

Abstract
A number of cyclic peptides including [FR]4, [FK]4, [WR]4, [CR]4, [AK]4, and [WK]n (n = 3-5) containing L-amino acids were produced using solid-phase peptide synthesis. We hypothesized that an optimal balance of hydrophobicity and charge could generate self-assembled nanostructures in aqueous solution by intramolecular and/or intermolecular interactions. Among all the designed peptides, [WR]n (n = 3-5) generated self-assembled vesicle-like nanostructures at room temperature as shown by transmission electron microscopy (TEM), scanning electron microscopy (SEM), and/or dynamic light scattering (DLS). This class of peptides represents the first report of surfactant-like cyclic peptides that self-assemble into nanostructures. A plausible mechanistic insight into the self-assembly of [WR]5 was obtained by molecular modeling studies. Modified [WR]5 analogues, such as [WMeR]5, [WR(Me)2]5, [WMeR(Me)2]5, and [WdR]5, exhibited different morphologies to [WR]5 as shown by TEM observations. [WR]5 exhibited a significant stabilizing effect for generated silver nanoparticles and glyceraldehyde-3-phosphate dehydrogenase activity. These studies established a new class of surfactant-like cyclic peptides that self-assembled into nanostructures and could have potential applications for the stabilization of silver nanoparticles and protein biomolecules.
1. Introduction
Well-defined nanostructures have drawn significant attention in the fields of biomedicine and biotechnology, materials science, and nanotechnology.1,2 The formation of ordered structures is facilitated by different types of noncovalent interactions, including electrostatic, hydrogen bonding, hydrophobic, and aromatic stacking.3–5 In this context, self-assembly of peptides at the nanoscale has emerged as a subject of considerable interest in recent years. Various types of peptide nanostructures have been reported with applications in diverse fields, such as antibacterial agents,6 neurological regeneration,7 and biosensors.8,9 Ghadiri et al.10 showed that cyclic D,L-peptides composed of alternating D- and L- amino acid residues can self-assemble into nanotubes by forming antiparallel hydrogen bonds between homochiral amino acid residues of adjacent rings.
Peptides have attracted much attention as an alternative to traditional surfactants for studies of membrane proteins, because of their tendency to augment the formation of ordered supramolecular assemblies.11 There is an urgent need to develop more suitable surfactants that can provide natural alternatives for studying the membrane environment. Linear surfactant-like peptides bearing hydrophilic and hydrophobic residues have been shown to act as membrane protein stabilizers.12 Moreover, it has been reported that these peptides can spontaneously undergo self-assembly to form dynamic nanostructures in a pure solution, similar to other lipids and surfactants12. Compared to linear peptides, cyclic peptides have greater potential as stabilizing agents due to their enhanced chemical and enzymatic stability.13 Microbial cyclic peptide surfactants have been previously reported in the literature.14,15 Random discovery by screening and challenges in the production of these natural surfactants by synthetic or genetic engineering methods have hindered the further development of these analogs. Thus, the applications of natural cyclic peptides as surfactants and protein stabilizers remain less explored. We hypothesized that self-assembled cyclic peptides with stabilizing and surfactant properties can be designed rationally. Herein, we report the discovery of nanosized surfactant-like L-cyclic peptides and their potential application as a new class of silver nanoparticles and biomolecule stabilizers. To the best of our knowledge, this class of peptides represents the first report of surfactant-like cyclic peptides that self-assemble into nanostructures. Furthermore, this is the first application of any surfactant-like synthetic cyclic peptide as a stabilizer for silver nanoparticles and an enzyme.
2. Results and Discussion
2.1. Self-Assembly of Peptides
A number of cyclic peptides including [FR]4, [FK]4, [WR]4, [CR]4, [AK]4, and [WK]5 (Fig. 1) containing L-amino acids were produced using solid-phase peptide synthesis. These peptide sequences were selected to consist of alternating hydrophobic and positively charged residues or alternating neutral and positively charged residues as controls. We hypothesized that an optimal balance of hydrophobicity and charge could generate self-assembled nanostructures in aqueous solution by intramolecular and/or intermolecular interactions. We have previously reported that among all the synthesized cyclic peptides, the amphipathic L-cyclic peptide containing tryptophan and arginine efficiently delivered cargoes intracellularly.16
Fig. 1.

Chemical structures of synthesized cyclic peptides (F = phenylalanine, R = arginine, K = lysine, W = tryptophan, C = cysteine, A = alanine).
All the cyclic peptides (2 mM) were dissolved in water and their structural morphologies were investigated using transmission electron microscopy (TEM) after a few days. Among all the designed peptides, [WR]4 (Fig. 1) generated self-assembled nanostructures after a specific incubation time at room temperature. Six cyclic peptides had the combination of positively charged residues and other amino acids. The indole ring of tryptophan in [WR]4 appears to play a crucial role in self-assembly because of high hydrophobicity and the generation of potential hydrophobic interactions. Moreover, the guanidine group of arginine residues in [WR]4 could take part in hydrogen bonding interactions. The other 5 cyclic peptides did not have an optimal combination of hydrophobic and positively charged residues required for the self-assembly process.
Initial conventional-TEM imaging of [WR]4 (2 mM aqueous solution) showed vesicle-like structures after 4 days (Fig. S1, Supplementary Information). Larger particles were observed by using a conventional TEM microscope after prolonged incubation.
Further evidence for the aggregation of [WR]4 was observed in a concentration-dependent fluorescence study. At lower concentrations, the fluorescence intensity was proportionally increased (excitation 280 nm, emission 360 nm). However, the intensity remained constant at a higher concentration of 1 mM (Fig. S2). The results suggest that fluorescence-quenching occurs at a concentration that is sufficiently high enough for aggregate formation. Since the hydrophobicity of Trp is weak, this peptide self-assembly occurs at a high concentration (i.e. ≥1 mM) possibly through intermolecular hydrophobic interactions between Trp residues.
As a result of the unusual behavior of [WR]4, other derivatives in this class ([WR]3 and [WR]5, (Fig. 1)) were synthesized and evaluated to investigate whether the ring size has any effect on the self-assembly process. Structural investigations of analogues of [WR]3 and [WR]5 were accomplished by TEM. Vesicle-like structures were formed for all compounds in this class. TEM of [WR]3 demonstrated small structure sizes of ~100 nm after 14 days of incubation. [WR]4 and [WR]5 exhibited larger structures (~ 550 and 930 nm, respectively) with much higher frequencies (Fig. S1).
Systematic studies on the self-assembly behavior of [WR]5 were carried out using different tools. Direct dissolution of the peptide in water did not reveal any specific structures after 24 h. TEM images showed the formation of a few micelle-like structures after 48 h. Nanovesicle structures with diameters between 50 and 100 nm were clearly observed after 12 days (Fig. S4). The formation of vesicles was also confirmed by scanning electron microscopy (SEM) after 20 days of incubation (Fig. 2a,b). SEM results clearly show self-assembly by aggregation of small particles to the larger particles.
Fig. 2.

(a) and (b) FE-SEM images of the self-assembled structure of [WR]5 (2 mM) in water after 20 days; (c) CD spectra of [WR]n (100 μM) in water, where n = 3-5; (d) TEM images of the self-assembly of [WR]5 (100 μM) in PBS after 48 h; (e) TEM images of [WR]5 (100 μM) in PBS after 12 days.
Zeta-potential measurements by dynamic light scattering (DLS) showed that self-assembled [WR]5 and [WR]4 had positive ζ potential values (+56.2 ± 7.62 mV) and (+31.4 ± 6.83 mV), respectively, which further demonstrate the formation of the positively charged surfaces by the guanidinium of R residues.
It is well known that the secondary structure of the peptides plays a critical role in the self-assembly pattern. Thus, circular dichroism (CD) was used to assess the secondary structure of these peptides. CD spectra of the aqueous solution of the peptides [WR]n (n = 3-5, 100 μM) at 25 °C (Fig. 2c) revealed two negative bands at approximately 202-205 nm and 214-216 nm and a positive band around 229 nm. To estimate the secondary structure content, CD spectra were analyzed using an estimation program.17 The analysis showed that peptides [WR]n n = 3-5 have 37.6-38.8% random and 31.6-35.7% sheet components with the remaining assigned to turn (12.4-12.5%) and helix (11.5-13.1%) (Table S1). The CD spectra of [WR]n (n = 3-5) displayed a distinct minimum of ellipticity around 202-205 nm, indicating that the peptides exist mostly in random coil states. The appearance of a negative band around 214-216 nm and a positive band at 229 nm is attributed to the strong exciton coupling produced by the aromatic chromophores of indole tryptophan residues stacking against one another.18 In addition, all peptides showed strong negative bands around 214-216 nm and a positive band at around 190, which are characteristic of the formation of distorted β-sheet structures. In general, the backbone-backbone intermolecular hydrogen bonding interactions proceed through parallel or antiparallel β-sheet-like stacking arrangements in linear peptides.10b The results indicate that the self-assembly of these cyclic peptides is driven mostly by the hydrophobic force, hydrogen bonding by partial β-structures, and/or the π-π stacking interactions between tryptophan residues. These data are consistent with those obtained with the previously reported cyclic peptides19 where the peptides did not show any specific secondary structure, and they self-assembled into bilayer vesicular nanostructures. The CD spectra demonstrated a significant increase in the spectral ellipticity of [WR]5 compared to those of [WR]3 and [WR]4, suggesting stronger intramolecular/intermolecular interactions in this peptide. These data are consistent with TEM exhibiting the formation of a high population of nanostructures by [WR]5. Considering these facts, [WR]5 was chosen as a model peptide for further self-assembly studies in detail.
Dynamic light scattering (DLS) was employed to identify the self-assembled structures in solution by determining the size distribution of the structures. DLS results showed that the majority (80%) of the nanostructures of [WR]5 (100 μM) were found in the 150-250 nm size range after 14 days (Fig. S3, Supplementary Information). Furthermore, it was observed that the size of the vesicle structures became larger over time as confirmed by TEM and SEM studies. This can be attributed to the fusion or aggregation of two or more vesicles as observed in the SEM image (Fig. S4, Supporting Information). This pattern of aggregation resembled that of traditional surfactants.
To get a better understanding of the impact of time on the aggregation process, the time-dependent studies by DLS were conducted for [WR]n (n = 3-5). The samples were prepared in water at a concentration of 1 mM and the size distribution was evaluated on the first, third, and fourteenth day. The first day presents the immediate size of the formed nanoparticles after dissolving in water. Initially, the peptides will have interactions with water. Additionally, intramolecular interactions contribute to the size distribution at this stage. The intramolecular and water hydrogen bonding interactions are generated mainly by the appropriate functional groups, such as guanidine residues. Subsequently, the size of the nanoparticles was compared after 3 days. The self-assembly trend in a short period is mainly related to the initial intramolecular interactions of peptide functional groups followed by the intermolecular interactions. However, the size distribution was also investigated after a relatively longer period (14 days) to determine the behavior of the nanoparticles after a prolonged incubation time. At a longer period, it is expected that the intermolecular interactions are the dominant driving force of self-assembly.
As depicted in Fig. 3, the majority of [WR]5 nanoparticles were found to be 319 ± 54 nm after 1 day. However, after 3 days, the self-assembly triggered, and [WR]5 nanoparticles grew larger (395 ± 53 nm). This trend continued, and after 2 weeks, [WR]5 nanoparticles of 543 ± 41 nm were observed, suggesting that time is a critical element for the size distribution of nanoparticles. A similar pattern was observed for [WR]4 nanoparticles. The sizes of [WR]4 nanoparticles were found to be 306 ± 102 nm, 424 ± 46 nm, and 641 ± 175 nm on days 1, 3, and 14, respectively. However, [WR]3 displayed a completely different pattern. [WR]3 nanoparticles started increasing from day 1 (241 ± 100 nm) to day 3 (352 ± 7 nm). However, the size of the nanoparticles was decreased to 216 ± 0.01 nm after 2 weeks. Several factors could be responsible for the collapsing of [WR]3 nanoparticles, such as the instability of nanoparticles in the long term due to the small ring size and insufficient intramolecular or intermolecular interactions due to the high strain in the cycle, or the inappropriate orientation of amino acids in the structure of peptide for interactions. The smaller ring size in [WR]3 leads to a more puckered and more cup-like nonplanar structure of the monomer which is less suited for self-assembly. The conformation of the monomer dictates the differences in the shapes of aggregated supramolecules. These results suggest that although time is a critical element in the size of the nanoparticles, the stability of the nanoparticles could affect the self-assembly process in the long period.
Fig. 3.

DLS studies of [WR]n (n = 3-5) (1 mM) on days 1, 3, and 14.
These data demonstrated the effect of the ring size on the nanoparticle size. On day 1, [WR]3, [WR]4, and [WR]5 were found to be 241 ± 100, 306 ± 102, and 319 ± 54 nm, respectively. This part of the result showed that the number of involved amino acids is an important element in the immediate size distribution of nanoparticles since [WR]5 led to a larger size of nanoparticles compared to that of [WR]3. However, [WR]4 nanoparticles were measured to be 424 ± 46 nm in size compared to 424 ± 46 nm for [WR]3 and 395 ± 53 nm for [WR]5 after 3 days of incubation, suggesting that the ring size with 8 amino acids is optimal for self-assembly. This trend was similar after 2 weeks as [WR]4 nanoparticles (641 ± 175 nm) were distinctly larger than [WR]3 (215 ± 0.01 nm) and [WR]5 (543 ± 41 nm). These results indicate that a [WR] cyclic peptide with eight amino acids has an optimal ring size for the maximal intermolecular and intramolecular interactions after 14 days. However, the size of primary nanoparticles on day 1 was found to be ring size dependent.
Additional characterization data were provided for these peptides, such as fluorescence spectroscopy (Fig. S17) and FTIR. Because of the highly sensitive indole chromophore in the structure of the tryptophan, this amino acid was used for its intrinsic fluorescence studies. Three cyclic peptides such as [WR]3, [WR]4, and [WR]5 (100 μM in H2O) were used for fluorescence studies. The data showed higher fluorescence intensity for the peptide containing a higher number of tryptophan residues [WR]5.
ATR-FTIR was used to further characterize the peptide amide bond in [WR]4. The spectrum of the dry powder of the peptide mixture revealed a two amide-I stretching at 1654 and 1640 cm−1. In general, antiparallel β-structures exhibit a principle β-sheet amide-I stretching vibration maximum at 1630–1640 cm−1 with a secondary absorption at a frequency normally 50–70 cm−1 higher. The absence of the secondary absorption shows that [WR]4 adopts partially a parallel β-structure orientation. This result is consistent with the stacking observed from the self-assembly simulations (Fig. 3).
Furthermore, the effect of salts and pH was investigated in the self-assembly process. [WR]5 showed self-assembled structures in phosphate buffered saline (PBS, pH 7.0) (Fig. 2e). The aggregation was faster in PBS compared to an aqueous solution resulting in the formation of larger vesicles (500-1000 nm) after 12 days (Fig. 2f). This could be due to the presence of different salts that can facilitate self-assembly. CD also revealed the development of an additional positive band at 196 nm in PBS. The pattern of the structure formation has a resemblance to surfactant-like linear peptides reported by Nagai et al.20 Moreover, it is well known that NaCl favors surfactant aggregation.21 When PBS containing NaCl (167 mM) was examined in the self-assembly of this peptide, the aggregation of [WR]5 was accelerated. NaCl decreases the repulsive force among the charged head groups, resulting in the enhancement of hydrophobic interactions between the peptides. However, in acidic medium (0.02 N HCl), some irregular structures were observed initially but a longer incubation time led to the degradation of the structures (Fig. S5, Supporting Information).
In order to assess the different self-assembly behaviors of [WR]5 in water and PBS, turbidity measurements22 were carried out to determine vesicle formation at 350 nm. A difference in turbidity was clearly visible from the results. A sudden increase in absorbance was observed above certain concentrations in both cases. Critical aggregation constant (CAC) values were determined from the intersection point obtained by extrapolating the lines. The CAC values for this peptide in water and PBS were found to be 0.9 mM and 0.45 mM, respectively (Fig. S6, Supporting Information), suggesting the surfactant-like nature of the peptide in the presence of salts.
Fluorescence studies were performed for [WR]4 to show the aggregation of the peptide at concentrations above 1 mM (Fig. S2), consistent with the data described in Fig. S6 for [WR]5. The aggregation of [WR]4 was observed in a concentration-dependent fluorescence study. At lower concentrations, the fluorescence intensity was proportionally increased (excitation 280 nm, emission 360 nm). However, the intensity remained constant at a higher concentration of 1 mM (Fig. S2, Supplementary Information).
As shown above, the self-assembly was promoted in the presence of salts. Molecular modeling studies were used to gain better insight into the nanostructure formation with [WR]5 in the presence of counterions. Initial coordinates for [WR]5 were obtained using GAUSSVIEW.23 Ab initio Hartree-Fock molecular orbital calculations with complete geometry optimizations were performed with the 6-31G24 basis set on the monomer [WR]5 utilizing GAUSSIAN’03 software. Two conformations were obtained (Fig. S7, Supporting Information). The lowest energy conformation did not allow any intramolecular H-bonding. The higher energy conformation (25 kCal/mol) shows intramolecular H-bonding between cationic and neutral side chains. Side chains have to reorganize for efficient intramolecular H-bonding resulting in a higher energy conformation. In the lowest energy conformation, tryptophan side chains are disposed perpendicular to the backbone. The higher energy conformation being more planar seems more suited for stacking. Stacking efficiency was explored utilizing supermolecule type intermolecular interaction calculations. Stacking efficiency = Estacked supermolecule – (Emonomer × no. of monomers + Ecounterion × no. of counterions).
[WR]5 is an amphiphilic peptide. Cationic side chains may facilitate counterion assisted self-assembly. First, the interaction of cationic side chains with small fluoride ions was studied (Fig. S8, Supporting Information). Fluoride counterions assisted the self-assembly propensity of the [WR]5 system and resulted in nanotube formation (Fig. S9, Supporting Information). Then, the interaction of trifluoroacetate ions (CF3COO−) with the cationic side chain was calculated, and counterion-mediated stacking was explored. The data indicated that [WR]5 can undergo self-assembly spontaneously in the presence of trifluoroacetate counterions. Trifluoroacetate counterions are larger in size. Thus, nanotubular stacking was not preferred. Instead, the system presented a spiral noncovalent association (Fig. 4), giving rise to nanostructure formation. A longer period of self-assembly simulation generated structures with larger size as observed in TEM and SEM experiments. We assume that intramolecular along with intermolecular interactions, such as hydrogen bonding, π-π interactions, and counterion interactions, work cooperatively during the self-assembly process of [WR]5. Intramolecular hydrogen bonding between W and R residues results in the high energy conformation which is suited for stacking; hence, it indirectly helps. Intermolecular interaction is purely electrostatic between cationic R residues and counteranions which may be basically any anion small enough to squeeze between monomers and interact efficiently. In [WR]5, self-assembly does not depend mainly on the diffusion of the peptide, but certainly depends on the diffusion of counterions. In the absence of any medium i.e. without any counterions, [WR]5 shows only minimal aggregation as it is not thermodynamically favored.
Fig. 4.

Self-assembled nanostructure formation of [WR]5 in the presence of CF3COO− ions. Stacking efficiency is given in kCal/mol.
We propose that the hydrophobic segments in the peptide initialize the supramolecular self-assembly and drive the peptide to form nanospherical water-filled structures. The positively charged portion of the peptide containing arginine residues orients itself to face the aqueous and counterion environment. Moreover, the hydrophobic residues stabilize the nanostructure as the particles become larger through inter- and intra-molecular hydrogen bonding. Additionally, the relatively weaker hydrogen bonded segments in the secondary structures allow the peptide to undergo assembly at extremely low concentrations (e.g. 100 μM). As shown in Fig. 2c, increasing the hydrophobic character by increasing the number of tryptophan residues from 3 to 5 for [WR]5 induced the formation of a completely different secondary structure, as shown by CD. However, as exhibited in Fig. 3, the size distribution of [WR]5 is relatively smaller than that of [WR]4 after 14 days, indicating that other parameters are also involved in the self-assembly process.25
Additional molecular modeling studies were then conducted in the presence of a 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) membrane bilayer using the SPC water model. The membrane was set up by a system builder module. Then, water was added. The whole system was subjected to simulated annealing followed by molecular dynamics at room temperature for 5 ns. When the side view is observed carefully, it does show two distinct surfaces. The results of the 5 ns trajectory showed that the [WR]5 dimer aligned at the oil (mimicked by membrane)-water interface with the hydrophobic surface containing tryptophan residues towards the hydrophobic residues in the membrane, suggesting the surfactant property of the peptide (Fig. S10). The stability of the system at the interface and low RMSD for all heavy atoms with time indicated successful simulation of the mode of action for the [WR]5 system as a biosurfactant or peptide surfactant (pepfactant). At the oil-water interface mimicked by the membrane-water interface in these simulations, this aggregated supramolecular form remains intact with hydrophobic groups towards the membrane and hydrophilic towards water, indicating the type of micellar formation.
In general, the intrinsic properties of a peptide sequence including hydrophobicity, charge, secondary structure propensity, and sequence patterning are responsible for controlling the self-assembly process.26,27 Thus, in order to investigate the effect of hydrophobicity and charge on the self-assembly process and to gain further mechanistic insight into the packing of the self-assembly of the [WR] series, a class of cyclic peptide analogues of [WR]5 was designed rationally by using modified building blocks of arginine and tryptophan (Fig. 5), and their self-assembly pattern was investigated.
Fig. 5.

Synthesized [WR]5 analogues.
Modified [WR]5 analogues exhibited different morphologies depending on the hydrophobicity and charge of the amino acids in the structure of the peptides as shown by TEM observations. To investigate the effect of hydrophobicity of tryptophan in the self-assembly, a new building block, N-methyl-L-tryptophan (WMe), was used as the hydrophobic residue in place of tryptophan. The resulting cyclic peptide [WMeR]5 showed nanotubular structures instead of spherical vesicles as evidenced by TEM (Fig. 6). Here, the enhanced hydrophobicity of the peptide and the absence of the NH moiety of tryptophan played critical roles in the self-assembly process.
Fig. 6.

TEM images of the self-assembled structures of [WMeR]5 (2 mM in water) after (A) 6 days and (B) 12 days. An individual nanotube structure is shown in the inset (scale: 100 nm).
To determine the role of positively charged arginine in the self-assembly process, another analogue, [WR(Me)2]5v was synthesized by replacing the arginine with N,N-dimethylarginine keeping tryptophan as a hydrophobic residue unchanged. Thus, the hydrophilicity of the charged residues was reduced which resulted in poor water solubility. Unlike the previous peptides, this compound (1 mM aqueous solution) generated micelle-like spherical nanostructures (Fig. S11, Supporting Information). It was found that the compound did not exhibit any large vesicles like [WR]5 even after prolonged incubation (25 days), presumably due to the absence of one of the free amine groups of guanidine. These data suggest that this free amino group contributes to the self-assembly process of [WR]5 possibly through hydrogen bonding. On the other hand, a peptide containing both N-methyltryptophan (WMe) and N,N-dimethylarginine (R(Me)2) ([WMeR(Me)2]5) did not produce any specific structures at 2 mM or 4 mM concentration. At a higher concentration (4 mM), [WMeR(Me)2]5 exhibited a few micelle-like nanostructures along with patches of membrane-like sheets between the nanostructures and the copper grid during the sample preparation (Fig. S12, Supporting Information). This peptide might have undergone self-assembly on the surface of the copper grid by hydrophobic interactions to form monolayer membranes, possibly due to the highly hydrophobic nature of the amino acids.
The formation of nanotubes through the self-assembly of D,L-cyclic peptides has been previously reported.10 The backbone-backbone interactions between peptides were found to be critical for the self-assembly of D,L-cyclic peptides. Our results indicated that hydrophobicity and charge were crucial for the self-assembled structure formation for these cyclic peptides, as they were in the case of linear surfactant-like peptides.11 Thus, we compared the nanostructure formation between homochiral [WR]5 with the corresponding heterochiral one to evaluate the effect of stereochemistry. Thus, D,L-cyclic peptide [WdR]5, where dR is d-arginine, was designed and synthesized for the comparative stereochemistry studies. Unlike the nanotube formation by D,L-cyclic peptides, [WdR]5 exhibited micelle-like nanostructures after prolonged incubation at room temperature (Fig. S13, Supporting Information). The detailed mechanism of the self-assembly for these cyclic peptides requires further investigation with modeling and simulation experiments.
Qualitative analysis of the conformation of these peptide analogues was obtained by CD. CD spectra of the aqueous solution of the peptides [WMeR]5 and [WR(Me)2]5 at room temperature (Fig. S14, Supporting Information) revealed two negative bands at approximately 203 and 217 nm. This does not resemble either a classic β-sheet or α-helical structure. Another analogue, [WMeR(Me)2]5, also showed similar bands, but with a lower ellipticity. These three peptide analogues, namely [WMeR]5, [WR(Me)2]5 and [WMeR(Me)2]5, showed negative bands at around 217 nm and positive bands at around 229 nm, which are characteristic of exciton coupling of tryptophan residues, indicating the presence of stacking interactions. However, [WdR]5 did not exhibit any characteristic signatures of exciton coupling of the tryptophan side chain, suggesting the absence of the stacking of indole rings.
2.2. Stabilization of Silver Nanoparticles by [WR]5 and Modified Analogues
We have previously reported the application of [WR]5 as a nuclear targeting molecular transporter for a wide range of cargos.16 It was found that a physical mixture of [WR]5 and HAuCl4 led to the formation of peptide-capped gold nanoparticles and enhanced the cellular uptake of drugs significantly.28 We investigated to determine whether [WR]5 can be used for the stabilization of silver nanoparticles and proteins. Several methods for the synthesis of silver nanoparticles have been previously reported. However, the stabilization of silver nanoparticles is required for various applications. The stabilizing agents (e.g., surfactants, polymer) prevent the particle aggregation by protecting these particles by steric stabilization. Cationic surfactants are commonly used as stabilizing agents in silver nanoparticle synthesis.29 Accounting for the above facts, we postulated that [WR]5 could behave as a cationic surfactant.
In order to explore whether [WR]5 can mimic the role of cationic surfactants in the synthesis of nanoparticles, a model experiment was performed using silver nitrate (AgNO3) as a source of Ag+ ions and sodium borohydride (NaBH4) as a reducing agent in the presence and absence of the cyclic peptide. Sodium borohydride was added into the equal volume mixture of the cyclic peptide [WR]5 (1 mM) and the AgNO3 (2 mM) solution. To demonstrate the role of the cyclic peptide as a stabilizer, sodium borohydride was used in excess amount (8 mM). The resulting mixture produced a yellow color immediately, indicating the formation of silver nanoparticles. UV-Vis spectra showed the characteristic plasmon band of the silver nanoparticle formation with a band at 410 nm (Fig. 7A). The formation of spherical silver nanoparticles was confirmed by TEM (Fig. 7B). In order to monitor the stability of the silver colloid, absorption of the colloid was measured after four weeks. The evolution of UV-Vis spectra is shown in Fig. 7A (curve b). There was only a minor red shift in peak position even after four weeks (curve c), confirming the stability of the silver colloids. No extra peaks or shoulders developed even after one month, eliminating the possibility of aggregation. In contrast, when sodium borohydride was added to the AgNO3 (2 mM) solution in the absence of the peptide (Fig. 7A, curve a), stable silver nanoparticles were not formed. These data confirm the surfactant-like nature of the cyclic peptide [WR]5. The stability of silver nanoparticles was also examined in the presence of the corresponding linear peptide. The linear peptide was not efficient in stabilizing silver nanoparticles (Fig. S18).
Fig. 7.
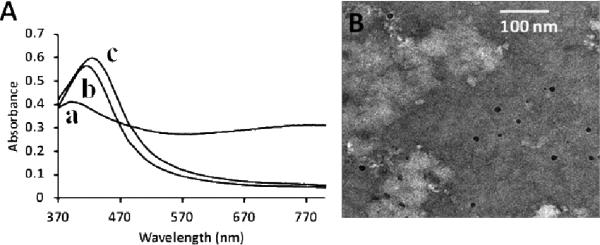
(A) UV-Vis spectra of the yellow color silver nanoparticle solution in the absence (a) and presence (b, c) of the [WR]5 peptide and comparison of the stability of silver nanoparticles in the presence of a peptide after 30 min of reaction (curve b) and one month (curve c); (B) TEM image of silver nanoparticles synthesized using sodium borohydride in the presence of [WR]5.
The stabilization of silver nanoparticles with [WR]5 was compared with a traditional surfactant (cetyltrimethylammonium bromide, CTAB). CTAB is a well known cationic surfactant which is used as a stabilizer in nanoparticle synthesis. It is already reported in the literature that CTAB shows a better stabilizing effect at low concentration.30 The stability of silver nanoparticles in the presence of CTAB was checked after 4 days, and it showed aggregation of nanoparticles. The decrease in band (~410 nm) intensity and the development of an extra band approximately at 610 nm indicate particle aggregation (Fig. S19). A traditional surfactant or some polymeric compounds form micelles which are used to stabilize the metal nanoparticles.31,32 The micelles are porous clusters with deep water filled cavities. In the case of traditional surfactants, micelles control the shape of the nanoparticles by trapping the metal ion and reagent in the small cavities. Here, micelles formed by peptides are water rich. Due to the self-assembly process, cyclic peptides initially form micelles which stabilize the nanoparticles. Thus, when AgNO3 and peptide solution are mixed, Ag+ ions may reside in the micelles. Once NaBH4 is added, Ag+ ions are converted into Ag0 that remains stable without any aggregation. Small surfactant-like peptides surround the metal nanoparticles and protect them from aggregation. TEM images show a clear layer of the peptide on the nanoparticle surface (Fig. S20). This follows a similar model to that of a traditional surfactant.
The effect of the peptide concentration in stabilizing silver nanoparticles was investigated using different concentrations of the peptide. Visual observation showed that the typical color of silver nanoparticles changed from pale yellow to dark yellow and brown, respectively, indicating the alteration of nanoparticle morphology in the presence of different concentrations of the peptide. This observation was consistent with UV-Vis spectroscopy (Fig. S15, Supporting Information) that showed a narrow band at 401 nm when 100 μM of the peptide was used. As the concentration of the peptide was increased from 100 to 200 μM, the absorbance curve did not change significantly. However, the absorbance band became short and shifted towards longer wavelengths when a peptide concentration of 500 μM was employed, indicating the aggregation and formation of larger particles. The absorbance band became broad and significantly shorter at a peptide concentration of 1 mM. The broadening of the band suggests the wide distribution (higher dispersity) of particle size. These results revealed that the optimization of the peptide concentration is necessary to achieve narrower particle size distribution. This pattern is similar to other synthesized surfactant-stabilized silver nanoparticles.
To investigate whether the other synthesized cyclic peptide analogues can show surfactant-like properties, they were explored as stabilizing agents in the silver nanoparticle synthesis. Among them, [WMeR]5 showed great potential as a stabilizing agent. Although it shows a slight red shift (6 nm) in the band after one week, there is no sign of aggregation of silver nanoparticles (Fig. 8A). This result supports the surfactant-like nature of [WMeR]5. However, the intensity of absorbance was decreased in the presence of [WR(Me)2]5 after 7 days (Fig. 8B), indicating the aggregation of nanoparticles. Furthermore, in the presence of [WMeR(Me)2]5, the plasmon band of silver nanoparticles shifted (46 nm) significantly to longer wavelengths, indicating the formation of large particles (Fig. 8C). Similar results were obtained for [WdR]5 (Fig. 8D). This could be attributed to the irregular structure formation by these peptides.
Fig. 8.

UV-vis spectra of silver nanoparticles in the presence of (A) [WMeR]5, (B) [WR(Me)2]5, (C) [WMeR(Me)2]5, and (D) [WdR]5 at room temperature after 15 min (a) and 7 days (b).
2.3. Stabilization of Glyceraldehyde-3-Phosphate Dehydrogenase (GAPDH) by [WR]5
It is already known that surfactants influence the enzymatic activities depending upon the interaction with the surface of the enzymes.33 Thus, the capability of this surfactant-like peptide in stabilizing biomacromolecules was investigated using glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as a model enzyme. The activity of the GAPDH enzyme was determined using the KDalert GAPDH assay kit. The KDalert GAPDH assay determines the conversion of NAD+ into NADH by GAPDH in the presence of phosphate and glyceraldehyde-3-phosphate (G-3-P). Under the suggested assay conditions, the rate of NADH production is proportional to the amount of the GAPDH enzyme present. To evaluate the effectiveness of the peptide in stabilizing enzymatic activity, experiments were carried out by mixing the enzyme with various peptide concentrations in phosphate buffered saline (PBS, NaCl (167 mM), pH 7.0). GAPDH was mixed in PBS as a control. The activity of the enzyme at different time intervals in the presence and absence of the peptide was measured using the fluorescence-based method. Plots were made to show the increase in fluorescence due to NAD+ reduction vs time. It was observed that the enzyme containing 100 μM of [WR]5 showed significantly enhanced activity compared to the control enzyme (Fig. 9A), suggesting the surfactant-like nature of the peptide. Moreover, the enzyme activity was reduced at a higher peptide concentration (500 μM), presumably due to the denaturation of the enzyme, indicating the ionic nature of the peptide-surfactant. A control experiment was also performed without the enzyme to rule out any effect of the peptide in the reduction process. Negligible fluorescence was recorded when the peptide solution was mixed with other components in the absence of the enzyme.
Fig. 9.
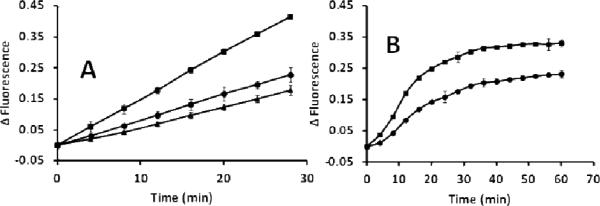
Enzymatic activity of GAPDH. Changes in fluorescence due to NAD+ reduction are the result of GAPDH activity. (A) Enzymatic activity shown till 30 min at room temperature; (B) enzymatic activity shown till 60 min at 50 °C. (●) GAPDH enzyme in PBS, (■) GAPDH enzyme in PBS in the presence of [WR]5 (100 μM), and (▲) GAPDH enzyme in PBS in the presence of [WR]5 (500 μM).
The presence of the peptide-surfactant enhanced the activity of the enzyme as described. Thus, it was essential to verify the influence of the peptide on the stability of the enzyme. To this end, the activity of GAPDH was determined in the presence of the peptide at 37 °C and 50 °C. The enzyme in the presence of the peptide showed 2-fold enhanced activity compared to controls at 50 °C (Fig. 9B) and 37 °C (Fig. S16, Supporting Information), indicating the enhanced stability of the GAPDH enzyme in the presence of the peptide-surfactant. These results suggest that this surfactant peptide could be a promising additive for stabilizing other biomacromolecules, such as membrane proteins.
3. Experimental
3.1. Synthesis of Cyclic Peptides
The synthesis of cyclic peptides was carried out according to the procedure previously reported by us.16 All reactions were carried out in Bio-Rad polypropylene columns by shaking and mixing using a Glas-Col small tube rotator in dry conditions at room temperature unless otherwise stated. In general, all linear peptides were synthesized by the solid-phase synthesis strategy employing N-(9-fluorenyl)methoxycarbonyl (Fmoc)-based chemistry and Fmoc-L-amino acid building blocks. 2-(1H–Benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU) and N,N-diisopropylethylamine (DIPEA) in N,N-dimethylformamide (DMF) were used as coupling and activating reagents, respectively. Fmoc-amino acid trityl chloride resins, 2-chlorotrityl chloride resin, coupling reagents, and Fmoc-amino acid building blocks were purchased from ChemPep (Miami, FL). Other chemicals and reagents were purchased from Sigma-Aldrich Chemical Co. (Milwaukee, WI). Fmoc deprotection at each step was carried out using piperidine in DMF (20% v/v). Side chain protected peptides were cleaved from the resins by shaking the resins with a mixture of trifluoroethanol (TFE)/acetic acid/dichloromethane (DCM) (2:2:6, v/v/v, 15 mL) for 2 h. The resins were filtered off, and the liquid was evaporated to dryness to get a side-chain protected linear peptide. The cyclization of the peptides was carried out in the presence of a mixture of 1-hydroxy-7-azabenzotriazole (HOAt, 163.3 mg, 1.2 mmol) and N,N′-diisopropylcarbodiimide (DIC, 187 μL, 1.2 mmol) in dry DMF (100 mL), and dry DCM (40 mL) for 24 h. DMF and DCM were evaporated. The side chain deprotection was carried out with trifluoroacetic acid (TFA)/thioanisole/anisole/1,2-ethanedithiol (EDT) (90:5:2:3 v/v/v/v) for 2 h. The crude peptides were precipitated by the addition of cold diethyl ether (75 mL, Et2O) and purified by reversed-phase Hitachi HPLC (L-2455) on a Phenomenex Prodigy 10 μm ODS reversed-phase column (2.1 cm × 25 cm) with a gradient system. The peptides were separated by eluting the crude peptides at 10.0 mL/min using a gradient of 0-100% acetonitrile (0.1% TFA) and water (0.1% TFA) over 60 min, and then, they were lyophilized. The chemical structure of [WR]4 was characterized by nuclear magnetic resonance spectra (1H NMR) determined on a Varian NMR spectrometer (500 MHz) as described previously.16 The chemical structures of all final products were confirmed using a high-resolution Applied Biosystems QStar Elite time-of-flight electrospray mass spectrometer. The purity of the final products (>95%) was confirmed by analytical HPLC. The analytical HPLC was performed on the Hitachi analytical HPLC system on a C18 Shimadzu Premier 3 μm column (150 cm × 4.6 mm).
3.2. Synthesis of Fmoc-Trp(Me)-OH
N-(9-Fluorenylmethoxycarbonyloxy)succinimide (Fmoc-ONSu, 11.55 g, 0.034 mol) was added to a round bottom flask, dissolved with 75 mL of DMF with constant stirring at 0 °C. 1-Methyl-L-trypthophan (5 g, 0.22 mol) was added to sodium carbonate solution (10%, 75 mL), and the mixture was stirred at room temperature to form a suspension. The suspension prepared above was added dropwise at 0 °C with constant stirring to the solution of Fmoc-ONSu in DMF. The precipitate formed immediately. The mixture was stirred for another 1 h and made at room temperature. Acetone (80 mL) was added to the mixture and the stirring continued overnight. The precipitate was removed by filtration, and the pH of the filtrate was adjusted to 7.0 with HCl. The solution was concentrated in a rotary evaporator. A 200 mL of ethyl acetate was added to the mixture and the resulting solution was stirred for 6 h at room temperature. The ethyl acetate layer was washed with 100 mL of 0.1 N HCl two times and then with brine (75 mL), and dried over anhydrous MgSO4. The solvent was removed using a rotary evaporator to yield crude creamy solid compounds. The compound was re-crystallized in ethyl acetate to obtain 4 g of pure material, which was used without further purification for the synthesis of cyclic peptides [WMeR]5, [WR(Me)2]5, and [WMeR(Me)2]5 according to the previously reported procedure.16
[FK]4: HR-MS (ESI-TOF, m/z): [M]+ calcd for C60H84N12O8, 1100.6535; found, 1100.5836 [M]+, 551.2918 [M + 2H]2+, 367.8752 [M + 3H]3+.
[AK]4: HR-MS (ESI-TOF, m/z): [M]+ calcd for C36H68N12O8, 796.5283; found, 796.4868 [M]+, 399.2434 [M + 2H]2+.
[CR]4: HR-MS (ESI-TOF) (m/z): [M + H]+ calcd for C36H69N20O8S4, 1037.4490; found, 1037.4096 [M + H]+, 345.1323 [M + 3H]3+, 259.1024 [M + 4H]4+.
[FR]4: HR-MS (ESI-TOF) (m/z): [M]+ calcd for C60H84N20O8, 1212.6781; found, 1212.6052 [M]+, 607.3026 [M + 2H]2+, 405.5428 [M + 3H]2+, 304.1588 [M + 4H]3+.
[WR]3: HR-MS (ESI-TOF) (m/z): [M + H]+ calcd for C51H67N18O6, 1027.5491; found, 1027.6030 [M + H]+.
[WR]4: HR-MS (ESI-TOF) (m/z): [M + H]+ calcd for C68H89N24O8, 1369.7295; found, 1369.7934 [M + H]+, 685.3967 [M + 2H]2+, 457.2696 [M + 3H]3+, 343.2066 [M + 4H]4+; FT-IR of [WR]4 was carried out using a Thermo-Nicolet 380 FT-IR spectrophotometer using OMNIC software. FT IR (cm−1) (ATR): 1654, 1640.36, 1531.22, 1494.39, 1431.63, 1198.35, 1170.79, 1119.02, 837.31, 799.83, 745.55, 719.71.
[WR]5: HR-MS (ESI-TOF) (m/z): [M + H]+ calcd for C85H111N30O10, 1711.9099; found, 1711.6974 [M + H]+, 571.5658 [M + 3H]3+, 428.9286 [M + 4H]4+, 343.1493 [M + 5H]5+.
[WMeR]5: HR-MS (ESI-TOF) (m/z): [M + H]+ calcd for C90H121N30O10, 1781.9882; found, 1782.0165 [M + H]+, 594.6633 [M + 3H]3+, 446.4739 [M + 4H]4+, 357.4033 [M + 5H]5+.
[WR(Me)2]5: HR-MS (ESI-TOF) (m/z): [M + H]+ calcd for C95H131N30O10, 1852.0664; found, 1852.1065 [M + H]+, [M + 2H]2+, 618.3545 [M + 3H]3+, 463.9962 [M + 4H]4+, 371.4213 [M + 5H]5+.
[WMeR(Me)2]5: HR-MS (ESI-TOF) (m/z): [M + H]+ calcd for C100H141N30O10, 1922.1447; found, 1921.5000 [M + H]+, 483.9131 [M + 4H]4+, 383.6360 [M + 5H]5+.
[WdR]5: HR-MS (ESI-TOF) (m/z): [M]+ calcd for C85H110N30O10, 1710.9021; found, 571.6383 [M + 3H]3+, 428.9571 [M + 4H]4+, 343.3879 [M + 5H]5+.
3.3. Transmission Electron Microscopy (TEM)
3.3.1. Conventional TEM
The TEM sample was prepared by drop casting a 2 mM aqueous solution (10 μL) onto the formvar coated carbon grid of mesh size 300, which was allowed to rest for 2 min. Excess solvent was carefully removed by capillary action (filter paper). The grids were allowed to dry at room temperature overnight and images were taken using a JEOL transmission electron microscope (Tokyo, Japan) operated at 80 kV.
3.3.2. Negative Staining
After drop casting of peptide solution, the grids were then stained with uranyl acetate (20 μL) for 2 min. Excess stain was removed, and the grids were allowed to dry overnight. Images were taken using a JEOL transmission electron microscope (Tokyo, Japan) operated at 80 kV.
3.4. Scanning Electron Microscopy (SEM)
The SEM sample was prepared by drop casting a 2 mM aqueous solution (10 μL) onto the mica surface. The mica surface was allowed to dry at room temperature overnight and analyzed using a Nova NanoSEM 450 using the directional backscatter (DBS) electron detector. All samples were imaged un-coated, in high vacuum mode.
3.5. Circular Dichroism
CD spectra were recorded on a JASCO J-810 spectropolarimeter using 1 mm path length cuvettes. The scan speed was 100 nm/min, and spectra were averaged over 8 scans. All samples were tested at 25 °C. The sample concentration was 100 μM in deionized water. The CD for background reference (water) was measured and subtracted from the sample.
3.6. Dynamic Light Scattering (DLS)
Dynamic light scattering and zeta potential experiments were conducted on a Malvern Instruments Zetasizer Nano ZS (Worcestershire, UK). The machine was calibrated using a 60 nm polystyrene standard. Each sample (1 mL, 100 μM) was loaded into a cell, and the particle size and zeta potential were measured simultaneously in triplicate. To determine the size distribution, 1 mL samples were placed in PCS1115 glass cuvettes and DLS was performed at 25 °C at a backscatter angle of 173° with an equilibration time of 120 s. The intensity-based z-averaged hydrodynamic diameters were reported based on 11 scans. To measure zeta potential via electrophoretic mobility, the samples were added to a cuvette equipped with a copper chip. The measurement was conducted at 25 °C and 3 cycles were performed (30 data points per cycle). DLS and zeta potential were conducted with 100 μM [WR]4 as a stock solution.
3.7. Synthesis of Silver Nanoparticles
A stock solution of silver nitrate was mixed with NaBH4 in the presence of cyclic peptides. Typically, 100 μL of 1 mM peptide solution was mixed with 100 μL of 2 mM silver nitrate solution at room temperature. Then, 100 μL of 8 mM solution of NaBH4 was added into the mixture to yield colloidal silver with a light yellowish color. The UV-Vis spectra of the solution were taken using a SpectraMax M2 fluorescence spectrophotometer (Molecular Devices, CA) at different time intervals.
3.8. Enzyme Activity Assay
The activity of the GAPDH enzyme was measured using the KDalert GAPDH assay kit (Ambion, Austin, Texas). Briefly, 1 μL of the GAPDH enzyme was mixed with 99 μL of PBS (pH 7.0) in an Eppendorf tube to obtain a control enzyme solution of activity 0.26 U/ml. The same proportion of the enzyme was mixed with different concentrations of peptide solution (100 μM, 500 μM and 1 mM) in PBS. Then, 10 μL of enzyme solutions from each tube was taken in 96 well plates followed by the addition of 90 μL of KDalert master mix into it using a multichannel pipettor. To determine the activity of the enzyme, fluorescence of the solutions was measured (Excitation: 560 nm, Emission: 590 nm). The reduction of NAD+ to NADH was continuously monitored on a SpectraMax M2 fluorescence spectrophotometer (Molecular Devices, CA) for 30 min at room temperature. Fluorescence measurements of NADH production with the control enzyme solution and the enzyme in 100 μM and 500 μM peptide solutions were carried out in triplicate. For the thermal stability study, the fluorescence of the solutions was measured for 60 min at 50 °C.
3.9. Molecular Modeling
Ab initio Hartree-Fock molecular orbital calculations were performed with the 6-31G basis set. The geometry of the monomers was completely optimized (all bond lengths, bond angles and dihedral angles were allowed to relax without any constraint). Intermolecular interaction calculations using the supermolecule approach were utilized to study stacking efficiency and interaction with counterions present in the reaction medium. Stacking efficiency = Estacked supermolecule – (Emonomer × no. of monomers + Ecounterion × no. of counterions). Molecular modeling studies without anions were conducted in the presence of a 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) membrane bilayer using the SPC water model. The simulation was performed at 300 K, and the NPT ensemble was taken using the Martyna-Tobias-Klein barostat method. Molecular dynamics simulations were performed by Shaw's Desmond software. The membrane was set up by a system builder module. Then, water was added. The whole system was subjected to simulated annealing followed by molecular dynamics at room temperature for 5 ns.
3.10. Fluorescence Spectroscopy
The steady-state fluorescence spectra were recorded using a SpectraMax M2 fluorescence spectrophotometer (Molecular Devices, CA) in a 96 well plate. The fluorescence of tryptophan residues in the samples was measured at excitation and emission wavelengths of 280 nm and 360 nm, respectively. The measurement was performed by excitation and emission slits with a nominal band pass of 1 nm.
3.11. ATR-FTIR
FT-IR of [WR]4 was carried out using a Thermo-Nicolet 380 FT-IR spectrophotometer using OMNIC software. ATR-FTIR was used to collect the spectra of the sample.
4. Conclusions
In summary, a new class of surfactant-like cyclic peptides was reported. Among all the designed peptides, [WR]n (n = 3-5) generated self-assembled vesicle-like nanostructures at room temperature as shown by TEM, SEM, and/or DLS. A plausible mechanistic insight into the self-assembly of [WR]5 was investigated by molecular modeling studies. Modified [WR]5 analogues, such as [WMeR]5, [WR(Me)2]5, [WMeR(Me)2]5, and [WdR]5, exhibited different morphologies to [WR]5 as shown by TEM. The surfactant-like cyclic peptide [WR]5 exhibited a significant stabilizing effect for generated silver nanoparticles and GAPDH activity. The data suggested that surfactant cyclic peptides, [WR]5 and [WMeR]5, could be promising materials for stabilization of silver nanoparticles. These studies established a new class of surfactant-like cyclic peptides that self-assembled into nanostructures and could have potential application for the stabilization of silver nanoparticles and protein biomolecules.
Supplementary Material
Acknowledgements
We acknowledge the financial support from the American Cancer Society Grant # RSG-07-290-01-CDD and the US National Science Foundation, Grant Number CHE 0748555 for the financial support. We thank the National Center for Research Resources, NIH, and Grant Number 1 P20 RR16457 for sponsoring the core facility.
Footnotes
dElectronic Supplementary Information (ESI) available: Additional supporting data and figures for TEM, DLS, SEM, turbidity measurements, molecular modeling, CD, and stabilization studies included. See DOI: 10.1039/b000000x/
Notes and references
- 1.Zhang S. Nat. Biotechnol. 2003;21:1171–1178. doi: 10.1038/nbt874. [DOI] [PubMed] [Google Scholar]
- 2.Whitesides GM, Mathias JP, Set CT. Science. 1991;254:1312–1315. doi: 10.1126/science.1962191. [DOI] [PubMed] [Google Scholar]
- 3.Bryson JW, Betz SF, Lu HS, Suich DJ, Zhou HX, O'Neil KT, DeGrado WF. Science. 1995;270:935–941. doi: 10.1126/science.270.5238.935. [DOI] [PubMed] [Google Scholar]
- 4.Dill KA. Biochem. 1990;29:7133–7155. doi: 10.1021/bi00483a001. [DOI] [PubMed] [Google Scholar]
- 5.Brooks CL. Acc. Chem. Res. 2002;35:447–454. doi: 10.1021/ar0100172. [DOI] [PubMed] [Google Scholar]
- 6.Ghadiri MR, Granja JR, Buehler LK. Nature. 1994;369:301–304. doi: 10.1038/369301a0. [DOI] [PubMed] [Google Scholar]
- 7.Ellis-Behnke RG, Liang YX, You SW, Tay DK, Zhang S, So KF, Schneider GE. Proc. Natl. Acad. Sci. U. S. A. 2006;103:5054–5059. doi: 10.1073/pnas.0600559103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nuraje N, Banerjee IA, IMacCuspie R, Yu L, Matsui H. J. Am. Chem. Soc. 2004;126:8088–8089. doi: 10.1021/ja048617u. [DOI] [PubMed] [Google Scholar]
- 9.Yemini M, Reches M, Rishpon J, Gazit E. Nano Lett. 2005;5:183–186. doi: 10.1021/nl0484189. [DOI] [PubMed] [Google Scholar]
- 10.a Ghadiri MR, Granja JR, Milligan RA, McRee DE, Khazanovich N. Nature. 1993;366:324–327. doi: 10.1038/366324a0. [DOI] [PubMed] [Google Scholar]; b Hartgerink JD, Granja JR, Milligan RA, Ghadiri MR. J. Am. Chem. Soc. 1996;118:43–50. [Google Scholar]; c Clark TD, Buriak JM, Kobayashi K, Isler MP, McRee DE, Ghadiri MR. J. Am. Chem. Soc. 1998;120:8949–8962. [Google Scholar]; d Fernandez-Lopez S, Kim HS, Choi EC, Delgado M, Granja JR, Khasanov A, Kraehenbuehl K, Long G, Weinberger DA, Wilcoxen KM, Ghadiri MR. Nature. 2001;412:452–455. doi: 10.1038/35086601. [DOI] [PubMed] [Google Scholar]
- 11.a Vauthey S, Santoso S, Gong H, Watson N, Zhang S. Proc. Natl. Acad. Sci. USA. 2002;99:5355–5360. doi: 10.1073/pnas.072089599. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Santoso S, Hwang W, Hartman H, Zhang S. Nano Lett. 2002;2:687–691. [Google Scholar]; c Von Maltzahn G, Vauthey S, Santoso S, Zhang S. Langmuir. 2003;19:4332–4337. [Google Scholar]
- 12.Yeh J, Du S, Tordajada A, Paulo J, Zhang S. Biochemistry. 2005;44:16912–16919. doi: 10.1021/bi051357o. [DOI] [PubMed] [Google Scholar]
- 13.Katsara M, Selios TT, Deraos S, Deraos G, Matsoukas MT, Lazoura E, Matsoukas J, Apostolopoulos V. Curr. Med. Chem. 2006;13:2221–2232. doi: 10.2174/092986706777935113. [DOI] [PubMed] [Google Scholar]
- 14.Deleu M, Razafindralambo H, Popineau Y, Jacques P, Thonart P, Paquot M. Colloids Surf., A. 1999;152(1–2):3–10. [Google Scholar]
- 15.Raaijmakers JM, de Bruijn I, de Kock MJD. Mol. Plant-Microbe Interact. 2006;19(7):699–710. doi: 10.1094/MPMI-19-0699. [DOI] [PubMed] [Google Scholar]
- 16.Mandal D, Nasrolahi Shirazi A, Parang K. Angew. Chem. Int. Ed. 2011;50:9633–9637. doi: 10.1002/anie.201102572. [DOI] [PubMed] [Google Scholar]
- 17.Raussens V, Ruysschaert JM, Goormaghtigh E. Anal Biochem. 2003:114–121. doi: 10.1016/s0003-2697(03)00285-9. [DOI] [PubMed] [Google Scholar]
- 18.a Dempsey CE, Mason PE, Jungwirth P. J. Am. Chem. Soc. 2011;133:7300–7303. doi: 10.1021/ja201349g. [DOI] [PubMed] [Google Scholar]; b Grishina IB, Woody RW. Faraday Discuss. 1994;99:245–262. doi: 10.1039/fd9949900245. [DOI] [PubMed] [Google Scholar]
- 19.Choi SJ, Jeong WJ, Kang SK, Lee M, Kim E, Ryu du Y, Lim YB. Biomacromolecules. 2012;13:1991–1995. doi: 10.1021/bm3005947. [DOI] [PubMed] [Google Scholar]
- 20.Nagai A, Nagai Y, Qu H, Zhang S. J. Nanosci. Nanotechnol. 2007;7:2246–2252. doi: 10.1166/jnn.2007.647. [DOI] [PubMed] [Google Scholar]
- 21.Zhou X, Hao J. J. Chem. Eng. Data. 2011;56:951–955. [Google Scholar]
- 22.Guo X, Cui B, Li H, Gong Z, Guo R. J. Poly. Sci. A. 2009;47:434–449. [Google Scholar]
- 23.Dennington R, II, Keith T, Millan J, Eppinnett K, Hovell WL, Gilliand R. Gaussview, Version 3.09. Semichem Inc; S. Mission KS: 2003. [Google Scholar]
- 24.Ditchfield R, Hehre WJ, Pople JA. J. Chem. Phys. 1971;54:724–728. [Google Scholar]
- 25.Gudlur S, Sukthankar P, Gao J, Avila LA, Hiromasa Y, Chen J, Iwamoto T, Tomich JM. PLoS One. 2012;7:e45374. doi: 10.1371/journal.pone.0045374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bemporad F, Calloni G, Campioni S, Plakoutsi G, Taddei N, Chiti F. Acc. Chem. Res. 2006;39:620–627. doi: 10.1021/ar050067x. [DOI] [PubMed] [Google Scholar]
- 27.Chiti F, Stefani M, Taddei N, Ramponi G, Dobson CM. Nature. 2003;424:805–808. doi: 10.1038/nature01891. [DOI] [PubMed] [Google Scholar]
- 28.a Nasrolahi Shirazi A, Mandal D, Tiwari RK, Guo L, Lu W, Parang K. Mol. Pharm. 2013;10:488–499. doi: 10.1021/mp3004034. [DOI] [PubMed] [Google Scholar]; b Nasrolahi Shirazi A, Tiwari RK, Chhikara BS, Mandal D, Parang K. Mol. Pharm. 2013;10:500–511. doi: 10.1021/mp300448k. [DOI] [PubMed] [Google Scholar]
- 29.Pal T, Sau TK, Jana NR. Langmuir. 1997;13:1481–1485. [Google Scholar]
- 30.Ashkarran AA, Iraji zad A, Ahadian MM, Hormozi Nezhad MR. Eur. Phy. J. Appl. Phys. 2009;48:10601. [Google Scholar]
- 31.Khan Z, Singh T, Hussain JI, Hashmi AA. Coll. Surf. B: Bio. 2013;104:11–17. doi: 10.1016/j.colsurfb.2012.11.017. [DOI] [PubMed] [Google Scholar]
- 32.Chen S, Guo C, Hu G-H, Wang J, Ma J-H, Liang X-F, Zheng L, Liu H-Z. Langmuir. 2006;22:9704–9711. doi: 10.1021/la061093m. [DOI] [PubMed] [Google Scholar]
- 33.Martinek K, Levashov AV, Klyachko N, Khmelniteki YL, Berezin IV. Eur. J. Biochem. 1986;155:453–468. doi: 10.1111/j.1432-1033.1986.tb09512.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.