

Abstract
N-(Chloro-3-methoxyphenyl)-2-picolinamide (3, ML128, VU0361737) is an mGlu4 positive allosteric modulator (PAM), which is potent and centrally penetrating. 3 is also the first mGlu4 PAM to show efficacy in a preclinical Parkinson disease model upon systemic dosing. As a noninvasive medical imaging technique and a powerful tool in neurological research, positron emission tomography (PET) offers a possibility to investigate mGlu4 expression in vivo under physiologic and pathological conditions. We synthesized a carbon-11 labeled ML128 ([11C]3) as a PET radiotracer for mGlu4, and characterized its biological properties in Sprague Dawley rats. [11C]3 was synthesized from N-(4-chloro-3-hydroxyphenyl)-2-picolinamide (2) using [11C]CH3I. Total synthesis time was 38±2.2 min (n = 7) from the end of bombardment to the formulation. The radioligand [11C]3 was obtained in 27.7±5.3% (n = 5) decay corrected radiochemical yield based on the radioactivity of [11C]CO2. The radiochemical purity of [11C]3 was >99%. Specific activity was 188.7±88.8 GBq/μmol (n = 4) at the end of synthesis (EOS).
PET images were conducted in 20 normal male Sprague Dawley rats including 11 control studies, 6 studies blocking with an mGlu4 modulator (4) to investigate specificity and 3 studies blocking with an mGlu5 modulator (MTEP) to investigate selectivity. These studies showed fast accumulation of [11C]3 (peak activity between 1-3 min) in several brain areas including striatum, thalamus, hippocampus, cerebellum, and olfactory bulb following with fast washout. Blocking studies with the mGlu4 modulator 4 showed 22-28 % decrease of [11C]3 accumulation while studies of selectivity showed only minor decrease supporting good selectivity over mGlu5. Biodistribution studies and blood analyses support fast metabolism. Altogether this is the first PET imaging ligand for mGlu4, in which the labeled ML128 was used for imaging its in vivo distribution and pharmacokinetics in brain.
Keywords: [11C]ML128, PET, Metabotropic glutamate receptor subtype 4 (mGlu4), Positive allosteric modulator
1. Introduction
In contrast to ionotropic glutamate receptors, which relay excitatory glutamate signal to open ion channels directly and instantly, metabotropic glutamate receptors (mGluRs) transfer glutamate signal indirectly through GTP-protein (G-protein) to various effector systems such as adenylate cyclase, phospholipase C, mitogen-activated protein kinase (MAPK), and phosphoinositide 3 kinase (PI3K)1. Their signal effects are prolonged and influence on many signal pathways in normal physiological or pathological conditions. mGluRs belong to family C of G-protein coupled receptors, and have three subgroups and eight subtypes2. mGlu receptor subtype 4 (mGlu4) belongs to group III mGluRs along with mGlu6, mGlu7, and mGlu8, while group I mGluRs include mGlu1 and mGlu5 and group II mGluRs comprise mGlu2 and mGlu3. While group I mGluRs are positively coupled to activate various downstream effectors and protein kinase pathways, group II and III mGluRs are negatively coupled, and inhibit the activity of adenylate cyclase. But, they activate other signal pathways such as MAPK and PI3K pathways3,4.
mGluRs contain extracellular region, transmembrane region and intracellular regions. Extracellular region consists of the Venus flytrap domain (VFD), the orthosteric binding domain of glutamate, and cysteine-rich domain which relays the conformational change induced by glutamate binding at VFD to transmembrane region5. The transmembrane region contains seven helical domains, and these domains deliver the conformational change in extracellular region to intracellular region of mGluRs. The binding locations at the transmembrane domain render the non-competitive binding sites for most allosteric modulators of mGluRs6. Intracellular region comprises C-terminus where signal transfers are occurred by G-protein coupling, kinase pathways, and alternative splicing.
mGlu4 is preferentially localized on presynaptic site. Expression of mGlu4 based on mRNA studies is most intensive in cerebellar cortex, and moderately expressed in hippocampus, striatum, olfactory bulb and cerebellum7-10. mGlu4 participates in presynaptic neurotransmission of excitatory glutamate signal, and has received much attention lately due to its implication for various neuronal diseases such as Parkinson's disease (PD) and movement disorders11-14. Imaging of presynaptic mGlu4 function might provide valuable insight to the synaptic transmission of glutamate. More profoundly, by combining the studies of presynaptic mGlu4 and postsynaptic mGlu5 it can provide in deep information of synaptic transmission of glutamate, which will be useful for evaluating neurotransmission related pathological processes. Thus, compounds that are potent and selective for mGlu4 could provide a valuable tool to investigate the involvement of these receptors in various diseases to develop new therapeutics.
Much effort has focused on development of mGluR allosteric modulators, which target the seven-transmembrane spanning domain. Compared to orthosteric compounds, allosteric modulators often display better selectivity among mGluR subtypes and adequate drug-like properties. Since PHCCC was identified as a selective mGlu4 PAM, there has been substantial progress in identifying positive allosteric modulators (PAM) for mGlu415. The Vanderbilt University group developed a selective mGlu4 PAM, N-(4-chloro-3-methoxyphenyl)picolinamide (3, ML128, VU0361737), which is potent (rEC50 = 110 nM & hEC50 = 240 nM) and centrally penetrate16. It is the first mGlu4 PAM to demonstrate efficacy in a preclinical rodent model of motor impairments associated with PD16, 17. This compound has been recommend as a molecular probe to investigate the role of selective allosteric activation of mGlu4 in vitro and in vivo.
To better understand the role of mGlu4 in normal and disease conditions, we were interested in developing an mGlu4 selective radiotracer for in vivo study. As a noninvasive medical imaging technique and a powerful tool in neurological research, positron emission tomography (PET) offers the possibility to visualize and analyze the target receptor expression under physiological and pathophysiological conditions. Moreover, PET tracers serve as invaluable biomarker during the chemical development of potential therapeutic drugs. Many PET radioligands have been developed for group I mGlus (e.g. [18F]FPEB, [11C]ABP688, etc. for mGlu518, 19 and [11C]YM202074 and [18F]MK1312 for mGlu120, 21), however, no PET tracer is available for imaging mGlu4. ML128 is a good candidate to be radiolabeled as a PET tracer for mGlu4. Although low microsomal stability, high clearance and short half-life, ML128 showed some favorable properties for developing a potential PET radiotracer, which include rapid penetration into rat brain following intraperitoneally injection (Tmax for brain: 0.5 h), high brain:plasma (B/P) partition coefficients (B/P=4.1), good selectivity over other mGluRs subtypes and the chemical structure of the precursor to allow fast labeling. On the other hand, the labeled ML128 can be used to imaging its in vivo distribution and pharmacokinetics in brain, which should give further insight into the underlying pharmacological processes.
Here we report on the radiosynthesis of a carbon-11 labeled mGlu4 PAM (3, ML128, VU0361737) as a PET tracer for mGlu4 and its preliminary biological evaluation in Sprague-Dawley rats to image mGlu4.
2. Results
2.1 Chemistry and radiochemistry
Compounds 2 - 4 were prepared by the amide coupling reaction between 1 and the corresponding aniline derivatives with 28%, 53%, and 68% yield, respectively. The radioligand [11C]3 was synthesized by methylation of the precursor 2 with [11C]iodomethane ([11C]CH3I) mediated by 5 M potassium hydroxide (KOH) in N,N-dimethylformamide (DMF) at 90°C for 5 min (Scheme 1), in which [11C]CH3I was obtained from [11C]carbon dioxide ([11C]CO2) via [11C]methane ([11C]CH4). The reaction mixture was purified by a semi-preparative HPLC, in which an isocratic method (see 2.3.1) was developed to achieve good separation between the desired product [11C]3 (9.8-10.6 min) and the precursor 2 (5.4-6.0 min) as well as [11C]CH3I (4.8-5.2 min) (Fig. 1(a)). Total synthesis time was 38±2.2 min (n = 7) from the EOB to the formulation. The radioligand [11C]3 was obtained in 27.7±5.3% (n = 5) decay corrected radiochemical yield (RCY) based on the radioactivity of [11C]CO2.
Scheme 1. Syntheses of reference ML128 (3), [11C]ML128 ([11C]3), its precursor (2), and blocking agent (4).
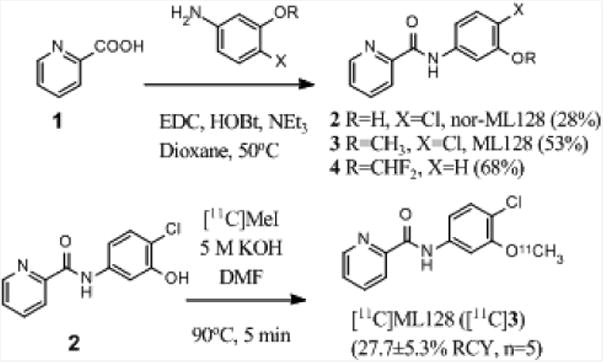
Figure 1.
(a) UV and radioactivity traces of semi-preparative HPLC (b) The analytical HPLC chromatogram for co-injection of [11C]3 with the cold reference 3.
The chemical identity of the purified product ([11C]3) was confirmed by co-injection with the cold reference 3 into radio-HPLC, and it showed the same peak on the UV chromatogram for both compounds (Fig. 1(b)). The time difference between the UV peak (4.7 min) and the radioactivity peak (5.0 min) in the chromatogram arose from the line setting between the radioactivity detector and the UV detector. Radio-TLC also confirmed that reference standard 3 and radioactive spot had same Rf value (Rf = 0.6 with ethyl aceate:hexane = 1:1). The radiochemical purity of [11C]3 was >99%. Specific activity was 188.7±88.8 GBq/μmol (n = 4) at the end of synthesis (EOS). Experimentally determined partition coefficient (logD) between pH7.4 phosphate buffered saline (PBS) and octanol was 3.09±0.14 (n = 5).
2.2. The selectivity to mGlu5 and mGlu822
The selectivity to mGlu5 and mGlu8 were studied by intracellular inositol phosphates (IPs) accumulation assay using CHO cells that expressed stably mGlu5a or mGlu8. 3 was tested at a single concentration (10 μM) for activity as an agonist or an antagonist. Testing for antagonism was performed in presence of the EC50 concentration of glutamate (5 μM). The results showed there was no agonist activity for mGlu5 (-15% maximal activity as obtained with maximal agonist concentrations for mGlu5), but slight agonist activity for mGlu8 (13% of maximal activity). The results showed 51% of inhibition of receptor activity for mGlu5 and -5% for mGlu8 in presence of an EC50 concentration of glutamate.
2.3. In vivo characterization of [11C]3
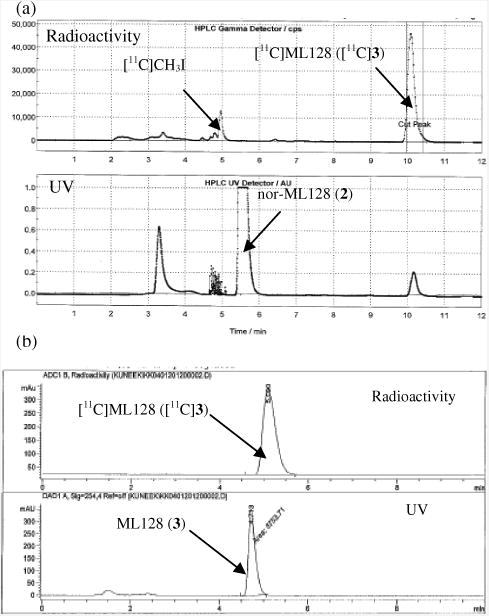
The PET images indicate that the radiotracer crosses blood brain barrier (BBB), and high accumulation is observed in hippocampus, striatum, thalamus, olfactory bulb, and cerebellum (Fig 2(a)). The time-activity curves (TACs) showed fast uptake (peak activity between 1 and 3 min) followed by fast wash-out within 20 min throughout the brain. Fast clearance of the radioligand [11C]3 from the brain agrees with the previous study indicating pharmacokinetic half-life of 0.15 h for 3 in the brain16 (Fig 2(b)).
Figure 2.
Pharmacokinetics of [11C]3. (a) Representative cumulative PET/CT images of rat brain at the time interval 2-15 min after administration of [11C]3 (24.1 MBq i.v.). Slice thickness is 0.625 mm. Str= striatum, Hp=hippocampus, Th=thalamus and Cer=cerebellum. (b) TACs show fast uptake (peak activity between 1 and 3 min) followed by fast washout throughout the brain.
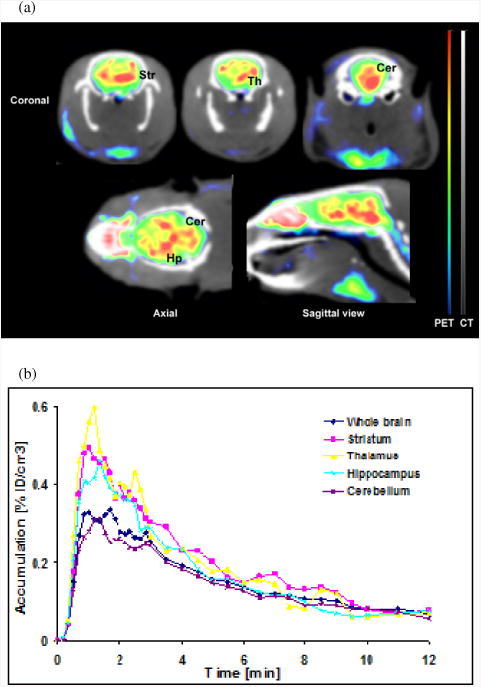
Comparison of [11C]3 distribution in rat brain at the time interval 2-15 min in a control study and after blocking of mGlu4 with 4·HCl or mGlu5 with 3-((2-methyl-1,3-thiazol-4-yl)ethynyl)-pyridine hydrochloride (MTEP·HCl) is presented in Fig 3(a). mGlu4 blocking conducted by administration of 4·HCl (10 mg/kg i.p.) as a blocking agent 30 min before injection of [11C]3 radioactivity showed 23-28% reduced uptake of [11C]3 in the striatum, hippocampus, cerebellum and averaged whole brain. Another blocking was done to investigate the radioligand's selectivity to mGlu5 by using an mGlu5 NAM, MTEP, as a blocking agent (5 mg/kg i.v. 2 min before injection of [11C]3). The results show that the uptake of [11C]3 was reduced 1-2% in the striatum and hippocampus and 12% averaged from the whole brain (Fig 3(b)).
Figure 3.
PET studies of specificity and selectivity of [11C]3. (a) Comparison of [11C]3 distribution in rat brain at the time interval 2-15 min in a control study, after blocking of mGlu4 with 4.HCl (10 mg/kg i.p. 30 min before [11C]3 injection) and after blocking of mGlu5 with MTEP (5 mg/kg i.v. 2 min before [11C]3). Coronal images are at the striatal and hippocampal level while axial and sagittal views at the midstriatal level. (b) The uptake of [11C]3 was reduced by 23-28% in all brain areas with 4.HCl while MTEP induced blocking was minor.
The concentration of the radioligand in blood plasma was measured by withdrawing blood from three male rats at three time points (5, 25, 55 min) after injection of [11C]3. The analysis of blood plasma revealed fast in vivo metabolism of [11C]3. The results show that the intact radioligand was rapidly reduced to 30% of initial amount in 5 min after the i.v. injection, and then gradually reduced to 10% over next 50 min (Fig 4).
Figure 4.
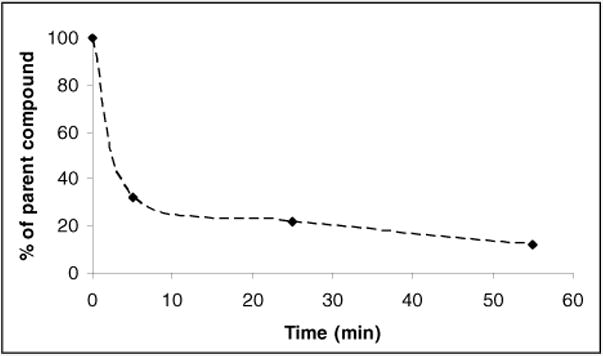
Metabolite corrected blood curve after [11C]3 injection.
2.4. Ex vivo characterization
The pharmacokinetic property of the radioligand was further evaluated by ex vivo biodistribution study. Total 15 male rats were sacrificed in a group of three at five different time points (1, 5, 10, 20, and 40 min) after injection of [11C]3, in which major organs were harvested and radio-counted. The amount of activity was reported as percentage of injected dose (ID) in unit weight of tissue. As shown in Fig 5, the radioactivity was distributed with less than 1% of ID over major organs. However, the uptake in liver and kidney was relatively higher than in other organs what also relates to fast metabolism.
Figure 5.
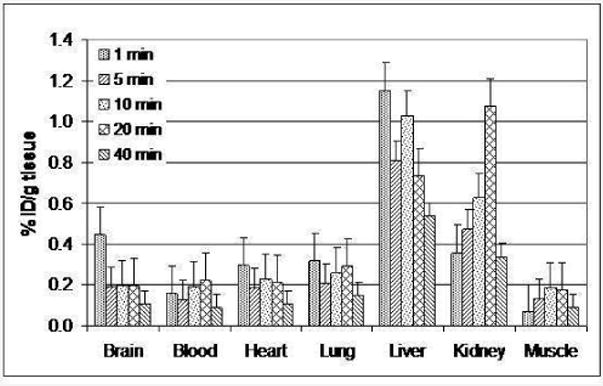
Biodistribution study of [11C]3.
3. Discussion
The current work describes the first PET imaging ligand what can be used to investigate a role of mGlu4 in glutaminergic neurotransmission. Compounds 2-4 were prepared by one-step syntheses from commercially available chemicals using amide bond formation reaction. A rapid methylation on phenolic oxygen was achieved with [11C]CH3I generated by two step gas-phase reaction from [11C]CO2. The radiochemistry was completed with two half-life cycle from the end of bombardment (EOB) to the formulation including HPLC purification. The high RCY was achieved without significant side reaction under a mild condition. There was enough separation between 2 and [11C]3 on HPLC, so no impurities from side product or precursor affected radiochemical purity. Radio-TLC and radio-HPLC indicated that the collected product was radiochemically pure. Co-injection with cold reference 3 demonstrated that the collected product was authentic radiolabeled target compound.
The PET image of brain with [11C]3 indicated that 3 crossed BBB which was consistent with previous study16. The positive brain permeability of 3 was also predicted by many theoretical properties such as clogP, molecular weight (MW), and topological polar surface area (tPSA). The clogP, MW, and tPSA of 3 were 2.80, 262.7, and 50.7, respectively23, and these theoretical properties satisfy the requirements as a central nervous system (CNS) drug presented by previous literatures24, 25. 3 also satisfies the number of hydrogen bond (HB) donors (n = 1), and HB acceptors (n = 3). Actual experimental logD (3.09±0.14) also indicated that 3 could be a CNS drug24, 25.
The PET images of rat brain showed that the radiotracer mainly occupied hippocampus, striatum, thalamus, olfactory bulb, and cerebellum. This result agrees with previous studies that report in vitro mGlu4 distribution in brain7-10. This is first study to verify in vivo mGlu4 distribution in the brain with mGlu4 active PET radiotracer.
TACs of regions of interest (ROIs) revealed transient uptake of [11C]3 followed by rapid clearance in the brain. Furthermore, no particular regions displayed either retention or slow wash-out in the brain. Such phenomena can be explained that efflux from brain was more dominant than actual specific binding to mGlu4. In spite of fast removal from brain regions, blocking study with 4·HCl (10 mg/kg i.p.) proved moderate reduction of binding in the brain, in which 4 had been reported to have high PAM activity (rEC50 = 80 nM) against mouse mGlu4 and to be centrally penetrating19. Because the dose of blocking agent was not optimized in this study the partial blocking effect might be attributed to insufficient saturation in target binding site. Moreover, since 3 has fast metabolism, it might occupy the target site only short period. Therefore, blocking effect might not be prolonged after PET radiotracer was injected.
The in-vivo study with MTEP·HCl demonstrated minor blocking effect with [11C]3. According to our in-vitro study with mGlu5, 3 had no agonist activity, but minor antagonist activity which was agreed with previous literature16. Therefore, [11C]3 can be considered to bind specifically to mGlu4.
As it was expected from previous experiment, 3 was featured by fast metabolism16. The previous literature showed that the half-life of 3 in plasma was 1.9 hours after i.p. injection, while our blood metabolite study indicated that [11C]3 was reduced to 30% within 5 min after i.v. injection (Fig. 4)16. Difference in pharmacokinetic characteristics might come from different injection methods. Fast metabolism was also observed in biodistribution studies (Fig. 5). The relative high ratio of carbon-11 species in liver and kidney reflected high metabolic activity. The metabolically unstable methoxy group might contribute to fast pharmacokinetic half-life of 3.
Based on above results, further development based on 3 is essential to improve pharmacokinetic features and binding properties to mGlu4. Such a structural modification must accommodate following features. (i) Considering carbon-11 half-life and PET scanning time, the metabolic half-life has to be at least 30 min. (ii) New compound should display better binding property toward mGlu4, and increased selectivity to other mGluRs subtypes. (iii) The structural modification does not affect the brain penetrability. It will be useful to evaluate [11C]3 with animal models of different neurological disorders.
4. Conclusion
This paper reports the radiosynthesis of [11C]3, a new carbon-11 labeled PET radiotracer for mGlu4 and its characterization in normal rats. This is the first report of in vivo PET imaging of mGlu4. 3 can serve as an invaluable lead compound for future innovations of imaging ligands for mGlu4 to investigate different neurological conditions.
5. Experimental
5.1. General
MTEP·HCl was purchased from Abcam PLC (Cambridge, MA). All other reagents and solvents utilized for experiments were purchased from commercial suppliers (Sigma-Aldrich, TCI America, VWR and J.T. Baker) and were used without further purification. NMR spectra were recorded on Varian 500 MHz spectrometer (Agilent Technology, Santa Clara, CA). All chemical shifts (δ) were reported in parts per million (ppm) downfield or upfield from the standard (tetramethylsilane for 1H and 13C; trichlorofluoromethane for 19F). 1H NMR data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, dd = double-doublet, ddd = double-double-doublet, t = triplet, br = broad,) coupling constant (Hz), and integration. High resolution mass spectrum (HRMS) was obtained by The Small Molecule Mass Spectrometry Facility at Harvard University using Agilent 6210 Time-of-Flight LC/MS. The melting point was measured by MP50 melting point system (Mettler Toledo LLC, Columbus, OH) and was uncorrected. LC-MS spectra were obtained on Agilent 1200 series HPLC coupled with 6310 ion trap mass spectrometer (Agilent Technology, Santa Clara, CA), in which an Agilent Eclipse XDB C8 analytical column (150 × 4.6 mm, 5 μm) was used. Flash chromatography was conducted on CombiFlash® Companion (Teledyne Isco, Lincoln, NE).
5.2. Chemistry
5.2.1. N-(4-Chloro-3-hydroxyphenyl)-2-picolinamide (2)
To a solution of 2-picolinic acid (1, 0.857 g, 6.97 mmol), N-hydroxybenzotriazole hydrate (HOBt.H2O, 1.066 g, 6.97 mmol), diisopropylethylamine (DIPEA, 1.80 g, 13.93 mmol) and N-(3-methylaminopropyl)-N'-ethylcarbodiimide hydrochloride (EDC·HCl, 2.0 g, 10.45 mmol) in dry 1,4-dioxane (80 mL) was added 5-amino-2-chlorophenol (1.20 g, 8.36 mmol) at room temperature. The mixture was heated to 50°C and stirred overnight. To the reaction mixture was added 300 mL of dichloromethane (DCM), and sequentially washed with water (3 × 100 mL). After the organic layer was separated and concentrated, the reaction mixture was purified by silica-gel chromatography (hexane/ethyl acetate: from 95/5 to 30:/70) to give the product as a pale-yellow solid (0.482 g, 1.94 mmol, 27.8% yield). mp 140-142°C. 1H NMR (500 MHz, CDCl3) δ ppm 10.02 (s, 1H), 8.62 (d, J = 5.0 Hz, 1H), 8.30 (d, J = 8.0 Hz, 1H), 7.92 (ddd, J = 7.6, 7.6, 1.6 Hz, 1H), 7.64 (d, J = 1.5 Hz, 1H), 7.50 (dd, J = 7.0, 5.0 Hz, 1H), 7.31 (d, J = 8.5 Hz, 1H), 7.27 (d, J = 2.5 Hz, 1H), 5.68 (bs, 1H); 13C NMR (125 MHz, DMSO-d6) δ ppm 163.02, 154.39, 150.91, 149.32, 139.37, 138.96, 130.71, 127.82, 123.15, 116.13, 112.88, 109.11. LC-MS, calculated for C12H9N2O2Cl: 248.04; observed: 249.0 [MH]+. HRMS m/z calcd for C12H9ClN2O2 (MH+), 249.0425; found 249.0437.
5.2.2. N-(4-Chloro-3-methoxyphenyl)-2-picolinamide (3)
3 was synthesized in a similar procedure as described for 2 by using 4-chloro-3-methoxyaniline (400 mg, 2.54 mmol), 1 (312 mg, 2.54 mmol), HOBt·H2O (389 mg, 2.54 mmol), DIPEA (0.66 g, 5.08 mmol), EDC·HCl (730 mg, 3.81 mmol) and anhydrous 1,4-dioxane (30 mL). 3 was obtained as a pale yellow solid (325 mg, 1.34 mmol, 53% yield); mp: 120 °C. 1H NMR (500 MHz, CDCl3) δ ppm 10.07 (s, 1H), 8.63 (d, J = 4.0 Hz, 1H), 8.29 (d, J = 8.0 Hz, 1H), 7.92 (ddd, J = 7.6, 7.6, 1.6 Hz, 1H), 7.81 (d, J = 2.0 Hz, 1H), 7.50 (dd, J = 7.0 Hz, 5.0 Hz, 1H), 7.34 (d, J = 8.5 Hz, 1H), 7.09 (dd, J = 9, 2.5 Hz, 1H), 3.97 (s, 3H). 13C NMR (125 MHz, CDCl3) δ ppm 162.41, 155.68, 149.85, 148.37, 138.17, 137.97, 130.47, 127.01, 122.72, 117.80, 112.45, 104.37. LC-MS, calculated for C13H11N2O2Cl: 262.05; observed: 263.0 [M+H]+.
5.2.3. N-(3-(Difluoromethoxy)phenyl)-2-picolinamide (4)
4 was synthesized in a similar procedure as described for 2 by using 3-(difluoromethoxy)aniline (1.0 g, 6.28 mmol), 1 (0.773 g, 6.28 mmol), HOBt.H2O (0.962 g, 6.28 mmol), DIPEA (1.62 g, 12.56 mmol), EDC·HCl (1.81 g, 9.42 mmol) and anhydrous 1,4-dioxane (80 mL). 4 was obtained as an off-white solid (1.12 g, 4.27 mmol, 68.0% yield). mp: 84°C. 1H NMR (500 MHz, CDCl3) δ ppm 10.09 (s, 1H), 8.62 (d, J = 4.9 Hz, 1H), 8.30 (d, J = 7.8 Hz, 1H), 7.91 (t, J = 8.8 Hz, 1H), 7.76 (s, 1H), 7.49 (m, 2H), 7.36 (t, J = 8.3 Hz, 1H), 6.90 (d, J = 1.5 Hz, 1H), 6.57 (t, J = 73.85 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ ppm 162.22, 152.18 (t, J = 2.94 Hz), 148.09, 139.51, 138.47, 130.54, 127.13, 123.07, 116.78, 116.33 (t, J = 257.8 Hz), 115.42, 111.20; 19F NMR (470 MHz, CDCl3) δ ppm -80.0; LC-MS, calculated for C13H10F2N2O2: 264.07; observed: 265.0 [M+H]+.
5.2.4.N-(3-(Difluoromethoxy)phenyl)-2-picolinamide hydrochloride salt (4·HCl)
To a solution of 4 (0.100 g, 0.379 mmol) in DCM at 0°C was added 2 M HCl in ether (0.95 ml, 1.90 mmol) dropwise, and stirred for 15 min. The solution was warmed up to room temperature, and stirred for 30 min before the solvent was removed by filtration. The corresponding salt (4·HCl) was obtained as white solid (0.100 g, 79% yield).
5.3. Radiochemistry
[11C]CO2 was produced by Siemens Eclipse HP 11 MeV cyclotron (Malvern, PA) using nitrogen target containing 2% oxygen with proton bombardment. [11C]CH3I was synthesized by TRACERlab FX MeI (GE Healthcare, Waukesha, WI), and [11C]3 was synthesized by TRACERlab FX M synthesis module (GE Healthcare). The radiochemical purity and specific activity of final radioactive product was measured by Agilent 1200 Series HPLC equipped with UV spectrometer monitored at 254 nm and Carroll and Ramsey Model 105S Single Channel Radiation Detector (Berkeley, CA) in which an Agilent Eclipse XDB C18 analytical column (150 × 4.6 mm, 5 μm) or a Gemini-NX C18 semi-preparative column (250 × 10 mm, 5 μm, Phenomenex Inc., Torrance, CA) was used. The radiochemical purity was also verified by Bioscan Inc AR-2000 radio-TLC imaging scanner (Washington DC) using Silicycle silica gel 60-F254 TLC plates (Quebec, Canada). Radioactivity was quantified with a CRC®-25PET dose calibrator (Capintec Inc., Ramsey, NJ).
5.3.1. Synthesis of N-(4-Chloro-3-[11C]methoxyphenyl)-2-picolinamide ([11C]3)
[11C]CO2 produced by cyclotron was reduced to [11C]methane ([11C]CH4) by Shimalite® nickel (Shinwa Chemical, Japan) at 390°C, which was converted into [11C]CH3I in gaseous iodination condition at 720°C. The resultant [11C]CH3I was transferred in a stream of nitrogen and trapped in a reaction vessel containing the precursor 2 (1.5 mg) and 5 M KOH (3.2 μL) in DMF (0.3 mL) at 0°C. After completion of the [11C]CH3I transfer, the reaction vessel was sealed and heated at 90°C for 5 min. After heating, the reaction mixture was diluted with 1 mL of HPLC eluent, and injected into radio-HPLC (Phenomenex Gemini-NX C18 semi-preparative column, 250 × 10 mm, 5 μm) using CH3CN/0.1 M HCO2NH4 solution (45/55) as the mobile phase at a flow rate of 5 mL/min. The desired product was eluted between 10.4 and 11.0 min. The fraction containing [11C]3 was diluted with 20 mL of water and passed through a C18 Sep-Pak® Plus cartridge (Waters Corp.), and the cartridge was washed with additional 5 mL of water. The [11C]3 was then eluted from cartridge with 1 mL of ethanol and passed through a sterile filter (0.22 μm, EMD Millipore Corp.). The final product was diluted with 9 mL of 0.9% saline to make 10% ethanol solution in saline for animal injection. Total synthesis time was 38±2.2 min (n = 7) from the EOB to the formulation. The radioligand [11C]3 was obtained in 27.7±5.3% (n = 5) decay corrected RCY based on [11C]CO2.
5.3.2. Quality Control (QC) Analysis
The chemical identity of [11C]3 was confirmed by co-injection with the cold (i.e nonradioactive) reference compound 3 into radio-HPLC (Column: Agilent Eclipse XDB C18 analytical column, 150 × 4.6 mm, 5 μm; eluent: CH3CN/0.1 M HCO2NH4 solution = 60:40; flow rate: 1 mL/min) and it showed the same peaks on the UV and the radioactive chromatograms. The purity of [11C]3 was obtained by the radio-HPLC analysis in the same condition described above. The radiochemical purity of [11C]3 was >99%. Radio-TLC was also performed. An aliquot of [11C]3 and the cold reference compound 3 were spotted on a TLC plate, and developed in a TLC chamber with eluent (ethyl acetate:hexane = 1:1). Their Rf values were checked by radio-TLC scanner and UV lamp, respectively.
5.3.3. Measurement of Specific Activity
Two aliquots (10 μL) of [11C]3 were taken for determination of the specific activity. An aliquot of [11C]3 was injected into analytical HPLC using the same method as in QC analysis, and the other aliquot was saved and its radioactivity was measured and corrected to end of synthesis (EOS). The amount of injected [11C]3 was calculated and expressed as μmol using a HPLC calibration curve developed from the reference 3 based on the five different concentrations (5, 10, 50, 100, and 500 ppm). Specific activity of [11C]3 was 188.7±88.8 GBq/μmol (n = 4) at the EOS.
5.4. Measurement of logD
The logD value of [11C]3 was determined by using previously published method26. One test tube containing 2.5 mL of PBS and 2.5 mL of octanol and five test tubes containing 2.5 mL of PBS and 0.5 mL of octanol were prepared. After aliquot of [11C]3 was added to first test tube containing 2.5 mL of PBS and 2.5 mL of octanol, the test tube was vortexed for 2 min, and centrifuged over 8000 rpm for 2 min. After 0.1 mL of octanol and 1.0 mL of PBS were taken from the test tube, 2.0 mL of octanol was transferred to next test tube. Same procedure was repeated until six sets of samples were obtained. Each set of samples was counted by 2480 Wizard2 Automatic Gamma Counter (PerkinElmer, Waltham, MA). The logD value was calculated by the equation: log(decay corrected radioactivity in octanol layer × 10/decay corrected radioactivity in PBS).
5.5. Intracellular Inositol Phosphate Accumulation Assay for mGlu5 and mGlu822
mGlu5 and mGlu8 were stably expressed in CHO cells. Cultured in 96-well plates to confluency (3 to 4 days), the cells were incubated overnight in glutamine-free culture medium supplemented with 18.5 kBq myo-[3H]inositol (NEN) to label the cell membrane phosphoinositides. After washing the cultures, incubations with 3 (10 μM) were carried out for 45 min at 37°C in Locke's buffer (156 mM NaCl, 5.6 mM KCl, 3.6 mM NaHCO3, 1 mM MgCl2, 1.3 mM CaCl2, 5.6 mM glucose, and 20 mM HEPES, pH 7.4) containing 20 mM LiCl to block the degradation of IPs. The reaction was terminated by aspiration, and IPs were extracted with 0.1 M HCl. The separation of [3H]IPs was performed by anion exchange chromatography and determined by a liquid scintillation counter. 3 was tested at a single concentration (10 μM) for activity as an agonist or an antagonist. Testing for antagonism was performed in presence of the EC50 concentration of glutamate (5 μM). Each compound was tested in duplicate in two separate experiments performed on different cell passages. In addition to the tested compounds each 96-well plate contained points for determination of basal activity, maximal agonist stimulation, agonist EC50 concentrations (i.e., concentration-response isotherm), and the IC50 concentration of a known antagonist for purposes of positive control and for activity calculations. The reported results for each compound were calculated for agonists as the percent of maximal activity (as obtained with maximal agonist concentrations) and for antagonist as the percent of inhibition of receptor activity (in presence of an EC50 concentration of glutamate)
5.6. In Vivo Characterization of [11C]3
Altogether twenty normal Sprague Dawley rats (male, 300– 500 g) were used to investigate in vivo imaging characteristics of [11C]3. Eleven rats were used for control studies, six rats were used to investigate specificity and three rats to investigate selectivity of [11C]3. All animal studies were performed by the guidance of the National Institute of Health guide for the care and use of laboratory animals and, approved and supervised under the subcommittee on research animals of the Harvard Medical School and Massachusetts General Hospital. For the imaging studies rats were anesthetized with isoflurane/nitrous oxide (1.0-1.5% isoflurane, with oxygen flow of 1-1.5 L/min) and the tail vein was catheterized for administration of the imaging ligand ([11C]3). The rats were adjusted into the scanner for imaging position (Triumph II Preclinical Imaging System, Gamma Medica-IDEAS, Northridge, CA). The vital signs such as heart rate and/or breathing were monitored throughout the imaging. Data acquisition of 60 min was started from the injection of radioligand [11C]3 (22.2–37 MBq i.v.). To investigate specificity of [11C]3 for the mGlu4, 4·HCl salt (10 mg/kg i.p.) was administered in 10% ethanol and 10% Tween-20 in water solution 30 min before the radioactivity. To investigate the selectivity of [11C]3, MTEP·HCl (5 mg/kg, an mGlu5 NAM) in sterile water solution was injected into the tail vein 2 min before the [11C]3 injection27. CT scan was performed after PET imaging to obtain anatomical information and correction for attenuation. The PET imaging data were corrected for uniformity, scatter, and attenuation and processed by using maximum-likelihood expectation-maximization (MLEM) algorithm with 30 iterations to dynamic volumetric images (18×10″, 14×30″, 20×60″, 10×180″). CT data were reconstructed by the modified Feldkamp algorithm using matrix volumes of 512×512×512 and pixel size of 170 μm. The ROIs, i.e. whole brain, cerebellum, striatum and hippocampus, were drawn onto coronal slices according to the brain outlines as derived from the rat brain atlas28 and corresponding TACs were created by PMOD 3.2 (PMOD Technologies Ltd., Zurich, Switzerland). Percent changes between the control and blocking studies were calculated in all brain areas.
5.7. Determination of [11C]3 in Blood Plasma
Blood samples were collected from the femoral artery in three Sprague Dawley rats at 5, 25, and 55 min after [11C]3 injection. The blood samples were centrifuged for 3 min, and plasma (0.3 mL) was retrieved, and mixed with 0.3 mL of acetonitrile. The mixture solution was vortexed, and centrifuged for 3 min to separate plasma protein, and supernatant (0.3 mL) was collected, and loaded to Strata-X Phenyl cartridge (500 mg, Phenomenex Inc.) saturated with water, and eluted with acetonitrile-0.1% TFA/water solution co-solvent by gradually increasing acetonitrile from 0% to 100%. The fractions of each sample were collected in eight test tubes. The radioactivity of supernatant and eluted portion (seventh test tube) of [11C]3 were counted by 2480 Wizard2 Automatic Gamma Counter.
5.8. Biodistribution Study
Total of 15 Sprague Dawley rats (male, weight 300-500 g) were used in this study. [11C]3 (27.4-48.1 MBq/animal) was injected into the 15 rats, and three of them were sacrificed at each of five different time points (1, 5, 10, 20, and 40 min after administration of [11C]3). Blood, brain, heart, lung, liver, muscle, and kidney were harvested, weighed, and counted in 2480 Wizard2 Automatic Gamma Counter. Radioactivity of the samples was corrected for decay and injected activity and expressed as percent injection dose per gram tissue (%ID/g tissue).
Acknowledgments
The authors like to thank NIMH Psychoactive Drug Screening Program and the research team at the Chapel Hill, North Carolina for functional assays of 322. The authors like to thank also the cyclotron personnel at Martinos Biomedical Imaging Center at Massachusetts General Hospital for technical support for radiosynthesis. No other potential conflict of interest was declared.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.Hur EM, Kim KT. Cell signal. 2002;14:407–418. doi: 10.1016/s0898-6568(01)00258-3. [DOI] [PubMed] [Google Scholar]
- 2.Schoepp DD, Jane DE, Monn JA. Neuropharmacology. 1999;38:1431–1476. doi: 10.1016/s0028-3908(99)00092-1. [DOI] [PubMed] [Google Scholar]
- 3.Pin JP, Acher F. Curr Drug Targets CNS Neurol Disord. 2002;1:297–317. doi: 10.2174/1568007023339328. [DOI] [PubMed] [Google Scholar]
- 4.Conn PJ, Pin JP. Ann Rev Pharmacol Toxicol. 1997;37:205–237. doi: 10.1146/annurev.pharmtox.37.1.205. [DOI] [PubMed] [Google Scholar]
- 5.Muto T, Tsuchiya D, Morikawa A, Jingami H. Proc Natl Acad Sci USA. 2007;104:3759–3764. doi: 10.1073/pnas.0611577104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hampson DR, Rose EM, Antflick JE. In: The Glutamate Receptors. Gereau RW, Swanson GT, editors. Totowa: Human Press; 2008. pp. 363–386. [Google Scholar]
- 7.Shigemoto R, Mizuno N. In: Handbook of chemical neuroanatomy Otterson OP. Storm-Mathisen J, editor. Vol. 18. Elsevier; 2000. pp. 63–98. [Google Scholar]
- 8.Bradley SR, Standaert DG, Rhodes KJ, Rees HD, Testa CM, Levey AI, Conn PJ. J Comp Neurol. 1999;407:33–46. [PubMed] [Google Scholar]
- 9.Corti C, Aldegheri L, Somogyi P, Ferraguti F. Neuroscience. 2002;110:403–420. doi: 10.1016/s0306-4522(01)00591-7. [DOI] [PubMed] [Google Scholar]
- 10.Kinoshita A, Ohishi H, Nomura S, Shigemoto R, Nakanishi S, Mizuno N. Neurosci Lett. 1996;207:199–202. doi: 10.1016/0304-3940(96)12519-2. [DOI] [PubMed] [Google Scholar]
- 11.Marino MJ, Williams DL, Jr, O'Brian JA, Valenti O, McDonald TP, Clements MK, Wang R, DiLella AG, Hess JF, Kinney GG, Conn PJ. Proc Natl Acad Sci USA. 2003;100:13668–13673. doi: 10.1073/pnas.1835724100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marino MJ, Valenti O, Conn PJ. Glutamate receptors and Parkinson's disease: Opportunities for intervention. Drugs Aging. 2003;20:377–397. doi: 10.2165/00002512-200320050-00006. [DOI] [PubMed] [Google Scholar]
- 13.Blandini F, Nappi G, Tassorelli C, Martignoni E. Prog Neurobiol. 2000;62:63–88. doi: 10.1016/s0301-0082(99)00067-2. [DOI] [PubMed] [Google Scholar]
- 14.Hopkins CR, Lindsley CW, Niswender CM. Future Med Chem. 2009;1:501–513. doi: 10.4155/fmc.09.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maj M, Bruno V, Dragic Z, Yamamoto R, Battaglia G, Inderbitzin W, Stoehr N, Stein T, Gasparini F, Vranesic I, Kuhn R, Nicoletti F, Flor PJ. Neuropharmacology. 2003;45:895–906. doi: 10.1016/s0028-3908(03)00271-5. [DOI] [PubMed] [Google Scholar]
- 16.Engers DW, Niswender CM, Weaver CD, Jadhav S, Menon UN, Zamorano R, Conn PJ, Lindsley CW, Hopkins CR. J Med Chem. 2009;52:4115–4118. doi: 10.1021/jm9005065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang JQ, Tueckmantel W, Zhu A, Pellegrino D, Brownell AL. Synapse. 2007;67:951–961. doi: 10.1002/syn.20445. [DOI] [PubMed] [Google Scholar]
- 18.Robichaud AJ, Engers DW, Lindsley CW, Hopkins CR. ACS Chem Neurosci. 2011;2:433–449. doi: 10.1021/cn200043e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ametamey SM, Kessler LJ, Honer M, Wyss MT, Buck A, Hintermann S, Auberson YP, Gasparini F, Schubiger PA. J Nucl Med. 2006;47:698–705. [PubMed] [Google Scholar]
- 20.Yanamoto K, Konno F, Odawara C, Yamasaki T, Kawamura K, Hatori A, Yui J, Wakizaka H, Nengaki N, Takei M, Zhang MR. Nucl Med Biol. 2010;37:615–624. doi: 10.1016/j.nucmedbio.2010.03.002. [DOI] [PubMed] [Google Scholar]
- 21.Hostetler ED, Eng W, Joshi AD, Sanabria-Bohórquez S, Kawamoto H, Ito S, O'Malley S, Krause S, Ryan C, Patel S, Williams M, Riffel K, Suzuki G, Ozaki S, Ohta H, Cook J, Burns HD, Hargreaves R. Synapse. 2011;65:125–135. doi: 10.1002/syn.20826. [DOI] [PubMed] [Google Scholar]
- 22.The functional assay was generously provided by the National Institute of Mental Health's Psychoactive Drug Screening Program, Contract # HHSN-271-2008-00025-C (NIMH PDSP). The PDSP is directed by Bryan L. Roth, MD, PhD at the University of North Carolina at Chapel Hill and Project Officer Jamie Driscol at NIMH, Bethesda, MD, USA.
- 23.clogP and tPSA were obtained by calculation program installed in ChemBioDraw 12.0.
- 24.Ghose AK, Herbertz T, Hudkins RL, Dorsey BD, Mallamo JP. ACS Chem Neurosci. 2012;3:50–68. doi: 10.1021/cn200100h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pajouhesh H, Lenz GR. NeuroRx. 2005;2:541–553. doi: 10.1602/neurorx.2.4.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kil KE, Biegon A, Ding YS, Fischer A, Ferrieri RA, Kim SW, Pareto D, Schueller MJ, Fowler JS. Nucl Med Biol. 2009;36:215–223. doi: 10.1016/j.nucmedbio.2008.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cosford NDP, Tehrani L, Roppe J, Schweiger E, Smith ND, Anderson J, Bristow L, Brodkin J, Jiang X, McDonald I, Rao S, Washburn M, Varney MA. J Med Chem. 2003;46:204–206. doi: 10.1021/jm025570j. [DOI] [PubMed] [Google Scholar]
- 28.Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates – The New Coronal Set. Academic press; 2004. [Google Scholar]