

Summary
The twin-arginine translocation system (Tat) transports folded proteins across the cytoplasmic membrane and is critical to virulence in Salmonella and other pathogens. Experimental and bioinformatic data indicate that 30 proteins are exported via Tat in Salmonella Typhimurium. However, there are no data linking specific Tat substrates with virulence. We inactivated every Tat-exported protein and determined the virulence phenotype of mutant strains. Though a tat mutant is highly attenuated, no single Tat-exported substrate accounts for this virulence phenotype. Rather, the attenuation is due primarily to envelope defects caused by failure to translocate three Tat substrates, the N-acetylmuramoyl-L-alanine amidases, AmiA and AmiC, and the cell division protein, SufI. Strikingly, neither the amiA amiC nor the sufI mutations alone conferred any virulence defect. Although AmiC and SufI have previously been localized to the divisome, the synthetic phenotypes observed are the first to suggest functional overlap. Many Tat substrates are involved in anaerobic respiration, but we show that a mutant completely deficient in anaerobic respiration retains full virulence in both the oral and systemic phases of infection. Similarly, an obligately aerobic mutant is fully virulent. These results suggest that in the classic mouse model of infection, S. Typhimurium is replicating only in aerobic environments.
Introduction
Salmonella enterica serovar Typhimurium is a facultative intracellular pathogen that is a leading cause of foodborne illness. Disease ranges from self-limiting gastroenteritis to acute systemic infection in susceptible hosts (Pegues and Miller, 2010). Following ingestion, S. Typhimurium travels through the small intestine until it reaches the distal ileum, where environmental signals trigger the expression of the type three secretion system (T3SS) encoded on Salmonella pathogenicity island 1 (SPI1). Injection of effector proteins into host epithelial cells triggers invasion by Salmonella of the intestinal mucosa and induction of inflammatory diarrhea (Ellermeier and Slauch, 2007; Winter et al., 2010a; Golubeva et al., 2012). Following invasion, S. Typhimurium is taken up by macrophages and disseminated throughout the body (Carter and Collins, 1974; Jones et al., 1994). Survival in macrophages requires the T3SS encoded on Salmonella pathogenicity island 2 (Hensel et al., 1995; Shea et al., 1996) and several additional virulence regulons (Linehan et al., 2005; Kato and Groisman, 2008; Fass and Groisman, 2009).
The envelope structure of Salmonella is the interface between pathogen and host and serves to protect the bacterium from many of the immune defenses. Assembly of the envelope structure requires the coordinated and delicate interplay of numerous machineries in the cell. The export of fully folded proteins across the cytoplasmic membrane is carried out by the Twin Arginine Transport (Tat) system (Settles et al., 1997; Weiner et al., 1998). Tat substrates have unusually long N-terminal signal sequences that contain a twin-arginine consensus sequence (S/TRRXFLK) (Chaddock et al., 1995; Berks, 1996). In E. coli and Salmonella, the components necessary for a functional Tat translocase are TatA, TatB, and TatC (Palmer et al., 2010; Palmer and Berks, 2012). Substrates associate with complexes of TatBC, which then results in assembly of a translocation channel formed by multiple subunits of TatA (Bolhuis et al., 2001; Alami et al., 2003; Gohlke et al., 2005; Orriss et al., 2007; Lange et al., 2007; Leake et al., 2008). Many anaerobic respiratory chain proteins contain complex centers or cofactors, such as iron-sulfur clusters or molybdopterin, and must fold and assemble in the cytoplasm before crossing the inner membrane (Berks, 1996; Rodrigue et al., 1999; Berks et al., 2003). Hence, anaerobic respiratory chain proteins are a sizable fraction of predicted Tat-exported proteins in S. Typhimurium (Dilks et al., 2003; Palmer et al., 2010). Additional proteins exported by Tat include proteins involved in cell septation. N-acetylmuramyl-L-alanine amidases are enzymes that remove cross-links in peptidoglycan and are involved in separating daughter cells. Two of the three enzymes of this type, AmiA and AmiC, are Tat substrates (Heidrich et al., 2001; Bernhardt and de Boer, 2003). The Tat-exported protein SufI (FtsP) is thought to play a structural role in assembly of the divisome (Samaluru et al., 2007; Tarry et al., 2009; Peters et al., 2011)
The physiological consequences of mutations in the Tat transport system have been most extensively studied in E. coli. Since the Tat system transports several anaerobic respiratory chain proteins, tat mutants are unable to utilize certain electron acceptors (Dilks et al., 2003; Lee et al., 2006). Also, tat mutants have impaired motility, septation defects, and are sensitive to detergents and bile (Stanley et al., 2001; Ize et al., 2003; Reynolds et al., 2011). The Tat system is conserved in numerous pathogens (Yen et al., 2002; Dilks et al., 2003), but the role of Tat in virulence has been studied only in a limited number of organisms. In some cases, the contribution of Tat export to virulence is at least partially direct. For example, the phospholipase toxins in Pseudomonas aeruginosa (Voulhoux et al., 2001; Ochsner et al., 2002) and Pseudomonas syringae (Bronstein et al., 2005) rely on the Tat system for transport to the periplasm, where these proteins can interact with the type II secretion apparatus. In other pathogenic organisms in which tat mutations attenuate, the role of this system in virulence is likely indirect. In enterohemorrhagic E. coli, loss of the Tat system led to a decrease in Shiga toxin 1 secretion, despite the fact that the toxin proteins do not have Tat-dependent signal sequences (Pradel et al., 2003). The phospholipase A of Legionella pneumophila is Tat-dependent, but this does not explain the virulence defect of Legionella tat mutants (Rossier and Cianciotto, 2005). In addition to lacking phospholipase activity, Pseudomonas syringae pv. tomato DC3000 tatC mutants display a slight decrease in type III secretion (Bronstein et al., 2005). A tatC mutant of Yersinia pseudotuberculosis is non-motile and is highly attenuated in mice when administered both orally and intraperitoneally, but the specific defect that decreases virulence was not determined. Type III secretion was shown to be unaffected in this mutant, and only a slight sensitivity to low pH was observed (Lavander et al., 2006). A tat mutant of the plant pathogen Agrobacterium tumefaciens was shown to be highly attenuated, but again, no molecular basis for this loss of virulence was described (Ding and Christie, 2003). Similarly, when the effect of tatBC mutations on Salmonella Enteriditis virulence was examined, it was found that this organism had many of the physiological defects observed for E. coli tat mutants. These mutants were also shown to be impaired for survival in polarized epithelial cells and in chickens (Mickael et al., 2010). The Tat system of S. Typhimurium has also been shown to be important for virulence in mice (Reynolds et al., 2011). However, the mechanism by which loss of the Tat export system impaired virulence in Salmonella serovars has not been determined.
In this study, we show that the virulence defect of a tat mutant of S. Typhimurium is due primarily to envelope defects associated with failure to translocate three Tat substrates: AmiA, AmiC, and SufI. Loss of all three is required to see the effect and the triple mutant recapitulates many of the envelope defects of the tat mutant. Although tat mutants show decreased type III secretion in vitro, we provide evidence that defects conferred by loss of Tat during oral infection are apparently independent of SPI1, while SPI2-dependent secretion must be partially functional during systemic infection in the tat mutant. In contrast, the Tat-exported proteins involved in respiration are not required for infection. Indeed, we show that a mutant completely deficient in anaerobic respiration, moaDE nrfA frdA, retains full virulence in both the oral and systemic phases of infection in BALB/c mice. Similarly, an obligate aerobic mutant, nrdDG, retains full virulence in both the oral and systemic phases of infection, while strains containing mutations in the aerobic respiratory chain components cytochrome oxidases bo3 and bd3, encoded by cyoABCD and cydAB, are attenuated. These results suggest that in the normal course of infection in the classic mouse model of infection, S. Typhimurium is replicating only in aerobic environments.
Results
A small subset of Tat-exported proteins contribute to virulence of S. Typhimurium
It has been shown that tatC mutants of S. Typhimurium are attenuated in both oral and i.p. competition assays and show impaired survival in J774 macrophages (Reynolds et al., 2011). We confirmed that a tatC deletion mutant is >100-fold attenuated after either oral or i.p. inoculation in BALB/c mice (Table 1), consistent with the previous results (Reynolds et al., 2011). The attenuation of a tat mutant is not surprising since loss of this system should lead to mislocalization of numerous envelope proteins (Dilks et al., 2003; Palmer et al., 2010). We wanted to more precisely define the virulence defect(s) conferred by the tat mutation.
Table 1.
A tatC mutant is attenuated in mice.
| Genotype a | Route of Inoculation | Organ b | Median CI c | Number of mice | P value d |
|---|---|---|---|---|---|
| tatC | oral | small intestine | 0.0065 | 11 | <0.0005 |
| oral | spleen | 0.0077 | 16 | <0.0005 | |
| i.p. | spleen | 0.0045 | 5 | <0.0005 |
Strains used were JS135 and JS1195 for oral infections, and 14028 and JS1194 for i.p. infections.
The entire small intestine was dissected and homogenized.
The competitive index (CI) was calculated as described in the experimental procedures.
The student’s t test was used to compare the output and the inoculums.
Experimental and bioinformatic data reveal 30 Tat-exported proteins in S. Typhimurium (Berks et al., 2000; Dilks et al., 2003) (Table S1). Sixteen of these proteins are involved in some aspect of anaerobic respiration, most of which either have a molybdopterin cofactor or are in a complex that requires a molybdopterin cofactor for function. Therefore, we took two approaches to inactivate these various Tat substrates. For the molybdopterin-independent proteins, we constructed deletions of the individual genes. In the case of the molybdopterin-dependent proteins, we inactivated molybdopterin biosynthesis, along with the additional genes required for anaerobic respiration, as described below.
Strains containing single deletion mutations in individual Tat substrates or multiple mutations in genes with similar functions were competed against the isogenic wild type strain after i.p. infection in BALB/c mice. As shown in Table 2, only the triple hydrogenase mutant and a mutant lacking the putative outer membrane protein YcbK were significantly attenuated relative to the wild type strain, 5 fold and 3 fold, respectively. The former result is consistent with that of Maier et al. (Maier et al., 2004), who previously showed that the hydrogenases were important for Salmonella virulence. However, in neither mutant is the virulence defect severe enough to explain the phenotype conferred by the tatC deletion (>200 fold attenuated i.p.).
Table 2.
Virulence characteristics of strains with mutations in specific Tat substrates.
| Relevant genotype a | Function | Median CI b | Number of mice | P value c |
|---|---|---|---|---|
| hyaAB hybABC hydBC | Hydrogenases | 0.20 | 9 | 0.039 |
| ycbK | Putative outer membrane protein; Peptidase M15 family | 0.32 | 4 | 0.018 |
| sufI | Suppressor of ftsI; cell division | 0.71 | 8 | NS |
| STM0084 | Putative sulfatase | 0.43 | 5 | NS |
| cueO | Multicopper oxidase | 0.61 | 5 | NS |
| PSLT46 | Putative carbonic anhydrase | 0.73 | 3 | NS |
| amiA amiC | N-acetylmuramoyl-L- alanine amidases | 1.09 | 11 | NS |
| fhuD | Fe3+ Hydroxamate siderophore transporter | 1.72 | 3 | NS |
| wcaM | Colanic acid biosynthesis | 2.23 | 5 | NS |
The listed mutant was competed against the isogenic wild type strain 14028 with an i.p. inoculation. Bacteria were recovered from spleens. The strains used were 14028, JS1196, JS1197, JS1198, JS1199, JS2000, JS2001, JS2002, JS2003, and JS2004.
The competitive index (CI) was calculated as described in the experimental procedures.
The student’s t test was used to compare the output and the inoculums. NS, not significant.
Salmonella does not grow in anaerobic environments of BALB/c mice
The Tat system is required for export of numerous cofactor-containing anaerobic respiratory chain proteins (Table S1), and E. coli tat mutants are unable to grow anaerobically on non-fermentable carbon sources in the presence of the terminal electron acceptors TMAO or DMSO (Sargent et al., 1998; Weiner et al., 1998). In addition to these compounds, Salmonella Typhimurium is capable of utilizing thiosulfate and tetrathionate for anaerobic respiration (Unden and Dunnwald, 2008). Enzymes involved in reduction of thiosulfate, TMAO, DMSO, tetrathionate and nitrate utilize a molybdenum-containing cofactor, and they are therefore inactive in a strain that does not produce molybdopterin (Amy, 1981; McNicholas et al., 1998). By deleting genes that encode nitrite reductase and fumarate reductase, along with genes in the molybdopterin biosynthesis pathway, we constructed a mutant that cannot perform anaerobic respiration, moaDE nrfA frdA. This mutant should, therefore, be defective in all 15 Tat-dependent protein complexes listed in the second part of Table S1 as well as the Nrf nitrite reductase complex. The inability of this strain to grow anaerobically on glycerol with several of the above listed terminal electron acceptors was confirmed (Figure S1). Note that the tatC mutant is actually leaky and did show some growth on DMSO and nitrate, whereas the moaDE nrfA frdA mutant did not grow under these conditions. The tatC mutant, as expected, was able to utilize fumarate as a terminal electron acceptor when grown on glycerol, in contrast to the triple mutant.
To examine the role of anaerobic respiration during infection, competitive virulence assays were performed in BALB/c mice both orally and intraperitoneally. The mutant deficient in anaerobic respiration, moaDE nrfA frdA, was fully virulent and recovered at levels equal to the wild type from either the small intestine or the spleen (Table 3). This suggests that S. Typhimurium is not utilizing anaerobic respiration during growth in the host in this classic mouse model of infection.
Table 3.
Competition assays with mutants affected in anaerobic growth.
| Relevant genotype a | Route of Inoculation | Organb | Median CI c | Number of mice | P value d |
|---|---|---|---|---|---|
| moaDE nrfA frdA | oral | spleen | 1.30 | 5 | NS |
| small intestine | 4.07 | 5 | NS | ||
| i.p. | spleen | 1.96 | 10 | NS | |
| fnr | oral | spleen | 1.29 | 11 | NS |
| small intestine | 0.54 | 11 | 0.029 | ||
| i.p. | spleen | 1.22 | 8 | NS | |
| nrdDG | oral | spleen | 2.65 | 5 | 0.003 |
| small intestine | 0.85 | 6 | NS | ||
| cecum | 1.24 | 6 | NS | ||
| large intestine | 0.75 | 7 | NS | ||
| i.p. | spleen | 1.28 | 9 | NS |
The indicated mutant was competed against the isogenic wild type strain. Strains used were JS135, JS2006, JS2008, and JS2010 for oral infections, and 14028, JS2005, JS2007, and JS2009 for i.p. infections.,.
In each case, the entire tissue was dissected and homogenized.
The competitive index (CI) was calculated as described in the experimental procedures.
The student’s t test was used to compare the output and the inoculums. NS, not significant.
To further examine the physiology of Salmonella in the host, we asked if other mutations that affected growth under anaerobic conditions were required for virulence. FNR is a global regulator that controls the transition to anaerobic metabolism (Kang et al., 2005; Green et al., 2009). An fnr mutant was constructed, and growth of this strain relative to the wild type was tested in laboratory medium using competition assays. The fnr mutant grew well under aerobic in vitro conditions, but a significant growth defect was observed under anaerobic conditions in vitro competition assays performed in LB (Table S2). When similar competitions were performed in BALB/c mice intraperitoneally, the fnr mutant was fully virulent. An oral infection showed loss of FNR did confer a slight virulence defect in the small intestine (Table 3).
To further test if Salmonella is growing in an anaerobic environment in the intestine, an obligately aerobic mutant was constructed. Ribonucleotide reductase is required for the last step of deoxynucleotide biosynthesis. The enzyme encoded by nrdAB functions only aerobically, leaving the enzyme encoded by nrdDG to function anaerobically (Fontecave et al., 1989; Garriga et al., 1996). An nrdDG mutant has no in vitro phenotype aerobically, but it does not grow anaerobically in rich medium (Table S2). In both orally and intraperitoneally administered competitive virulence assays, the nrdDG mutant was recovered equally well as wild type, even in the large intestine, the lumen of which is anaerobic (Table 3). Purine or pyrimidine auxotrophs of Salmonella cannot survive in the host, indicating that de novo synthesis is required for survival (Fields et al., 1986; Mahan et al., 1993). Therefore, since the nrdDG mutant does not have a virulence defect in the host, Salmonella must be growing solely in aerobic environments in BALB/c mice; bacteria not associated with aerobic host tissue are not replicating.
Aerobic respiratory chain components are required for full virulence
The above data indicate that Salmonella is growing aerobically in the murine host. If so, loss of components of the aerobic respiratory chain would negatively impact virulence. We tested this hypothesis in both the intestine and during the systemic phase of infection. NADH dehydrogenase I is encoded by nuoA-N, and NADH dehydrogenase II is encoded by ndh (Unden and Dunnwald, 2008). Both are used under aerobic conditions, but NADH dehydrogenase II predominates in fully aerobic conditions (Spiro et al., 1989; Spiro and Guest, 1990). Table S2 shows that both a nuoA-N mutant and an ndh mutant have an in vitro growth defect compared to wild type when grown aerobically, but not anaerobically in LB medium. We then asked what the effects of these mutations would be in the host. The data in Table 4 show that, in an orally administered competitive virulence assay, a nuoA-N mutant is attenuated 10-fold compared to wild type in the small intestine, while an ndh mutant is 4-fold attenuated. This suggests that both NADH dehydrogenase I and II are important for growth in the small intestine.
Table 4.
Competition assays with mutants affected in aerobic respiration.
| Relevant genotype a | Method of Inoculation | Organ | Median CI b | Number of mice | P value c |
|---|---|---|---|---|---|
| nuoA-N | oral | spleen | 0.11 | 7 | 0.010 |
| small intestine | 0.093 | 6 | <0.0005 | ||
| i.p. | spleen | 0.008 | 3 | <0.0005 | |
| ndh | oral | spleen | 0.15 | 6 | NS |
| small intestine | 0.25 | 4 | NS | ||
| i.p. | spleen | 1.29 | 7 | NS | |
| cyoABCD | oral | spleen | 0.15 | 4 | 0.042 |
| small intestine | 0.74 | 7 | NS | ||
| i.p. | spleen | 0.097 | 4 | <0.0005 | |
| cydAB | oral | spleen | 0.0009 | 7 | 0.023 |
| small intestine | 0.043 | 4 | <0.0005 | ||
| i.p. | spleen | 0.050 | 5 | <0.0005 |
The indicated mutant was competed against the isogenic wild type strain. The strains used were JS135, JS2013, JS2014, JS2016, JS2018 in oral competition and 14028, JS2011, JS2012, JS2015, and JS2017 in i.p. competitions.
The competitive index (CI) was calculated as described in the experimental procedures.
The student’s t test was used to compare the output and the inoculums. NS, not significant.
During aerobic respiration, the quinol oxidases transfer electrons to oxygen. Quinol oxidase bo3 has a lower affinity for oxygen and is maximally expressed under aerobic conditions (Rice and Hempfling, 1978; D’Mello et al., 1995), while quinol oxidase bd3 has a higher affinity for oxygen and is maximally expressed under microaerophilic conditions (D’Mello et al., 1996; Tseng et al., 1996). We deleted the cydAB genes encoding bd3 or the cyoABCD genes encoding bo3 and determined the effects in vitro and in vivo. Both the cyoABCD and cydAB mutants displayed growth defects in aerobic in vitro competitions, but not in anaerobic competitions (Table S2), consistent with the role these enzymes play in aerobic respiration. In in vivo competitions in mice, the cyoABCD mutant did not have an appreciable virulence defect in the intestine when administered orally, but it did display a defect systemically (Table 4). The cydAB mutant was significantly attenuated in the small intestine. This is consistent with the hypothesis that Salmonella is replicating in an aerobic environment in the small intestine, albeit at an oxygen tension where quinol oxidase bd3 is critical (He et al., 1999).
Loss of AmiA, AmiC, and SufI accounts for most of the virulence defect observed in tat mutants
The above results indicate that loss of neither the entire set of molybdopterin-dependent Tat substrates nor any single Tat-exported molybdopterin-independent protein accounts for the role of Tat during infection. It seemed likely that multiple factors in the latter class are contributing to the virulence defect observed in a tatC mutant. Therefore, we began constructing strains with multiple mutations based on perceived common function. Loss of AmiA and AmiC, two N-acetylmuramyl-L-alanine amidases involved in septation, has been shown in E. coli to cause pleiotropic cell envelope defects (Stanley et al., 2001; Ize et al., 2003). It is also know that amiA amiC mutants of Salmonella show increased sensitivity to antimicrobial peptides (Weatherspoon-Griffin et al., 2011). SufI (also called FtsP) is a cell-division protein that also localizes to the septal ring (Reddy, 2007; Tarry et al., 2009). Given the fact that all three of these proteins were implicated in cell septation, we constructed an amiA amiC sufI triple mutant and tested its phenotypes in vivo and in vitro.
We tested the relative virulence of an amiA amiC sufI mutant in mice. As shown above, the amiA amiC double mutant and the sufI single mutant competed evenly with wild type in i.p. competition assays (Table 2). In contrast, the triple mutant amiA amiC sufI was about 60-fold attenuated compared to wild type in an i.p. competition assay (Table 5). This is a synthetic genetic interaction, suggesting that these three gene products are acting in a common process. Indeed, this phenotype is approaching that observed for a tatC mutant, which is approximately 220-fold down compared to wild type after i.p. infection. To confirm that the majority of the tatC phenotype is due to mislocalization of AmiA, AmiC, and SufI, we asked what effect a tatC mutation would have in the amiA amiC sufI background. In the i.p. competition of amiA amiC sufI tatC versus amiA amiC sufI, the quadruple mutant is 4-fold down compared to the triple mutant. After oral infection, the two strains competed evenly in the intestine, but the amiA amiC sufI tatC was still slightly attenuated compared to the triple mutant in the spleen. These data indicate that most of the tatC virulence phenotype in systemic infection and all of the virulence defect in the intestine can be attributed to loss of AmiA, AmiC, and SufI.
Table 5.
Competition assays with mutants impaired in septation.
| Strain A a | Strain B a | Route of Inoculation | Organ | Median CI b | Number of Mice | P value c |
|---|---|---|---|---|---|---|
| amiA amiC sufI | WT | i.p. | spleen | 0.016 | 5 | 0.0029 |
| amiA amiC sufI tatC | amiA amiC sufI | i.p. | spleen | 0.26 | 9 | 0.036 |
| oral | spleen | 0.32 | 9 | 0.033 | ||
| small intestine | 1.30 | 9 | NS | |||
| amiA amiC sufI +amiA+ | WT | i.p. | spleen | 1.49 | 8 | NS |
| amiA amiC sufI +amiC+ | WT | i.p. | spleen | 0.36 | 8 | 0.034 |
| amiA amiC sufI +sufI+ | WT | i.p. | spleen | 0.47 | 8 | NS |
| +amiA+ | WT | i.p. | spleen | 1.09 | 5 | NS |
| +amiC+ | WT | i.p. | spleen | 1.02 | 10 | NS |
| +sufI+ | WT | i.p. | spleen | 0.96 | 5 | NS |
The strains used were 14028, JS2019, JS2020, JS2034, JS2035, JS2036, JS2031, JS2032, and JS2033 for i.p. infections, and JS2019 and JS2020 for oral infections.
The competitive index (CI) was calculated as described in the experimental procedures.
The student’s t test was used to compare the output and the inoculums.
To further confirm the above phenotypes, we complemented the triple mutant with each of the amiA, amiC, and sufI genes. Each gene was PCR amplified along with downstream Kan resistance cassette and integrated downstream of the purA gene. The data in Table 5 show that these constructs conferred no phenotype in the animal in an otherwise wild type background, indicating that neither gene dosage nor insertion downstream of purA had any effect on virulence. Each of these constructs was moved into the amiA amiC sufI mutant and the complemented strains were competed against wild type. Interestingly, amiA and sufI complemented the virulence defect of the triple mutant, whereas amiC partially complemented; the amiC complemented strain was still statistically attenuated compared to wild type.
The results above show that the phenotype conferred by loss of Tat can be largely explained by mislocalization of the AmiA, AmiC and SufI proteins. However, it is formally possible that in the absence of AmiA, AmiC and SufI, the Tat export system is itself nonfunctional and, thus, the amiA amiC sufI triple mutant is defective in proper localization of additional proteins. To test this hypothesis, we directly assayed for Tat-dependent export using a GFP construct encoding the TorA Tat signal sequence on the N-terminus. In the wild type background, GFP is localized to the periplasm, apparently concentrated at the cell poles (Figure S2). This is consistent with previous data (Berthelmann and Bruser 2004). In the tatC deletion mutant, this localization is lost and the GFP is found diffusely in the cytoplasm. The amiA amiC sufI triple mutant resembles the wild type. This indicates that the Tat export system remains functional in this background, strongly suggesting that the remaining Tat-dependent proteins are appropriately exported in the triple mutant background.
Loss of Tat confers a number of in vitro phenotypes, including impaired septation, decreased motility, and sensitivity to detergents (Stanley et al., 2001; Ize et al., 2003; Reynolds et al., 2011). We therefore compared the relative phenotypes of the tatC mutant with the amiA amiC sufI triple mutant under various conditions. As has been observed previously in S. Enteriditis (Mickael et al., 2010) and E. coli (Stanley et al., 2001), tat mutants of S. Typhimurium had an increased percentage of cells that were apparently undivided when grown in low salt rich medium (Figure 1); this phenotype was exacerbated at higher temperatures (not shown). The amiA amiC double mutant showed a slight but significant septation defect compared to the wild type. The sufI mutant showed a phenotype indistinguishable from the tat mutant, whereas the amiA amiC sufI triple mutant showed more pronounced phenotype, even compared to the tatC mutant.
Figure 1.

Microscopic analysis of strains following growth in no salt LB. Strains were grown for 6 h. Cells were washed before fixation and Gram staining and observation by light microscopy at a magnification of ×600. Scale bar=10 μm. At least 250 cells in at least 5 random non-overlapping microscopic fields were counted for calculation of an average number of bacilli in a chain and % single cells. Lines designate statistical comparisons of Avg cells/chain using a Student’s T test. *, p<0.05; **, p<0.005; NS, Not significant. Strains used were 14028, JS1194, JS2002, JS1198, and JS2019.
Motility is also affected in tatC mutants and less flagellin was detected on the surface of tatC cells (Reynolds et al., 2011). Figure 2 shows the relative motility of the tat, amiA amiC, sufI, and amiA amiC sufI mutants. The tat mutants were partially defective in this motility assay. Both the amiA amiC mutant and sufI mutant also showed a motility defect, but not as severe as the tat mutant. Interestingly, combining the amiA amiC sufI mutations did not seem to confer a more severe phenotype. Production of flagella is down-regulated during systemic infection and dispensable for S. Typhimurium virulence in the mouse model, so this decreased motility is unlikely to explain the i.p. virulence defect of tatC mutants (Schmitt et al., 2001).
Figure 2.
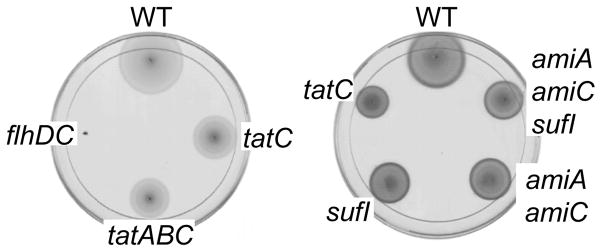
Motility in LB with 0.3% agar. The plates were grown at 37° for 3 hours. The strains tested are clockwise: (Left plate)14028, JS1194, JS1185, and JS557; (Right plate) 14028, JS2019, JS2002, JS1198, and JS1194.
We also tested our tatC mutant for sensitivity to detergents using in vitro competition assays. Both the tatC mutant and the amiA amiC sufI mutant grew equally with the wild type strain in the absence of detergents (Figure 3A). Thus, the virulence phenotypes cannot be explained by simple growth defects. However, our data show that both the tatC mutant and amiA amiC sufI triple mutant were equally sensitive to SDS compared to wild type; approximately 500-fold more wild type cells were recovered in competition assays performed in LB containing 5% SDS. Addition of the amiA, aimC, or sufI genes into the triple mutant partially complemented the SDS phenotype, indicating that all three gene products are contributing to the SDS resistance. As has been shown previously, we observed that tat mutants are also sensitive to bile (Reynolds et al., 2011). When grown in aerated LB supplemented with 1% or 2% bile salts, fewer tatC mutant cells are recovered compared to wild type after 18 hours growth. Again, the amiA amiC sufI triple mutant was indistinguishable from the tatC mutant in this assay (Figure 3B). But interestingly, while complementation with amiC completely rescued the bile sensitivity phenotype, addition of neither amiA nor sufI had any significant effect. Thus, the sensitivity of the tatC mutant to SDS can be explained by mislocalization of AmiA, AmiC and SufI, whereas the sensitivity to bile seems to be solely due to loss of AmiC. These data suggest first that SDS and bile act via different mechanisms to damage cells, and that, second, AmiA and AmiC are not simply redundant.
Figure 3.

SDS and bile sensitivity of mutants in aerobic LB. Mixed cultures of the indicated mutant and isogenic wild type strain were grown for 18 hours shaking in the presence of SDS (N=2) or bile (N=4) as indicated. Competitive indices is calculated as % mutant output/ % mutant input. Strain used were 14028, JS1194, JS2019, JS2034, JS2035, and JS2036.
Loss of Tat affects type III secretion but this is independent of virulence
In the plant pathogen Pseudomonas syringae, a tatC mutation conferred a 30% decrease in translocation of a type III secreted effector (Bronstein et al., 2005). Since type III secretion is important for S. Typhimurium survival in the host, both in the intestine and systemically (Mills et al., 1995; Hensel et al., 1995; Shea et al., 1996), we asked what effect a tatC mutation has on type III secretion and whether the amiA amiC sufI mutations recapitulate these phenotypes. First, we determined the effect of a tatC deletion on hilA transcription. The level of expression of the SPI1 type III secretion system, involved in invasion in the intestine, is directly dependent on the level of hilA expression (Ellermeier et al., 2005), and the SPI1 regulatory system responds to a number of envelope defects (Golubeva et al., 2012). The data in Figure 4A show that deletion of tatC decreased hilA expression nearly 4-fold, and the triple amiA amiC sufI mutations decreased hilA expression approximately 2-fold. This level of effect on hilA expression might be enough to impact type III secretion and virulence, since deletion of fliZ, a regulator of SPI1 type III secretion, has a 4-fold effect on hilA expression and the mutant is approximately 60-fold down in oral competition assays in mice (Chubiz et al., 2010).
Figure 4.

Role of Tat in type III secretion systems. Loss of TatC or the exported substrates AmiA AmiC and SufI decreases expression of a hilA-lac fusion (A) and an ssaG-lac fusion (B). β-galactosidase activity units are defined as (μmol of ONP formed/min) × 106/ (OD600 × ml of cell suspension) and are reported as mean ± standard deviation where n=4. Loss of TatC affects translocation of the SPI2 effector SspH2 (C). Strains were grown in SPI2 inducing conditions and then assayed as described in Materials and Methods. The LacZ-CyaA fusion is not competent for type III secretion. Type III secretion is also non-functional in a dsbA mutant background (Ellermeier and Slauch, 2004; Lin et al., 2008). Strains uses were JS749, JS2023, JS2024, JS2025, JS2026, JS2027, 14028 pCE39, 14028 pSG161, JS1194 pSG161, and JS362.
To test directly if the virulence defect of a tatC mutant can be explained by loss of SPI1 type III secretion, we examined the virulence phenotype of a tatC mutation in a strain deleted for the SPI1 locus. If the virulence defect conferred by the tatC mutation is due to loss of SPI1 type III secretion, then it should not confer any additional phenotype in the SPI1 deletion background. As a control, we tested the phenotype conferred by the SPI1 locus deletion after oral infection and recovery of bacteria from the small intestine. The ΔSPI1 mutant was 67-fold attenuated in this assay (Table 6). Deletion of tatC in this background conferred an additional 63-fold attenuation. This is not significantly different than the phenotype conferred by loss of a functional Tat export system in the wild type background (Table 1). Importantly, introduction of the amiA amiC sufI deletions into the ΔSPI1 background conferred a phenotype statistically indistinguishable from that conferred by the tatC mutation. Thus, the virulence defects conferred by the tatC or the amiA amiC sufI mutations are largely independent of any effect on SPI1 type III secretion.
Table 6.
Competition assays with SPI1 mutants.
| Strain A a | Strain B a | Median CI b | Number of Mice | P value c |
|---|---|---|---|---|
| Δspi1 | wt | 0.015 | 7 | <0.005 |
| ΔtatC Δspi1 | Δspi1 | 0.016 | 8 | <0.005 |
| ΔamiAC ΔsufI Δspi1 | Δspi1 | 0.0065 | 5 | <0.005 |
The strains used were JS135, JS2028, JS2029, and JS2030. Mice were infected orally with 109 bacteria/mouse. Bacteria were recovered from the distal portion of the small intestine.
The competitive index (CI) was calculated as described in the experimental procedures.
The student’s t test was used to compare the output and the inoculums. NS, not significant.
We also examined the effect of a tatC deletion on SPI2 type III secretion, necessary for survival in macrophages, by measuring the level of ssaG transcription. SsaG encodes a structural protein in the SPI2 type III secretion apparatus and has been used to monitor expression of SPI2 (Walthers et al., 2007). Figure 4B shows that deletion of tatC decreased ssaG expression nearly 5-fold, and the triple amiA amiC sufI mutations decreased ssaG expression approximately 2-fold. This is similar to the effect seen on hilA expression.
We were unable to perform in vivo tests of epistasis with the SPI2 mutant because the strain is severely attenuated (> 100,000-fold in an i.p. competition assay). Therefore, we directly measured secretion of a tagged effector protein in vitro. We used a fusion of an N-terminal portion of SspH2, a known SPI2 type III secreted effector (Miao and Miller, 2000), to the catalytic domain of the adenylate cyclase toxin of Bordetella perusssis, CyaA, which converts ATP to cAMP in the presence of calmodulin in host cells (Hanski, 1989). cAMP is only produced if the fusion protein is translocated into host cells. We infected J774 macrophages with strains expressing this fusion and measured the level of cAMP after 6 hours. Figure 4C shows that translocation of the SspH2-CyaA fusion protein is decreased in the tatC background, but it is not abolished, since more cAMP is detected in this strain than in the negative controls. Importantly, however, SPI2 type III secretion must be largely functional in the tat mutant in vivo because the level of attenuation in the tat mutant is not nearly as severe as in a SPI2 mutant.
Discussion
The Twin Arginine Transport system (Tat) translocates folded proteins across the cytoplasmic membrane (Settles et al., 1997; Weiner et al., 1998). The Tat system has been shown to be important for virulence in several organisms, and where a molecular mechanism has been offered, attenuation has been attributed to mislocalization of a specific secreted toxin (Voulhoux et al., 2001; Ochsner et al., 2002; Pradel et al., 2003; Bronstein et al., 2005; Rossier and Cianciotto, 2005). Since Salmonella is not known to secrete any Tat-dependent toxins, the specific role of the Tat pathway in virulence was less obvious.
To more precisely define the nature of attenuation in a Salmonella tat mutant, we inactivated each of the genes encoding Tat-exported proteins and determined the virulence phenotype of each mutant. No single mutation caused the virulence phenotype equivalent to the tat deletion. Indeed, only loss of the outer membrane protein YcbK caused any measurable (2-fold) virulence defect. This putative outer membrane protein is a peptidase in the M15 family. As previously shown (Maier et al., 2004), deletion of the genes encoding three periplasmic hydrogenases hya, hyb, and hyd, also conferred a significant virulence defect, but only 5-fold compared to the 220-fold attenuation conferred by the tat mutation; deletion of the individual hydrogenase genes does not attenuate Salmonella (Maier et al., 2004).
Deletion of the two N-acetylmuramyl-L-alanine amidases, amiA and amiC, also did not significantly attenuate virulence. However, combining these mutations with deletion of sufI (also called ftsP), encoding a divisome protein, conferred a significant virulence defect approaching that observed in the tat mutant. Indeed, the tat mutation conferred only a 4- fold defect in the amiA, amiC, sufI background during systemic infection. This remaining attenuation is easily explained by the loss of the hydrogenases and YcbK.
AmiA, AmiB, and AmiC are periplasmic enzymes that cleave the peptide from the N-acetylmuramyl moiety of the glycan chain of peptidoglycan, thereby breaking the crosslink between glycan strands (Heidrich et al., 2001; Typas et al., 2012). These enzymes have been implicated primarily in cell septation and separation. It is not clear why AmiA and AmiC require Tat for export, whereas AmiB does not (Bernhardt and de Boer, 2003; Ize et al., 2003). In some cases, they would appear to be redundant. In E. coli, significant phenotypes, including biochemical changes in peptidoglycan structure, are observed in double or triple mutants. Moreover, over-expression of AmiB will suppress the SDS sensitivity phenotype of an amiA amiC double mutant in E. coli (Ize et al., 2003). However, AmiC and AmiB are localized to the septal ring in dividing cells, whereas AmiA is distributed throughout the periplasm (Bernhardt and de Boer, 2003; Peters et al., 2011), although AmiA activity requires the divisome associated FtsEX (Yang et al., 2011)(see below). Our results with complemented strains suggest that both AmiA and AmiC contribute to SDS resistance, whereas only AmiC can confer resistance to bile.
In E. coli, tat mutants, amiA amiC mutants, and sufI mutants, all show septation defects and elongated chains of cells (Stanley et al., 2001; Heidrich et al., 2001; Samaluru et al., 2007). Cultures of Salmonella Enteritidis tat mutants similarly showed a significant fraction of elongated cells (Mickael et al., 2010). We also observed septation defects in S. Typhimurium strain 14028 mutants lacking Tat, AmiA and AmiC, or AmiA, AmiC and SufI. The amiA amiC sufI triple mutant had a slightly exacerbated phenotype that was significantly different than the tatC mutant, suggesting that, similar to what we observed for anaerobic respiration, the tat mutant could be slightly leaky. Perhaps a small fraction of Tat-dependent proteins can be exported via the Sec system. This could be particularly true for these divisome proteins that do not have any complex centers that need assembly in the cytoplasm. However, the septation defects do not strictly correlate with attenuation, suggesting that either this phenotype is not manifest during infection in the amiA amiC or sufI mutants or that impaired septation per se does not have a significant impact in the host.
Loss of Tat or AmiA and AmiC in E. coli or Salmonella affects membrane integrity and confers sensitivity to SDS, bile, certain antimicrobial peptides, and vancomycin (Stanley et al., 2001; Ize et al., 2003; Weatherspoon-Griffin et al., 2011). Indeed, the amiA amiC sufI mutant shows equal sensitivity to SDS and bile as the tat mutant (Fig 3). It is not mechanistically clear why the inability to break peptidoglycan crosslinks should affect outer membrane permeability. Interestingly, amiA and amiC are transcriptionally induced above the basal level of expression by CpxR, apparently in response to periplasmic stress (Weatherspoon-Griffin et al., 2011), further connecting these enzymes with overall envelope integrity. One can easily imagine that this membrane permeability and increased sensitivity to various compounds could explain attenuation in the animal. However, if true, it would seem likely that the mutant is sensitive to different compounds in the various niches in the animal, given that the mutants are attenuated both orally and i.p. Perhaps the triple mutant has additional envelope defects directly or indirectly caused by loss of the divisome proteins. Although we show that in vitro Type III secretion (T3S) could be affected in the tat mutant and, to a lesser extent, the triple mutant, loss of SPI1 T3S does not account for the oral defect conferred by loss of Tat, or AmiA, AmiC, and SufI (Table 6), while SPI2 T3S must remain at least partially functional in the tat mutant during systemic infection.
SufI (FtsP), like AmiC, is localized to the septal ring (Tarry et al., 2009). The protein is structurally similar to the multicopper oxidase CueO, another Tat-exported protein, but SufI does not bind copper (Tarry et al., 2009). Rather, it is proposed to play a structural role in the divisome. Localization of SufI and AmiC requires FtsN, and, as such, both proteins are thought to be assembled into the divisome complex only during cell constriction to restrict their activities to cell separation and prevent cell rupture that could occur if the amidases acted at inappropriate times or places (Peters et al., 2011).
SufI was identified as an overproduction suppressor of an ftsI mutation (Samaluru et al., 2007). FtsI is a transpeptidase that is also localized to the divisome (Weiss et al., 1999). Overproduction of SufI also suppresses loss of FtsEX and mutations in sufI and ftsEX or sufI and envC are synthetically lethal (Samaluru et al., 2007). Yang et al. (Yang et al., 2011) have provided evidence that the ABC-transporter like complex, FtsEX, controls EnvC via ATP-dependent conformational changes. EnvC then activates both AmiA and AmiB, the latter being localized to the divisome (Peters et al., 2011). Thus, although the exact role of SufI in the divisome complex is unknown, there are genetic and structural ties between SufI and the amidases. Our data provide an additional functional connection between SufI, AmiA and AmiC in that loss of the three, although not synthetically lethal in vitro, does confer a synthetic virulence defect. But what is the nature of this synthetic defect? Perhaps AmiB or EnvC are not localized or functioning appropriately in the sufI mutant. This defect would be exacerbated by loss of AmiA and AmiC.
A large fraction of Tat-exported proteins are involved in anaerobic respiration. To test the role of these systems during infection, we created a strain that is incapable of using any alternative electron acceptor (moaDE nrfA frdA). Interestingly, complete loss of anaerobic respiration had no effect on the virulence of S. Typhimurium in the classic BALB/c mouse model, either in the intestine or during systemic infection. As a further test, we deleted the genes encoding the anaerobic ribonucleotide reductase, nrdDG (Fontecave et al., 1989; Garriga et al., 1996), creating an obligately aerobic mutant. This mutant was also fully virulent, even in the intestine after oral infection. In contrast, mutants lacking components of the aerobic respiratory chain are significantly attenuated. These data are consistent with previous reports that the full TCA cycle, not the reductive path, is needed for virulence (Yimga et al., 2006). Taken together, our results strongly suggest that Salmonella replicates only in aerobic tissues in the animal, both during systemic infection and in the intestine. Although there are data suggesting a thin layer of oxygen is present at the surface of intestinal epithelial cells (Marteyn et al., 2010), it is likely, in the classic mouse model, that the bacteria colonize the Peyer’s patches, and this constitutes the primary site of replication (Carter and Collins, 1974; Jones et al., 1994). Salmonella cells are clearly present in the lumen (and can be easily isolated from stool samples, for example), but our data suggest that the bacteria are not significantly replicating in the lumen, certainly not in the large intestine. The bacteria are also likely sensing the environment of the intestine to control gene expression, for example, to regulate the SPI1 T3SS invasion apparatus (Golubeva et al., 2012). An FNR mutant is slightly attenuated orally, which could reflect effects on virulence gene expression in the intestine (Fink et al., 2007). But these potential changes in gene expression are seemingly independent of replication.
Baumler and associates (Winter et al., 2010b; Thiennimitr et al., 2011) have shown that inflammatory diarrhea provides both a carbon source (ethanolamine) and an alternative electron acceptor, namely tetrathionate, which Salmonella uses to replicate in the anaerobic lumen of the intestine. But this inflammatory response occurs in mice only after pre-treated with streptomycin. This mouse colitis model likely mimics the inflammatory diarrhea conferred by non-typhoid Salmonella in humans. The classic mouse model, in contrast, is more indicative of typhoid fever in humans. Neither ethanolamine utilization (Stojiljkovic et al., 1995; Thiennimitr et al., 2011) nor tetrathionate respiration (Hensel et al., 1999; Winter et al., 2010b); Table 3) contribute to the virulence of S. Typhimurium in normal mice. Interestingly, in the human-specific Salmonella serovars, S. Paratyphi A strain ATCC 9150 has lost the ability to respire tetrathionate (Liu et al., 2009) and S. Paratyphi C strain RKS4594s has a mutation in the ethanolamine utilization operon (McClelland et al., 2004). S. Typhi strain CT18 has a mutation in the regulator of tetrathionate operon (Parkhill et al., 2001). Several additional Tat-dependent systems, including anaerobic respiratory systems contain pseudogenes in these serovars. These data suggest that, like S. Typhimurium in normal mice, the human-specific pathogens are replicating in solely aerobic tissues and not in the lumen of the intestine (Winter and Baumler, 2011).
Experimental procedures
Bacterial strains and growth conditions
Bacterial strains and plasmids are described in Table S3. Except for JS198, all Salmonella enterica serovar Typhimurium strains used in this study are isogenic derivatives of strain 14028 (American Type Culture Collection) and were constructed using P22 HT105/1 int-201 (P22) mediated transduction (Maloy et al., 1996). Deletion of various genes and concomitant insertion of an antibiotic resistance cassette was carried out using lambda Red-mediated recombination (Datsenko and Wanner, 2000) as described (Ellermeier et al., 2002). Primers were purchased from IDT Inc. The endpoints of each deletion are indicated in Table S3. In all cases, the appropriate insertion of the antibiotic resistance marker was checked by PCR analysis and the construct was moved into a clean wild type background (14028) by P22 transduction. In some cases, antibiotic resistance cassettes were removed using the temperature sensitive plasmid pCP20 encoding the FLP recombinase (Cherepanov and Wackernagel, 1995). The TorA-GFPuv expression plasmid (pAT100) was constructed as described in the supporting information.
For complementation studies, a fragment containing the gene of interest and a downstream Kan resistance cassette (from pKD4 but without FRT sites) (Datsenko and Wanner, 2000) was integrated as a single copy via lambda Red-mediated recombination 40 bp downstream of the purA stop codon and 116 bp upstream of the yjeB gene start codon, resulting in a deletion of 20 bp. We knew from previous studies that insertions at this site did not affect virulence. The amiA gene was complemented with a fragment corresponding to 459 bp upstream of the start codon to 50 bp downstream of the stop codon. The amiC gene fragment corresponded to 500 bp upstream of the start codon to 46 bp downstream of the stop codon. The sufI gene was complemented with a fragment extending from 220 bp upstream of the start codon to 45 bp downstream of the stop codon. DNA sequencing showed that all constructs were wild type.
Luria-Bertani (LB) medium (Silhavy et al., 1984) was routinely used for growth of bacteria, except where noted. Bacterial strains were grown at 37°C except for strains containing the temperature sensitive plasmids, pCP20or pKD46 (Datsenko and Wanner, 2000), which were grown at 30°C. Antibiotics were used at the following concentrations: 20 μg ml−1 chloramphenicol; 50 μg ml−1 kanamycin; and 12 μg ml−1 tetracycline.
For tests of anaerobic respiration, we used a supplemented MOPS defined medium (Neidhardt et al., 1974), called EZ (Teknova). Plates were made with either 0.4% glucose or 0.4% glycerol and supplemented with 1μM Na2SeO3 and 1μM (NH4)6Mo7O24·4H2O. Alternative electron acceptors were added, when indicated, at 0.4% nitrate, 0.4% fumarate or 0.4% DMSO. Plates were incubated at 37° in an anaerobic glove box.
Competition assays
BALB/c mice (BALB/cAnNHsd) were purchased from Harlan Sprague Dawley, Inc. For most intraperitoneal infections, isogenic strains were constructed in 14028; for most oral experiments, isogenic strains were constructed starting with the tetracycline-resistant strain JS135 (Stanley et al., 2000). Bacterial strains were grown overnight (16 h) in LB medium. Cultures of the two strains of interest were mixed 1:1 and the mixture was washed and diluted in sterile 0.15 M NaCl (for intraperitoneal infections) or 0.1 M sodium phosphate buffer, pH 8.0 (for oral infections). Female mice were inoculated in groups of 4 to 6 either intraperitoneally (i.p.) with approximately 1000 total bacteria or orally with approximately 109 total bacteria. Inocula were plated on LB and then replica plated onto the appropriate selective medium to determine the total number and percentage of the two strains used for the infection. Mice were sacrificed after 3 to 5 days of infection and the indicated organs were removed. Unless otherwise indicated, the entire tissue was homogenized and the homogenate was diluted and plated on LB medium (for i.p.) or LB/Tetracycline (for oral infections). The resulting colonies were replica plated onto selective medium to determine the relative percent of each strain recovered. The competitive index (CI) was calculated as (percent strain A recovered/percent strain B recovered)/(percent strain A inoculated/percent strain B inoculated). The CI of each set of assays was analyzed statistically using the Student’s t test. In most cases, the strains were rebuilt by P22 transduction, and the mouse assay was repeated to ensure that the virulence phenotypes were the result of the designated mutations. All animal work was reviewed and approved by the University of Illinois IACUC and performed under protocols 04137 and 07070.
For aerobic in vitro competitions, bacteria were grown as above for inoculation, but 0.1 ml of the bacterial mixture was introduced into 5 ml of LB in a 50 ml flask. If SDS or bile were part of the experiment, they were added at this time. Flasks were incubated at 37°C on a platform shaker rotating at 225 RPM for 18 hr. For anaerobic competitions, bacteria were inoculated into 18 mm tubes containing 5 ml LB with 0.2% glucose which had been in the anaerobic chamber for at least two days. The cells were incubated without agitation at 37°C for 24 hr in an anaerobic chamber. In both cases, cells were subsequently diluted and plated on LB medium, and the resulting colonies were replica plated onto the selective medium to determine the relative percent of each strain recovered. Competitive index was calculated as above and the Student’s t-test was used for statistical analyses.
Morphology analysis
S. Typhimurium cells were observed by light microscopy using an Olympus IX70 microscope at a magnification of 600x. All strains were grown overnight at 37°C in LB. The next day cells were washed with dH2O twice and then diluted to OD600 0.05 and grown in 5 ml LB with no NaCl in 18 mm tubes, at 37°C, 200 rpm. After 6 hr, cells were washed twice with dH2O and diluted to 0.01 OD ml−1. A 10 μl aliquot of cells was placed on a slide to dry. The cells were fixed with cold Methanol (−20°C) for 30 minutes. Gram staining was performed using an HT90A kit (Sigma-Aldrich) according to the manufacturer’s instructions. At least 250 S. Typhimurium cells in at least 5 random non-overlapping microscopic fields were counted for calculation of an average number of bacilli in a chain.
Motility assay
Strains were tested for ability to swim on 0.3% agar Luria-Bertani (LB) plates supplemented with 0.5% glucose. Bacteria were inoculated into motility agar by transferring 2 μl of overnight culture with a pipette tip and incubated 3 hours at 37° C.
Assay of β-galactosidase activity
β-galactosidase assays were performed using a microtiter plate assay as previously described (Slauch and Silhavy, 1991) on strains grown under the indicated conditions. β-galactosidase activity units are defined as (μmol of ONP formed min−1) × 106/ (OD600 × ml of cell suspension) and are reported as mean ± standard deviation where n = 4. For log phase cultures, bacteria were grown overnight in LB, diluted 1 to 100 in the indicated medium and, upon reaching OD600 of 0.2, diluted 1 to 4 and grown to OD600 of 0.2–0.3. Cultures grown in SPI1-inducing conditions were initially inoculated into LB (0.5% NaCl), grown for 8–12 hours, then subcultured 1 to 100 and grown statically for 18–22 hours in 3 ml LB with 1% NaCl (high salt LB) in a 13 × 100 mm tubes. For SPI2 inducing conditions, cells were grown overnight in LB with aeration then diluted 1 to 100 in 2 ml of N minimal medium with 0.2% glycerol and 8 mM MgCl2 and grown 16 h with aeration.
Type III secretion system assays
To assay SPI2-dependent type III secretion, cultures of serovar Typhimurium strains producing a SspH2-CyaA fusion were grown under SPI2-inducing conditions (described above). Bacteria were washed in 0.15 M NaCl and opsonized with 50% mouse serum (Equitech-Bio) for 20 min at 37°C. The opsonized bacteria were then used to infect RAW264.7 cells at a 10:1 ratio. After 1 h, the macrophages were washed three times with phosphate buffered saline and 1 ml of RPMI 1640 containing 10% fetal bovine serum, and 6.25 μg of gentamicin per ml was added. The infection was allowed to proceed for 5 h. Infected macrophages were then washed three times with phosphate buffered saline. The cells were lysed with 200 μl of 0.1 M HCl and heated for 10 min at 95°C. The levels of cAMP were determined using a Direct cAMP Correlate-EIA kit (Assay Designs). The protein content of each sample was determined by a BCA assay (Pierce) relative to a bovine serum albumin standard curve. All cAMP assays were performed in triplicate and repeated at least two times; the results of a representative experiment are shown.
Fluorescence microscopy
Strains were grown overnight in LB at 37°C, diluted 1:100 in fresh LB medium and grown for 8 hours to an OD600 of ~3.5. Aliquots equivalent to O.2 OD600 of cells were collected by centrifugation and washed twice with dH2O and re-suspended in 1 ml of dH2O. Cells were diluted 1 to 10 and 10 μl were applied to a microscope slide (FisherFinest). The slides were dried at room temperature, fixed in cold methanol (−20°C) for 30 minutes, dried and covered with a cover glass mounted with Fluoromount-G (SouthernBiotech). The slides were examined using a Zeiss LSM 510 Meta confocal fluorescence microscope. Emission was detected with a filter specific for GFP (488 nm).
Supplementary Material
Acknowledgments
This work was supported by NIH grants AI080705 and AI063230 to J.M.S. We thank Lea Slauch for technical assistance and Jim Imlay for helpful discussions and suggestions.
References
- Alami M, Luke I, Deitermann S, Eisner G, Koch HG, Brunner J, Muller M. Differential interactions between a twin-arginine signal peptide and its translocase in Escherichia coli. Mol Cell. 2003;12:937–946. doi: 10.1016/s1097-2765(03)00398-8. [DOI] [PubMed] [Google Scholar]
- Amy NK. Identification of the molybdenum cofactor in chlorate-resistant mutants of Escherichia coli. J Bacteriol. 1981;148:274–282. doi: 10.1128/jb.148.1.274-282.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berks BC. A common export pathway for proteins binding complex redox cofactors? Mol Microbiol. 1996;22:393–404. doi: 10.1046/j.1365-2958.1996.00114.x. [DOI] [PubMed] [Google Scholar]
- Berks BC, Palmer T, Sargent F. The Tat protein translocation pathway and its role in microbial physiology. Adv Microb Physiol. 2003;47:187–254. doi: 10.1016/s0065-2911(03)47004-5. [DOI] [PubMed] [Google Scholar]
- Berks BC, Sargent F, Palmer T. The Tat protein export pathway. Mol Microbiol. 2000;35:260–274. doi: 10.1046/j.1365-2958.2000.01719.x. [DOI] [PubMed] [Google Scholar]
- Bernhardt TG, de Boer PA. The Escherichia coli amidase AmiC is a periplasmic septal ring component exported via the twin-arginine transport pathway. Mol Microbiol. 2003;48:1171–1182. doi: 10.1046/j.1365-2958.2003.03511.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berthelmann F, Bruser T. Localization of the Tat translocon components in Escherichia coli. FEBS Lett. 2004;569:82–88. doi: 10.1016/j.febslet.2004.05.054. [DOI] [PubMed] [Google Scholar]
- Bolhuis A, Mathers JE, Thomas JD, Barrett CM, Robinson C. TatB and TatC form a functional and structural unit of the twin-arginine translocase from Escherichia coli. J Biol Chem. 2001;276:20213–20219. doi: 10.1074/jbc.M100682200. [DOI] [PubMed] [Google Scholar]
- Bronstein PA, Marrichi M, Cartinhour S, Schneider DJ, DeLisa MP. Identification of a twin-arginine translocation system in Pseudomonas syringae pv. tomato DC3000 and its contribution to pathogenicity and fitness. J Bacteriol. 2005;187:8450–8461. doi: 10.1128/JB.187.24.8450-8461.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter PB, Collins FM. The route of enteric infection in normal mice. J Exp Med. 1974;139:1189–1203. doi: 10.1084/jem.139.5.1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaddock AM, Mant A, Karnauchov I, Brink S, Herrmann RG, Klosgen RB, Robinson C. A new type of signal peptide: central role of a twin-arginine motif in transfer signals for the delta pH-dependent thylakoidal protein translocase. EMBO J. 1995;14:2715–2722. doi: 10.1002/j.1460-2075.1995.tb07272.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherepanov PP, Wackernagel W. Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene. 1995;158:9–14. doi: 10.1016/0378-1119(95)00193-a. [DOI] [PubMed] [Google Scholar]
- Chubiz JE, Golubeva YA, Lin D, Miller LD, Slauch JM. FliZ regulates expression of the Salmonella pathogenicity island 1 invasion locus by controlling HilD protein activity in Salmonella enterica serovar Typhimurium. J Bacteriol. 2010;192:6261–6270. doi: 10.1128/JB.00635-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Mello R, Hill S, Poole RK. The oxygen affinity of cytochrome bo′ in Escherichia coli determined by the deoxygenation of oxyleghemoglobin and oxymyoglobin: Km values for oxygen are in the submicromolar range. J Bacteriol. 1995;177:867–870. doi: 10.1128/jb.177.3.867-870.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Mello R, Hill S, Poole RK. The cytochrome bd quinol oxidase in Escherichia coli has an extremely high oxygen affinity and two oxygen-binding haems: implications for regulation of activity in vivo by oxygen inhibition. Microbiology. 1996;142 (Pt 4):755–763. doi: 10.1099/00221287-142-4-755. [DOI] [PubMed] [Google Scholar]
- Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dilks K, Rose RW, Hartmann E, Pohlschroder M. Prokaryotic utilization of the twin-arginine translocation pathway: a genomic survey. J Bacteriol. 2003;185:1478–1483. doi: 10.1128/JB.185.4.1478-1483.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Z, Christie PJ. Agrobacterium tumefaciens twin-arginine-dependent translocation is important for virulence, flagellation, and chemotaxis but not type IV secretion. J Bacteriol. 2003;185:760–771. doi: 10.1128/JB.185.3.760-771.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellermeier CD, Ellermeier JR, Slauch JM. HilD, HilC and RtsA constitute a feed forward loop that controls expression of the SPI1 type three secretion system regulator hilA in Salmonella enterica serovar Typhimurium. Mol Microbiol. 2005;57:691–705. doi: 10.1111/j.1365-2958.2005.04737.x. [DOI] [PubMed] [Google Scholar]
- Ellermeier CD, Janakiraman A, Slauch JM. Construction of targeted single copy lac fusions using lambda Red and FLP-mediated site-specific recombination in bacteria. Gene. 2002;290:153–161. doi: 10.1016/s0378-1119(02)00551-6. [DOI] [PubMed] [Google Scholar]
- Ellermeier CD, Slauch JM. RtsA coordinately regulates DsbA and the Salmonella pathogenicity island 1 type III secretion system. J Bacteriol. 2004;186:68–79. doi: 10.1128/JB.186.1.68-79.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellermeier JR, Slauch JM. Adaptation to the host environment: regulation of the SPI1 type III secretion system in Salmonella enterica serovar Typhimurium. Curr Opin Microbiol. 2007;10:24–29. doi: 10.1016/j.mib.2006.12.002. [DOI] [PubMed] [Google Scholar]
- Fass E, Groisman EA. Control of Salmonella pathogenicity island-2 gene expression. Curr Opin Microbiol. 2009;12:199–204. doi: 10.1016/j.mib.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields PI, Swanson RV, Haidaris CG, Heffron F. Mutants of Salmonella typhimurium that cannot survive within the macrophage are avirulent. Proc Natl Acad Sci U S A. 1986;83:5189–5193. doi: 10.1073/pnas.83.14.5189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink RC, Evans MR, Porwollik S, Vazquez-Torres A, Jones-Carson J, Troxell B, et al. FNR is a global regulator of virulence and anaerobic metabolism in Salmonella enterica serovar Typhimurium (ATCC 14028s) J Bacteriol. 2007;189:2262–2273. doi: 10.1128/JB.00726-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontecave M, Eliasson R, Reichard P. Oxygen-sensitive ribonucleoside triphosphate reductase is present in anaerobic Escherichia coli. Proc Natl Acad Sci U S A. 1989;86:2147–2151. doi: 10.1073/pnas.86.7.2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garriga X, Eliasson R, Torrents E, Jordan A, Barbe J, Gibert I, Reichard P. nrdD and nrdG genes are essential for strict anaerobic growth of Escherichia coli. Biochem Biophys Res Commun. 1996;229:189–192. doi: 10.1006/bbrc.1996.1778. [DOI] [PubMed] [Google Scholar]
- Gohlke U, Pullan L, McDevitt CA, Porcelli I, de LE, Palmer T, et al. The TatA component of the twin-arginine protein transport system forms channel complexes of variable diameter. Proc Natl Acad Sci U S A. 2005;102:10482–10486. doi: 10.1073/pnas.0503558102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golubeva YA, Sadik AY, Ellermeier JR, Slauch JM. Integrating global regulatory input into the Salmonella pathogenicity island 1 type III secretion system. Genetics. 2012;190:79–90. doi: 10.1534/genetics.111.132779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green J, Crack JC, Thomson AJ, LeBrun NE. Bacterial sensors of oxygen. Curr Opin Microbiol. 2009;12:145–151. doi: 10.1016/j.mib.2009.01.008. [DOI] [PubMed] [Google Scholar]
- Hanski E. Invasive adenylate cyclase toxin of Bordetella pertussis. Trends Biochem Sci. 1989;14:459–463. doi: 10.1016/0968-0004(89)90106-0. [DOI] [PubMed] [Google Scholar]
- He G, Shankar RA, Chzhan M, Samouilov A, Kuppusamy P, Zweier JL. Noninvasive measurement of anatomic structure and intraluminal oxygenation in the gastrointestinal tract of living mice with spatial and spectral EPR imaging. Proc Natl Acad Sci U S A. 1999;96:4586–4591. doi: 10.1073/pnas.96.8.4586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidrich C, Templin MF, Ursinus A, Merdanovic M, Berger J, Schwarz H, et al. Involvement of N-acetylmuramyl-L-alanine amidases in cell separation and antibiotic-induced autolysis of Escherichia coli. Mol Microbiol. 2001;41:167–178. doi: 10.1046/j.1365-2958.2001.02499.x. [DOI] [PubMed] [Google Scholar]
- Hensel M, Nikolaus T, Egelseer C. Molecular and functional analysis indicates a mosaic structure of Salmonella pathogenicity island 2. Mol Microbiol. 1999;31:489–498. doi: 10.1046/j.1365-2958.1999.01190.x. [DOI] [PubMed] [Google Scholar]
- Hensel M, Shea JE, Gleeson C, Jones MD, Dalton E, Holden DW. Simultaneous identification of bacterial virulence genes by negative selection. Science. 1995;269:400–403. doi: 10.1126/science.7618105. [DOI] [PubMed] [Google Scholar]
- Ize B, Stanley NR, Buchanan G, Palmer T. Role of the Escherichia coli Tat pathway in outer membrane integrity. Mol Microbiol. 2003;48:1183–1193. doi: 10.1046/j.1365-2958.2003.03504.x. [DOI] [PubMed] [Google Scholar]
- Jones BD, Ghori N, Falkow S. Salmonella typhimurium initiates murine infection by penetrating and destroying the specialized epithelial M cells of the Peyer’s patches. J Exp Med. 1994;180:15–23. doi: 10.1084/jem.180.1.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Y, Weber KD, Qiu Y, Kiley PJ, Blattner FR. Genome-wide expression analysis indicates that FNR of Escherichia coli K-12 regulates a large number of genes of unknown function. J Bacteriol. 2005;187:1135–1160. doi: 10.1128/JB.187.3.1135-1160.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato A, Groisman EA. The PhoQ/PhoP regulatory network of Salmonella enterica. Adv Exp Med Biol. 2008;631:7–21. doi: 10.1007/978-0-387-78885-2_2. [DOI] [PubMed] [Google Scholar]
- Lange C, Muller SD, Walther TH, Burck J, Ulrich AS. Structure analysis of the protein translocating channel TatA in membranes using a multi-construct approach. Biochim Biophys Acta. 2007;1768:2627–2634. doi: 10.1016/j.bbamem.2007.06.021. [DOI] [PubMed] [Google Scholar]
- Lavander M, Ericsson SK, Broms JE, Forsberg A. The twin arginine translocation system is essential for virulence of Yersinia pseudotuberculosis. Infect Immun. 2006;74:1768–1776. doi: 10.1128/IAI.74.3.1768-1776.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leake MC, Greene NP, Godun RM, Granjon T, Buchanan G, Chen S, et al. Variable stoichiometry of the TatA component of the twin-arginine protein transport system observed by in vivo single-molecule imaging. Proc Natl Acad Sci U S A. 2008;105:15376–15381. doi: 10.1073/pnas.0806338105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee PA, Tullman-Ercek D, Georgiou G. The bacterial twin-arginine translocation pathway. Annu Rev Microbiol. 2006;60:373–395. doi: 10.1146/annurev.micro.60.080805.142212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin D, Rao CV, Slauch JM. The Salmonella SPI1 type three secretion system responds to periplasmic disulfide bond status via the flagellar apparatus and the RcsCDB system. J Bacteriol. 2008;190:87–97. doi: 10.1128/JB.01323-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linehan SA, Rytkonen A, Yu XJ, Liu M, Holden DW. SlyA regulates function of Salmonella pathogenicity island 2 (SPI-2) and expression of SPI-2-associated genes. Infect Immun. 2005;73:4354–4362. doi: 10.1128/IAI.73.7.4354-4362.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu WQ, Feng Y, Wang Y, Zou QH, Chen F, Guo JT, et al. Salmonella paratyphi C: genetic divergence from Salmonella choleraesuis and pathogenic convergence with Salmonella typhi. PLoS ONE. 2009;4:e4510. doi: 10.1371/journal.pone.0004510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahan MJ, Slauch JM, Mekalanos JJ. Selection of bacterial virulence genes that are specifically induced in host tissues. Science. 1993;259:686–688. doi: 10.1126/science.8430319. [DOI] [PubMed] [Google Scholar]
- Maier RJ, Olczak A, Maier S, Soni S, Gunn J. Respiratory hydrogen use by Salmonella enterica serovar Typhimurium is essential for virulence. Infect Immun. 2004;72:6294–6299. doi: 10.1128/IAI.72.11.6294-6299.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maloy SR, Stewart VJ, Taylor RK. Genetic Analysis of pathogenic bacteria: a laboratory manual. Plainview, NY: Cold Spring Harbor Laboratory Press; 1996. [Google Scholar]
- Marteyn B, West NP, Browning DF, Cole JA, Shaw JG, Palm F, et al. Modulation of Shigella virulence in response to available oxygen in vivo. Nature. 2010;465:355–358. doi: 10.1038/nature08970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClelland M, Sanderson KE, Clifton SW, Latreille P, Porwollik S, Sabo A, et al. Comparison of genome degradation in Paratyphi A and Typhi, human-restricted serovars of Salmonella enterica that cause typhoid. Nat Genet. 2004;36:1268–1274. doi: 10.1038/ng1470. [DOI] [PubMed] [Google Scholar]
- McNicholas PM, Chiang RC, Gunsalus RP. Anaerobic regulation of the Escherichia coli dmsABC operon requires the molybdate-responsive regulator ModE. Mol Microbiol. 1998;27:197–208. doi: 10.1046/j.1365-2958.1998.00675.x. [DOI] [PubMed] [Google Scholar]
- Miao EA, Miller SI. A conserved amino acid sequence directing intracellular type III secretion by Salmonella typhimurium. Proc Natl Acad Sci U S A. 2000;97:7539–7544. doi: 10.1073/pnas.97.13.7539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mickael CS, Lam PK, Berberov EM, Allan B, Potter AA, Koster W. Salmonella enterica serovar Enteritidis tatB and tatC mutants are impaired in Caco-2 cell invasion in vitro and show reduced systemic spread in chickens. Infect Immun. 2010;78:3493–3505. doi: 10.1128/IAI.00090-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills DM, Bajaj V, Lee CA. A 40 kb chromosomal fragment encoding Salmonella typhimurium invasion genes is absent from the corresponding region of the Escherichia coli K- 12 chromosome. Mol Microbiol. 1995;15:749–759. doi: 10.1111/j.1365-2958.1995.tb02382.x. [DOI] [PubMed] [Google Scholar]
- Neidhardt FC, Bloch PL, Smith DF. Culture medium for enterobacteria. J Bacteriol. 1974;119:736–747. doi: 10.1128/jb.119.3.736-747.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochsner UA, Snyder A, Vasil AI, Vasil ML. Effects of the twin-arginine translocase on secretion of virulence factors, stress response, and pathogenesis. Proc Natl Acad Sci U S A. 2002;99:8312–8317. doi: 10.1073/pnas.082238299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orriss GL, Tarry MJ, Ize B, Sargent F, Lea SM, Palmer T, Berks BC. TatBC, TatB, and TatC form structurally autonomous units within the twin arginine protein transport system of Escherichia coli. FEBS Lett. 2007;581:4091–4097. doi: 10.1016/j.febslet.2007.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer T, Berks BC. The twin-arginine translocation (Tat) protein export pathway. Nat Rev Microbiol. 2012;10:483–496. doi: 10.1038/nrmicro2814. [DOI] [PubMed] [Google Scholar]
- Palmer T, Sargent F, Berks BC. The Tat protein export pathway. In: Böck A, Curtiss R3, Kaper J, Karp PD, Neidhardt FC, Nystrom T, et al., editors. EcoSal-Escherichia coli and Salmonella: Cellular and Molecular Biology. Washington, DC: ASM Press; 2010. ecosal.org. [Google Scholar]
- Parkhill J, Dougan G, James KD, Thomson NR, Pickard D, Wain J, et al. Complete genome sequence of a multiple drug resistant Salmonella enterica serovar Typhi CT18. Nature. 2001;413:848–852. doi: 10.1038/35101607. [DOI] [PubMed] [Google Scholar]
- Pegues DA, Miller SI. Salmonella species, including Salmonella Typhi. In: Mandell GL, Bennett JE, Dolin R, editors. Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases. Philadelphia, PA: Churchill Livingstone; 2010. pp. 2887–2903. [Google Scholar]
- Peters NT, Dinh T, Bernhardt TG. A fail-safe mechanism in the septal ring assembly pathway generated by the sequential recruitment of cell separation amidases and their activators. J Bacteriol. 2011;193:4973–4983. doi: 10.1128/JB.00316-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradel N, Ye C, Livrelli V, Xu J, Joly B, Wu LF. Contribution of the twin arginine translocation system to the virulence of enterohemorrhagic Escherichia coli O157:H7. Infect Immun. 2003;71:4908–4916. doi: 10.1128/IAI.71.9.4908-4916.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy M. Role of FtsEX in cell division of Escherichia coli: viability of ftsEX mutants is dependent on functional SufI or high osmotic strength. J Bacteriol. 2007;189:98–108. doi: 10.1128/JB.01347-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds MM, Bogomolnaya L, Guo J, Aldrich L, Bokhari D, Santiviago CA, et al. Abrogation of the twin arginine transport system in Salmonella enterica serovar Typhimurium leads to colonization defects during infection. PLoS ONE. 2011;6:e15800. doi: 10.1371/journal.pone.0015800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice CW, Hempfling WP. Oxygen-limited continuous culture and respiratory energy conservation in Escherichia coli. J Bacteriol. 1978;134:115–124. doi: 10.1128/jb.134.1.115-124.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigue A, Chanal A, Beck K, Muller M, Wu LF. Co-translocation of a periplasmic enzyme complex by a hitchhiker mechanism through the bacterial tat pathway. J Biol Chem. 1999;274:13223–13228. doi: 10.1074/jbc.274.19.13223. [DOI] [PubMed] [Google Scholar]
- Rossier O, Cianciotto NP. The Legionella pneumophila tatB gene facilitates secretion of phospholipase C, growth under iron-limiting conditions, and intracellular infection. Infect Immun. 2005;73:2020–2032. doi: 10.1128/IAI.73.4.2020-2032.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samaluru H, SaiSree L, Reddy M. Role of SufI (FtsP) in cell division of Escherichia coli: evidence for its involvement in stabilizing the assembly of the divisome. J Bacteriol. 2007;189:8044–8052. doi: 10.1128/JB.00773-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sargent F, Bogsch EG, Stanley NR, Wexler M, Robinson C, Berks BC, Palmer T. Overlapping functions of components of a bacterial Sec-independent protein export pathway. EMBO J. 1998;17:3640–3650. doi: 10.1093/emboj/17.13.3640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt CK, Ikeda JS, Darnell SC, Watson PR, Bispham J, Wallis TS, et al. Absence of all components of the flagellar export and synthesis machinery differentially alters virulence of Salmonella enterica serovar Typhimurium in models of typhoid fever, survival in macrophages, tissue culture invasiveness, and calf enterocolitis. Infect Immun. 2001;69:5619–5625. doi: 10.1128/IAI.69.9.5619-5625.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Settles AM, Yonetani A, Baron A, Bush DR, Cline K, Martienssen R. Sec-independent protein translocation by the maize Hcf106 protein. Science. 1997;278:1467–1470. doi: 10.1126/science.278.5342.1467. [DOI] [PubMed] [Google Scholar]
- Shea JE, Hensel M, Gleeson C, Holden DW. Identification of a virulence locus encoding a second type III secretion system in Salmonella typhimurium. Proc Natl Acad Sci U S A. 1996;93:2593–2597. doi: 10.1073/pnas.93.6.2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silhavy TJ, Berman ML, Enquist LW. Experiments with gene fusions. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 1984. [Google Scholar]
- Slauch JM, Silhavy TJ. cis-acting ompF mutations that result in OmpR-dependent constitutive expression. J Bacteriol. 1991;173:4039–4048. doi: 10.1128/jb.173.13.4039-4048.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiro S, Guest JR. FNR and its role in oxygen-regulated gene expression in Escherichia coli. FEMS Microbiol Rev. 1990;6:399–428. doi: 10.1111/j.1574-6968.1990.tb04109.x. [DOI] [PubMed] [Google Scholar]
- Spiro S, Roberts RE, Guest JR. FNR-dependent repression of the ndh gene of Escherichia coli and metal ion requirement for FNR-regulated gene expression. Mol Microbiol. 1989;3:601–608. doi: 10.1111/j.1365-2958.1989.tb00207.x. [DOI] [PubMed] [Google Scholar]
- Stanley NR, Findlay K, Berks BC, Palmer T. Escherichia coli strains blocked in Tat-dependent protein export exhibit pleiotropic defects in the cell envelope. J Bacteriol. 2001;183:139–144. doi: 10.1128/JB.183.1.139-144.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley TL, Ellermeier CD, Slauch JM. Tissue-specific gene expression identifies a gene in the lysogenic phage Gifsy-1 that affects Salmonella enterica serovar Typhimurium survival in Peyer’s patches. J Bacteriol. 2000;182:4406–4413. doi: 10.1128/jb.182.16.4406-4413.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stojiljkovic I, Baumler AJ, Heffron F. Ethanolamine utilization in Salmonella typhimurium: nucleotide sequence, protein expression, and mutational analysis of the cchA cchB eutE eutJ eutG eutH gene cluster. J Bacteriol. 1995;177:1357–1366. doi: 10.1128/jb.177.5.1357-1366.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarry M, Arends SJ, Roversi P, Piette E, Sargent F, Berks BC, et al. The Escherichia coli cell division protein and model Tat substrate SufI (FtsP) localizes to the septal ring and has a multicopper oxidase-like structure. J Mol Biol. 2009;386:504–519. doi: 10.1016/j.jmb.2008.12.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiennimitr P, Winter SE, Winter MG, Xavier MN, Tolstikov V, Huseby DL, et al. Intestinal inflammation allows Salmonella to use ethanolamine to compete with the microbiota. Proc Natl Acad Sci U S A. 2011;108:17480–17485. doi: 10.1073/pnas.1107857108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas JD, Daniel RA, Errington J, Robinson C. Export of active green fluorescent protein to the periplasm by the twin-arginine translocase (Tat) pathway in Escherichia coli. Mol Microbiol. 2001;39:47–53. doi: 10.1046/j.1365-2958.2001.02253.x. [DOI] [PubMed] [Google Scholar]
- Tseng CP, Albrecht J, Gunsalus RP. Effect of microaerophilic cell growth conditions on expression of the aerobic (cyoABCDE and cydAB) and anaerobic (narGHJI, frdABCD, and dmsABC) respiratory pathway genes in Escherichia coli. J Bacteriol. 1996;178:1094–1098. doi: 10.1128/jb.178.4.1094-1098.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Typas A, Banzhaf M, Gross CA, Vollmer W. From the regulation of peptidoglycan synthesis to bacterial growth and morphology. Nat Rev Microbiol. 2012;10:123–136. doi: 10.1038/nrmicro2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unden G, Dunnwald P. The aerobic and anaerobic respiratory chain of Escherichia coli and Salmonella enterica: enzymes and energetics. In: Böck A, Curtiss R3, Kaper J, Karp PD, Neidhardt FC, Nystrom T, et al., editors. EcoSal-Escherichia coli and Salmonella: Cellular and Molecular Biology. Washington, DC: ASM Press; 2008. Ecosal.org. [Google Scholar]
- Voulhoux R, Ball G, Ize B, Vasil ML, Lazdunski A, Wu LF, Filloux A. Involvement of the twin-arginine translocation system in protein secretion via the type II pathway. EMBO J. 2001;20:6735–6741. doi: 10.1093/emboj/20.23.6735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walthers D, Carroll RK, Navarre WW, Libby SJ, Fang FC, Kenney LJ. The response regulator SsrB activates expression of diverse Salmonella pathogenicity island 2 promoters and counters silencing by the nucleoid-associated protein H-NS. Mol Microbiol. 2007;65:477–493. doi: 10.1111/j.1365-2958.2007.05800.x. [DOI] [PubMed] [Google Scholar]
- Weatherspoon-Griffin N, Zhao G, Kong W, Kong Y, Morigen, Andrews-Polymenis H, et al. The CpxR/CpxA two-component system up-regulates two Tat-dependent peptidoglycan amidases to confer bacterial resistance to antimicrobial peptide. J Biol Chem. 2011;286:5529–5539. doi: 10.1074/jbc.M110.200352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner JH, Bilous PT, Shaw GM, Lubitz SP, Frost L, Thomas GH, et al. A novel and ubiquitous system for membrane targeting and secretion of cofactor-containing proteins. Cell. 1998;93:93–101. doi: 10.1016/s0092-8674(00)81149-6. [DOI] [PubMed] [Google Scholar]
- Weiss DS, Chen JC, Ghigo JM, Boyd D, Beckwith J. Localization of FtsI (PBP3) to the septal ring requires its membrane anchor, the Z ring, FtsA, FtsQ, and FtsL. J Bacteriol. 1999;181:508–520. doi: 10.1128/jb.181.2.508-520.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter SE, Baumler AJ. A breathtaking feat: to compete with the gut microbiota, Salmonella drives its host to provide a respiratory electron acceptor. Gut Microbes. 2011;2:58–60. doi: 10.4161/gmic.2.1.14911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter SE, Keestra AM, Tsolis RM, Baumler AJ. The blessings and curses of intestinal inflammation. Cell Host Microbe. 2010a;8:36–43. doi: 10.1016/j.chom.2010.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter SE, Thiennimitr P, Winter MG, Butler BP, Huseby DL, Crawford RW, et al. Gut inflammation provides a respiratory electron acceptor for Salmonella. Nature. 2010b;467:426–429. doi: 10.1038/nature09415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang DC, Peters NT, Parzych KR, Uehara T, Markovski M, Bernhardt TG. An ATP-binding cassette transporter-like complex governs cell-wall hydrolysis at the bacterial cytokinetic ring. Proc Natl Acad Sci U S A. 2011;108:E1052–E1060. doi: 10.1073/pnas.1107780108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen MR, Tseng YH, Nguyen EH, Wu LF, Saier MH., Jr Sequence and phylogenetic analyses of the twin-arginine targeting (Tat) protein export system. Arch Microbiol. 2002;177:441–450. doi: 10.1007/s00203-002-0408-4. [DOI] [PubMed] [Google Scholar]
- Yimga MT, Leatham MP, Allen JH, Laux DC, Conway T, Cohen PS. Role of gluconeogenesis and the tricarboxylic acid cycle in the virulence of Salmonella enterica serovar Typhimurium in BALB/c mice. Infect Immun. 2006;74:1130–1140. doi: 10.1128/IAI.74.2.1130-1140.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.