedures. Sections were cut, deparaffinized, and stained as previously described (34). Rabbit anti-4-hydroxyl-2-nonenal (anti-HNE) (1:500 dilution prepared in blocking solution; Alpha Diagnostic International, San Antonio, TX) and rabbit anti-Postn (Abcam, Cambridge, MA) were used. Samples were examined by using a Zeiss fluorescence microscope (AxioCam HRm; Zeiss, Germany), Zeiss objective lenses (Plan-Neofluar 5×/0.15 and 40×/0.75), and AxioVision Rel 4.8 acquisition software. The fluorescence source was obtained from Chrome Photofluro (Chrome Technology Corp., Rockingham, VT), and Texas Red was applied as the filter. Fluorescence intensities were quantified using ImageJ (NIH [http://rsbweb.nih.gov/ij/]).
Isolation and culture of TM cells.
Trabecular meshwork cells were isolated from Cyp1b1+/+ and Cyp1b1−/− Immorto mice, as previously described (35). Briefly, trabecular meshwork was collected from one litter (4 to 5 pups, 4 weeks old). The eyes were bisected along the equator, and anterior chambers were harvested. After removal of the lens and iris, the cilliary body was grasped and cut away. The outflow tissue was then distinguished. The band was cut and placed into a 35-mm sterile petri dish coated with 2 μg/ml fibronectin in Dulbecco's PBS (Sigma, St. Louis, MO). One hundred microliters of low-glucose Dulbecco's modified Eagle's medium (LG-DMEM; Sigma) containing 20% fetal bovine serum (FBS), 2 mM l-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 1% nonessential amino acids, and murine recombinant gamma interferon (R&D, Minneapolis, MN) at 44 U/ml was added. A sterile coverslip was placed on top of the TM tissue to keep it attached. One milliliter of additional medium was added, and the medium was changed every 3 days. Cells and tissues were maintained at 33°C with 5% CO2. The explants were carefully monitored to prevent contamination by other cell types. The growth pattern and morphology of cultured TM cells were different from those of neighboring cell types. Keratocytes and corneal endothelial cells, which usually grew separately from TM cells, were removed with a sterile cotton swab so that only the TM cells were left on the dish (36). Confluent cultures were trypsinized with 0.25% trypsin and 1 mM EDTA and transferred onto 1% gelatin-coated 60-mm tissue culture plates at split ratios of 1:3. Human TM-1 cells were cultured at 37°C under similar growth conditions.
Cell proliferation assays.
To determine cell proliferation rates, Cyp1b1+/+ and Cyp1b1−/− TM cells were plated onto multiple sets of plates and counted for 2 weeks, as previously described (37). For DNA synthesis rates, Cyp1b1+/+ and Cyp1b1−/− TM cells undergoing DNA synthesis were labeled with 5-ethynyl-2′-deoxyuridine (EdU) from a Clik-iT-EdU flow cytometry kit (Invitrogen, Carlsbad, CA), followed by fluorescence-activated cell sorter (FACS) analysis, as recommended by the supplier.
Apoptosis assays.
Cyp1b1+/+ and Cyp1b1−/− TM cells were plated at 1.25 × 104 cells/well in a 96-well plate. Cells were pretreated with 1 mM hydrogen peroxide (H2O2; Fisher Scientific, Fair Lawn, NJ) or 10 nM staurosporine (an apoptosis stimulus) (Enzo Life Sciences, Plymouth Meeting, PA) for 8 h, and levels of apoptosis were compared with the basal level of apoptosis. Caspase-3 and -7 activities of apoptotic cells were then evaluated with a Caspase-Glo 3/7 assay (Promega, Madison, WI), using a luminescent microplate reader (Victa2 1420 multilabel counter; PerkinElmer, Waltham, MA).
Cell viability assays.
Cells of each genotype were plated at 1.5 × 104 cells per well on a 96-well plate and incubated with 1.25 mM H2O2 for 24 h at 33°C. The percentage of live cells was determined by using the CellTiter 96 AQueous nonradioactive cell proliferation assay kit (Promega, Madison, WI).
Dihydroethidium measurements of oxidative stress.
TM cells (5 × 104) were plated onto glass coverslips (Corning, NY) 1 day before staining. They were then challenged with 0.85 mM H2O2 for 40 min, rinsed with normal medium twice, and incubated with 5 mM dihydroethidium (DHE; Invitrogen, Carlsbad, CA) for 20 min at 33°C. After aspiration, cells were incubated with normal medium at 33°C for 30 min twice to recover. Cells on the coverslip were mounted with PBS and photographed as described above. Four fields for each sample/treatment from four independent experiments were selected and analyzed for fluorescence density with ImageJ from the NIH (http://rsbweb.nih.gov/ij/). Values were obtained from 5 to 6 cells captured within each field.
Cell adhesion assays.
Adhesion assays were performed as previously described (37). Briefly, cells of each genotype were plated into wells of a 96-well plate previously coated with various concentrations of ECM proteins, including fibronectin, collagen I, collagen IV, and vitronectin. Following 2 h of incubation, cells that were not able to adhere were washed away. Attached cells were then lysed and quantified for intracellular acid phosphatase levels by using a 96-well plate reader at 405 nm.
Western blot analysis.
Collection of cell lysates and analysis with 4 to 20% SDS-PAGE gels were performed as previously described (13). Proteins were transferred onto nitrocellulose membranes and blotted with the following specific antibodies: anti-myocilin 7.1 (a generous gift of Michael P. Fautsch, Mayo Clinic, Rochester, MN [38]), anti-CYP1B1 (developed in our laboratory), anti-thrombospondin-1 (anti-TSP1) (1:1,000, A6.1; Neo Markers), anti-TSP2 (1:1,000; BD Transduction), antifibronectin, anti-ADAMTS10 (Santa Cruz Biotechnology), anti-CHI3L1, SPARC, osteopontin, Postn (1:1,000; R&D), and anti-tenascin-C (1:1000, Millipore AB19013). All blots were stripped and incubated with anti-β-actin (Sigma) antibody for a loading control. For analysis of secreted proteins, cells were incubated with serum-free growth medium for 48 h. Conditioned medium was collected and clarified by centrifugation, and the levels of various ECM proteins were determined by Western blotting as described above.
FACScan analysis.
Flow cytometry was performed as previously described (39). Cells were incubated with anti-integrin α2, α3, α4, α5, β1, and β8 (Santa Cruz); anti-integrin α5β1 (MAB1999; Millipore), α6 (MAB1378; Millipore), αV (AB1930; Millipore), αVβ3 (MAB1976; Millipore), and β3 (MAB1957; Millipore); and anti-integrin β4 (BD Pharmingen) primary antibodies at a 1:200 dilution. Cells were then washed and incubated with appropriate fluorescein isothiocyanate (FITC)-conjugated secondary antibodies (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA). Cells were washed with Tris-buffered saline (TBS), resuspended in TBS, and analyzed by using a FACScan caliber cytometer (Becton Dickinson, Franklin Lakes, NJ). For detection of α-smooth muscle actin, a FITC-conjugated anti-α-smooth muscle actin antibody (Sigma) was used.
Adenovirus infection of TM cells.
Replication-deficient adenoviruses expressing full-length mouse Cyp1b1 (GenBank accession no. NM_009994), under the control of the cytomegalovirus (CMV) promoter, were generated by using the pAdTrack-CMV vector and AdEasy system as previously described (13, 40). TM cells were plated onto 60-mm dishes and were incubated with either 1.5 PFU/cell {for MTS [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium] assays} or 3 PFU/cell (for adhesion assays) recombinant adenovirus expressing Cyp1b1 sense cDNA or the vector control in 1.5 ml Opti-MEM together with 15 μl Lipofectin (Invitrogen) in a tissue culture incubator at 33°C overnight. Cells were fed on the next day with normal growth medium, recovered for 2 days (for MTS assays) or 4 days (for adhesion assays), and collected for analyses.
CYP1B1 knockdown in TM cells.
Silencing of Cyp1b1 in Cyp1b1+/+ TM cells were performed with various lentiviruses targeting different regions of mouse Cyp1b1 (Mission lentiviral transduction particles [GenBank accession no. NM_009994], clone identification no. TRCN0000250434, -35, -36, -37, and -38; Sigma). Briefly, cells were plated onto wells of a 12-well plate at 75% confluence prior to transduction. On the next day, an appropriate amount of viral particles at a multiplicity of infection (MOI) of 2 transforming units (TU)/cell was added together with 8 μg/ml hexadimethrine bromide, to enhance transduction, and was incubated with cells at 33°C overnight. Mission pLKO.1-puro nonmammalian small interfering RNA (siRNA) control transduction particles (product no. SHC002V; Sigma) were used as the nontargeting negative control. Cells were fed with fresh growth medium on day 3. Selection began on day 4 with medium containing 5 μg/ml puromycin to select the transfected cells with the specific siRNA construct. Medium was replaced with fresh medium containing puromycin every 4 days until resistant colonies were identified. Colonies were further expanded and assayed for knockdown of mouse Cyp1b1 by Western blotting as described above. Stable cell populations expressing the specific siRNAs TRCN0000250434 and TRCN0000250438 were used for further analysis. Similarly, silencing of CYP1B1 in human TM-1 cells was performed with Mission lentiviral particles from Sigma (GenBank accession no. NM_000104) (clone identification no. TRCN0000062323, -24, -25, -26, and -27). A stable population expressing a specific siRNA, TRCN0000062324, was used for further analysis.
RNA purification and quantitative real-time PCR.
Total RNA extraction, cDNA synthesis, amplification, and quantification of DNA were performed as previously described (41). RpL13A was used as a housekeeping gene for normalization. The primers used are as follows: mAQP1-F (CTTGTCTGTGGCCCTTGGA), mAQP1-R (GTTGATACCGCAGCCAGTGTAG), RpL13A-F (TCTCAAGGTTGTTCGGCTGAA), and RpL13A-R (GCCAGACGCCCCAGGTA).
Statistical analysis.
Data are reported as means ± standard errors of the means (SEM) unless indicated otherwise. One-way analysis of variance (ANOVA) with Tukey's multiple-comparison test (see Fig. 5) or two-tailed unpaired Student's t test (for all the other data statistics) was used. Differences were regarded as significant when the P value was <0.01 or when the P value was <0.05.
Fig 5.
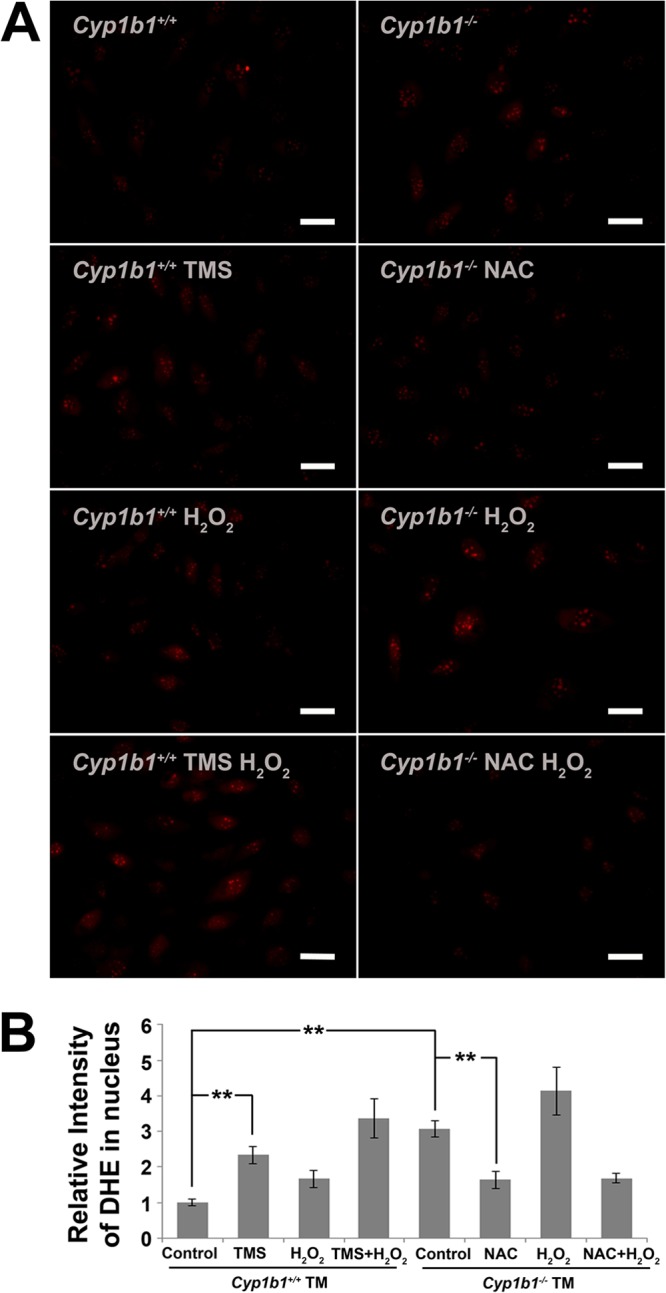
Increased intracellular oxidative stress in Cyp1b1−/− TM cells. (A) Increased DHE staining in Cyp1b1−/− TM cells (∗∗, P < 0.0001) (n = 4), which was prevented by the antioxidant NAC (∗∗, P = 0.003) (n = 4). Cyp1b1+/+ TM cells showed increased DHE staining when incubated with TMS (∗∗, P = 0.005) (n = 3). (B) Quantitative assessment of fluorescence intensities.
RESULTS
Elevation of intraocular pressure in Cyp1b1−/− mice.
Given the fact that CYP1B1 mutations contribute to pathogenesis of glaucoma, we determined the impact that a lack of Cyp1b1 has on intraocular pressure (IOP) in C57BL/6J mice. We systematically measured the IOPs of Cyp1b1+/+ and Cyp1b1−/− mice at various ages, including 3 weeks, 3 to 6 weeks, 6 to 12 weeks, 3 to 6 moths, and 6 to 10 months of age. Cyp1b1−/− mouse IOPs at all time points were significantly elevated compared with those of Cyp1b1+/+ mice. The results from mice 6 to 12 weeks of age are shown as representative results (Fig. 1). Figure 1 shows a modest but statistically significant elevation of IOP in Cyp1b1−/− mice compared with that in Cyp1b1+/+ mice. The mean IOP of Cyp1b1+/+ mice was 10.14 ± 0.07 mm Hg, which is consistent with the previously reported value of 10.4 ± 2.9 mm Hg (42). Correspondingly, the mean IOP of Cyp1b1−/− mice was 11.45 ± 0.20 mm Hg. The 10% difference in IOP was statistically significant and agrees with the current view that a 10% change in IOP is considerable and not to be neglected (43).
Fig 1.
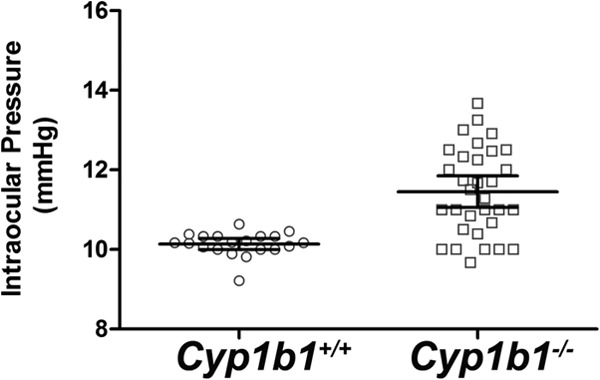
Increased intraocular pressure (IOP) in 6- to 12-week-old Cyp1b1−/− mice (Cyp1b1+/+, n = 20; Cyp1b1−/−, n = 32) (P < 0.0001). Bars indicate means ± 95% confidence intervals.
Disruption of collagen fibers and increased oxidative stress in Cyp1b1−/− TM tissue.
Cyp1b1−/− albino mice exhibit severe ocular drainage structure abnormalities resembling those reported for human PCG patients, yet those observed in pigmented Cyp1b1−/− mice were much more modest (21). Here we investigated the abnormalities of TM tissues in pigmented Cyp1b1−/− mice on the C57BL/6J background using TEM. Figure 2A shows TM tissues collected from 3-week-old Cyp1b1+/+ (left) and Cyp1b1−/− (right) mice. Cyp1b1+/+ TM normally consisted of multiple trabecular beams composed of a core of collagen fibers interspersed with small amounts of elastic tissues surrounded by a single layer of flattened endothelial cells (TM cells). Collagen fibers (Fig. 2A, bottom left, asterisk) occupied the core of the beams and were oriented parallel to the long axis. The periodicity of collagen fibers was homogenous. The TM cells presented normal cell morphology with homogenous cytoplasm, regular cell membranes, and close contact with the collagenous core (Fig. 2A, bottom left, arrow).
Fig 2.

Irregular collagen distribution and increased oxidative stress in Cyp1b1−/− TM tissue. (A) TM tissues of Cyp1b1+/+ mice show highly organized trabecular beams with robust collagen (bottom left, asterisk) and healthy TM cells (arrow). Cyp1b1−/− mouse TM presented irregular collagen distribution (bottom right, asterisk) and TM cells under stress with a distorted cytoplasm (arrow) (n = 15). Scale bar, 2 μm. Five sectors per eye were evaluated. (B) Quantitative assessment of the amount of collagen occupying the spaces in the trabecular beams (∗∗∗, P = 0.0004) (Cyp1b1+/+ TM, n = 4; Cyp1b1−/− TM, n = 3). (C) Severity grades of abnormal collagen fibers in Cyp1b1+/+ and Cyp1b1−/− TM tissues (P = 0.0006) (Cyp1b1+/+ TM, n = 4; Cyp1b1−/− TM, n = 3). (D) Severity grades of abnormal TM cells in Cyp1b1+/+ and Cyp1b1−/− TM tissues (P = 0.0088) (Cyp1b1+/+ TM, n = 4; Cyp1b1−/− TM, n = 3). (E) Significant increase in HNE staining in Cyp1b1−/− TM tissue. S, Sclera; CB, cilliary body; I, iris. Scale bar, 25 μm. (F) Quantitative assessment of the fluorescent intensity (∗, P = 0.0327) (Cyp1b1+/+, n = 5; Cyp1b1−/−, n = 6). (G) Administration of NAC in Cyp1b1−/− pups (P2) successfully prevented disorganization of collagen fibrils in TM (asterisks) and improved the health status of TM cells (arrows) (n = 8). (H) Quantitative assessment of the amount of collagen occupying the spaces in the trabecular beams (∗∗, P = 0.001) (Cyp1b1−/− with saline, n = 3; Cyp1b1−/− with NAC, n = 3). (I) Severity grades of abnormal collagen fibers in TM tissues (P = 0.0474) (Cyp1b1−/− with saline, n = 3; Cyp1b1−/− with NAC, n = 3). (J) Severity grades of abnormal TM cells in TM tissues (P = 0.0075) (Cyp1b1−/− with saline, n = 3; Cyp1b1−/− with NAC, n = 3).
In contrast, Cyp1b1−/− animals presented a marked disruption of the anterior and posterior TM, characterized by multifocal atrophy of the trabecular beams with accentuation of the intertrabecular spaces. The collagen fibers were markedly fragmented and irregularly distributed (Fig. 2A, bottom right, asterisk). TM cells presented irregularities of the cellular contour associated with a loss of contact with the basement membrane and extracellular matrix (Fig. 2A, bottom right, arrow) as well as variable cell swelling with cytoplasmic vacuolization and intracytoplasmic accumulation of organelle debris. Figure 2B shows a quantitative assessment of the respective amounts of collagen fibers occupying Cyp1b1+/+ and Cyp1b1−/− TM tissues. Figure 2C and D show the respective severity grades of abnormal collagen morphology and TM cell morphology presented in Cyp1b1+/+ and Cyp1b1−/− TM tissues determined as described in Materials and Methods. All the above-described quantitative assessments indicated severe morphological changes in the collagen fibers and TM cells of Cyp1b1−/− TM tissue.
The normal tissue arrangement in Cyp1b1+/+ mice provides tensile strength and flexibility of the TM tissue (22). The TM needs to maintain the physical properties of elasticity, tension, and softness to ensure the physiological outflow facility (25). Fibrillar collagen, consisting of closely packed thick collagen fibrils, is a supporting framework of the trabecular meshwork (23). Meanwhile, the general extracellular matrix (ECM) proteins in TM tissue also appear to control patterns of fluid outflow and alter TM tissue usage based on demand (44). The breakdown or abnormal presence of collagen fibrils and ECM proteins in Cyp1b1−/− TM can change the viscoelastic properties of these tissues and affect their responses to injury. It can also change the stiffness of the surrounding microenvironment, alter the shape and cytoskeleton organization of Cyp1b1−/− TM cells, and affect the proper expansion of intercellular spaces and the allowance-of-outflow pathway, which consequently leads to compromised aqueous humor outflow in Cyp1b1−/− eyes.
Given the nature of CYP1B1-catalyzed reactions, we hypothesized that Cyp1b1 is closely related to cellular oxidative homeostasis. Oxidative stress usually results in lipid peroxidation, especially in the eye, which is rich in polyunsaturated fatty acids. 4-Hydroxyl-2-noneal (HNE), one of the major aldehyde products of this reaction, contributes to oxidative stress-mediated damages and provides a direct measure of this process (45). We next determined oxidative stress in Cyp1b1+/+ and Cyp1b1−/− TM tissue by staining paraffin sections with anti-HNE antibody. Figure 2E and F show a significant increase in the amount of HNE staining in TM tissues (arrows) from Cyp1b1−/− mice compared with Cyp1b1+/+ mice.
To confirm that the changes in the collagen distribution of TM tissue from Cyp1b1−/− mice are indeed due to increased oxidative stress, NAC, a free radical scavenger with antioxidant properties, was administered to Cyp1b1−/− mice starting from postnatal day 2 (P2). Cyp1b1−/− pups received daily intraperitoneal injections of 1.5 mg/g body weight of NAC in saline for 3 weeks. Control pups were injected with saline. Figure 2G shows that while the Cyp1b1−/− mice receiving the saline control still presented the above-described abnormalities of collagen (bottom left, asterisk) and TM cells (bottom left, arrow), Cyp1b1−/− pups receiving NAC injections presented normal trabecular meshwork architecture with well-organized collagen (bottom right, asterisk) and TM cell morphology (bottom right, arrow). Figure 2H is a quantitative assessment of the respective amounts of collagen fibers occupying the TM from Cyp1b1−/− pups receiving the saline control and those receiving the antioxidant NAC. Figure 2I and J are the respective severity grades of abnormal collagen morphology and TM cell morphology presented for TM tissues from Cyp1b1−/− pups receiving the saline control and those receiving NAC. These quantitative assessments indicated that the antioxidant NAC could prevent the disruption of TM tissue morphology in Cyp1b1−/− mice and confirmed a role for increased oxidative stress in the abnormal TM structure in Cyp1b1−/− mice.
Isolation of mouse TM cells.
In order to delineate the cell-autonomous mechanisms of Cyp1b1 action in TM function, we prepared TM cells from Cyp1b1+/+ and Cyp1b1−/− Immorto mice. Figure 3A (a1 to a4) shows the morphology of subconfluent Cyp1b1+/+ and Cyp1b1−/− TM cells. They are both very similar to mouse TM cells that are widely acknowledged (35), and a lack of Cyp1b1 minimally affected cell morphology. Importantly, when confluent, both Cyp1b1+/+ and Cyp1b1−/− cells formed a cobblestone-like monolayer, which is a specific characteristic of TM cells (35) (Fig. 3Aa5 and a6). This specific growth pattern in vitro helps distinguish TM cells from neighboring cell types such as sclera fibrocytes, keratocytes, and corneal endothelium (46).
Fig 3.

Isolation of Cyp1b1+/+ and Cyp1b1−/− TM cells. (A) Phase micrograph of TM cells. Shown are subconfluent TM cells (a1 to a4) and confluent TM cells with a specific cobblestone-like monolayer morphology (a5 and a6). Scale bars, 250 μm (a1 and a2) and 25 μm (a3 to a6). (B to D) Cyp1b1+/+ and Cyp1b1−/− TM cells were positive for α-smooth muscle actin (α-SMA) (B), ADAMTS10 and CHI3L1 (C), and AQP1 (D). (E) Expression of myocilin in Cyp1b1+/+ and Cyp1b1−/− TM conditioned medium (CM). An increase in myocilin levels was detected in CM from both Cyp1b1+/+ and Cyp1b1−/− TM cells incubated with dexamethasone (DEX) (∗∗, P = 0.0036; #, P > 0.05) (n = 3). EtOH, ethyl alcohol. (F) Quantitative assessment of data in panel E. (G) Expression of mouse CYP1B1 in Cyp1b1+/+ TM cells and not in Cyp1b1−/− TM cells. TCDD induced the expression of CYP1B1 in Cyp1b1+/+ TM cells. Human TM-1 cells constitutively expressed CYP1B1. β-Actin was used as a loading control.
To further confirm the identity of these cells as TM cells, we detected the expression of TM cell markers, including alpha-smooth muscle actin (47, 48), ADAMTS10 (49), CHI3L1 (50–52), and AQP1 (50, 53, 54). Our FACS, Western blot, and quantitative PCR (qPCR) analyses confirmed positive expression of all these markers in Cyp1b1+/+ and Cyp1b1−/− TM cells (Fig. 3B to D).
Myocilin is a protein closely linked to the development of glaucoma (55), of which the highest expression levels are observed in TM cells (56) at both intra- and extracellular sites (57). Treatment of TM cells with dexamethasone elicits increased expression of myocilin only in TM cells (58) and not in other neighboring cell types, including corneal fibroblasts (59). Figure 3E shows that Cyp1b1+/+ and Cyp1b1−/− TM cells expressed myocilin (55 to 57 kDa) (arrow) in conditioned medium (CM). The upper shadow band at around 60 kDa is likely albumin in samples, as previously reported (38). The conditioned media from these cells were normalized and loaded according to the protein concentration. Importantly, following incubation with 500 nM dexamethasone, the secretion of myocilin was enhanced in both Cyp1b1+/+ and Cyp1b1−/− TM cells. A quantitative assessment of the data is shown in Fig. 3F. The increase in the myocilin expression level in Cyp1b1+/+ TM cells was significant (P = 0.0036). However, the increased expression level in Cyp1b1−/− TM cells did not reach statistical significance (P > 0.05). Overall, the combined expression of all the above-described TM markers supports that the Cyp1b1+/+ and Cyp1b1−/− cell lines described here are indeed TM cells.
The in vitro expression of CYP1B1 was assessed by Western blotting of the cell lysates prepared from Cyp1b1+/+ and Cyp1b1−/− TM cells as well as those incubated with 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) (10 nM for 24 h). Mouse TM cells constitutively expressed CYP1B1, which was further induced in the presence of TCDD, an AhR (aryl hydrocarbon receptor) agonist and a known inducer of CYP1B1 expression (Fig. 3G) (5, 60). As expected, the expression of CYP1B1 was not detected in Cyp1b1−/− TM cells. Thus, mouse TM cells constitutively express CYP1B1. Importantly, human TM-1 cells, an immortalized human TM cell line that resembles human TM cultures from a 30-year-old nonglaucomatous individual (61), also constitutively expressed CYP1B1 (Fig. 3G).
Enhanced apoptosis of Cyp1b1−/− TM cells in response to stress.
We observed similar growth rates between Cyp1b1+/+ and Cyp1b1−/− TM cells by counting cell numbers for 2 weeks (Fig. 4A). We also evaluated the percentage of Cyp1b1+/+ and Cyp1b1−/− TM cells undergoing active DNA synthesis using 5-ethynyl-2′-deoxyuridine (EdU) incorporation (Fig. 4B). The percentage of Cyp1b1−/− TM cells that were EdU positive was also similar to that of Cyp1b1+/+ TM cells. Thus, Cyp1b1 deficiency did not influence the proliferation of TM cells.
Fig 4.
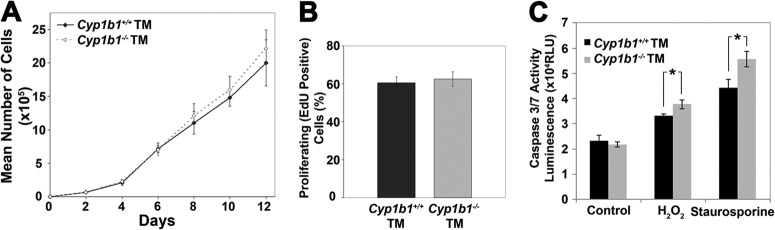
Enhanced apoptosis in Cyp1b1−/− TM cells under stress. (A and B) Cyp1b1+/+ and Cyp1b1−/− TM cells proliferated at similar rates. (C) A significant increase in the rate of apoptosis was observed for Cyp1b1−/− TM cells incubated with H2O2 (∗, P = 0.0139) (n = 3) or staurosporine (∗, P = 0.0129) (n = 3) compared with Cyp1b1+/+ cells. RLU, relative light units.
Increased TM cell loss and concurrent fusion and thickening of trabecular lamellae are some of the major characteristics of glaucoma and can further cause decreased outflow capacity (62). We next determined the rates of apoptosis in Cyp1b1+/+ and Cyp1b1−/− TM cells both at steady state and under exogenous stress. Although we observed similar basal rates of apoptosis in Cyp1b1+/+ and Cyp1b1−/− TM cells, Cyp1b1−/− TM cells were significantly more apoptotic than Cyp1b1+/+ TM cells when incubated with 1 mM hydrogen peroxide (H2O2) or 10 nM staurosporine for 8 h (Fig. 4C).
Increased intracellular oxidative stress in Cyp1b1−/− TM cells.
The increase in lipid peroxidation in Cyp1b1−/− TM tissue (Fig. 2B) and an enhanced sensitivity toward H2O2 (Fig. 4C) both suggested compromised oxidative homeostasis in Cyp1b1−/− TM cells. To directly illustrate intracellular oxidative stress, immunofluorescence of dihydroethidium (DHE) staining was performed. DHE is a cell-permeable compound, which, upon oxidization, forms “ethidium” of red fluorescence and intercalates into the DNA in the nucleus. Figure 5A shows a group of representative samples stained with DHE. At the basal level, Cyp1b1−/− TM cells presented a significantly higher level of DHE staining than did Cyp1b1+/+ TM cells, indicating more intracellular oxidative stress. Inhibition of CYP1B1 activity by 2,3′,4,5′-tetramethoxystilbene (TMS) (a CYP1B1-specific inhibitor, at 5 μM for 24 h) (63) in Cyp1b1+/+ TM cells also resulted in enhanced oxidative stress. Importantly, the enhanced DHE staining in Cyp1b1−/− TM cells was partially prevented when Cyp1b1−/− cells were incubated with the antioxidant NAC. In general, these results indicated a higher-level intracellular oxidative state with Cyp1b1 deficiency in TM cells that was alleviated by the administration of NAC (Fig. 5B).
When challenged with 0.85 mM H2O2 for 40 min, all cell types showed increased levels of DHE staining compared to basal levels. The fact that DHE fluorescence significantly increased with H2O2 and decreased with the antioxidant NAC validated the suitability and sensitivity of DHE staining for assaying cellular oxidative stress in this study. Thus, Cyp1b1−/− TM cells exhibited an increase in intracellular oxidative stress.
Decreased viability of Cyp1b1−/− TM cells.
Altered cell viability and a subsequent increased TM cell loss with the presence of oxidative insult have been reported for glaucomatous TM tissues (64). To test viability, Cyp1b1+/+ and Cyp1b1−/− TM cells were plated into a 96-well plate and incubated with 1.25 mM H2O2 for 24 h. As a result, the H2O2 challenge decreased the viability of Cyp1b1+/+ TM cells by 20%, while the viability of Cyp1b1−/− TM cells was decreased by 70% (Fig. 6B and D). This is consistent with the increased rate of apoptosis in Cyp1b1−/− TM cells under stress (Fig. 4C).
Fig 6.

Decreased cell viability of Cyp1b1−/− TM cells. (A) Expression of CYP1B1 in Cyp1b1−/− TM cells expressing the empty vector or Cyp1b1 with various virus inputs. (B) Cyp1b1−/− TM cells were significantly less viable under conditions of oxidative challenge (∗∗, P = 0.0013) (n = 3), which was reversed with restoration of Cyp1b1 expression (∗∗, P = 0.0084) (n = 3). (C) Expression levels of CYP1B1 in Cyp1b1+/+ TM cells expressing a Cyp1b1-specific siRNA or nontargeted control siRNA. (D) Cyp1b1+/+ TM cells were significantly more viable under conditions of oxidative stress (∗∗, P = 0.0028) (n = 3), which was prevented by knockdown of Cyp1b1 (∗∗, P = 0.0002) (n = 3). (E) Expression level of CYP1B1 in human TM-1 cells expressing a CYP1B1-specific siRNA or control siRNA. (F) Human TM-1 cells infected with a human CYP1B1-specific siRNA were significantly less viable than those infected with a control siRNA (∗, P = 0.0117) (n = 3). (G) Cyp1b1+/+ TM cells were more sensitive to oxidative stress in the presence of TMS (∗∗, P = 0.0023) (n = 3). Cyp1b1−/− TM cells were significantly more viable when preincubated with NAC (∗∗, P = 0.0005) (n = 3).
To investigate whether the changes in cell viability described above were specifically due to a lack of Cyp1b1, we restored expression of Cyp1b1 in Cyp1b1−/− TM cells by infecting cells with adenoviruses expressing mouse Cyp1b1. Control cells were infected with a green fluorescent protein (GFP)-expressing vector. Figure 6A shows that Cyp1b1 was effectively expressed in TM cells in a virus input-dependent manner. The significant improvement of TM cell viability upon Cyp1b1 reexpression compared with the vector control is shown in Fig. 6B. Similar results were observed in Cyp1b1+/+ TM cells whose Cyp1b1 level was knocked down (Fig. 6C). Infection of Cyp1b1+/+ TM cells with lentiviral transduction particles expressing a mouse-specific Cyp1b1 siRNA resulted in a loss of cell viability under oxidative challenge (Fig. 6D).
Similar phenotypic changes were also observed in human TM-1 cells. CYP1B1 was silenced in human TM-1 cells using lentiviral transduction particles expressing a human-specific CYP1B1 siRNA (Fig. 6E). Figure 6F shows that the silencing of CYP1B1 significantly decreased the viability of human TM cells under stress conditions. These studies further confirmed a protective role of CYP1B1 in maintaining human TM-1 cell viability under conditions of oxidative stress. Thus, TM cells are sensitive to oxidative stress due to a lack of Cyp1b1. Consistently, inhibition of CYP1B1 activity in Cyp1b1+/+ TM cells by TMS (5 μM for 24 h) significantly reduced their survival rate to a level similar to that of Cyp1b1−/− TM cells (Fig. 6G). Importantly, the enhanced cytotoxicity of Cyp1b1−/− TM cells was completely prevented when they were incubated with NAC (5 mM for 24 h). These results further confirmed a role for oxidative stress in the decreased viability of Cyp1b1−/− TM cells.
Cyp1b1−/− TM cells are more adherent.
Adhesion of TM cells to ECM proteins is another critical function in determining the fluid outflow through the TM, and its changes can affect trabecular outflow resistance by altering the dimensions or directions of flow pathways (65). We next examined the properties of adhesion of Cyp1b1+/+ and Cyp1b1−/− TM cells to various ECM proteins. We observed enhanced adhesion of Cyp1b1−/− TM cells to fibronectin, vitronectin, collagen I, and collagen IV compared with Cyp1b1+/+ TM cells (Fig. 7A).
Fig 7.

Cyp1b1−/− TM cells were more adherent on various ECM proteins. (A) Cyp1b1−/− TM cells were significantly more adhesive on fibronectin (P = 0.0226) (n = 3), vitronectin (P = 0.0379) (n = 3), collagen I (P = 0.0093) (n = 3), and collagen IV (P = 0.0007) (n = 3) than Cyp1b1+/+ TM cells. (B) Reexpression of Cyp1b1 in Cyp1b1−/− TM cells resulted in a significant decrease in adhesion to fibronectin compared to the control (P = 0.0012) (n = 3). (C) Knockdown of Cyp1b1 in Cyp1b1+/+ TM cells resulted in a significant increase in adhesion to fibronectin compared to the control (P = 0.0172) (n = 3). (D) Knockdown of Cyp1b1 in human TM-1 cells also resulted in a significant increase in adhesion to fibronectin compared to the control (P = 0.0146) (n = 3). (E) Cyp1b1−/− TM cells became less adhesive on fibronectin when they were pretreated with NAC (P = 0.0389) (n = 3). Inhibition of CYP1B1 activity by TMS significantly increased the adhesion of Cyp1b1+/+ TM cells to fibronectin (P = 0.0168) (n = 3). OD, optical density.
To demonstrate that the changes in cell adhesion described above are due specifically to a lack of Cyp1b1, we restored Cyp1b1 in Cyp1b1−/− TM cells by expressing mouse Cyp1b1 or a control GFP-expressing vector. Figure 7B shows that restoration of Cyp1b1 significantly reversed the adhesion of Cyp1b1−/− TM cells back to normal. Those cells infected with the control construct showed no change in their adhesion properties. Similar results were observed for Cyp1b1+/+ TM cells infected with lentiviral transduction particles expressing a Cyp1b1 siRNA. Silencing of Cyp1b1 in Cyp1b1+/+ TM cells resulted in enhanced adhesion to fibronectin (Fig. 7C). Similarly, human TM-1 cells with their CYP1B1 gene silenced also showed increased adhesion to fibronectin (Fig. 7D). These results confirmed an essential role of CYP1B1 in modulation of TM cell adhesion.
Inhibition of CYP1B1 activity in Cyp1b1+/+ TM cells by TMS (5 μM for 48 h) also increased their adhesion to fibronectin (Fig. 7E), similar to what was observed for Cyp1b1−/− TM cells. Importantly, the enhanced adhesion of Cyp1b1−/− TM cells was drastically reversed when the cells were incubated with the antioxidant NAC (5 mM for 48 h) (Fig. 7E), indicating a role for oxidative stress in the abnormal adhesion of Cyp1b1−/− TM cells. Abnormal adhesion of TM cells may exert an important negative impact on their proper expansion of intercellular spaces and allowance of the outflow pathways (66, 67). These dysregulated adhesions are consistent with the proposed TM dysfunction that adversely affects aqueous humor outflow and IOP in Cyp1b1−/− mice.
Decreased production of Postn in TM of Cyp1b1−/− mice and humans with glaucoma.
TM tissue is composed of trabecular beams with various ECM proteins which are in a balance of synthesis and degradation. ECM proteins in TM tissue play important roles in controlling patterns of fluid outflow and altering TM tissue usage based on demand (44), and its remodeling is also an essential function of TM for maintenance of the outflow facility. Various ECM proteins, including fibronectin, tenascin-C, osteopontin, and thrombospondins, exert their effect through a cell-mediated process modulating IOP (3, 64). We next examined the production of various ECM proteins in CM and cell lysates prepared from Cyp1b1+/+ and Cyp1b1−/− TM cells. Figure 8A shows that Cyp1b1+/+ and Cyp1b1−/− TM cells produced comparable levels of fibronectin, thrombospondin-1, thrombospondin-2, osteopontin, and osteonectin. However, we observed significant decreases in the levels of Postn and tenascin-C in the CM and lysates of Cyp1b1−/− TM cells. Postn, a matricellular protein essential for collagen fibrillogenesis (27), functions to bridge and stabilize adjacent collagen fibrils during fibril fusion (27). Interestingly, when Cyp1b1−/− TM cells were incubated with NAC (5 mM for 48 h), the secretion of Postn was restored to normal levels (Fig. 8B). These results strongly suggest that the altered secretion of Postn in Cyp1b1−/− TM cells was a result of increased oxidative stress.
Fig 8.

Altered expression of ECM proteins in Cyp1b1−/− TM cells. (A) Decreased tenascin-C and Postn secretion into the CM of Cyp1b1−/− TM cells. (B) The decreased Postn secretion from Cyp1b1−/− TM cells was completely prevented in the presence of NAC. (C) Cyp1b1−/− TM tissue showed lower levels of Postn staining (arrows) than did Cyp1b1+/+ TM tissue. Scale bar, 25 μm. (D) Quantitative assessment of the fluorescent density shown in panel C (∗, P = 0.0332) (Cyp1b1+/+, n = 3; Cyp1b1−/−, n = 4). (E) Human glaucomatous TM (GTM) tissues also showed decreased Postn staining compared to normal tissue (NTM). SC, Schlemm's canal. Scale bars, 200 μm (top) and 25 μm (bottom). (F) Quantitative assessment of data in panel E (∗, P = 0.0291) (n = 4).
We next investigated whether the in vivo Postn level was affected in Cyp1b1−/− TM tissue. TM tissues from Cyp1b1−/− mice had significantly lower levels of Postn than did TM tissues from Cyp1b1+/+ mice (Fig. 8C, arrows, and D). This was also consistent with our above-described TEM results showing that Cyp1b1−/− TM had abnormal collagen production and organization (Fig. 2A). Immunohistochemical staining of human TM tissues also revealed that human glaucomatous TM tissues had decreased levels of Postn compared with normal human TM tissues (Fig. 8E and F). Details about the eye donors are summarized in Table S1 in the supplemental material. To our knowledge, this is the first report of altered Postn levels in glaucomatous human TM tissue, and it demonstrates an important role for Postn in TM integrity and function.
Abnormal collagen organization in TM of Postn−/− mice.
To confirm that a lack of Postn plays a critical role in disorganization of collagens in TM tissue, we examined TM tissues collected from adult Postn+/+ and Postn−/− mice by TEM. Figure 9A shows that the wild-type Postn+/+ TM presented a normal ultrastructural architecture with a well-organized trabecular cytoplasm containing regular collagen fibers (left, asterisk) and normal TM cells similar to those of the wild-type Cyp1b1+/+ TM shown in Fig. 2A. In contrast, TM tissues from Postn−/− mice (Fig. 9A, right) were markedly atrophied, with accentuation of the intertrabecular spaces and marked disruption of the collagen fibers (asterisk). In addition, due to the lack of support from regular collagen fibers, the trabecular cytoplasm became uneven and presented severe distortions. Figure 9B is a quantitative assessment of the respective amounts of collagen fibers occupying TM from Postn+/+ and Postn−/− mice. Figure 9C and D are the respective severity grades of abnormal collagen morphology and TM cell morphology presented in the Postn+/+ and Postn−/− TM tissue. These quantitative assessments indicate that a lack of Postn negatively impacts the structural integrity of mouse TM tissue.
Fig 9.

Abnormal collagen organization in TM tissue from Postn-deficient (Postn−/−) mice. (A) Postn+/+ TM tissue contained normal collagen fibrils (bottom left, asterisk). In contrast, TM tissues from Postn−/− mice showed large empty spaces indicative of defective collagen organization (bottom right, asterisk) (n = 5). Scale bar, 2 μm. Five sectors per eye were evaluated. Postn deficiency did not affect the collagens in sclera (S). (B) Quantitative assessment of the amount of collagens occupying the spaces in the trabecular beams (∗∗, P = 0.0063) (n = 4). (C) Severity grades of abnormal collagen fibers in Postn+/+ and Postn−/− TM tissues (P = 0.0004) (n = 4). (D) Severity grades of abnormal TM cell morphology in Postn+/+ and Postn−/− TM tissues (P = 0.0011) (n = 4).
DISCUSSION
Here we demonstrated an important role for Cyp1b1 as a modulator of oxidative homeostasis that contributes to the ultrastructural organization and function of TM tissue. We showed increased IOP in Cyp1b1−/− mice. We observed increased lipid peroxidation and irregular collagen fibril distribution in the TM from Cyp1b1−/− mice, which were prevented by administration of NAC. Cyp1b1−/− TM cells were less viable under oxidative challenge, were more adhesive to matrix proteins, and secreted significantly less Postn. In addition, inhibition of CYP1B1 activity or silencing of Cyp1b1 with siRNAs successfully recapitulated aberrant Cyp1b1−/− phenotypes. All the aberrant cellular responses were reversed in the presence of NAC. Our study is the first to provide evidence demonstrating a significant role for Postn in the structural integrity of TM tissue. We showed that Cyp1b1−/− TM tissue and cells express decreased levels of Postn compared with those expressed by the Cyp1b1+/+ TM. In addition, human glaucomatous TM tissue also expressed decreased levels of Postn compared with those expressed by normal human TM tissue. Furthermore, the TM tissue was atrophic with disorganized collagen in Postn−/− mice. Together, our results indicate that the metabolic activity of Cyp1b1 modulates the oxidative status of the TM, such that in its absence, increased oxidative stress leads to irregular collagen fibril distribution and abnormal functions of the TM through decreased Postn production.
Elevated IOP in Cyp1b1−/− mice.
Our measurements of IOPs are reliable. All measurements were done within 4 to 7 min after anesthetization to avoid the effect of anesthesia on IOP (68). The IOPs of Cyp1b1+/+ mice that we measured were consistent with the previously reported IOP of wild-type C57BL/6 mice (42). There are several possibilities for the modest (10%) increase in IOP of Cyp1b1−/− mice. First, similar to what was reported by Chang et al. for their murine model, the defects may be only focally present (69) in Cyp1b1−/− eyes. IOP has been reported to be normal even when 50% of the iridocorneal angle circumference was severely abnormal, and only eyes with abnormalities extending over 80% had drastically elevated IOP (69). Similar compensatory mechanisms in humans have also been reported when angle recession extends for over 67% of the circumference, and a drastically higher IOP develops (70). Second, mice on the C57BL/6 background do not spontaneously have their IOP elevated much above 21 mm Hg, the level defined as an abnormal IOP for humans. Commonly, a 7% to 12% (P < 0.05) difference in IOP is considered significant in transgenic mice on the C57BL/6 background (71, 72). Third, the ratio of drainage through the uveoscleral pathway to that through the conventional pathway may also change in transgenic mice (71). However, whether Cyp1b1 deficiency impacts the uveoscleral pathway remains to be explored.
Cyp1b1 deficiency may also affect the metabolic status of the cilliary body and its consequent production of aqueous humor. It is possible that an increase in the utilization of the uveoscleral drainage pathway and/or a decrease in the formation of the aqueous humor partially compensated for the compromised conventional outflow in Cyp1b1−/− mice, resulting in only a moderate increase in IOP. More efforts will be made to assess and compare aqueous humor dynamics of Cyp1b1+/+ and Cyp1b1−/− mice in the future. Collectively, even if IOP did not increase to a large extent, Cyp1b1−/− mice as well as other transgenic mice (43, 71–73) are still qualified as suitable models for studying anterior segment anomalies.
Postnatal development of the trabecular meshwork in mice.
One of the major findings in the present study is that administration of the antioxidant NAC at early stages after birth alleviated the developmental dysgenesis of TM in Cyp1b1−/− mice. This result closely agreed with the postnatal development processes in mouse eyes. At birth, mouse TM has a primitive appearance and remains undeveloped until postnatal day 4 (P4). TM undergoes differentiation but is not fully developed by P10. Major developmental changes do not occur until as late as P18. Mouse TM does not reach full maturity until P35 to P42 (74).
Based on this important timeline for murine angle development, we decided to administer NAC to P2 pups prior to the start of TM development. The administration of the antioxidant successfully prevented the development of disorganized collagen in the Cyp1b1−/− mouse TM. Thus, we hypothesize that the Cyp1b1−/− TM is already exposed to relevant oxidative stress early in life. This also coincides with the common knowledge that prevention of oxidative stress is an important mechanism to protect the outflow facility. It is appealing to speculate that Cyp1b1−/− mice serve as a model for oxidative stress-induced glaucoma. The prevention of the lesion with the antioxidant NAC in Cyp1b1−/− mice during TM development suggests a possible clinical intervention for those individuals genetically at high risk of developing glaucoma.
Cyp1b1 deficiency and TM cell dysfunction.
We found that Cyp1b1−/− mouse TM cells are more sensitive to oxidative stress challenge. Cellular responses to reactive oxygen species (ROS) depend mainly on the basal redox status of the cell. The decreased viability of Cyp1b1−/− TM cells under H2O2 indicated an insufficient cellular antioxidant capacity in Cyp1b1−/− TM cells to detoxify ROS. This is consistent with previous reports of the association of TM cell loss and oxidative insults in primary open angle glaucoma (44, 64). This notion is further supported by the fact that the antioxidant NAC completely reversed Cyp1b1−/− cell viability back to normal. The CYP1B1 metabolism of endogenous cellular products contributes to the maintenance of the intracellular oxidative status in the TM. While Cyp1b1+/+ TM tissue neutralizes the endogenous generation of ROS to some degree, Cyp1b1 deficiency may deprive the TM tissue of its ability to counteract ROS, rendering it less protected and much more susceptible to the onset of glaucoma. This notion is further supported by the reported severity of TM development in Cyp1b1-deficient albino mice (21).
Tyrosine can be hydroxylated to l-Dopa via either tyrosinase (Tyr) or tyrosine hydroxylase (TH). l-Dopa can be oxidized further by Tyr to dopaquinone, which then undergoes several cascades and finally forms melanin (75). l-Dopa has critical roles in the early development of retina (76) and heart (77). Libby et al. further described that a pathway involving l-Dopa (or an l-Dopa metabolite) participates in angle formation and that disturbances in this pathway can be treated by l-Dopa administration (21). Currently, there is no report of a direct relationship between Cyp1b1 and l-Dopa production. According to the hints provided (21), Cyp1b1 deficiency may affect the activity of TH/Tyr or bind to a transcriptional repressor of the Th/Tyr gene, which would reduce the level of production of l-Dopa from tyrosine. Besides affecting l-Dopa levels, Cyp1b1 may also affect the metabolism of l-Dopa to other developmentally important molecules or affect the signaling of these molecules in developmental and physiologic pathways.
Alternatively, both human and mouse Cyp1b1 orthologs oxidize all-trans-retinol to all-trans-retinal and then to all-trans-retinoic acid (9), which promotes the proliferation of a subset of avian neural crest cells that express TH (78). Thus, the deficiency of Cyp1b1 may impair the proliferation of TH-positive neural crest cells and decrease the production of l-Dopa. From l-Dopa, Tyr triggers the cascade of the production of melanin (75), which has antioxidant properties. If the levels of l-Dopa can indeed be downregulated by Cyp1b1 deficiency, the consequent decrease in the production of melanin will further cause increased oxidative stress in relevant cells and tissues, which in our case is the TM. This is also consistent with our result that Cyp1b1−/− mice have increased oxidative stress in TM cells and tissues.
TM cells are mechanosensitive (79). It is appealing to speculate that changes in the stiffness of the surrounding microenvironment (including collagen fibrils and other ECM proteins) alter cytoskeleton organization and the adhesion property of Cyp1b1−/− TM cells, rendering them more adhesive. Such an increase in TM cell adhesion to the ECM proteins of trabecular beams further narrows the passageways for aqueous humor outflow in Cyp1b1−/− mice. Meanwhile, cytoskeletal structure and cell-matrix interactions in TM cells are also known to be directly affected by oxidative stress (80). The fact that the antioxidant NAC successfully prevented the enhanced adhesion of Cyp1b1−/− TM cells also suggests a direct role for oxidative stress in the altered adhesion function with Cyp1b1 deficiency.
The fact that adenoviral reexpression of Cyp1b1, siRNA-mediated knockdown of Cyp1b1, and inhibition of CYP1B1 activity all successfully reversed these phenotypical changes confirmed a notable effect of Cyp1b1 on the maintenance of the TM cell function discussed above. Importantly, similar functional changes were observed when CYP1B1 was inhibited in a human TM cell line (TM-1), which further confirms the importance of CYP1B1 in TM cell function, which determines aqueous humor outflow.
ECM secretion and the microenvironment of TM cells were also changed by Cyp1b1 deficiency. Postn is an ECM protein essential for collagen fibril assembly and maturation (27). Cyp1b1−/− TM tissue was revealed to express less Postn than normal Cyp1b1+/+ TM tissue (Fig. 8B to D) and contains irregularly distributed collagens (Fig. 2A). Likewise, human glaucomatous TM tissues were shown to express decreased levels of Postn compared with the levels expressed in normal TM tissues (Fig. 8E and F). The donors of normal tissues were females aged 36, 69, 93, and 96 years. None had any history of ocular disease. Some glaucomatous donors did not have IOP measurements just prior to death, due to more serious medical concerns.
Critical information, including donor age, the kind of ocular and overall medical treatments received, IOP measurements, and the expiration-to-enucleation time of eye balls from each patient, is summarized in Table S1 in the supplemental material. None of the four glaucomatous donors had ever received medical treatments that would likely affect the expression levels of Postn. In detail, glaucomatous tissue donors GL62L and GL113L did not receive any ocular treatment before death, and the therapies that they received for cancer treatment were unlikely to affect Postn expression in their TM tissue. The drugs pilocarpine (a muscarinic receptor agonist), timolol (Timoptic) (a β-adrenergic receptor inhibitor), dorzolamide (Trusopt) (a carbonic anhydrase inhibitor), apraclonidine (Iopidine) (an α-adrenergic receptor agonist), and betaxolol (Betoptics) (a β-adrenergic receptor inhibitor), which patients GL70LM and GL118R had received before death, have not yet been reported in the literature to impact Postn expression. The cataract surgery or intraocular lens implant surgery that these patients received should not affect Postn expression in the TM. Regarding the IOP before death, although the IOPs of patients GL62L and GL113L were not recorded, patient GL70LM was suffering from an IOP of 22 mm Hg for 18 years, and patient GL113R had an IOP of 26 mm Hg, indicating defective TM tissue functions in at least these two patients. Moreover, three out of the four eye samples were collected within 2 to 4 h of death, and all were fixed in formalin. The sample collection and fixation processes involved should not affect the expression of most proteins in ocular tissues. In general, the details provided in Table S1 in the supplemental material confirm the reliability of the immunohistochemical analysis of human TM samples shown in Fig. 8E.
Abnormal collagen alignment and large empty spaces, indicating collagen loss, were observed in TM tissues from Postn−/− mice by electron microscopy (Fig. 9). Overall, these results substantiate a role for Cyp1b1 in dictating the mechanical strength and structural integrity of the collagen-containing TM tissue. Importantly, the antioxidant NAC restored the expression of Postn in Cyp1b1−/− TM cells (Fig. 8B) and supported the critical role of oxidative stress in the abnormal microenvironment and dysfunctions of Cyp1b1−/− TM cells.
In summary, our results demonstrate increased oxidative stress and decreased production of Postn with Cyp1b1 deficiency and consequent TM tissue dysgenesis and dysfunctions. A disintegrated TM in Cyp1b1−/− mice impairs aqueous humor outflow and shifts the balance toward the initiation of glaucoma. An understanding of the mechanisms through which Cyp1b1 modulates TM functions will provide important insights into angle abnormalities in Cyp1b1−/− mice and preventive approaches for glaucoma patients with a CYP1B1 mutation. The novel finding of Postn as an important regulator of TM tissue development and function and its modulation by oxidative stress may provide a novel biomarker for detection and treatment efficacy of antiglaucoma drugs.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by support grants EY018179, DK090249, EY16995, EY021357, P30-EY016665, and P30 CA014520 to the UW Paul P. Carbone Cancer Center from the National Institutes of Health and by an unrestricted departmental award from Research To Prevent Blindness. N.S. is a recipient of a research award from the American Diabetes Association, 1-10-BS-160, and the Retina Research Foundation. C.M.S. is supported by a grant from the American Heart Association, 0950057G.
Footnotes
Published ahead of print 26 August 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.00856-13.
REFERENCES
- 1.Quigley HA, Broman AT. 2006. The number of people with glaucoma worldwide in 2010 and 2020. Br. J. Ophthalmol. 90:262–267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weinreb RN, Khaw PT. 2004. Primary open-angle glaucoma. Lancet 363:1711–1720 [DOI] [PubMed] [Google Scholar]
- 3.Tamm ER. 2009. The trabecular meshwork outflow pathways: structural and functional aspects. Exp. Eye Res. 88:648–655 [DOI] [PubMed] [Google Scholar]
- 4.Savas U, Bhattacharyya KK, Christou M, Alexander DL, Jefcoate CR. 1994. Mouse cytochrome P-450EF, representative of a new 1B subfamily of cytochrome P-450s. Cloning, sequence determination, and tissue expression. J. Biol. Chem. 269:14905–14911 [PubMed] [Google Scholar]
- 5.Sutter TR, Tang YM, Hayes CL, Wo YY, Jabs EW, Li X, Yin H, Cody CW, Greenlee WF. 1994. Complete cDNA sequence of a human dioxin-inducible mRNA identifies a new gene subfamily of cytochrome P450 that maps to chromosome 2. J. Biol. Chem. 269:13092–13099 [PubMed] [Google Scholar]
- 6.Coon MJ. 2005. Cytochrome P450: nature's most versatile biological catalyst. Annu. Rev. Pharmacol. Toxicol. 45:1–25 [DOI] [PubMed] [Google Scholar]
- 7.Jansson I, Stoilov I, Sarfarazi M, Schenkman JB. 2001. Effect of two mutations of human CYP1B1, G61E and R469W, on stability and endogenous steroid substrate metabolism. Pharmacogenetics 11:793–801 [DOI] [PubMed] [Google Scholar]
- 8.Chambers D, Wilson L, Maden M, Lumsden A. 2007. RALDH-independent generation of retinoic acid during vertebrate embryogenesis by CYP1B1. Development 134:1369–1383 [DOI] [PubMed] [Google Scholar]
- 9.Choudhary D, Jansson I, Stoilov I, Sarfarazi M, Schenkman JB. 2004. Metabolism of retinoids and arachidonic acid by human and mouse cytochrome P450 1b1. Drug Metab. Dispos. 32:840–847 [DOI] [PubMed] [Google Scholar]
- 10.Boutin JA, Audinot V, Ferry G, Delagrange P. 2005. Molecular tools to study melatonin pathways and actions. Trends Pharmacol. Sci. 26:412–419 [DOI] [PubMed] [Google Scholar]
- 11.Ma X, Idle JR, Krausz KW, Gonzalez FJ. 2005. Metabolism of melatonin by human cytochromes p450. Drug Metab. Dispos. 33:489–494 [DOI] [PubMed] [Google Scholar]
- 12.Young IM, Leone RM, Francis P, Stovell P, Silman RE. 1985. Melatonin is metabolized to N-acetyl serotonin and 6-hydroxymelatonin in man. J. Clin. Endocrinol. Metab. 60:114–119 [DOI] [PubMed] [Google Scholar]
- 13.Tang Y, Scheef EA, Wang S, Sorenson CM, Marcus CB, Jefcoate CR, Sheibani N. 2009. CYP1B1 expression promotes the proangiogenic phenotype of endothelium through decreased intracellular oxidative stress and thrombospondin-2 expression. Blood 113:744–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Choudhary D, Jansson I, Rezaul K, Han DK, Sarfarazi M, Schenkman JB. 2007. Cyp1b1 protein in the mouse eye during development: an immunohistochemical study. Drug Metab. Dispos. 35:987–994 [DOI] [PubMed] [Google Scholar]
- 15.Hakkola J, Pasanen M, Pelkonen O, Hukkanen J, Evisalmi S, Anttila S, Rane A, Mantyla M, Purkunen R, Saarikoski S, Tooming M, Raunio H. 1997. Expression of CYP1B1 in human adult and fetal tissues and differential inducibility of CYP1B1 and CYP1A1 by Ah receptor ligands in human placenta and cultured cells. Carcinogenesis 18:391–397 [DOI] [PubMed] [Google Scholar]
- 16.Stoilov I, Rezaie T, Jansson I, Schenkman JB, Sarfarazi M. 2004. Expression of cytochrome P4501b1 (Cyp1b1) during early murine development. Mol. Vis. 10:629–636 [PubMed] [Google Scholar]
- 17.Panicker SG, Reddy AB, Mandal AK, Ahmed N, Nagarajaram HA, Hasnain SE, Balasubramanian D. 2002. Identification of novel mutations causing familial primary congenital glaucoma in Indian pedigrees. Invest. Ophthalmol. Vis. Sci. 43:1358–1366 [PubMed] [Google Scholar]
- 18.Soley GC, Bosse KA, Flikier D, Flikier P, Azofeifa J, Mardin CY, Reis A, Michels-Rautenstrauss KG, Rautenstrauss BW. 2003. Primary congenital glaucoma: a novel single-nucleotide deletion and varying phenotypic expression for the 1,546-1,555dup mutation in the GLC3A (CYP1B1) gene in 2 families of different ethnic origin. J. Glaucoma 12:27–30 [DOI] [PubMed] [Google Scholar]
- 19.Vincent AL, Billingsley G, Buys Y, Levin AV, Priston M, Trope G, Williams-Lyn D, Heon E. 2002. Digenic inheritance of early-onset glaucoma: CYP1B1, a potential modifier gene. Am. J. Hum. Genet. 70:448–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stoilov I, Akarsu AN, Sarfarazi M. 1997. Identification of three different truncating mutations in cytochrome P4501B1 (CYP1B1) as the principal cause of primary congenital glaucoma (buphthalmos) in families linked to the GLC3A locus on chromosome 2p21. Hum. Mol. Genet. 6:641–647 [DOI] [PubMed] [Google Scholar]
- 21.Libby RT, Smith RS, Savinova OV, Zabaleta A, Martin JE, Gonzalez FJ, John SW. 2003. Modification of ocular defects in mouse developmental glaucoma models by tyrosinase. Science 299:1578–1581 [DOI] [PubMed] [Google Scholar]
- 22.Gabelt BT, Kaufman PL. 2005. Changes in aqueous humor dynamics with age and glaucoma. Prog. Retin. Eye Res. 24:612–637 [DOI] [PubMed] [Google Scholar]
- 23.Aihara M, Lindsey JD, Weinreb RN. 2003. Ocular hypertension in mice with a targeted type I collagen mutation. Invest. Ophthalmol. Vis. Sci. 44(4):1581–1585 [DOI] [PubMed] [Google Scholar]
- 24.Kupfer C, Kaiser-Kupfer MI. 1979. Observations on the development of the anterior chamber angle with reference to the pathogenesis of congenital glaucomas. Am. J. Ophthalmol. 88:424–426 [DOI] [PubMed] [Google Scholar]
- 25.Borras T, Comes N. 2009. Evidence for a calcification process in the trabecular meshwork. Exp. Eye Res. 88:738–746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Horiuchi K, Amizuka N, Takeshita S, Takamatsu H, Katsuura M, Ozawa H, Toyama Y, Bonewald LF, Kudo A. 1999. Identification and characterization of a novel protein, periostin, with restricted expression to periosteum and periodontal ligament and increased expression by transforming growth factor beta. J. Bone Miner. Res. 14:1239–1249 [DOI] [PubMed] [Google Scholar]
- 27.Norris RA, Damon B, Mironov V, Kasyanov V, Ramamurthi A, Moreno-Rodriguez R, Trusk T, Potts JD, Goodwin RL, Davis J, Hoffman S, Wen X, Sugi Y, Kern CB, Mjaatvedt CH, Turner DK, Oka T, Conway SJ, Molkentin JD, Forgacs G, Markwald RR. 2007. Periostin regulates collagen fibrillogenesis and the biomechanical properties of connective tissues. J. Cell. Biochem. 101:695–711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yoshiba N, Yoshiba K, Hosoya A, Saito M, Yokoi T, Okiji T, Amizuka N, Ozawa H. 2007. Association of TIMP-2 with extracellular matrix exposed to mechanical stress and its co-distribution with periostin during mouse mandible development. Cell Tissue Res. 330:133–145 [DOI] [PubMed] [Google Scholar]
- 29.Snider P, Hinton RB, Moreno-Rodriguez RA, Wang J, Rogers R, Lindsley A, Li F, Ingram DA, Menick D, Field L, Firulli AB, Molkentin JD, Markwald R, Conway SJ. 2008. Periostin is required for maturation and extracellular matrix stabilization of noncardiomyocyte lineages of the heart. Circ. Res. 102:752–760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shimazaki M, Nakamura K, Kii I, Kashima T, Amizuka N, Li M, Saito M, Fukuda K, Nishiyama T, Kitajima S, Saga Y, Fukayama M, Sata M, Kudo A. 2008. Periostin is essential for cardiac healing after acute myocardial infarction. J. Exp. Med. 205:295–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sorocos K, Kostoulias X, Cullen-McEwen L, Hart AH, Bertram JF, Caruana G. 2011. Expression patterns and roles of periostin during kidney and ureter development. J. Urol. 186:1537–1544 [DOI] [PubMed] [Google Scholar]
- 32.Rios H, Koushik SV, Wang H, Wang J, Zhou HM, Lindsley A, Rogers R, Chen Z, Maeda M, Kruzynska-Frejtag A, Feng JQ, Conway SJ. 2005. Periostin null mice exhibit dwarfism, incisor enamel defects, and an early-onset periodontal disease-like phenotype. Mol. Cell. Biol. 25:11131–11144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pease ME, Cone FE, Gelman S, Son JL, Quigley HA. 2011. Calibration of the TonoLab tonometer in mice with spontaneous or experimental glaucoma. Invest. Ophthalmol. Vis. Sci. 52(2):858–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Filla MS, David G, Weinreb RN, Kaufman PL, Peters DM. 2004. Distribution of syndecans 1-4 within the anterior segment of the human eye: expression of a variant syndecan-3 and matrix-associated syndecan-2. Exp. Eye Res. 79:61–74 [DOI] [PubMed] [Google Scholar]
- 35.Tamm ER, Russell P, Piatigorsky J. 1999. Development of characterization of a immortal and differentiated murine trabecular meshwork cell line. Invest. Ophthalmol. Vis. Sci. 40:1392–1403 [PubMed] [Google Scholar]
- 36.Begley CG, Yue BY, Hendricks RL. 1991. Murine trabecular meshwork cells in tissue culture. Curr. Eye Res. 10:1015–1030 [DOI] [PubMed] [Google Scholar]
- 37.Park S, DiMaio TA, Scheef EA, Sorenson CM, Sheibani N. 2010. PECAM-1 regulates proangiogenic properties of endothelial cells through modulation of cell-cell and cell-matrix interactions. Am. J. Physiol. Cell Physiol. 299:C1468–C1484. 10.1152/ajpcell.00246.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ezzat MK, Howell KG, Bahler CK, Beito TG, Loewen N, Poeschla EM, Fautsch MP. 2008. Characterization of monoclonal antibodies against the glaucoma-associated protein myocilin. Exp. Eye Res. 87:376–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Su X, Sorenson CM, Sheibani N. 2003. Isolation and characterization of murine retinal endothelial cells. Mol. Vis. 9:171–178 [PubMed] [Google Scholar]
- 40.Kondo S, Scheef EA, Sheibani N, Sorenson CM. 2007. PECAM-1 isoform-specific regulation of kidney endothelial cell migration and capillary morphogenesis. Am. J. Physiol. Cell Physiol. 292:C2070–C2083. 10.1152/ajpcell.00489.2006 [DOI] [PubMed] [Google Scholar]
- 41.Sheibani N, Morrison ME, Gurel Z, Park S, Sorenson CM. 2012. BIM deficiency differentially impacts the function of kidney endothelial and epithelial cells through modulation of their local microenvironment. Am. J. Physiol. Renal Physiol. 302:F809–F819. 10.1152/ajprenal.00498.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cone FE, Steinhart MR, Oglesby EN, Kalesnykas G, Pease ME, Quigley HA. 2012. The effects of anesthesia, mouse strain and age on intraocular pressure and an improved murine model of experimental glaucoma. Exp. Eye Res. 99:27–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Haddadin RI, Oh DJ, Kang MH, Villarreal G, Jr, Kang JH, Jin R, Gong H, Rhee DJ. 2012. Thrombospondin-1 (TSP1)-null and TSP2-null mice exhibit lower intraocular pressures. Invest. Ophthalmol. Vis. Sci. 53:6708–6717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stamer WD. 2012. The cell and molecular biology of glaucoma: mechanisms in the conventional outflow pathway. Invest. Ophthalmol. Vis. Sci. 53:2470–2472 [DOI] [PubMed] [Google Scholar]
- 45.Esterbauer H, Schaur RJ, Zollner H. 1991. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic. Biol. Med. 11:81–128 [DOI] [PubMed] [Google Scholar]
- 46.Polansky JR, Wood IS, Maglio MT, Alvarado JA. 1984. Trabecular meshwork cell culture in glaucoma research: evaluation of biological activity and structural properties of human trabecular cells in vitro. Ophthalmology 91:580–595 [DOI] [PubMed] [Google Scholar]
- 47.de Kater AW, Shahsafaei A, Epstein DL. 1992. Localization of smooth muscle and nonmuscle actin isoforms in the human aqueous outflow pathway. Invest. Ophthalmol. Vis. Sci. 33:424–429 [PubMed] [Google Scholar]
- 48.Wiederholt M. 1998. Direct involvement of trabecular meshwork in the regulation of aqueous humor outflow. Curr. Opin. Ophthalmol. 9:46–49 [DOI] [PubMed] [Google Scholar]
- 49.Kuchtey J, Olson LM, Rinkoski T, Mackay EO, Iverson TM, Gelatt KN, Haines JL, Kuchtey RW. 2011. Mapping of the disease locus and identification of ADAMTS10 as a candidate gene in a canine model of primary open angle glaucoma. PLoS Genet. 7:e1001306. 10.1371/journal.pgen.1001306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Du Y, Roh DS, Mann MM, Funderburgh ML, Funderburgh JL, Schuman JS. 2012. Multipotent stem cells from trabecular meshwork become phagocytic TM cells. Invest. Ophthalmol. Vis. Sci. 53:1566–1575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gonzalez P, Epstein DL, Luna C, Liton PB. 2006. Characterization of free-floating spheres from human trabecular meshwork (HTM) cell culture in vitro. Exp. Eye Res. 82:959–967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liton PB, Liu X, Stamer WD, Challa P, Epstein DL, Gonzalez P. 2005. Specific targeting of gene expression to a subset of human trabecular meshwork cells using the chitinase 3-like 1 promoter. Invest. Ophthalmol. Vis. Sci. 46:183–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stamer WD, Peppel K, O'Donnell ME, Roberts BC, Wu F, Epstein DL. 2001. Expression of aquaporin-1 in human trabecular meshwork cells: role in resting cell volume. Invest. Ophthalmol. Vis. Sci. 42:1803–1811 [PubMed] [Google Scholar]
- 54.Baetz NW, Hoffman EA, Yool AJ, Stamer WD. 2009. Role of aquaporin-1 in trabecular meshwork cell homeostasis during mechanical strain. Exp. Eye Res. 89:95–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stone EM, Fingert JH, Alward WL, Nguyen TD, Polansky JR, Sunden SL, Nishimura D, Clark AF, Nystuen A, Nichols BE, Mackey DA, Ritch R, Kalenak JW, Craven ER, Sheffield VC. 1997. Identification of a gene that causes primary open angle glaucoma. Science 275:668–670 [DOI] [PubMed] [Google Scholar]
- 56.Resch ZT, Hann CR, Cook KA, Fautsch MP. 2010. Aqueous humor rapidly stimulates myocilin secretion from human trabecular meshwork cells. Exp. Eye Res. 91:901–908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wentz-Hunter K, Kubota R, Shen X, Yue BY. 2004. Extracellular myocilin affects activity of human trabecular meshwork cells. J. Cell. Physiol. 200:45–52 [DOI] [PubMed] [Google Scholar]
- 58.Lo WR, Rowlette LL, Caballero M, Yang P, Hernandez MR, Borras T. 2003. Tissue differential microarray analysis of dexamethasone induction reveals potential mechanisms of steroid glaucoma. Invest. Ophthalmol. Vis. Sci. 44:473–485 [DOI] [PubMed] [Google Scholar]
- 59.Wentz-Hunter K, Shen X, Yue BY. 2003. Distribution of myocilin, a glaucoma gene product, in human corneal fibroblasts. Mol. Vis. 9:308–314 [PubMed] [Google Scholar]
- 60.Brake PB, Zhang L, Jefcoate CR. 1998. Aryl hydrocarbon receptor regulation of cytochrome P4501B1 in rat mammary fibroblasts: evidence for transcriptional repression by glucocorticoids. Mol. Pharmacol. 54:825–833 [DOI] [PubMed] [Google Scholar]
- 61.Filla MS, Liu X, Nguyen TD, Polansky JR, Brandt CR, Kaufman PL, Peters DM. 2002. In vitro localization of TIGR/MYOC in trabecular meshwork extracellular matrix and binding to fibronectin. Invest. Ophthalmol. Vis. Sci. 43:151–161 [PubMed] [Google Scholar]
- 62.Tektas OY, Lutjen-Drecoll E. 2009. Structural changes of the trabecular meshwork in different kinds of glaucoma. Exp. Eye Res. 88:769–775 [DOI] [PubMed] [Google Scholar]
- 63.Chun YJ, Kim S, Kim D, Lee SK, Guengerich FP. 2001. A new selective and potent inhibitor of human cytochrome P450 1B1 and its application to antimutagenesis. Cancer Res. 61:8164–8170 [PubMed] [Google Scholar]
- 64.Tan JC, Peters DM, Kaufman PL. 2006. Recent developments in understanding the pathophysiology of elevated intraocular pressure. Curr. Opin. Ophthalmol. 17:168–174 [DOI] [PubMed] [Google Scholar]
- 65.Tian B, Gabelt BT, Geiger B, Kaufman PL. 2009. The role of the actomyosin system in regulating trabecular fluid outflow. Exp. Eye Res. 88:713–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tian B, Geiger B, Epstein DL, Kaufman PL. 2000. Cytoskeletal involvement in the regulation of aqueous humor outflow. Invest. Ophthalmol. Vis. Sci. 41:619–623 [PubMed] [Google Scholar]
- 67.Lutjen-Drecoll E, Gabelt BT, Tian B, Kaufman PL. 2001. Outflow of aqueous humor. J. Glaucoma 10(Suppl.1):S42–S44 [DOI] [PubMed] [Google Scholar]
- 68.Savinova OV, Sugiyama F, Martin JE, Tomarev SI, Paigen BJ, Smith RS, John SW. 2001. Intraocular pressure in genetically distinct mice: an update and strain survey. BMC Genet. 2:12. 10.1186/1471-2156-2-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chang B, Smith RS, Peters M, Savinova OV, Hawes NL, Zabaleta A, Nusinowitz S, Martin JE, Davisson ML, Cepko CL, Hogan BL, John SW. 2001. Haploinsufficient Bmp4 ocular phenotypes include anterior segment dysgenesis with elevated intraocular pressure. BMC Genet. 2:18. 10.1186/1471-2156-2-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Alper MG. 1963. Contusion angle deformity and glaucoma. Gonioscopic observations and clinical course. Arch. Ophthalmol. 69:455–467 [DOI] [PubMed] [Google Scholar]
- 71.Zhang Y, Davidson BR, Stamer WD, Barton JK, Marmorstein LY, Marmorstein AD. 2009. Enhanced inflow and outflow rates despite lower IOP in bestrophin-2-deficient mice. Invest. Ophthalmol. Vis. Sci. 50:765–770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Haddadin RI, Oh DJ, Kang MH, Filippopoulos T, Gupta M, Hart L, Sage EH, Rhee DJ. 2009. SPARC-null mice exhibit lower intraocular pressures. Invest. Ophthalmol. Vis. Sci. 50:3771–3777 [DOI] [PubMed] [Google Scholar]
- 73.Smith RS, Zabaleta A, Kume T, Savinova OV, Kidson SH, Martin JE, Nishimura DY, Alward WL, Hogan BL, John SW. 2000. Haploinsufficiency of the transcription factors FOXC1 and FOXC2 results in aberrant ocular development. Hum. Mol. Genet. 9:1021–1032 [DOI] [PubMed] [Google Scholar]
- 74.Smith R, John S, Nishina P, Sundberg J. 2001. Systematic evaluation of the mouse eye: anatomy, pathology and biomethods. CRC Press, Bocan Raton, FL [Google Scholar]
- 75.Slominski A, Tobin DJ, Shibahara S, Wortsman J. 2004. Melanin pigmentation in mammalian skin and its hormonal regulation. Physiol. Rev. 84:1155–1228 [DOI] [PubMed] [Google Scholar]
- 76.Ilia M, Jeffery G. 1999. Retinal mitosis is regulated by dopa, a melanin precursor that may influence the time at which cells exit the cell cycle: analysis of patterns of cell production in pigmented and albino retinae. J. Comp. Neurol. 405:394–405 [DOI] [PubMed] [Google Scholar]
- 77.Thomas SA, Matsumoto AM, Palmiter RD. 1995. Noradrenaline is essential for mouse fetal development. Nature 374:643–646 [DOI] [PubMed] [Google Scholar]
- 78.Rockwood JM, Maxwell GD. 1996. An analysis of the effects of retinoic acid and other retinoids on the development of adrenergic cells from the avian neural crest. Exp. Cell Res. 223:250–258 [DOI] [PubMed] [Google Scholar]
- 79.Clark AF. 2012. The cell and molecular biology of glaucoma: biomechanical factors in glaucoma. Invest. Ophthalmol. Vis. Sci. 53:2473–2475 [DOI] [PubMed] [Google Scholar]
- 80.Zhou L, Li Y, Yue BY. 1999. Oxidative stress affects cytoskeletal structure and cell-matrix interactions in cells from an ocular tissue: the trabecular meshwork. J. Cell. Physiol. 180:182–189 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.