

Abstract
The conversion of S-nitrosothiols to thiosulphonates by reaction with the sodium salt of benzenesulfinic acid (PhSO2Na) has been examined in detail with the exemplary substrates S-nitrosoglutathione (GSNO) and S-nitrosylated bovine serum albumin (SNO-BSA). The reaction stoichiometry (2:1, PhSO2Na:RSNO) and the rate law (first order in both PhSO2Na and RSNO) have been determined under mild acidic conditions (pH 4.0). The products have been identified as the corresponding thiosulphonates (GSSO2Ph and BSA-SSO2Ph) along with PhSO2NHOH obtained in a 1:1 ratio. GSH, GSSG, and BSA were unreactive to PhSO2Na.
Keywords: S-nitrosothiols, Benzenesulfinic acid, S-phenylthiosulphonates, Protein labeling
Protein S-nitrosothiols have emerged as an important post-translational modification involved in cellular signal transduction. Both hypo- and hyper-S-nitrosylation have been implicated in disease states.1 The importance of this functional group in biology motivates the development of analytical methods for identifying and quantifying protein S-nitrosothiols. The most prominent technique used today is the biotin-switch technique,2 which has facilitated the identification of many protein S-nitrosothiols. The complexity of experimental design, in particular the necessity of multi-step chemical processing, has led to interest in the development of probes that can simplify the procedure by reacting directly and selectively with protein S-nitrosothiols.3
Recently, several triphenylphosphine based probes have been studied that undergo Staudinger-like ligation of S-nitrosothiols to form sulfenamides,4 which can be further manipulated to form disulfides,5 thioethers,6 or dehydro-alanines.7 The requirement of organic solvents limits the potential of triphenylphosphine based probes. Water soluble triphenylphosphine probes derivatized with sulfonic acids have been developed that convert S-nitrosothiols into S-alkylphosphonium salts detectable by mass spectrometry.8
Over a quarter of a century ago a reaction (Scheme 1) was reported in the literature9 which transformed S-nitroso-glutathione (GSNO) into S-phenylsulphonylglutathione (GSSO2Ph) under highly acidic conditions (pH 0.5) in the presence of benzenesulfinic acid. This reaction has promising features as a viable detection strategy for S-nitrosylated proteins. However, the highly acidic pH conditions are not compatible with biological samples. The fundamental reaction properties that ultimately determine the utility of this approach, namely the reaction mechanism and rate, pH dependence, and utility towards protein S-nitrosothiol detection, remain unknown. These issues are fully addressed for the first time by studying the reaction of benzenesulfinic acid sodium salt with GSNO and SNO-BSA, and characterizing the products.
Scheme 1.
As a point of departure, the effect of pH on reaction rate (at ambient temperature) was assessed by measuring the disappearance of GSNO by UV/Vis spectroscopy (545 nm, 15 M−1cm−1, Varian Cary 6000i) upon addition of benzenesulfinic acid sodium salt (Fig 1). As a control, GSNO degradation was monitored over the same time frame and found to be negligible. GSNO (10 mM) was reacted with 2.0 equiv of PhSO2Na in aqueous solutions with pH ranging from 1.0 to 5.0. The initial reaction rate was observed to increase by 50% as the pH was varied from 1 to 2.5, where the rate reaches a maximum close to the reported pKa of PhSO2H.10 The rate declines as the pH increases. These results reveal that the reaction is feasible under modestly acidic conditions and does not require the harsh acidic reaction conditions previously employed,9 which are incompatible with protein samples.
Figure 1.

Rate of disappearance of GSNO (545 nm, 15 M−1cm−1) taken in the initial 2 min after mixing vs pH for reactions of PhSO2Na (20 mM) and GSNO (10 mM) in buffer (pH 1.0–2.0 KCl/HCl, 200 mM and pH 2.5–5.0, potassium hydrogen phthalate, 100 mM). Reactions were performed in triplicate and the length of the error bar represents the standard deviation.
Since a mild pH is desirable when working with protein samples, further investigations were conducted at pH 4.0 rather than at the optimum pH. Ammonium formate (100 mM) was chosen as the buffer because it is compatible with mass spectrometry analysis due to its volatility and low molecular weight ions. Reversed phase HPLC in tandem with mass spectrometry was used to monitor reaction progress and to identify all products (see supporting info, Fig SI-1). As a control, GSNO degradation was monitored over the same time frame and found to be negligible. Two products were identified by high resolution mass spectrometry and ion fragmentation as GSSO2Ph and PhSO2NHOH (see supporting info, Fig SI-2) in a 1:1 ratio. It is important to note that the nitroxyl group on GSNO becomes incorporated into PhSO2NHOH, otherwise known as Piloty’s acid, which is a poor HNO donor at neutral pH.11 Furthermore, the reaction profiles show that PhSO2NHOH is stable at pH 4.0 for ca. 2 h. These results stand in stark contrast to O-nitrosoascorbate, the byproduct of the switch assay, which is considerably less stable than S-nitrosothiols and releases NO in minutes at physiological pH.12
Reaction profiles were obtained by comparing absorbance peak areas to calibration curves made with authentic samples and simulated using Copasi integration software (Fig 2).13 By varying initial concentrations of GSNO and PhSO2Na, the rate law and reaction stoichiometry were determined. Evaluation of the kinetic parameters within the framework of this model (cf. Table 1) reveals the reaction to be first order in both GSNO and PhSO2Na; the first addition of PhSO2Na is the rate determining step with a rate constant of 0.041 M−1s−1.
Figure 2.
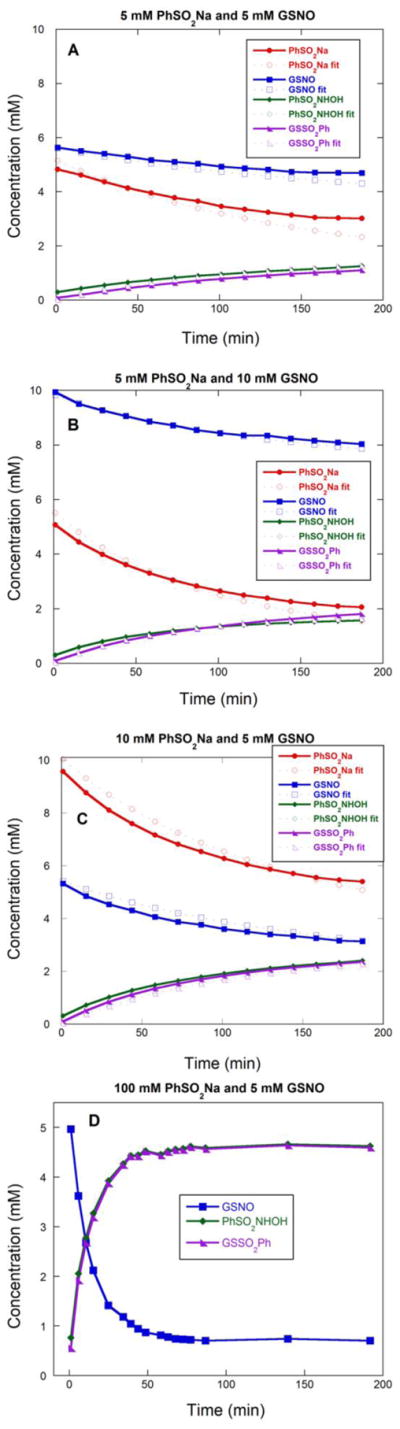
Reaction profiles obtained by comparing absorbance peak areas to calibration curves for three different initial conditions (A) 5 mM PhSO2Na, 5 mM GSNO, (B) 5 mM PhSO2Na, 10 mM GSNO, (C) 10 mM PhSO2Na, 5 mM GSNO (D) 100 mM PhSO2Na, 5 mM GSNO. All reactions were run in 100 mM ammonium formate, pH 4.0.
Table 1.
Rate constants calculated by Copasi modeling.
| k1 (M−1s−1) | k2 | ||
|---|---|---|---|
| R1 | GSNO + H+ = GSNOH+ | 2.72E+03 | 1.36 s−1 |
| R2 | GSNOH+ + PhSO2Na →B | 0.041a | 0 s−1 |
| R3 | B + H+ = BH+ | 2.79E+06 | 0 s−1 |
| R4 | BH+ + PhSO2Na = PhSO2NHOH + GSSO2Ph | 2.86E+02 | 1.26E+02 M−1s−1 |
Rate determining step.
When GSNO was treated with a large excess of PhSO2Na (20:1) at pH 4.0, the reaction is 85% complete in 1 h without the detection of byproducts (Fig 2D). The concentration of PhSO2NHOH and GSSO2Ph remained constant over the next 7 h. This experiment indicates that a large excess of PhSO2Na can be used to increase the rate of the reaction without creating undesirable byproducts.
The mechanism detailed in Scheme 2 is consistent with the Copasi simulations. The oxygen of the S-nitrosothiol is protonated in the first step, activating the addition by the benzenesulfinate anion. The protonation of the resulting intermediate activates the attack of the second benzenesulfinate thus furnishing the products in ratio of 1:1.
Scheme 2.

To further probe the suitability of this reaction as a S-nitrosothiol labeling strategy, control reactions of PhSO2Na with GSH and GSSG were assessed (Scheme 3). Reactions between GSH (5 mM) and PhSO2Na (10 mM) and GSSG (5 mM) and PhSO2Na (10 mM) were monitored using the same HPLC method in tandem with MS. GSSO2Ph concentration was quantified by UV-vis and remained undetectable over 3 h (see supporting info, Fig SI-3). The concentration of PhSO2Na was also quantified and remained unchanged over the course of the reaction. The mass spectra were also taken at each time point and were absent of byproducts. Since the absorbance of GSH is low, the MS signal of GSH while mixed with PhSO2Na was observed over time. The ratio of signal between the reaction and a standard remained unchanged over 3 h without the detection of byproducts (see supporting info, Fig SI-3).
Scheme 3.
With an understanding of the mechanism and selectivity of the reaction, the reactivity of PhSO2Na with a protein S-nitrosothiol was probed. As a proof of concept, a test protein, bovine serum albumin (BSA), was used because it contains a single reactive thiol (Cys34) that undergoes S-nitrosylation.14 BSA, moreover, contains 17 protein disulfides and is typically obtained with mixed disulfides at Cys34.
In a preliminary study, BSA (10 mg/mL, ammonium formate buffer, pH 4.0) with and without PhSO2Na (0.1 M) was examined and the deconvoluted mass spectra were virtually the same (Fig 3A). The free thiol content of BSA (Sigma) was found to be 49% by the DTDP method.15 It should be noted that in addition to BSA (66,430 amu), covalent adducts were detected including the mixed disulfide BSA-Cys (66,548 amu), and glycosylation adducts BSA(Glc) (66,592 amu) and BSA-Cys(Glc) (66,711 amu) which have been observed by others.16
Figure 3.
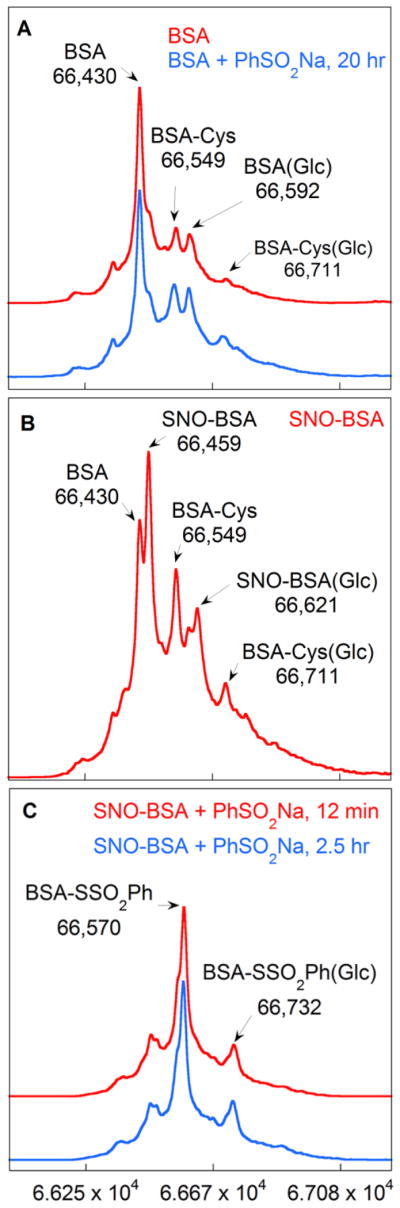
Deconvoluted mass spectra of (A) BSA (10mg/mL) and BSA with PhSO2Na (0.1 M), (B) SNO-BSA (100 μM total protein, 44% SNO) (C) SNO-BSA and PhSO2Na (0.1 M) after 12 min and after 2.5 h. All reactions were run in pH 4.0 ammonium formate buffer (100 mM).
In order to probe the viability of the reaction, BSA was nitrosylated employing 5 equiv S-nitrosocysteine in HEN buffer (100 mM HEPES, 1.0 mM EDTA, and 0.1 mM neocuprine, pH 8.0) for 1 h in the dark. The nitrosylated protein was purified by elution through a Sephadex G25 column which also served to exchange the HEN buffer with 100 mM ammonium formate (pH 4.0). The concentration of total protein was 100 μM (ε280 = 44,300 M−1cm−1)17 and a Greiss/Saville assay18 determined the SNO content to be 44%, leaving 5% free thiol content as determined by the DTDP method.15 The mass spectrum of SNO-BSA is seen in Fig 3B. The S-nitrosothiol adduct is seen as the major component (66,459 amu). BSA-Cys was unreactive while BSA(Glc) underwent nitrosylation as seen by the peak at 66,621 amu. The BSA starting material (66,430 amu) is likely due to the fragmentation of the labile nitroxide and is not from an incomplete reaction. Also, S-nitrosothiol fragmentation of the NO group is well documented even when using soft ionization methods such as ESI.19
In Figure 3C, the thiosulphonate adduct (66,570 amu) was dominant 12 min after the addition of PhSO2Na (0.1 M final concentration). The deconvoluted spectrum remained unaltered after 2.5 h, suggesting the reaction was near completion in the first 12 min. Notably, the essentially complete reaction of the S-nitrosothiol preclude recovery of the parent peak by fragmentation loss of NO, thus the thiol peak is sharply diminished as compared to Fig 3B.
In summary, the mechanism of thiosulphonate formation from the reaction of S-nitrosothiols and PhSO2Na has been elucidated. While the reaction rate is slow when using a 2:1 stoichiometric ratio of PhSO2Na to S-nitrosothiol, excess PhSO2Na can be used to increase the reaction rate which does not result in any unwanted side reactions. The reaction is selective for S-nitrosothiols and is tolerable to typical protein functionalities, including thiols and disulfides, as seen for the reactions of SNO-BSA and BSA. The byproduct, PhSO2NHOH, is stable over the duration of the labeling procedure.
Supplementary Material
Acknowledgments
We thank the National Institute of Health (CoBRE 5P20RR024237-04) for funding this work.
Footnotes
Supporting information (synthetic procedures for GSNO and GSSO2Ph and Figs SI-1-3) associated with this article can be found, in the online version at:
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.(a) Seth D, Stamler JS. Curr Opin Chem Biol. 2011;15:129–136. doi: 10.1016/j.cbpa.2010.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Foster MW, Hess DT, Stamler JS. Trends Mol Med. 2009;15:391–404. doi: 10.1016/j.molmed.2009.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Hess DT, Matsumoto A, Kim SO, Marshall HE, Stamler JS. Nat Rev Mol Cell Biol. 2005;6:150–166. doi: 10.1038/nrm1569. [DOI] [PubMed] [Google Scholar]; (d) Singel DJ, Stamler JS. Annu Rev Physiol. 2005;67:99–145. doi: 10.1146/annurev.physiol.67.060603.090918. [DOI] [PubMed] [Google Scholar]
- 2.(a) Jaffrey SR, Erdjument-Bromage H, Ferris CD, Tempst P, Snyder SH. Nat Chem Biol. 2001;3:193–197. doi: 10.1038/35055104. [DOI] [PubMed] [Google Scholar]; (b) Forrester MT, Foster MW, Benhar M, Stamler JS. Free Radical Bio Med. 2009;46:119–126. doi: 10.1016/j.freeradbiomed.2008.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang H, Xian M. Curr Opin Chem Biol. 2011;15:32–37. doi: 10.1016/j.cbpa.2010.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang H, Xian M. Angew Chem Int Ed. 2008;47:6598–6601. doi: 10.1002/anie.200801654. [DOI] [PubMed] [Google Scholar]
- 5.(a) Pan J, Xian M. Chem Commun. 2011;47:352–354. doi: 10.1039/c0cc02076a. [DOI] [PubMed] [Google Scholar]; (b) Zhang J, Li S, Zhang D, Wang H, Whorton AR, Xian M. Org Lett. 2010;12:4208–4211. doi: 10.1021/ol101863s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang D, Devarie-Baez O, Pan J, Wang H, Xian M. Org Lett. 2010;12:5674. doi: 10.1021/ol102491n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang H, Zhang J, Xian M. J Am Chem Soc. 2009;131:13238–13239. doi: 10.1021/ja905558w. [DOI] [PubMed] [Google Scholar]
- 8.(a) Bechtold E, Reisz JA, Klomsiri C, Tsang AW, Wright MW, Poole LB, Furdui CM, King SB. ACS Chem Biol. 2010;5:405–414. doi: 10.1021/cb900302u. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Seneviratne U, Godoy LC, Wishnok JS, Wogan GN, Tannenbaum SR. J Am Chem Soc. 2013;135:7693–7704. doi: 10.1021/ja401565w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Hart T. Tetrahedron Lett. 1985;26:2013–2016. [Google Scholar]; (b) Hart T, Vine MB, Walkden NR. Tetrahedron Lett. 1985;26:3879–3882. [Google Scholar]
- 10.Ali ST, Karamat S, Kona J, Fabian WMF. J Phys Chem A. 2010;114:12470–12478. doi: 10.1021/jp102266v. [DOI] [PubMed] [Google Scholar]
- 11.DuMond JF, King SB. Antioxid Redox Sign. 2011;14:1637–1648. doi: 10.1089/ars.2010.3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kirsch M, Buscher AM, Aker S, Schulz R, de Groot H. Org Biomol Chem. 2009;7(9):1954–1962. doi: 10.1039/b901046g. [DOI] [PubMed] [Google Scholar]
- 13.Hoops S, Sahle S, Gauges R, Lee C, Pahle J, Simus N, Singhal M, Xu L, Mendes P, Kummer U. Bioinformatics. 2006;22:3067–74. doi: 10.1093/bioinformatics/btl485. [DOI] [PubMed] [Google Scholar]
- 14.Stamler JS, Jaraki O, Simon D, Keaney J, Vita J, Singel D, Valeri CR, Loscalzo J. Proc Natl Acad Sci USA. 1992;89:7674–7677. doi: 10.1073/pnas.89.16.7674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Riener CK, Kada G, Gruber HJ. Anal Bioanal Chem. 2002;373:266–276. doi: 10.1007/s00216-002-1347-2. [DOI] [PubMed] [Google Scholar]
- 16.Beck JL, Ambahera S, Yong SR, Sheil MM, de Jersey J, Ralph SF. Anal Biochem. 2004;325:326–336. doi: 10.1016/j.ab.2003.10.041. [DOI] [PubMed] [Google Scholar]
- 17.Elgersma AV, Zsom RLJ, Norde W, Lyklema J. J Coll Interf Sci. 1990;138:145–156. [Google Scholar]
- 18.Basu S, Hill JD, Shields H, Huang J, King SB, Kim-Shapiro DB. Nitric Oxide-Biol Ch. 2006;15:1–4. doi: 10.1016/j.niox.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 19.Wang Y, Liu T, Wu C, Li H. J Am Soc Mass Spectr. 2008;19:1353–1360. doi: 10.1016/j.jasms.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.